Abstract
Phaeodactylum tricornutum is a marine diatom rich in omega-3 fatty acids, a nutraceutical-relevant product. Long chain-polyunsaturated fatty acids (LC-PUFAs) are crucial dietary components for human development and growth. With the availability of genome information and genetic engineering tools, the productivities of OMEGAs have improved, but the functional and organizational relationship of such protein-encoding genes associated with LC-PUFAs biosynthesis is still not clear. Henceforth, our study highlights the conservation pattern, functionality and interaction of LC-PUFAs protein-encoding genes via in silico analysis. The transcriptome and quantitative PCR analysis demonstrates downregulation of ACS4, ELO6b, PTD5a, and MYB106 genes associated with LC-PUFAs synthesis and upregulation of ECoAH and ACAT1 genes associated with β-oxidation in nitrogen-depleted conditions in P. tricornutum. Phylogenomics studies of LC-PUFAs protein-encoding genes show a highly conserved evolutionary pattern in various microalgal lineages. Further, for elucidating the interaction of LC-PUFA metabolic genes, subcellular networks were predicted and pathway enrichment analysis was performed, providing new insights on the crosstalk between LC-PUFA protein-encoding genes (ELO6, PTD5, ACS, and ACL1), regulatory elements (LEC2, MYB, WIN) and transporters (ABCD1). In conclusion, such extensive functional enrichment analysis will undoubtedly aid in the development of genetically engineered algal strains with enhanced production of biomolecules i.e., LC-PUFAs.
1. Introduction
Microalgae with distinct characteristics are increasingly being pursued for the production of beneficial high value compounds [1]. Long chain-polyunsaturated fatty acids (LC-PUFAs), such as eicosatetraenoic acid (EPA; C20:5Δ5, 8,11,14,17, n − 3) and docosahexaenoic acid (DHA; C22:6Δ4, 7, 10,13,16,19, n − 3), are one of the major vital constituents in human nutrition, having high commercial and nutraceutical values. Omega-3 fatty acids are majorly produced from marine fish sources such as salmon, mullet, and mackerel [2]. Excessive trawling and contamination of the water sources has led to serious constraints on extraction of fish oil, therefore there is a high demand for the identification of alternative resource for sustainable production of LC-PUFAs [3].
In microalgae, the LC-PUFA metabolism occurs in a constitutive manner and forms an integral component of the photosynthetic membranes providing integrity and stability along with aiding in the quenching of reactive oxygen species (ROS) [4,5]. LC-PUFA metabolisms in these photosynthetic cell factories is growth dependent and fluctuates across different species in a significant way based on their environmental conditions. Studies based on varying parameters such as nutrient deprivation, light saturation, high/low pH, salinity, temperature, etc. demonstrated their effect on biomass yields along with other biocommodities [6]. Stress-related conditions, especially nutrient depletion, has been well reported for halting growth rates, which results in accumulating storage compounds [7]. In such instances the fate of LC-PUFAs is triggered towards redistribution to triacylglycerols (TAGs) [8,9].
It has been demonstrated that biomass accumulation and the degree of saturation of fatty acids is highly influenced by the nitrogen in the medium. For example, it was observed that Botryococcus braunii produces a higher proportion of LC-PUFAs in nitrogen depleted conditions [10], whereas in Mortierella alpina excess nitrogen tends to enhance growth along with LC-PUFA production [11]. Studies related to the enhancement of LC-PUFAs by either media and/or genetic engineering strategies has been previously reported in various microalgae species (P. tricornutum, Nannochloropsis sp., Aurantiochytrium sp.) [9,12,13,14,15,16,17], however its commercial production is still a major concern in terms of cost and productivity [18]. Additionally, optimization of the production of LC-PUFAs through genetic engineering requires in depth analysis of the LC-PUFA metabolic pathways as well as the organizational and functional relationships of their functional genes.
The fate of LC-PUFAs in microalgae is still lacking in terms of distribution, transportation, and metabolic regulation [14,19]. Further, little information is available in the public databases about the functional genes involved in the LC-PUFA metabolism. The increasing number of sequenced microalgal genomes coupled with the application of the bioinformatics approach can facilitate functional annotation of genes and enzymes involved in LC-PUFA metabolism in various algal species across diverse phylogenetic groups. Recently, comparative genomics utilizing bioinformatics resources and algal omics data have enabled a better understanding of lipid biosynthetic pathways for biofuel production [20]. In the present study, we aimed to elucidate functional genes associated with LC-PUFA metabolism among microalgal repertoire employing in silico analysis.
A set of 13 functional genes well known to play a vital role in LC-PUFA metabolism were identified from 10 microalgae belonging to major classes including Chlorophyta (Chlamydomonas reinhardtii, Volvox carteri, Ostreococcus taurii, Monoraphidium neglectum, Coccomyxa subellipsoidea), Rhodophyta (Porphyridium purpureum), Heterokonta (Thalassiosira pseudonana, Nannochloropsis gaditana, Phaeodactylum tricornutum), and Haptophyta (Isochrysis galabana) (Supplementary Information Table S1), in order to unveil their evolutionary relationship, sub-cellular localization, and conserved domains and/or motifs patterns within these microalgal repertoires. Additionally, our focus was to understand the functional relevance of LC-PUFA metabolic genes in marine diatom P. tricornutum via assessing gene expression in nitrogen-repleted and nitrogen-depleted conditions. Thus, our computational analysis will further provide new insights to understand LC-PUFA metabolism in terms of finding novel targets, which could be employed for enhancing the production of these essential fatty acids in the photosynthetic cell factories.
2. Materials and Methods
2.1. Gene Retrieval and Metabolic Pathway Analysis
The proteins related to LC-PUFAs in microalgae was retrieved from the Kyoto Encyclopedia of Genes and Genomes (https://www.genome.jp/kegg/) (accessed on 1 July 2022) [21]. Genes encoding for regulatory and transporter proteins were obtained from ARALIP, the Arabidopsis acyl-lipid metabolism website (http://aralip.plantbiology.msu.edu/enzymes) (accessed on 8 July 2022). ARALIP provides all the genes, transcription factors and transporter proteins related to lipid biosynthesis in Arabidopsis thaliana (AT) [22]. The reference dataset from A. thaliana was further subjected to BLASTp (https://blast.ncbi.nlm.nih.gov/Blast.cgi) (accessed on 12 July 2022) [23] with a set threshold e-value of 1 × e−10. Homologs identified in different microalgal strains such as Chlamydomonas reinhardtii (CR), Volvox carteri (VC), Coccomyxa subellipsoidea (CS), Ostreococcus tauri (OT), Nannochloropsis gaditana (NG), Phaeodactylum tricornutum (PT), Thalassiosira pseudonana (TP), Isochrysis galbana (IG), Porphyridium purpureum (PP), and Monoraphidium neglectum (MN) were selected based on the percentage identity (above 30–40%), query coverage (above 60%) and e-value scores. The EggNOG-mapper 5.0 (http://eggnog5.embl.de/#/app/home) (accessed on 15 July 2022) [24] was used for assigning functions, simultaneously; all the genes present were assigned with gene ontology (GO) terms, EC number, and KEGG ID (KO number) (Table S1). Similarly, regulatory elements and transporter proteins were identified using BLASTp (with an e-value threshold set to 1 × e−10). A schematic representation of LC-PUFA biosynthesis and metabolism pathways in microalgae is illustrated in Figure 1 [21,22], depicting the presence of various functional genes involved in the production of LC-PUFAs among the microalgal lineage. Furthermore, proteins of LC-PUFA pathway are denoted as clusters I and II in the supplementary information (Table S1), facilitating better understanding of the individual components involved in the production of omega-3/6 fatty acids.
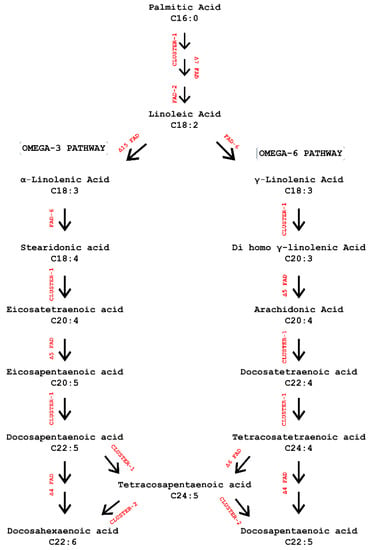
Figure 1.
