
Next-Generation Cancer Models for Drug Testing: Recent Advances in Immunocompetent Microphysiological Systems


Abstract
1. Introduction
2. Organ-on-a-Chip Models for Cancer Immunotherapy
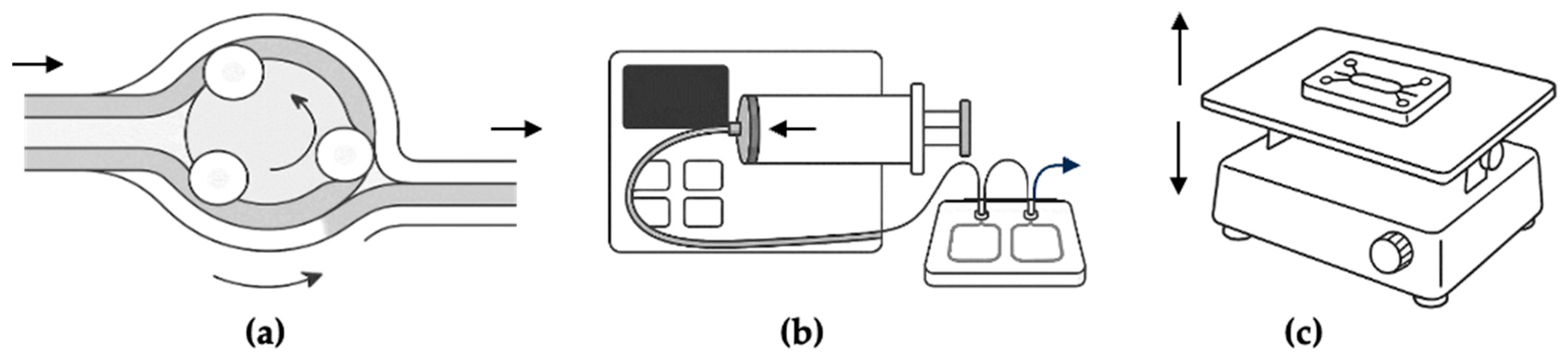
2.1. Technological Overview
2.2. Current Industrial Developments and Companies Commercializing MPSs in 2025
2.3. Application by Tumor Type
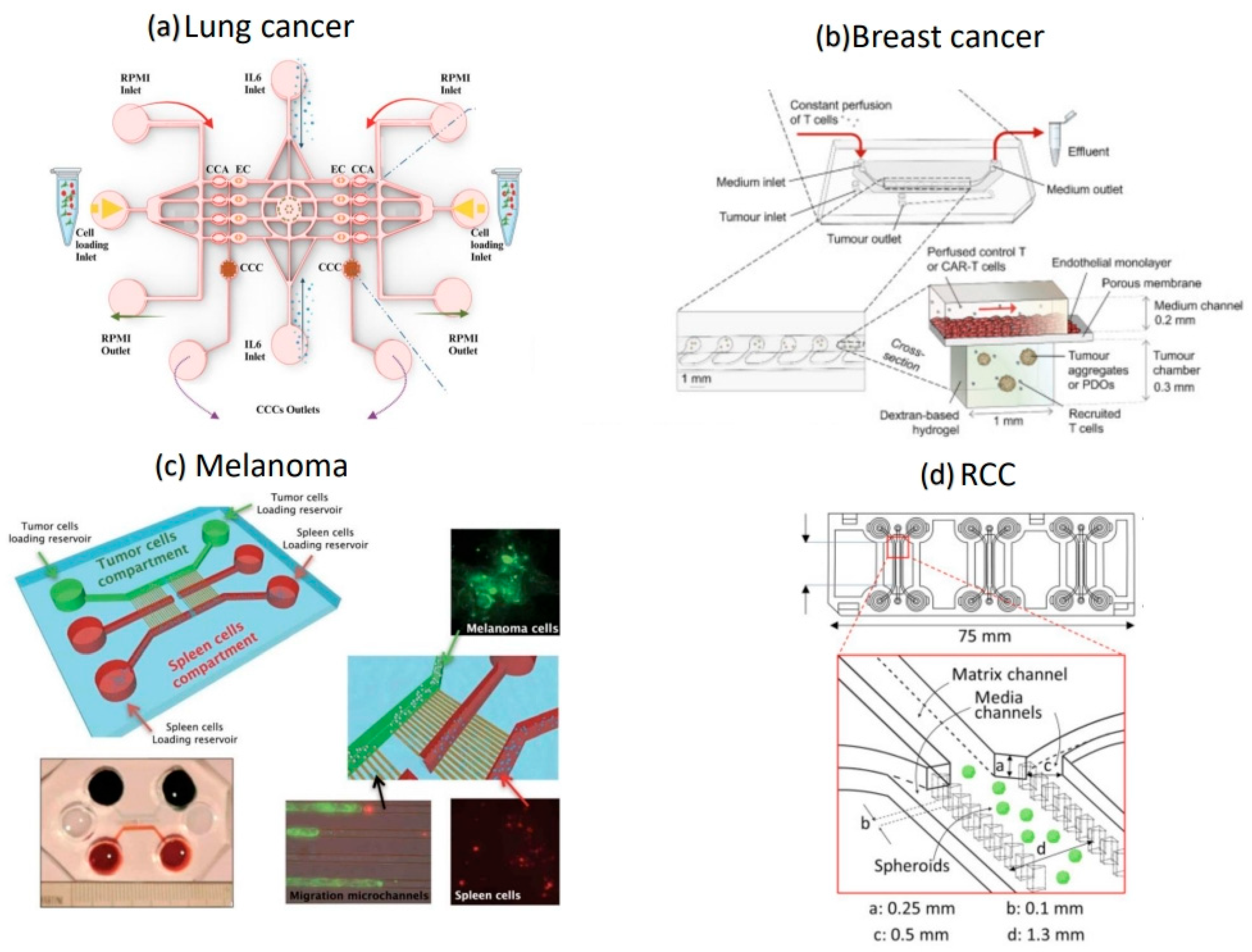
2.3.1. Lung Cancer
2.3.2. Breast Cancer
2.3.3. Melanoma