Schematic representation of LC-PUFAs biosynthesis in microalgal cells. Genes marked in red are the ones involved in elongation, desaturation, and β-oxidation of omega fatty acids in different cellular locations. Abbreviations: elongase 6, 2, 4 (ELO); 3-oxoacyl-CoA reductase (KCR); 3-hydroxyacyl-CoA-dehydratase (HACD); enoyl-CoA reductase (ECR); fatty acid desaturases (FAD); Acyl coA synthetase (ACS/LCFACS); acyl-coenzyme A oxidase 1 (ACDase); enoyl CoA hydratase 2 (ECoAH); acetyl-CoA acyltransferase (ACAT).
2.2. Transcriptome Analysis
For functional analysis, temporal expression profile of specifically LC-PUFAs genes was assessed in model diatom. RNAseq data were retrieved from the European Bioinformatics Institute (EMBL-EBI) and the European Nucleotide Archive (ENA) (www.ebi.ac.uk) (accessed on 8 July 2022) (Accession No. SRP040703); the reads were checked for sequence quality (Phred score > 20) and adapter content using FastQC (www.bioinformatics.babraham.ac.uk/projects/fastqc/) (accessed on 22 July 2022), followed by read trimming and adapter removal using bbduk.sh (www.jgi.doe.gov/data-and-tools/bbtools/bb-tools-user-guide/bbduk-guide/) (accessed on 25 July 2022) available in the BBtools package (www.jgi.doe.gov/data−and−tools/bbtools) (accessed on 28 July 2022).
Later, Salmon (www.combine-lab.github.io/salmon/getting_started/) (accessed on 30 July 2022) [25] was used to map reads to the annotated reference transcriptome of P. tricornutum CCAP 1055/1 (Ensembl Protists release 50-November 2020) followed by read counting using featureCounts [26]. For differential expression analysis, DESeq2 [27] was used by utilizing Wald’s test, the resulting p-values were adjusted using the Benjamini and Hochberg method [28]. In the heatmap normalization, the expression values were transformed using the variance stabilizing transformation method available in the DESeq2 package. To show significant genes on the heatmap, all genes with adjusted p-value < 0.05 were marked as YES. Genes which are marked as NO either had adjusted p-value > 0.05 or did not have enough data to perform any statistical testing. All the visualization of heatmaps were made using ClustVis (https://biit.cs.ut.ee/clustvis_large/) (accessed on 1 August 2022) [29].
2.3. Validation of RNA-Seq Data by Quantitative Real-Time PCR (qPCR)
To evaluate the functionality of target genes, we conducted experiments with the marine diatom P. tricornutum (UTEX 646). Initially, the culture was maintained axenically in sterile artificial seawater enriched in f/2 nutrients. Cells were cultured under nitrogen repleted (NR) and nitrogen depleted (ND) conditions. Around 2 × 108 cells, collected on the third day by centrifugation at 3500× g, 4 °C for 10 min, were snap frozen and stored at −80 °C. RNA extraction was performed using TRIzol reagent as described [30]. RT-PCR analyses was conducted using specific primers for the genes (Table S3). For each sample, 1 μg of total RNA was reverse-transcribed using oligoDT primers and a first strand RevertAid cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA, USA). Quantitative PCR amplification was performed using SYBR Green Kit (Bio-Rad Laboratories, Hercules, CA, USA) in 96-well optical reaction plates, following the manufacturer’s instructions, using CFX96 Touch Real-Time PCR detection system (Bio-Rad Laboratories, Hercules, CA, USA). The threshold cycle (Ct) for each well was measured, and the mRNA levels of the target genes in nitrogen repleted and depleted cells were quantified after normalization with β-actin as an internal standard.
2.4. Construction of Phylogenetic Trees
The evolutionary relationship existing within proteins related to LC-PUFAs biosynthesis and metabolism in selected microalgal species are illustrated here by constructing phylogenetic trees using MEGA X software employing maximum-likelihood [31,32], the neighbor-joining method [33], and the maximum-parsimony method [34]. All prediction methods were utilized to attain congruence in topology. There was a total of 3538 positions in the final dataset. Initially the sequences were aligned using ClustalW [35], and subsequently a bootstrapping method was employed with 1000 rounds of replication, to construct the phylogenetic trees.
2.5. Prediction of Subcellular Localization, Motifs and Domain Architecture
The subcellular location of the candidate proteins was predicted using four online prediction software programs, namely TargetP (www.cbs.dtu.dk/services/TargetP/) (accessed on 20 July 2022) [36], WolfPsort (wolfpsort.hgc.jp/) (accessed on 22 July 2022) [37], Euk-mPLoc2.0 (www.csbio.sjtu.edu.cn/bioinf/euk-multi-2/) (accessed on 24 July 2022) [38], and CELLO (cello.life.nctu.edu.tw/) (accessed on 26 July 2022) [39]. All these prediction tools were utilized to evaluate each protein sequence and the maximum consensus-predicted location was allocated to the respective proteins. Motif predictions used the MEME program with default parameters (https://meme-suite.org/meme/tools/meme) (accessed on 21 July 2022) [40]. Further domain arrangements were predicted using the HMMER 3.0 web server and PROSITE from ExPASy (https://prosite.expasy.org/) (accessed on 18 July 2022) for each of the gene families [40,41].
2.6. Subcellular Networking and Pathway Enrichment Analysis
A complete network of the LC-PUFA biosynthesis related proteins, regulatory elements and transporter proteins was constructed using the STRING database (https://string-db.org/version11.0) (accessed on 20 July 2022) [42] and visualized using cytoscape software [43,44]. For functional annotation based on gene ontology (GO) and analysis of KEGG pathway, enrichment was performed by the cytoscape plugin ClueGO [45]. Genes involved in LC-PUFA biosynthesis along with the associated regulatory genes and transporters retrieved from ARALIP were used as queries in ClueGO to find significantly enriched GO terms (BP, CC and MF) and pathways (KEGG, REACTOME) using an adjusted p-value cutoff of 0.05. All the enriched terms and pathways are shown in a single network where nodes correspond to enriched terms/pathways and edge weight corresponds to kappa score calculated by ClueGO [45].
2.7. Statistical Analysis
All of the studies were carried out in biological triplicate and the results are reported as average ± SE. To determine significance, statistical procedures such as ANOVA and t-test were carried out in Microsoft Excel.
3. Results
3.1. Gene Mining and Analysis in Microalgal Lineage
An overview of LC-PUFA biosynthetic pathway in microalgal cell is illustrated in Figure 1. The first set of enzymes elongase (ELO6, 2, 4), 3-oxoacyl-CoA reductase (KCR), 3-hydroxyacyl-CoA dehydratase (HACD), enoyl-CoA reductase (ECR), referred to as cluster 1 (Figure 1), were involved in de novo fatty acid synthesis in chloroplast where palmitic acid (C16:0) is transferred from chloroplast to endoplasmic reticulum as free fatty acid to further undergo elongations and desaturations. In the first step, C16:0 undergoes a stepwise addition of carbon atoms via enzymes of cluster 1, resulting in the synthesis of linoleic acid (C18:2ω-6, LA) [46]. In addition, C18:2ω-6, LA via Δ6 desaturation enters the omega-3 pathway, forming alpha-linolenic acid (C18:3ω-3, ALA). Further linoleic acid can be diverted towards omega-6 pathway by forming precursor gamma-linolenic acid (C18:3ω-g, GLA). Additionally, C18:3ω-3, ALA via Δ6 desaturation and cluster 1 reactions forms C20:5ω-3, EPA, which is further converted to C22:5ω-3, DPA-3 and C22:6ω-3, DHA via Δ5 elongase and Δ4 desaturased enzymes [47]. Degradation of synthesized LC-PUFAs further undergoes β-oxidation reactions (cluster 2) to reduce carbon chain length and interacts with long-chain acyl-CoA synthetases (ACS). These crucial enzymes are responsible for the esterification and activation of free FAs to CoA to form acyl-CoAs. Further, the LC-PUFAs undergo stepwise reactions involved in β-oxidation. Enzymes such as acyl-CoA oxidase (ACDase), enoyl-CoA hydratase (ECoAH), and acetyl-CoA acyltransferase (ACAT) are responsible for retro conversion of these LC-PUFAs. They utilize tetracosahexaenoic acid (C24:6) and tetracosapentaenoic acid (C24:5) as substrates for the synthesis of docosahexaenoic acid (22:6ω-3, DHA) and docosapentaenoic acid (DPA C22:5), respectively.
Comparative analysis among 10 different microalgal species resulted in the identification of nearly 125 putative orthologous genes, 87 regulatory elements and 90 transporter proteins. Protein IDs and relevant information on all the genes in the study is listed in the Supplementary Information (Table S1). The functions of each sequence were assigned and classified via similarity search and identification of conserved domains and motifs. Further, the GO terms, KO numbers, and EC numbers were allocated to all the genes via EggNOG-mapper 5.0 (http://eggnog5.embl.de/#/app/home) (accessed on 28 July 2022) [24].
3.2. Functional Characterization of LC-PUFAs
3.2.1. Temporal Expression Profile
The raw reads of genes, regulatory elements, and transporter proteins related to LC-PUFAs were selected from the publicly available transcriptome datasets (NCBI Accession number SRP040703) for analyzing their regulation under specific conditions. P. tricornutum was selected for the transcriptome analysis based on the previous literature [20,48,49,50], showing a high amount of LC-PUFA content, especially EPA content [51]. Our analysis reveals that genes such as 3-oxoacyl-CoA reductase (KCR), long chain fatty acyl-CoA synthetase (LC-FACS (ACL1, ACS4)), and elongase (ELO6b) are significantly downregulated in the ND condition (Figure 2A). Further, it was observed that the expression of two genes involved in β-oxidation, enoyl-CoA hydratase (ECoAH) and acetyl-CoA acyltransferase (ACAT1), were 1.59 and 1.57 log2-fold higher, respectively (Figure 2A). LC-PUFA biosynthesis in P. tricornutum in the ND conditions reported remodelling of membrane lipids [52].
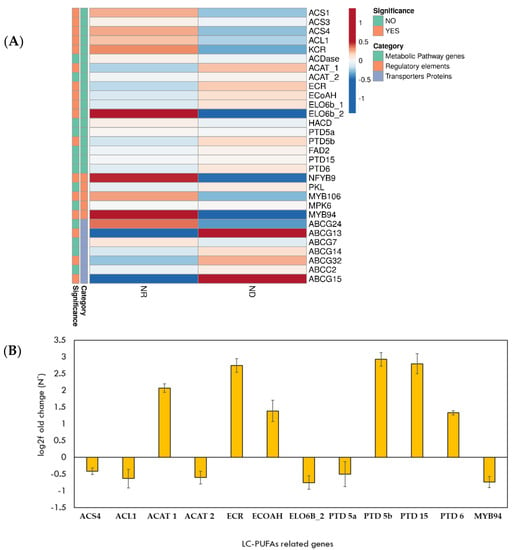
Figure 2.
Functional analysis of LC-PUFA metabolic genes in marine diatom P. tricornutum. (A) Heat map depicting expression profiles in NR and ND conditions; (B) log2-fold change of functional genes related to LC-PUFAs in P. tricornutum subjected to ND as compared with NR conditions.
In addition, other factors, such as regulatory elements and transporter proteins in P. tricornutum, provided some important cues in the regulation of LC-PUFA biosynthetic pathway. The MYB106 homolog present in P. tricornutum displayed (log2-fold = 1) decreased expression in ND conditions (Figure 2A). Moreover, homologs of ABCG transporter proteins (ABCG 13, 32, 15) showed an upregulation in the ND condition (Figure 2A). Basically, these transporters function as homodimers or heterodimers among A. thaliana for the cuticular lipid transport [53] and these comprise of long chain fatty acids, which may also be involved in the transport of LC-PUFAs in the microalgal cells.
3.2.2. Molecular Analysis of LC-PUFAs Genes Expression in P. tricornutum
The transcript abundance of selected LC-PUFA genes were validated by qPCR analysis (Figure 2B). Twelve homologs identified in P. tricornutum were selected for validation of the transcriptome analysis. The quantitative PCR results are consistent with the transcriptome data, confirming their relative expression profiles. Genes responsible for elongation (ELO6b_2), desaturation (PTD5a), and LC-PUFAs incorporation in membrane lipids (ACS4, ACL1) [54] were downregulated (i.e., 0.75, 0.50, 0.41, 0.63 log2-fold, respectively) in ND as compared to NR (Figure 2B). However, the expression profile of genes involved in β-oxidation (ACAT 1, ECoAH) and fatty acid elongation (C16–C18) (ECR) were upregulated (i.e., 2.06, 1.38, 2.73 log2-fold, respectively) in the ND condition (Figure 2B). One of the fatty acid desaturased genes, PTD-5b, showed high abundance (2.92 log2-fold) in the ND condition (known for neutral lipid accumulation), correlating with previous reports showing its function in the enhancement of the neutral lipid content when overexpressed in P. tricornutum [55]. Further, high expression of PTD6 and PTD15 (1.32. 2.79 log2-fold) in the ND condition show the substrate specificity of desaturases to maintain C18-PUFA pool in the cells for further incorporation of LC-PUFAs in TAGs [56]. Additionally, the relative expression of transcription factor MYB94 showed downregulation in ND conditions (0.73 log2-fold), correlating with the transcriptome expression levels.
3.3. Organization Studies
3.3.1. Phylogenomic Analysis
Phylogenomic analysis explains the evolutionary arrangements and identifies the conservation patterns of respective genes. Different methods were employed for phylogenomic tree construction to reduce the ambiguity (Figure 3 and Figure S1A–F), and results highlighted similar conservation patterns in all the identified homologs, including regulatory elements and transporter proteins in various microalgal species (Figure S1A–F).
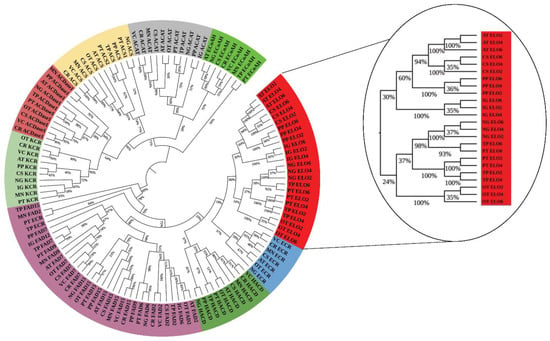
Figure 3.
Phylogenomics analysis of conventional LC-PUFA metabolic genes by maximum likelihood method (ML) method with evaluation of 1000 rounds of bootstrapping test using MEGA 10.0.
Results shows that majority of the proteins were clustered together due to their common ancestral origin. Proteins involved in fatty acid biosynthesis are highly conserved among microalgal and plant lineages [6]. Proteins belonging to the LC-PUFAs biosynthesis (ELO, FAD, ACDase, ECoAH, ACAT etc.) were clustered in a single clad within microalgal lineage and are conserved among plant and algal systems (Figure 3). Similarly, due to the shared ancestry of diatom lineage [57], most of the studied protein was present in a single clad in P. tricornutum and T. pseudonana. Moreover, the phylogenetic analysis of regulatory elements and transporter proteins (Figure S1C–F) such as ADAP, WRI4, NFYB9, ABCC2, and PXA1 showed a high degree of conservation among these selected microalgal species.
3.3.2. Subcellular Localization
Based on the subcellular locations, proteins are assigned with biological functions within that organelle [58]. Subcellular localization was predicted for all proteins involved in the LC-PUFA biosynthesis pathway using various tools to increase prediction specificity and eliminate false positives and negatives [59,60]. Our analysis predicts that LC-PUFAs synthesis-related proteins are variably distributed among the endoplasmic reticulum (ER) (27%), chloroplast (14%), peroxisomes (14%), plasma membrane (17%), mitochondria (2%), cytoplasm (2%), and others (extracellular space, golgi, nucleus, lysosome, and vacuoles) (24%) (Table S1). He et al. illustrated the role of different cellular compartment in the synthesis of LC-PUFAs in plants [61]. Among the identified protein dataset, ER harbors the most LC-PUFAs synthesis related proteins (27%) since the majority of elongation and desaturation reactions take place in the ER [62].