2.3.4. Renal Cell Carcinoma
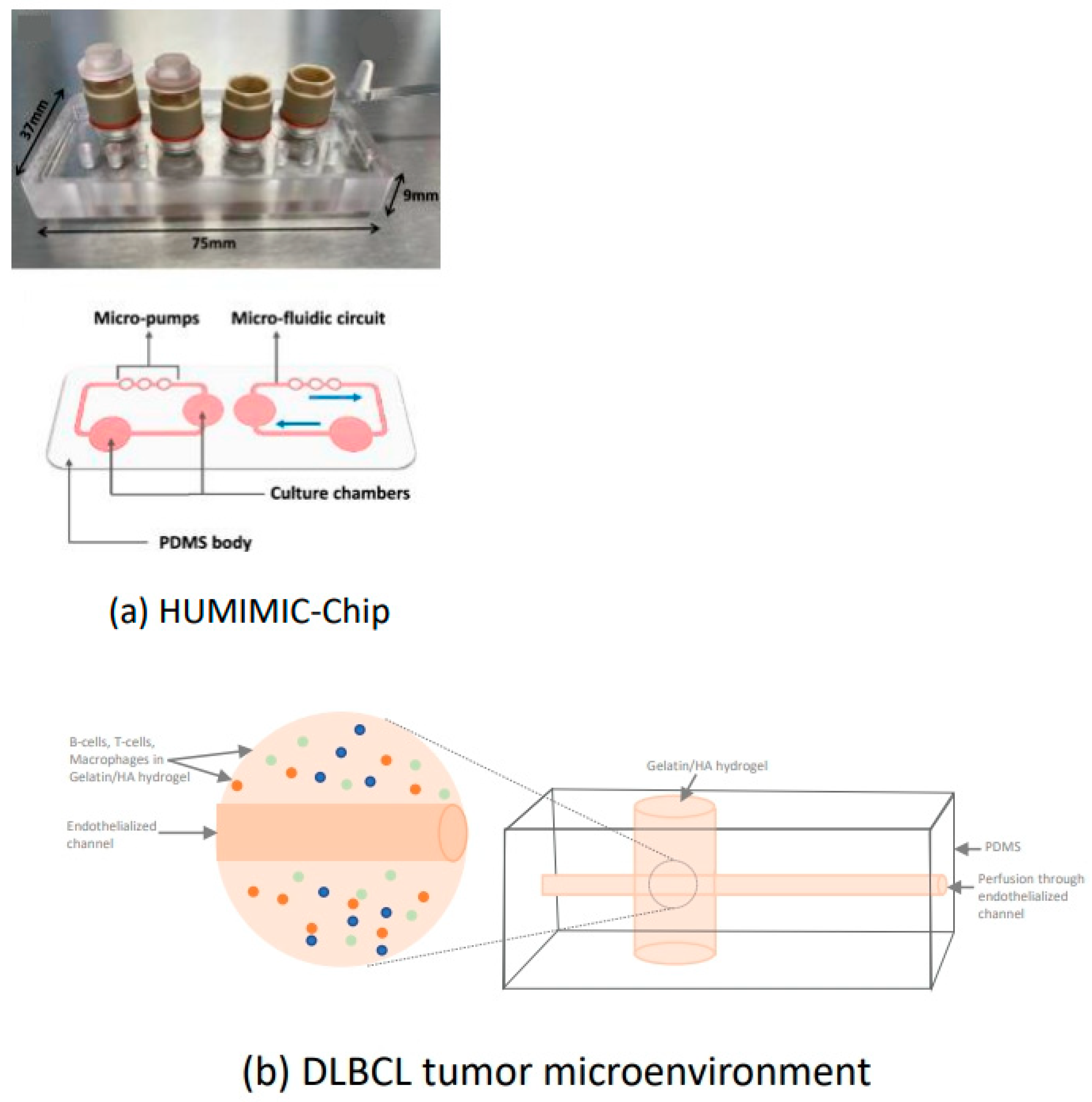
2.3.5. Diffuse Large B-Cell Lymphoma
2.3.6. Hepatocellular Carcinoma
2.3.7. Prostate Cancer
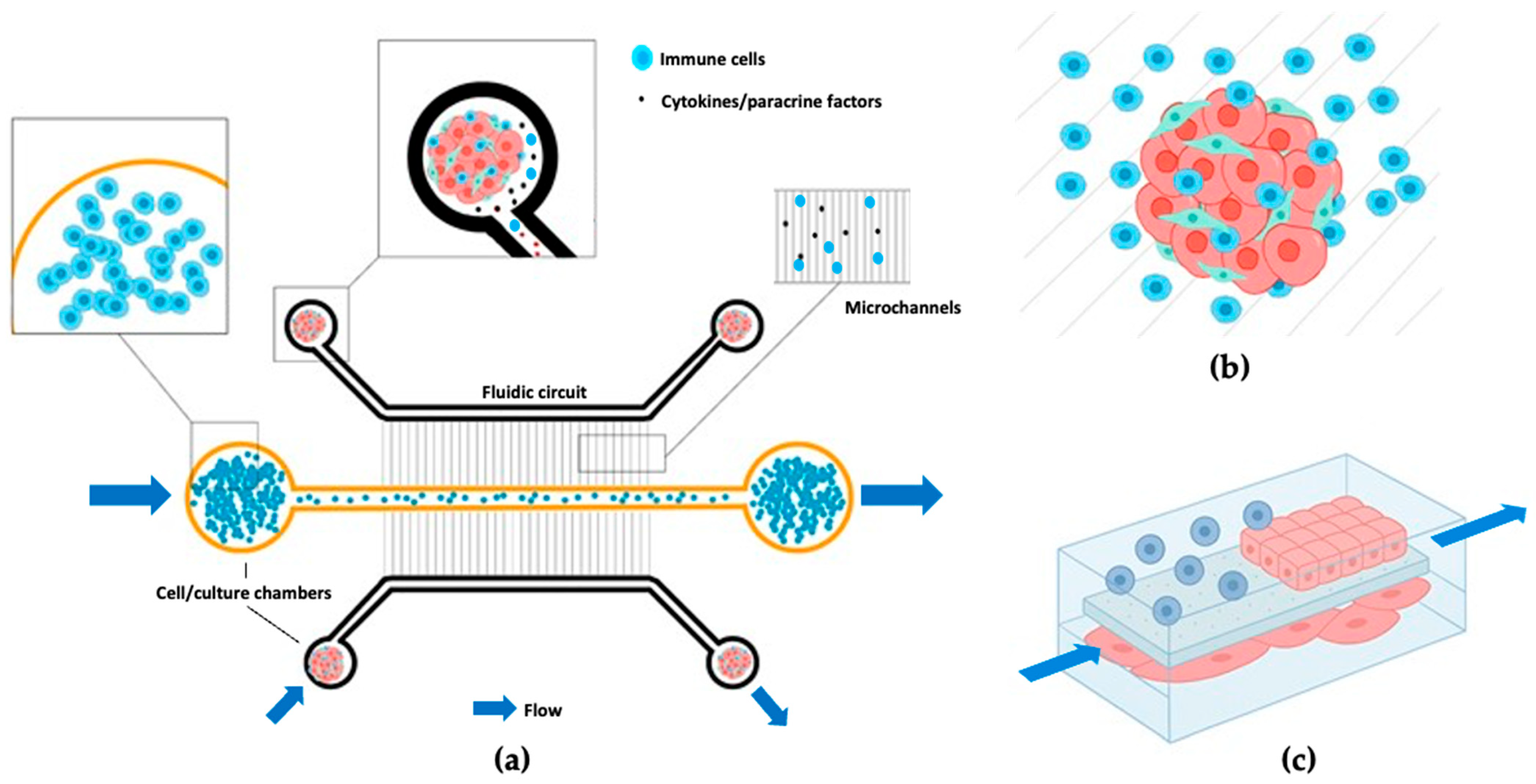
3. Key Experimental Approaches Across Models
3.1. Cell Sources and Cell Types
3.2. Immune Cell Integration Methods
3.3. Model Viability and Stability
3.4. Engineering Innovations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 27 June 2025).
- Sharma, P.; Allison, J.P. The Future of Immune Checkpoint Therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer Immunotherapy Using Checkpoint Blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Pardoll, D.M. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Oncology (Cancer)/Hematologic Malignancies Approval Notifications. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/oncology-cancerhematologic-malignancies-approval-notifications (accessed on 27 June 2025).