3.3.3. Identification of Motifs and Domains
Protein sequence characterization involves recognition of the motif and domain regions present as compressed structures within the cells. Since motifs and domains are highly conserved in nature compared to other protein parts, they tend to evolve with the least minimal changes. Among the LC-PUFA biosynthesis pathway, around 13 motif structures were identified (Figure 4) by MEME tool [40], which represents a high degree of conservation among the studied microalgal strains. The height of each stack represents the positional conservation of sequences and the height of symbols within each stack reflects the relative frequency of the corresponding amino acid (Figure 4).

Figure 4.
Motif and domain architecture of LC-PUFAs biosynthetic pathway genes using MEME and HMMER tool.
3.3.4. Subcellular Networking and Pathway Enrichment Analysis
The LC-PUFA genes, regulatory elements and transporter proteins within plant and microalgal lineage were further studied using the STRING interactions database [43,44]. The interactive network showed association of LC-PUFA genes with various regulatory elements (LEC 1 & 2, ABI4, MPK6, PKL, WIN1 and members of the MYB family MYB 30, 41,94,106) and with transporter proteins of ATP binding cassette subfamily G & D (ABCG) members (Figure 5A). In the present study, a new homolog in microalgal lineage similar to the LEC transcription factor was found, its overexpression studies showed increased LC-PUFA content in plants [63]. The string network showed interaction of the ABCD1 transporter protein with enoyl-CoA hydratase (an ECoAH-enzyme involved in β-oxidation), and other transporters and regulatory elements (LEC), highlighting crosstalk between LC-PUFA synthesis, β-oxidation, transporters, and regulatory elements.
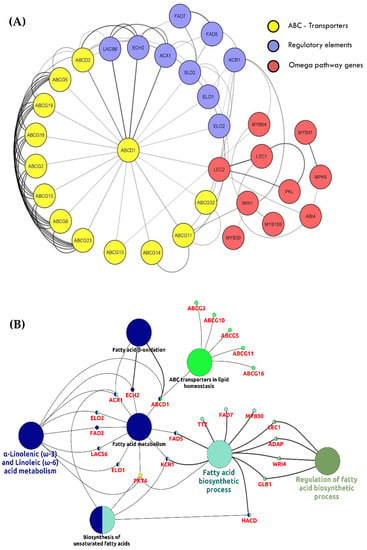
Figure 5.
Interactive network and pathway enrichment analysis of LC-PUFAs associated functional genes. (A) Sub-cellular network prediction for LC-PUFA metabolic genes interacting with transcription factors and transporter proteins using STRING database. (B) Pathway enrichment by Gene Ontology (GO) terms was visualized using the ClueGO/CluePedia plugin from Cytoscape.
Further analysis was carried out using the Cytoscape plugin ClueGO [43] to visualize the GO terms and pathways to identify a complex network (Figure 5B). The analysis shows connection between the β-oxidation pathway, long chain polyunsaturated fatty acid (omega-3 fatty acids) biosynthesis, some important members of ABC transporters (involved in lipid homeostasis) and regulatory elements based on GO and pathway enrichment. Our interaction analysis demonstrates that ABCD1 connects the fatty acid β-oxidation process, the fatty acid biosynthetic process, and α-linolenic acid metabolism. Genes involved in unsaturated fatty acid biosynthesis are the ones which are present in both metabolism as well as biosynthesis. This particularly highlights the fact that LC-PUFA biosynthesis is not just a crosstalk of elongases and desaturases, but multiple other potential targets are also involved for their regulation and synthesis in these microalgal cells.
4. Discussion
Comprehensive studies on different microalgal species belonging to eustigmatophyte, diatoms, and red alga have revealed that fatty acid production is substantially conserved across these strains [22,64,65,66]. Similarly, our studies with phylogenomic analysis reveal that all LC-PUFA functional genes associated with biosynthesis, degradation, regulatory elements, and transporter proteins are highly conserved among microalgal repertoire (Figure 3 and Figure S1A–F). Additionally, the homologs of transporter and regulatory proteins involved in the regulation of lipid biosynthesis and transport in plant species [67,68] were identified in the selected microalgal species (Figure S1C–F), and are expected to perform similar functions in these photosynthetic cell factories. This detailed study assisted in the identification of previously unknown genes associated with LC-PUFA metabolism in various microalgal species and their functional targets involved in metabolic regulation.
It was observed that fatty acid desaturases included highly conserved histidine-rich motifs with di-iron cores, serving as reserved sites for oxygen activation and substrate oxidation [69]. Similarly, the discovery of a cytochrome b5 domain in fatty acid desaturase corresponds to prior findings of a key regulatory domain governing desaturase production in Saccharomyces cerevisiae [70]. A comparable cytochrome b5 domain was discovered in Pavlova sp. Δ5-desaturase, which is entirely responsible for increased LC-PUFA biosynthesis [71].
Our findings on the subcellular localization indicate that the majority of genes involved in LC-PUFA metabolism are found in the ER (Supplementary Table S1). Protein-tagging investigations using green fluorescent protein in S. cerevisiae have been used to indicate the presence of elongases and desaturases in the ER [72]. Furthermore, fatty acid desaturases and long chain-fatty acyl-CoA synthetase (LC-FACS) were found in many locations (cytoplasm, plasma membrane, mitochondria, peroxisome, and chloroplast) in various microalgal lineages (Supplementary Table S1). This has previously been linked to the relocalization of proteins from one cellular membrane to another in yeast mutants [73,74]. Additionally, LC-FACS is recognized for transporting and activating free fatty acids within these cells, which validates their presence in the cytoplasm as well as other organelles [68]. Our findings demonstrate that the LC-PUFA functional genes are not only restricted to ER but are also present in multiple compartments and thus may be solely responsible for maintaining LC-PUFA pools within these cell factories. Perhaps such a mechanism has previously been highlighted in the other algae and higher plants [64,65]. Understanding the influence of environmental conditions on gene expression can further aid in elucidating molecular mechanisms in terms of the external regulators involved in the progression of a specific metabolic pathway; in this case, we investigated the effect of the most important component nitrogen on the LC-PUFA metabolism, which has received little attention in the microalgal lineage. The transcriptomics study demonstrated the relative expression pattern for LC-PUFA functional genes in the marine diatom P. tricornutum in NR and ND conditions.
In the present study, our findings suggest that enzymes involved for fatty acid elongation (KCR, ELO6b), desaturation (PTD5a, PTD5b, FAD2, PTD15, PTD6), and LC-PUFA incorporation in membrane lipids (ACS1, ACS3, ACS4, ACL1) have distinct regulatory patterns in NR and ND conditions. Lipid accumulation in ND is primarily mediated by a combination of increased de novo fatty acid synthesis and fatty acid remodeling from membrane lipids [6,75,76], which might be linked to a high abundance of ECR (responsible for fatty acid elongation) in the ND condition. Furthermore, fatty acids must be present in the acyl-CoA form during FA production, membrane glycerolipid formation, and the β-oxidation process [77]. The acyl-Coenzyme A (CoA) synthetases (ACS) in each compartment control the internal acyl-CoA pools by esterification of FAs to CoA. The ACS gene was shown to be significantly downregulated in the ND condition (Figure 2), although ECoAH and ACAT (β-oxidation enzymes) were increased, indicating a drop in the acyl-CoA pool for lipid synthesis (de novo LC-PUFAs). This clearly demonstrates that in P. tricornutum, ACS is related to membrane lipid synthesis, and high expression of genes involved with β-oxidation can be linked to the creation of an acyl-CoA pool via mitochondria and/or peroxisomal β-oxidation. Based on this information, the flux of LC-PUFAs within microalgal cells and its fate in NR and ND conditions has been illustrated in Figure 6.
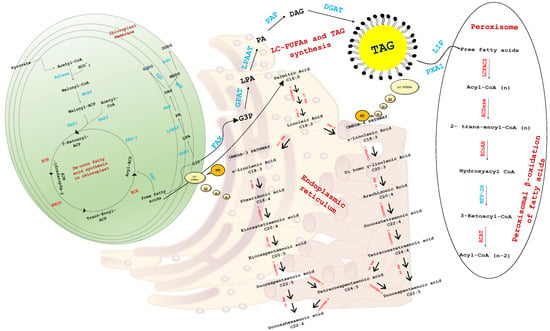
Figure 6.
An overview of LC-PUFAs flux within microalgal cells and its fate in NR and ND conditions. Genes marked in red are the one involved in elongation, desaturation and β-oxidation of omega fatty acids in different cellular locations. Abbreviations: elongase 6, 2, 4 (ELO); 3-oxoacyl-CoA reductase (KCR); 3-hydroxyacyl-CoA-dehydratase (HACD); enoyl-CoA reductase (ECR); fatty acid desaturases (FAD); Acyl-CoA synthetase (ACS/LCFACS); acyl-CoA oxidase 1 (ACDase); enoyl-CoA hydratase 2 (ECoAH); acetyl-CoA acyltransferase (ACAT); Acetyl-CoA Carboxylase (ACCase); Malonyl-CoA-acyl carrier protein transacylase (MCAT); ketoacyl-ACP synthase (KAS); Fatty acid synthase (FAS); Fatty acid thioesterase (FAT); 3-oxoacyl-CoA reductase (KCR); 3-hydroxyacyl-CoA-dehydratase (HACD), enoyl-CoA reductase (ECR), Coenzyme A (CoA); diacylglycerol (DAG); diacylglycerol acyltransferase (DGAT); digalactosyldiacylglycerol (DGDG); sulfoquinovosyl diacylglycerol (SQDG); diacylglyceryl-3-O-4′- (N,N,N-trimethyl)-homoserine (DGTS); fatty acid export (FAX1); glycerol-3-phosphate (G3P); glycerol 3-phosphate acyltransferase (GPAT); lysophosphatidic acid acyltransferase (LPAAT); monogalactosyldiacylglycerols (MGDG); phosphatidic acid (PA); phosphatidic acid phosphatase (PAP); lipase (LIP); triacylglycerol (TAG); multi-functional protein dehydrogenase (MFP-DH).
The cytosolic acyl-CoA pool produces C18-FAs, which are used to make extra-chloroplastic EPA. During EPA production, the membrane-bound desaturases PTD5, PTD6, and FAD2 appear to be positioned in the ER membrane and facing the cytoplasm [78]. Our transcriptome and qPCR analysis (Figure 2) revealed that PTD5a expression was significantly lower and PTD5b expression was higher in the ND condition, indicating that membrane lipid incorporation (which is typically absent in ND) is mediated by PTD5a, and EPA incorporation in DAGs is mediated by PTD5b [55,56].
Moreover, expression profiles of regulatory elements and transporter proteins highlight a coordinated regulation of LC-PUFA metabolism in microalgae. Transcription factors in Chlorella sp. belonging to the MYB family are reported to be involved in starch to lipid conversion [79] and MYB106 upregulates the expression of the wax inducer 1 (WIN1) transcription factor, which is directly linked to the expression of LC-FACS [80]. Our results are consistent with these findings, showing a down regulation in LC-FACS expression (Figure 2A) and the MYB106 under ND conditions in P. tricornutum. MYB106 may play a role as a positive regulator of LC-PUFA biosynthesis. Likewise, MYB family members such as MYB30 target the long chain acyl-CoA elongase complex for extracellular accumulation of very-long-chain fatty acids (VLC-FAs). Its activity is associated with the acyl-ACP thioesterase enzyme, responsible for the supply of free fatty acids for VLC-FA synthesis [81]. Moreover, transcription factor MYB94 has a role as a stress marker in plants and it is known for induction of cuticular wax synthesis gene in stress condition [82]; we observed downregulation of MYB94 in the ND condition. However, the link between these regulatory elements with LC-PUFAs biosynthesis needs to be further exploited in detail.
Transporter proteins such as ABCG 13, 32, and 15 showed a downregulation in the ND condition (Figure 2). A recent study showed that homologs of ABCD1 transporters in C. reinhardtii (CrFAX1 and CrFAX2) are involved in mediating fatty acid export in lipid biosynthesis. In addition, overexpression of both CrFAXs increases the intracellular TAG content and it increased omega-3/6 (LC-PUFAs) fatty acids content (C18:3) in the CrFAXs overexpressed cell lines by 69%, compared to that of wild type strain [83]. As a result, these regulatory elements and transporter proteins remotely govern LC-PUFA production and can be regarded as critical targets for its increase within the microalgal species.
Overall, the relative expression profiles via transcriptome and qPCR validation (Figure 2) reveal that genes related to LC-PUFAs and their incorporation in phospholipids are downregulated, while there is a shuffle of lipids from membrane to TAGs is observed in the ND condition (Figure 6). Nutrient repletion favors conditions for membrane lipid production [8,84]. The expression profiles of transporter proteins and regulatory elements supports the hypothesis of LC-PUFAs formation in NR conditions. In addition, it is evident that transporter proteins play a major role in free fatty acid transport and in maintaining the LC-PUFA pools in these cell factories.
The results of ClueGO and STRING analyses (Figure 5) show that the metabolic pathways are closely linked to each other and are under controlled transcriptional regulation, hence contributing towards the survival of cells. STRING analysis depicts a tight interactive network of LC-PUFA proteins. ClueGO analysis showed a clear picture of crosstalk between LC-PUFA biosynthesis and lipid metabolic pathways. These findings can be further explored in terms of genetic engineering studies for enhancement of LC-PUFAs in these photosynthetic cell factories.
In summary, combining in silico and gene expression analyses of LC-PUFA-associated functional genes resulted in the identification and characterization of putative homologs in marine diatom P. tricornutum. It contributed to the understanding of the complex regulatory mechanisms of LC-PUFA metabolism through functional and organisational investigations that identified the proteins involved in LC-PUFA synthesis, regulation, distribution, and degradation. As a result, a genome-wide analysis of individual genes and their encoded products across microalgal species has thus revealed insights to allow engineering for increased LC-PUFA biosynthesis. Thus, our findings may aid attempts to achieve industrial scale-up of LC-PUFAs, a novel approach in algal biorefineries.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/hydrobiology1040027/s1, S1-Materials and Methods, References [52,85,86,87,88,89] are cited in the Supplementary Materials; Table S1: List of candidate genes, regulatory elements, transporter proteins involved in LC-PUFAs synthesis along with their gene ontology terms among different species namely, Arabidopsis thaliana (AT), Chlamydomonas reinhardtii (CR), Volvox carteri (VC), Coccomyxa subellipsoidea (CS), Ostreococcus tauri (OT), Nannochloropsis gaditana (NG), Phaeodactylum tricornutum (PT), Thalassiosira pseudonana (TP), Isochrysis galbana (IG), Porphyridium purpureum (PP) and Monoraphidium neglectum (MN); Table S2: Total lipid profiles of Phaeodactylum tricornutum. Subjected to nitrogen replete and deplete conditions at 72 h of cultivation; Table S3: List of primers used for qPCR validation studies in Phaeodactylum tricornutum. Subjected to nitrogen replete (NR) and deplete (ND) conditions at 72 h of cultivation; Figure S1A. Phylogenetic tree of conventional LC-PUFAs metabolic biosynthetic pathway genes by Neighbor joining (NJ) method with evaluation of 1000 rounds of bootstrapping test using MEGA 10.0; Figure S1B: Phylogenetic tree of conventional LC-PUFAs metabolic biosynthetic pathway genes by Maximum parsimony method (MP) method with evaluation of 1000 rounds of bootstrapping test using MEGA 10.0, Phylogenetic tree of LC-PUFAs biosynthetic pathway related proteins; Figure S1C: Phylogenetic tree of regulatory elements/transcription factors related to LC-PUFAs biosynthetic pathway by Maximum likelihood method (ML) method; Figure S1D. Phylogenetic tree of regulatory elements/transcription factors related to LC-PUFAs biosynthetic pathway by Maximum parsimony method (MP) method; Figure S1E: Phylogenetic tree of transporter proteins related to LC-PUFAs biosynthetic pathway by Maximum likelihood method (ML) method; Figure S1F: Phylogenetic tree of transporter proteins related to LC-PUFAs biosynthetic pathway by Maximum parsimony method (MP) method.
Author Contributions
Conceptualization, M.R., P.P.J. and A.K.; methodology, M.R., A.K. and A.A.N.; software, M.R. and A.K., validation, M.R., A.K. and A.A.N.; formal analysis, M.R. and P.P.J., investigation, M.R.; resources, M.R. and P.P.J.; data curation, M.R. and A.K.; writing—original draft preparation, M.R., A.A.N. and P.P.J.; writing—review and editing, P.P.J., N.J.K. and T.F.; visualization, M.R.; supervision, P.P.J., A.A.N., N.J.K. and T.F.; project administration, P.P.J.; funding acquisition, P.P.J. All authors have read and agreed to the published version of the manuscript.