- Paul, J.; Mitchell, A.P.; Kesselheim, A.S.; Rome, B.N. Overlapping and Non-Overlapping Indications for Checkpoint Inhibitors in the US. J. Clin. Oncol. 2024, 42, 11057. [Google Scholar] [CrossRef]
- Tao, Z.; Chyra, Z.; Kotulová, J.; Celichowski, P.; Mihályová, J.; Charvátová, S.; Hájek, R. Impact of T Cell Characteristics on CAR-T Cell Therapy in Hematological Malignancies. Blood Cancer J. 2024, 14, 213. [Google Scholar] [CrossRef]
- Marchal, I. Cancer Vaccines: The Experts Speak. Nat. Biotechnol. 2025, 43, 477–481. [Google Scholar] [CrossRef]
- Park, J.S.; Withers, S.S.; Modiano, J.F.; Kent, M.S.; Chen, M.; Luna, J.I.; Culp, W.T.N.; Sparger, E.E.; Rebhun, R.B.; Monjazeb, A.M.; et al. Canine Cancer Immunotherapy Studies: Linking Mouse and Human. J. Immunother. Cancer 2016, 4, 97. [Google Scholar] [CrossRef]
- Mak, I.W.; Evaniew, N.; Ghert, M. Lost in Translation: Animal Models and Clinical Trials in Cancer Treatment. Am. J. Transl. Res. 2014, 6, 114–118. [Google Scholar]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T Cells in Cancer Immunosuppression—Implications for Anticancer Therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Fearon, D.T. T Cell Exclusion, Immune Privilege, and the Tumor Microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. Cancer Immunotherapy, Part 3: Challenges and Future Trends. Pharm. Ther. 2017, 42, 514–521. [Google Scholar]
- Mangani, S.; Kremmydas, S.; Karamanos, N.K. Mimicking the Complexity of Solid Tumors: How Spheroids Could Advance Cancer Preclinical Transformative Approaches. Cancers 2025, 17, 1161. [Google Scholar] [CrossRef]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, Ł.; Lamperska, K. 2D and 3D Cell Cultures—A Comparison of Different Types of Cancer Cell Cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef]
- Courau, T.; Bonnereau, J.; Chicoteau, J.; Bottois, H.; Remark, R.; Assante Miranda, L.; Toubert, A.; Blery, M.; Aparicio, T.; Allez, M.; et al. Cocultures of Human Colorectal Tumor Spheroids with Immune Cells Reveal the Therapeutic Potential of MICA/B and NKG2A Targeting for Cancer Treatment. J. Immunother. Cancer 2019, 7, 74. [Google Scholar] [CrossRef]
- Rodrigues, D.B.; Reis, R.L.; Pirraco, R.P. Modelling the Complex Nature of the Tumor Microenvironment: 3D Tumor Spheroids as an Evolving Tool. J. Biomed. Sci. 2024, 31, 13. [Google Scholar] [CrossRef]
- Hartung, T.; Smirnova, L. A Path Forward Advancing Microphysiological Systems. ALTEX 2025, 42, 183–203. [Google Scholar] [CrossRef]
- Bhatia, S.N.; Ingber, D.E. Microfluidic Organs-on-Chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef]
- Somova, M.; Simm, S.; Padmyastuti, A.; Ehrdardt, J.; Schoon, J.; Wolff, I.; Burchardt, M.; Roennau, C.; Pinto, P.C. Integrating Tumor and Healthy Epithelium in a Micro-Physiology Multi-Compartment Approach to Study Renal Cell Carcinoma Pathophysiology. Sci. Rep. 2024, 14, 9357. [Google Scholar] [CrossRef]
- Chernyavska, M.; Masoudnia, M.; Valerius, T.; Verdurmen, W.P.R. Organ-on-a-Chip Models for Development of Cancer Immunotherapies. Cancer Immunol. Immunother. 2023, 72, 3971–3983. [Google Scholar] [CrossRef] [PubMed]
- Avula, L.R.; Grodzinski, P. How Organ-on-a-Chip Is Advancing Cancer Research and Oncology—A Cancer Hallmarks’ Perspective. Front. Lab. A Chip Technol. 2024, 3, 1487377. [Google Scholar] [CrossRef]
- Leung, C.M.; de Haan, P.; Ronaldson-Bouchard, K.; Kim, G.-A.; Ko, J.; Rho, H.S.; Chen, Z.; Habibovic, P.; Jeon, N.L.; Takayama, S.; et al. A Guide to the Organ-on-a-Chip. Nat. Rev. Methods Primers 2022, 2, 33. [Google Scholar] [CrossRef]