Funding
The work was supported by the grants provided from the Department of Biotechnology (DBT) and Biotechnology Industry Research Assistance Council (BIRAC), India (Grant no. BT/PB/Center/03/2011-Phase II, BT/PR31155/PBD/26/725/2019, BT/SB0078/02/19). Fellowship(s) for M.R., A.K. and A.A.N. are supported from Department of Biotechnology (DBT), New Delhi, India, are duly acknowledged.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article or Supplementary Materials.
Acknowledgments
We would like to thank our laboratory colleagues Mukul Kareya (Postdoctoral Research Associate, IBPC, France) and Iqra Mariam (Research Associate, TIFR, Mumbai) for providing support with data analysis.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
NR; Nitrate replete, ND; Nitrate deplete, ELO; Elongase, PAP; Phosphatidic acid phosphatase, KCR;3-Oxoacyl-CoA reductase, LIP; Lipase, HACD; 3-Hydroxyacyl-CoA-dehydratase, TAG; Triacylglycerol, ECR; Enoyl-CoA reductase, PA; Phosphatidic acid, FAD; Fatty acid desaturases, ACS; Acyl-CoA synthetase, ACDase; Acyl-CoA oxidase 1, ECoAH; Enoyl CoA hydratase 2, ACAT; Acetyl-CoA acyltransferase, ACCase; Acetyl-CoA Carboxylase, MCAT; Malonyl-CoA-acyl carrier protein transacyclase, KAS; Ketoacyl-ACP synthase, FAS; Fatty acid synthase, FAT; Fatty acid thioesterase, KCR; 3-Oxoacyl-CoA reductase, HACD; 3-Hydroxyacyl-CoA-dehydratase, ECR; Enoyl-CoA reductase, CoA; CoenzymeA, DAG; Diacylglycerol, DGAT; Diacylglycerol acyltransferase, DGDG; Digalactosyldiacylglycerol, SQDG; Sulfoquinovosyl diacylglycerol, DGTS; Diacylglyceryl-3-O-4′-(N,N,N-trimethyl)-homoserine, FAX1; Fatty acid export 1, G3P; Glycerol-3-phosphate, GPAT; Glycerol 3-phosphate acyltransferase, LPAAT; Lysophosphatidic acid acyltransferase, MGDG; Monogalactosyldiacylglycerols, MFPDH; Multi-functional protein dehydrogenase.
References
- Chen, Y.; Xu, C.; Vaidyanathan, S. Microalgae: A robust green bio-bridge between energy and environment. Crit. Rev. Biotechnol. 2018, 38, 351–368. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M.L.; Haslam, R.P.; Napier, J.A.; Sayanova, O. Metabolic engineering of Phaeodactylum for the enhanced accumulation of omega-3 long chain polyunsaturated fatty acids. Metab. Eng. 2014, 22, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Vaezi, R.; Napier, J.A.; Sayanova, O. Identification and functional characterization of genes encoding omega-3 polyunsaturated fatty acid biosynthetic activities from unicellular microalgae. Mar. Drugs 2013, 11, 5116–5129. [Google Scholar] [CrossRef] [PubMed]
- Hultberg, M.; Jönsson, H.L.; Bergstrand, K.-J.; Carlsson, A.S. Impact of light quality on biomass production and fatty acid content in the microalga Chlorella vulgaris. Bioresour. Technol. 2014, 159, 465–467. [Google Scholar] [CrossRef]
- Meesapyodsuk, D.; Qiu, X. Biosynthetic mechanism of very long chain polyunsaturated fatty acids in Thraustochytrium sp. 26185. J. Lipid Res. 2016, 57, 1854–1864. [Google Scholar] [CrossRef]
- Hu, Q.; Sommerfeld, M.; Jarvis, E.; Ghirardi, M.; Posewitz, M.; Seibert, M.; Darzins, A. Microalgal triacylglycerols as feedstocks for biofuel production: Perspectives and advances. Plant Mol. Biol. 2008, 54, 621–639. [Google Scholar] [CrossRef]
- Goncalves, E.C.; Wilkie, A.C.; Kirst, M.; Rathinasabapathi, B. Metabolic regulation of triacylglycerol accumulation in the green algae: Identification of potential targets for engineering to improve oil yield. Plant Biotechnol. J. 2016, 14, 1649–1660. [Google Scholar] [CrossRef]
- Hulatt, C.J.; Smolina, I.; Dowle, A.; Kopp, M.; Vasanth, G.K.; Hoarau, G.G.; Wijffels, R.H.; Kiron, V. Proteomic and transcriptomic patterns during lipid remodeling in Nannochloropsis gaditana. Int. J. Mol. Sci. 2020, 21, 6946. [Google Scholar] [CrossRef]
- Remmers, I.M.; Martens, D.E.; Wijffels, R.H.; Lamers, P.P. Dynamics of triacylglycerol and EPA production in Phaeodactylum tricornutum under nitrogen starvation at different light intensities. PLoS ONE 2017, 12, e0175630. [Google Scholar] [CrossRef]
- Blifernez-Klassen, O.; Chaudhari, S.; Klassen, V.; Wördenweber, R.; Steffens, T.; Cholewa, D. Metabolic survey of Botryococcus braunii: Impact of the physiological state on product formation. PLoS ONE 2018, 13, e0198976. [Google Scholar] [CrossRef]
- Vadivelan, G.; Venkateswaran, G. Production and enhancement of omega-3 fatty acid from Mortierella alpina CFR-GV15: Its food and therapeutic application. Biomed. Res. Int. 2014, 2014, 657414. [Google Scholar] [CrossRef]
- Cerón García, M.C.; Sánchez Mirón, A.; Fernández Sevilla, J.M.; Molina Grima, E.; García Camacho, F. Mixotrophic growth of the microalga Phaeodactylum tricornutum: Influence of different nitrogen and organic carbon sources on productivity and biomass composition. Process Biochem. 2005, 40, 297–305. [Google Scholar] [CrossRef]
- Cui, Y.; Thomas-Hall, S.R.; Schenk, P.M. Phaeodactylum tricornutum microalgae as a rich source of omega-3 oil: Progress in lipid induction techniques towards industry adoption. Food Chem. 2019, 297, 124937. [Google Scholar] [CrossRef]
- Hamilton, M.L.; Powers, S.; Napier, J.A.; Sayanova, O. Heterotrophic Production of Omega-3 Long-Chain Polyunsaturated Fatty Acids by Trophically Converted Marine Diatom Phaeodactylum tricornutum. Mar. Drugs 2016, 14, 53. [Google Scholar] [CrossRef]
- Haslam, R.P.; Hamilton, M.L.; Economou, C.K.; Smith, R.; Hassall, K.L.; Napier, J.A.; Sayanova, O. Overexpression of an endogenous type 2 diacylglycerol acyltransferase in the marine diatom Phaeodactylum tricornutum enhances lipid production and omega-3 long-chain polyunsaturated fatty acid content. Biotechnol. Biofuels 2020, 13, 87. [Google Scholar] [CrossRef]
- Kadalag, N.L.; Pawar, P.R.; Prakash, G. Co-cultivation of Phaeodactylum tricornutum and Aurantiochytrium limacinum for polyunsaturated omega-3 fatty acids production. Bioresour. Technol. 2022, 346, 126544. [Google Scholar] [CrossRef]
- Zhu, B.-H.; Tu, C.-C.; Shi, H.-P.; Yang, G.-P.; Pan, K.-H. Overexpression of endogenous delta-6 fatty acid desaturase gene enhances eicosapentaenoic acid accumulation in Phaeodactylum tricornutum. Process Biochem. 2017, 57, 43–49. [Google Scholar] [CrossRef]
- Mühlroth, A.; Li, K.; Røkke, G.; Winge, P.; Olsen, Y.; Hohmann-Marriott, M.F.; Vadstein, O.; Bones, A.M. Pathways of lipid metabolism in marine algae, co-expression network, bottlenecks and candidate genes for enhanced production of EPA and DHA in species of Chromista. Mar. Drugs 2013, 11, 4662–4697. [Google Scholar] [CrossRef]