- Khot, M.I.; Levenstein, M.A.; de Boer, G.N.; Armstrong, G.; Maisey, T.; Svavarsdottir, H.S.; Andrew, H.; Perry, S.L.; Kapur, N.; Jayne, D.G. Characterising a PDMS Based 3D Cell Culturing Microfluidic Platform for Screening Chemotherapeutic Drug Cytotoxic Activity. Sci. Rep. 2020, 10, 15915. [Google Scholar] [CrossRef]
- Banik, S.; Uchil, A.; Kalsang, T.; Chakrabarty, S.; Ali, M.A.; Srisungsitthisunti, P.; Mahato, K.K.; Surdo, S.; Mazumder, N. The Revolution of PDMS Microfluidics in Cellular Biology. Crit. Rev. Biotechnol. 2023, 43, 465–483. [Google Scholar] [CrossRef]
- Peng, X.; Janićijević, Ž.; Loureiro, L.R.; Hoffmann, L.; Lee, P.S.; Cela, I.; Kruppke, B.; Kegler, A.; Feldmann, A.; Bachmann, M.; et al. 3D Microphysiological Tumor Model for Dual-Targeting CAR T Cell Immunotherapy. BioRxiv 2024. [Google Scholar] [CrossRef]
- Seo, J.; Ha, G.; Lee, G.; Nasiri, R.; Lee, J. Modeling Tumor-Immune Interactions Using Hybrid Spheroids and Microfluidic Platforms for Studying Tumor-Associated Macrophage Polarization in Melanoma. Acta Biomater. 2024, 190, 233–246. [Google Scholar] [CrossRef]
- Miller, C.P.; Fung, M.; Jaeger-Ruckstuhl, C.A.; Xu, Y.; Warren, E.H.; Akilesh, S.; Tykodi, S.S. Therapeutic Targeting of Tumor Spheroids in a 3D Microphysiological Renal Cell Carcinoma-on-a-Chip System. Neoplasia 2023, 46, 100948. [Google Scholar] [CrossRef]
- Berger Fridman, I.; Kostas, J.; Gregus, M.; Ray, S.; Sullivan, M.R.; Ivanov, A.R.; Cohen, S.; Konry, T. High-Throughput Microfluidic 3D Biomimetic Model Enabling Quantitative Description of the Human Breast Tumor Microenvironment. Acta Biomater. 2021, 132, 473–488. [Google Scholar] [CrossRef]
- Foxall, R.; Narang, P.; Glaysher, B.; Hub, E.; Teal, E.; Coles, M.C.; Ashton-Key, M.; Beers, S.A.; Cragg, M.S. Developing a 3D B Cell Lymphoma Culture System to Model Antibody Therapy. Front. Immunol. 2021, 11, 605231. [Google Scholar] [CrossRef]
- Agliari, E.; Biselli, E.; De Ninno, A.; Schiavoni, G.; Gabriele, L.; Gerardino, A.; Mattei, F.; Barra, A.; Businaro, L. Cancer-Driven Dynamics of Immune Cells in a Microfluidic Environment. Sci. Rep. 2014, 4, 6639. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.I.; Davies, S.P.; Hewett, P.W.; Wilkinson, A.L.; Oo, Y.H.; Lu, W.Y.; El Haj, A.J.; Shetty, S. Organ-on-a-Chip for Studying Immune Cell Adhesion to Liver Sinusoidal Endothelial Cells: The Potential for Testing Immunotherapies and Cell Therapy Trafficking. Front. Cell Dev. Biol. 2024, 12, 1359451. [Google Scholar] [CrossRef] [PubMed]
- Wisdom, K.M.; Suijker, J.; van den Broek, L.; Sridharan, B.; Grandhi, T.S.P.; Cheng, A.; Lamb, M.; Titus, S.A.; Gehman, A.; Poore, D.; et al. Lung Tumor Microphysiological System with 3D Endothelium to Evaluate Modulators of T-Cell Infiltration. BioRxiv 2022. [Google Scholar] [CrossRef]
- Padmyastuti, A.; Sarmiento, M.G.; Dib, M.; Ehrhardt, J.; Schoon, J.; Somova, M.; Burchardt, M.; Roennau, C.; Pinto, P.C. Microfluidic-Based Prostate Cancer Model for Investigating the Secretion of Prostate-Specific Antigen and MicroRNAs In Vitro. Sci. Rep. 2023, 13, 11623. [Google Scholar] [CrossRef]
- Boussommier-Calleja, A.; Atiyas, Y.; Haase, K.; Headley, M.; Lewis, C.; Kamm, R.D. The Effects of Monocytes on Tumor Cell Extravasation in a 3D Vascularized Microfluidic Model. Biomaterials 2019, 198, 180–193. [Google Scholar] [CrossRef]
- Mannino, R.G.; Santiago-Miranda, A.N.; Pradhan, P.; Qiu, Y.; Mejias, J.C.; Neelapu, S.S.; Roy, K.; Lam, W.A. 3D Microvascular Model Recapitulates the Diffuse Large B-Cell Lymphoma Tumor Microenvironment in Vitro. Lab Chip 2017, 17, 407–414. [Google Scholar] [CrossRef]