- Yang, F.; Yuan, W.; Ma, Y.; Balamurugan, S.; Li, H.-Y.; Fu, S.; Wu, L. Harnessing the lipogenic potential of δ6-desaturase for simultaneous hyperaccumulation of lipids and polyunsaturated fatty acids in Nannochloropsis oceanica. Front. Mar. Sci. 2019, 6, 682. [Google Scholar] [CrossRef]
- Diao, J.; Song, X.; Guo, T.; Wang, F.; Chen, L.; Zhang, W. Cellular engineering strategies toward sustainable omega-3 long chain polyunsaturated fatty acids production: State of the art and perspectives. Biotechnol. Adv. 2020, 40, 107497. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef]
- Li-Beisson, Y.; Shorrosh, B.; Beisson, F.; Andersson, M.X.; Arondel, V.; Bates, P.D.; Baud, S.; Bird, D.; Debono, A.; Durrett, T.P.; et al. Acyl-lipid metabolism. Arab. Book 2010, 8, e0133. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, 309–314. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general-purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res. 2015, 43, 566–570. [Google Scholar] [CrossRef]
- Rio, D.C.; Ares, M., Jr.; Hannon, G.J.; Nilsen, T.W. Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb. Protoc. 2010, 2010, 5439. [Google Scholar] [CrossRef]
- Verbruggen, H.; Theriot, E.C. Building trees of algae: Some advances in phylogenetic and evolutionary analysis. Eur. J. Phycol. 2008, 43, 229–252. [Google Scholar] [CrossRef]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. CABIOS 1992, 8, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Mount, D.W. Maximum parsimony method for phylogenetic prediction. CSH Protoc. 2008, 2008, 32. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Emanuelsson, O.; Brunak, S.; von Heijne, G.; Nielsen, H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2007, 2, 953–971. [Google Scholar] [CrossRef]
- Horton, P.; Park, K.-J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, 585–587. [Google Scholar] [CrossRef]
- Chou, K.-C.; Shen, H.-B. A new method for predicting the subcellular localization of eukaryotic proteins with both single and multiple sites: Euk-mploc 2.0. PLoS ONE 2010, 5, e9931. [Google Scholar] [CrossRef]
- Yu, C.S.; Chen, Y.C.; Lu, C.H.; Hwang, J.K. Prediction of protein subcellular localization. Proteins 2006, 64, 643–651. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, 202–208. [Google Scholar] [CrossRef]
- Sigrist, C.J.; de Castro, E.; Cerutti, L.; Cuche, B.A.; Hulo, N.; Bridge, A.; Bougueleret, L.; Xenarios, I. New and continuing developments at PROSITE. Nucleic Acids Res. 2013, 41, 344–347. [Google Scholar] [CrossRef]
- Liu, W.; Chen, D. Phylogeny, functional annotation, and protein interaction network analyses of the Xenopus tropicalis basic helix-loop-helix transcription factors. Biomed. Res. Int. 2013, 2013, 145037. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, 447–452. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef]
- Robertson, R.; Guihéneuf, F.; Schmid, M.; Stengel, D.; Fitzgerald, G.; Ross, P.; Stanton, C. Algae-Derived Polyunsaturated Fatty Acids: Implications for Human Health. In Polyunsaturated Fatty Acids: Sources, Antioxidant Properties and Health Benefits; Catalá, A., Ed.; Nova Sciences: Hauppauge, NY, USA, 2013; pp. 1–54. [Google Scholar]
- Shanab, S.M.M.; Hafez, R.M.; Fouad, A.S. A review on algae and plants as potential source of arachidonic acid. J. Adv. Res. 2018, 11, 3–13. [Google Scholar] [CrossRef]
- Harwood, J.L. Algae: Critical sources of very long-chain polyunsaturated fatty acids. Biomolecules 2019, 9, 708. [Google Scholar] [CrossRef]
- Molino, A.; Martino, M.; Larocca, V.; Di Sanzo, G.; Spagnoletta, A.; Marino, T.; Karatza, D.; Iovine, A.; Mehariya, S.; Musmarra, D. Eicosapentaenoic acid extraction from Nannochloropsis gaditana using carbon dioxide at super critical conditions. Mar. Drugs 2019, 17, 132. [Google Scholar] [CrossRef]
- Pudney, A.; Gandini, C.; Economou, C.K.; Smith, R.; Goddard, P.; Napier, J.A.; Spicer, A.; Sayanova, O. Multifunctionalizing the marine diatom Phaeodactylum tricornutum for sustainable co-production of omega-3 long chain polyunsaturated fatty acids and recombinant phytase. Sci. Rep. 2019, 9, 11444. [Google Scholar] [CrossRef]
- Yang, Z.K.; Niu, Y.F.; Ma, Y.H.; Xue, J.; Zhang, M.H.; Yang, W.D.; Liu, J.S.; Lu, S.H.; Guan, Y.; Li, H.Y. Molecular and cellular mechanisms of neutral lipid accumulation in diatom following nitrogen deprivation. Biotechnol. Biofuels 2013, 6, 67. [Google Scholar] [CrossRef]
- Levitan, O.; Dinamarca, J.; Zelzion, E.; Lun, D.S.; Guerra, L.T.; Kim, M.K.; Kim, J.; Van Mooy, B.A.S.; Bhattacharya, D.; Falkowski, P.G. Remodeling of intermediate metabolism in the diatom Phaeodactylum tricornutum; under nitrogen stress. Proc. Natl. Acad. Sci. 2015, 112, 412–417. [Google Scholar] [CrossRef]
- McFarlane, H.E.; Shin, J.J.H.; Bird, D.A.; Samuels, A.L. Arabidopsis abcg transporters, which are required for export of diverse cuticular lipids, dimerize in different combinations. Plant Cell 2010, 22, 3066–3075. [Google Scholar] [CrossRef]
- Mashek, D.G.; Li, L.O.; Coleman, R.A. Long-chain acyl-CoA synthetases and fatty acid channeling. Future Lipidol. 2007, 2, 465–476. [Google Scholar] [CrossRef]
- Peng, K.-T.; Zheng, C.-N.; Xue, J.; Chen, X.-Y.; Yang, W.-D.; Liu, J.-S.; Bai, W.; Li, H.-Y. Delta 5 fatty acid desaturase upregulates the synthesis of polyunsaturated fatty acids in the marine diatom Phaeodactylum tricornutum. J. Agric. Food Chem. 2014, 62, 8773–8776. [Google Scholar] [CrossRef]
- Degraeve-Guilbault, C.; Gomez, R.E.; Lemoigne, C.; Pankansem, N.; Morin, S.; Tuphile, K.; Joubès, J.; Jouhet, J.; Gronnier, J.; Suzuki, I.; et al. Plastidic Δ6 fatty-acid desaturases with distinctive substrate specificity regulate the pool of c18-PUFAs in the ancestral picoalga Ostreococcus tauri. Plant Physiol 2020, 184, 82–96. [Google Scholar] [CrossRef]
- Bowler, C.; Allen, A.E.; Badger, J.H.; Grimwood, J.; Jabbari, K.; Kuo, A.; Maheswari, U.; Martens, C.; Maumus, F.; Otillar, R.P.; et al. The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature 2008, 456, 239–244. [Google Scholar] [CrossRef]
- Dönnes, P.; Höglund, A. Predicting protein subcellular localization: Past, present, and future. Genom. Proteom. Bioinform. 2004, 2, 209–215. [Google Scholar] [CrossRef]
- Shen, Y.Q.; Burger, G. Unite and conquer: Enhanced prediction of protein subcellular localization by integrating multiple specialized tools. BMC Bioinform. 2007, 8, 420. [Google Scholar] [CrossRef]
- Misra, N.; Panda, P.K.; Parida, B.K. Genome-wide identification and evolutionary analysis of algal LPAT genes involved in TAG biosynthesis using bioinformatics approaches. Mol. Biol. Rep. 2014, 41, 8319–8332. [Google Scholar] [CrossRef]
- He, M.; Qin, C.-X.; Wang, X.; Ding, N.-Z. Plant unsaturated fatty acids: Biosynthesis and regulation. Front. Plant Sci. 2020, 11, 390. [Google Scholar] [CrossRef]
- Jonasdottir, S.H. Fatty acid profiles and production in marine phytoplankton. Mar. Drugs 2019, 17, 151. [Google Scholar] [CrossRef] [PubMed]
- Manan, S.; Ahmad, M.Z.; Zhang, G.; Chen, B.; Haq, B.U.; Yang, J.; Zhao, J. Soybean LEC2 regulates subsets of genes involved in controlling the biosynthesis and catabolism of seed storage substances and seed development. Front. Plant Sci. 2017, 8, 1604. [Google Scholar] [CrossRef] [PubMed]
- Li-Beisson, Y.; Beisson, F.; Riekhof, W. Metabolism of acyl-lipids in Chlamydomonas reinhardtii. Plant J. 2015, 82, 504–522. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Benning, C. Lipid metabolism in microalgae distinguishes itself. Curr. Opin. Biotechnol. 2013, 24, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Merchant, S.S.; Kropat, J.; Liu, B.; Shaw, J.; Warakanont, J. TAG, you’re it! Chlamydomonas as a reference organism for understanding algal triacylglycerol accumulation. Curr. Opin. Biotechnol. 2012, 23, 352–363. [Google Scholar]
- Lavell, A.A.; Benning, C. Cellular Organization and Regulation of Plant Glycerolipid Metabolism. Plant Cell Physiol. 2019, 60, 1176–1183. [Google Scholar] [CrossRef]
- Marchive, C.; Nikovics, K.; To, A.; Lepiniec, L.; Baud, S. Transcriptional regulation of fatty acid production in higher plants: Molecular bases and biotechnological outcomes. Eur. J. Lipid Sci. Technol. 2014, 116, 1332–1343. [Google Scholar] [CrossRef]
- Dehghan Nayeri, F.; Yarizade, K. Bioinformatics study of delta-12 fatty acid desaturase 2 (FAD2) gene in oilseeds. Mol. Biol. Rep. 2014, 41, 5077–5087. [Google Scholar] [CrossRef]
- Mitchell, A.G.; Martin, C.E. A novel cytochrome b5-like domain is linked to the carboxyl terminus of the Saccharomyces cerevisiae delta-9 fatty acid desaturase. J. Biol. Chem. 1995, 270, 29766–29772. [Google Scholar] [CrossRef]
- Thiyagarajan, S.; Arumugam, M.; Kathiresan, S. Identification and Functional Characterization of Two Novel Fatty Acid Genes from Marine Microalgae for Eicosapentaenoic Acid Production. Appl. Biochem. Biotechnol. 2020, 190, 1371–1384. [Google Scholar] [CrossRef]
- Li, N.; Xu, C.; Li-Beisson, Y.; Philippar, K. Fatty acid and lipid transport in plant cells. Trends Plant Sci. 2016, 21, 145–158. [Google Scholar] [CrossRef]
- Yu, S.Y.; Li, H.; Tong, M.; Ouyang, L.L.; Zhou, Z.G. Identification of a Δ6 fatty acid elongase gene for arachidonic acid biosynthesis localized to the endoplasmic reticulum in the green microalga Myrmecia incisa Reisigl. Gene 2012, 493, 219–227. [Google Scholar] [CrossRef]
- Tatzer, V.; Zellnig, G.; Kohlwein, S.D.; Schneiter, R. Lipid-dependent subcellular relocalization of the acyl chain desaturase in yeast. Mol. Biol. Cell 2002, 13, 4429–4442. [Google Scholar] [CrossRef]
- Wu, T.; Fu, Y.; Shi, Y.; Li, Y.; Kou, Y.; Mao, X.; Liu, J. Functional characterization of long-chain acyl-coa synthetase gene family from the oleaginous alga Chromochloris zofingiensis. J. Agric. Food Chem. 2020, 68, 4473–4484. [Google Scholar] [CrossRef]
- Li-Beisson, Y.; Thelen, J.J.; Fedosejevs, E.; Harwood, J.L. The lipid biochemistry of eukaryotic algae. Prog. Lipid Res. 2019, 74, 31–68. [Google Scholar] [CrossRef]
- Groot, P.H.; Scholte, H.R.; Hülsmann, W.C. Fatty acid activation: Specificity, localization and function. Adv. lipid Res. 1976, 14, 75–126. [Google Scholar]
- Domergue, F.; Abbadi, A.; Ott, C.; Zank, T.K.; Zähringer, U.; Heinz, E. Acyl carriers used as substrates by the desaturases and elongases involved in very long-chain polyunsaturated fatty acids biosynthesis reconstituted in yeast. J. Biol. Chem. 2003, 278, 35115–35126. [Google Scholar] [CrossRef]
- Fan, J.; Ning, K.; Zeng, X.; Luo, Y.; Wang, D.; Hu, J.; Li, J.; Xu, H.; Huang, J.; Wan, M.; et al. Genomic foundation of starch-to-lipid switch in oleaginous Chlorella spp. Plant Physiol. 2015, 169, 2444–2461. [Google Scholar] [CrossRef]
- Kannangara, R.; Branigan, C.; Liu, Y.; Penfield, T.; Rao, V.; Mouille, G.; Höfte, H.; Pauly, M.; Riechmann, J.L.; Broun, P. The transcription factor win1/shn1 regulates cutin biosynthesis in Arabidopsis thaliana. Plant Cell 2007, 19, 1278–1294. [Google Scholar] [CrossRef]
- Raffaele, S.; Vailleau, F.; Léger, A.; Joubès, J.; Miersch, O.; Huard, C.; Blée, E.; Mongrand, S.; Domergue, F.; Roby, D. A MYB transcription factor regulates very-long-chain fatty acid biosynthesis for activation of the hypersensitive cell death response in Arabidopsis. Plant Cell 2008, 20, 752–767. [Google Scholar] [CrossRef]
- Lee, S.B.; Suh, M.C. Cuticular wax biosynthesis is up-regulated by the MYB94 transcription factor in Arabidopsis. Plant Cell Physiol. 2015, 56, 48–60. [Google Scholar] [CrossRef]
- Li, N.; Zhang, Y.; Meng, H.; Li, S.; Wang, S.; Xiao, Z.; Chang, P.; Zhang, X.; Li, Q.; Guo, L.; et al. Characterization of fatty acid exporters involved in fatty acid transport for oil accumulation in the green alga Chlamydomonas reinhardtii. Biotechnol. Biofuels 2019, 12, 14. [Google Scholar] [CrossRef]
- Rehmanji, M.; Nesamma, A.A.; Khan, N.J.; Fatma, T.; Jutur, P.P. Media engineering in marine diatom Phaeodactylum tricornutum employing cost-effective substrates for sustainable production of high-value renewables. Biotechnol. J. 2022, 17, e2100684. [Google Scholar] [CrossRef]
- Kareya, M.S.; Mariam, I.; Shaikh, K.M.; Nesamma, A.A.; Jutur, P.P. Photosynthetic carbon partitioning and metabolic regulation in response to very-low and high CO2 in Microchloropsis gaditana NIES 2587. Front. Plant Sci. 2020, 11, 981. [Google Scholar] [CrossRef]
- Guerra, L.T.; Levitan, O.; Frada, M.; Sun, J.; Falkowski, P.; Dismukes, G. Regulatory branch points affecting protein and lipid biosynthesis in the diatom Phaeodactylum tricornutum. Biomass Bioenerg. 2013, 59, 306–315. [Google Scholar] [CrossRef]
- Shaikh, K.M.; Nesamma, A.A.; Abdin, M.Z.; Jutur, P.P. Molecular profiling of an oleaginous trebouxiophycean alga Parachlorella kessleri subjected to nutrient deprivation for enhanced biofuel production. Biotechnol. Biofuels 2019, 12, 182. [Google Scholar] [CrossRef]
- Singh, R.; Paliwal, C.; Nesamma, A.A.; Narula, A.; Jutur, P.P. Nutrient deprivation mobilizes the production of unique tocopherols as a stress-promoting response in a new indigenous isolate Monoraphidium sp. Front. Mar. Sci. 2020, 7, 575817. [Google Scholar] [CrossRef]
- Brown, M.R. The amino-acid and sugar composition of 16 species of microalgae used in mariculture. J. Exp. Mar. Biol. Ecol. 1991, 145, 79–99. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).