- Georgescu, A.; Oved, J.H.; Galarraga, J.H.; Cantrell, T.; Mehta, S.; Dulmovits, B.M.; Olson, T.S.; Fattahi, P.; Wang, A.; Candarlioglu, P.L.; et al. Self-Organization of the Hematopoietic Vascular Niche and Emergent Innate Immunity on a Chip. Cell Stem Cell 2024, 31, 1847–1864.e6. [Google Scholar] [CrossRef]
- Xie, Y.; Ning, K.; Sun, W.; Feng, L.; Chen, Y.; Sun, W.; Li, Y.; Yu, L. A Pump-Free Microfluidic Co-Culture System for Investigating NK Cell-Tumor Spheroid Interactions in Flow Conditions. J. Biotechnol. 2025, 397, 11–21. [Google Scholar] [CrossRef]
- van Os, L.; Yeoh, J.; Witz, G.; Ferrari, D.; Krebs, P.; Chandorkar, Y.; Zeinali, S.; Sengupta, A.; Guenat, O.T. Immune Cell Extravasation in an Organ-on-Chip to Model Lung Inflammation. Eur. J. Pharm. Sci. 2023, 187, 106485. [Google Scholar] [CrossRef]
- Sardarabadi, P.; Lee, K.Y.; Sun, W.L.; Kojabad, A.A.; Liu, C.H. Investigating T Cell Immune Dynamics and IL-6’s Duality in a Microfluidic Lung Tumor Model. ACS Appl. Mater. Interfaces 2024, 17, 4354–4367. [Google Scholar] [CrossRef]
- Maulana, T.I.; Teufel, C.; Cipriano, M.; Roosz, J.; Lazarevski, L.; van den Hil, F.E.; Scheller, L.; Orlova, V.; Koch, A.; Hudecek, M.; et al. Breast Cancer-on-Chip for Patient-Specific Efficacy and Safety Testing of CAR-T Cells. Cell Stem Cell 2024, 31, 989–1002.e9. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Mehrotra, S.; Zare-Eelanjegh, E.; Rodrigues, R.O.; Akbarinejad, A.; Ge, D.; Amato, L.; Kiaee, K.; Fang, Y.; Rosenkranz, A.; et al. A Heart-Breast Cancer-on-a-Chip Platform for Disease Modeling and Monitoring of Cardiotoxicity Induced by Cancer Chemotherapy. Small 2021, 17, 2004258. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.I.; Jäger, J.; de Winde, C.M.; Roest, H.P.; van der Laan, L.J.W.; Gibbs, S.; Koning, J.J.; Mebius, R.E. Integration of Lymphatic Vasculature to a Human Lymph Node-on-Chip Enhances Physiological Immune Properties. BioRxiv 2025. [Google Scholar] [CrossRef]
- Ehlers, H.; Nicolas, A.; Schavemaker, F.; Heijmans, J.P.M.; Bulst, M.; Trietsch, S.J.; van den Broek, L.J. Vascular Inflammation on a Chip: A Scalable Platform for Trans-Endothelial Electrical Resistance and Immune Cell Migration. Front. Immunol. 2023, 14, 1118624. [Google Scholar] [CrossRef]
- Sasserath, T.; Rumsey, J.W.; McAleer, C.W.; Bridges, L.R.; Long, C.J.; Elbrecht, D.; Schuler, F.; Roth, A.; Bertinetti-LaPatki, C.; Shuler, M.L.; et al. Differential Monocyte Actuation in a Three-Organ Functional Innate Immune System-on-a-Chip. Adv. Sci. 2020, 7, 2000323. [Google Scholar] [CrossRef]
- Michielon, E.; Boninsegna, M.; Waaijman, T.; Fassini, D.; Spiekstra, S.W.; Cramer, J.; Gaudriault, P.; Kodolányi, J.; de Gruijl, T.D.; Homs-Corbera, A.; et al. Environmentally Controlled Microfluidic System Enabling Immune Cell Flow and Activation in an Endothelialised Skin-On-Chip. Adv. Healthc. Mater. 2024, 13, e2400750. [Google Scholar] [CrossRef]
- Roldan, N.; Rapet, A.; Stucki, A.O.; Raggi, G.; Fytianos, K.; Geiser, T.; Hobi, N.; Guenat, O.T. Acute Lung Injury On-Chip: Building a Disease Predictive Model That Emulates Alveolar Biomechanics. Toxicol. Lett. 2018, 295, S126–S127. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Zhu, L.; Zhang, J.; Guo, Q.; Kuang, J.; Li, D.; Wu, M.; Mo, Y.; Zhang, T.; Gao, X.; Tan, J. Advanced Lung Organoids and Lung-on-a-Chip for Cancer Research and Drug Evaluation: A Review. Front. Bioeng. Biotechnol. 2023, 11, 1299033. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer (IARC). Press Release 361: Breast Cancer Cases and Deaths Are Projected to Rise Globally (PR 361_E.pdf). February 2025. Available online: https://www.iarc.who.int/wp-content/uploads/2025/02/pr361_E.pdf (accessed on 27 June 2025).
- Subhan, M.A.; Parveen, F.; Shah, H.; Yalamarty, S.S.K.; Ataide, J.A.; Torchilin, V.P. Recent Advances with Precision Medicine Treatment for Breast Cancer Including Triple-Negative Sub-Type. Cancers 2023, 15, 2204. [Google Scholar] [CrossRef]
- Sledge, G.W.; Mamounas, E.P.; Hortobagyi, G.N.; Burstein, H.J.; Goodwin, P.J.; Wolff, A.C. Past, Present, and Future Challenges in Breast Cancer Treatment. J. Clin. Oncol. 2014, 32, 1979–1986. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-Y.; Yu, T.-J.; Shao, Z.-M. Precision Medicine for Breast Cancer: Advances and Challenges. Transl. Breast Cancer Res. 2024, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Mu, Q.; Fung, M.; Xu, X.; Zhu, L.; Ho, R.J.Y. Challenges and Opportunities in Metastatic Breast Cancer Treatments: Nano-Drug Combinations Delivered Preferentially to Metastatic Cells May Enhance Therapeutic Response. Pharmacol. Ther. 2022, 236, 108108. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Singh, D.; Laversanne, M.; Vignat, J.; Vaccarella, S.; Meheus, F.; Cust, A.E.; de Vries, E.; Whiteman, D.C.; Bray, F. Global Burden of Cutaneous Melanoma in 2020 and Projections to 2040. JAMA Dermatol. 2022, 158, 495. [Google Scholar] [CrossRef]
- Waseh, S.; Lee, J.B. Advances in Melanoma: Epidemiology, Diagnosis, and Prognosis. Front. Med. 2023, 10, 1268479. [Google Scholar] [CrossRef]
- Möller, L.; Stang, A.; Kajüter, H. Epidemiologie Des Nierenzellkarzinoms in Deutschland 2019. Die Onkol. 2023, 29, 565–570. [Google Scholar] [CrossRef]
- Fiebig, J.; Kraywinkel, K. Epidemiologie Des Nierenzellkarzinoms in Deutschland. Die Onkol. 2019, 25, 483–487. [Google Scholar] [CrossRef]
- Onkopedia. Nierenzellkarzinom (Hypernephrom). Available online: https://www.onkopedia.com/de/onkopedia/guidelines/nierenzellkarzinom-hypernephrom (accessed on 27 June 2025).
- Somova, M.; Grosse, M.; Schulze, F.; Burchardt, M.; Pinto, P.C. Microfluidic Co-Culture of Renal Healthy and Tumor Epithelium to Model Kidney Cancer Progression. J. Vis. Exp. 2025, 215, e67456. [Google Scholar] [CrossRef]
- Ernst, M.; Dührsen, U.; Hellwig, D.; Lenz, G.; Skoetz, N.; Borchmann, P. Diffuse Large B-Cell Lymphoma and Related Entities—Diagnosis, Treatment, and Follow-Up. Dtsch. Arztebl. Int. 2023, 120, 289. [Google Scholar] [CrossRef]
- Onkopedia. Diffuses Großzelliges B-Zell-Lymphom. Available online: https://www.onkopedia.com/de/onkopedia/guidelines/diffuses-grosszelliges-b-zell-lymphom/@@guideline/html/index.html (accessed on 27 June 2025).
- Motola-Kuba, D.; Zamora-Valdés, D.; Uribe, M.; Méndez-Sánchez, N. Hepatocellular Carcinoma. An Overview. Ann. Hepatol. 2006, 5, 16–24. [Google Scholar] [CrossRef]
- Waller, L.P. Hepatocellular Carcinoma: A Comprehensive Review. World J. Hepatol. 2015, 7, 2648. [Google Scholar] [CrossRef] [PubMed]
- Gabbia, D.; De Martin, S. Insights into Hepatocellular Carcinoma: From Pathophysiology to Novel Therapies. Int. J. Mol. Sci. 2024, 25, 4188. [Google Scholar] [CrossRef] [PubMed]
- Ghouri, Y.; Mian, I.; Rowe, J. Review of Hepatocellular Carcinoma: Epidemiology, Etiology, and Carcinogenesis. J. Carcinog. 2017, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Samant, H.; Amiri, H.S.; Zibari, G.B. Addressing the Worldwide Hepatocellular Carcinoma: Epidemiology, Prevention and Management. J. Gastrointest. Oncol. 2021, 12, S361–S373. [Google Scholar] [CrossRef]
- McGlynn, K.A.; Petrick, J.L.; El-Serag, H.B. Epidemiology of Hepatocellular Carcinoma. Hepatology 2021, 73, 4–13. [Google Scholar] [CrossRef]
- He, P.; Wan, H.; Wan, J.; Jiang, H.; Yang, Y.; Xie, K.; Wu, H. Systemic Therapies in Hepatocellular Carcinoma: Existing and Emerging Biomarkers for Treatment Response. Front. Oncol. 2022, 12, 1015527. [Google Scholar] [CrossRef]
- Tang, W.; Chen, Z.; Zhang, W.; Cheng, Y.; Zhang, B.; Wu, F.; Wang, Q.; Wang, S.; Rong, D.; Reiter, F.P.; et al. The Mechanisms of Sorafenib Resistance in Hepatocellular Carcinoma: Theoretical Basis and Therapeutic Aspects. Signal Transduct. Target. Ther. 2020, 5, 87. [Google Scholar] [CrossRef]
- Siegel, R.L.; Kratzer, T.B.; Giaquinto, A.N.; Sung, H.; Jemal, A. Cancer Statistics, 2025. CA Cancer J. Clin. 2025, 75, 10–45. [Google Scholar] [CrossRef]
- Wirth, M.F.M. Lokal Begrenztes Prostatakarzinom: Therapie. In Uroonkologie; Springer Medizin: Berlin/Heidelberg, Germany, 2022. [Google Scholar]
- Leitlinienprogramm Onkologie. S3-Leitlinie Prostatakarzinom–Langversion 7.0. Available online: https://www.leitlinienprogramm-onkologie.de/fileadmin/user_upload/Downloads/Leitlinien/Prostatatkarzinom/Version_7/LL_Prostatakarzinom_Langversion_7.0.pdf (accessed on 27 June 2025).
- Mateo, J.; de Bono, J.S.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Agarwal, N.; Olmos, D.; Thiery-Vuillemin, A.; et al. Olaparib for the Treatment of Patients with Metastatic Castration-Resistant Prostate Cancer and Alterations in BRCA1 and/or BRCA2 in the PROfound Trial. J. Clin. Oncol. 2024, 42, 571–583. [Google Scholar] [CrossRef]
- Rahbar, K.; Ahmadzadehfar, H.; Kratochwil, C.; Haberkorn, U.; Schäfers, M.; Essler, M.; Baum, R.P.; Kulkarni, H.R.; Schmidt, M.; Drzezga, A.; et al. German Multicenter Study Investigating 177Lu-PSMA-617 Radioligand Therapy in Advanced Prostate Cancer Patients. J. Nucl. Med. 2017, 58, 85–90. [Google Scholar] [CrossRef]
- Namekawa, T.; Ikeda, K.; Horie-Inoue, K.; Inoue, S. Application of Prostate Cancer Models for Preclinical Study: Advantages and Limitations of Cell Lines, Patient-Derived Xenografts, and Three-Dimensional Culture of Patient-Derived Cells. Cells 2019, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, P.; Luo, R.; Wang, Y.; Li, Z.; Guo, Y.; Yao, Y.; Li, M.; Tao, T.; Chen, W.; et al. Biomimetic Human Disease Model of SARS-CoV-2-Induced Lung Injury and Immune Responses on Organ Chip System. Adv. Sci. 2021, 8, 2002928. [Google Scholar] [CrossRef] [PubMed]
- Kromidas, E.; Geier, A.; Weghofer, A.; Liu, H.Y.; Weiss, M.; Loskill, P. Immunocompetent PDMS-Free Organ-on-Chip Model of Cervical Cancer Integrating Patient-Specific Cervical Fibroblasts and Neutrophils. Adv. Healthc. Mater. 2024, 13, e2302714. [Google Scholar] [CrossRef]
- Sung, K.E.; Beebe, D.J. Microfluidic 3D Models of Cancer. Adv. Drug Deliv. Rev. 2014, 79–80, 68–78. [Google Scholar] [CrossRef]
- Gaebler, D.; Hachey, S.J.; Hughes, C.C.W. Microphysiological Systems as Models for Immunologically ‘Cold’ Tumors. Front. Cell Dev. Biol. 2024, 12, 1389012. [Google Scholar] [CrossRef]
- Gil, J.F.; Moura, C.S.; Silverio, V.; Gonçalves, G.; Santos, H.A. Cancer Models on Chip: Paving the Way to Large-Scale Trial Applications. Adv. Mater. 2023, 35, e2300692. [Google Scholar] [CrossRef]
- Ayuso, J.M.; Rehman, S.; Virumbrales-Munoz, M.; McMinn, P.H.; Geiger, P.; Fitzgerald, C.; Heaster, T.; Skala, M.C.; Beebe, D.J. Microfluidic Tumor-on-a-Chip Model to Evaluate the Role of Tumor Environmental Stress on NK Cell Exhaustion. Sci. Adv. 2021, 7, eabc2331. [Google Scholar] [CrossRef]
- Liu, X.; Fang, J.; Huang, S.; Wu, X.; Xie, X.; Wang, J.; Liu, F.; Zhang, M.; Peng, Z.; Hu, N. Tumor-on-a-Chip: From Bioinspired Design to Biomedical Application. Microsyst. Nanoeng. 2021, 7, 50. [Google Scholar] [CrossRef]
- Boussommier-Calleja, A.; Li, R.; Chen, M.B.; Wong, S.C.; Kamm, R.D. Microfluidics: A New Tool for Modeling Cancer–Immune Interactions. Trends Cancer 2016, 2, 6–19. [Google Scholar] [CrossRef]
- Ajikumar, A.; Lei, K.F. Microfluidic Technologies in Advancing Cancer Research. Micromachines 2024, 15, 1444. [Google Scholar] [CrossRef]
- Morrison, A.I.; Sjoerds, M.J.; Vonk, L.A.; Gibbs, S.; Koning, J.J. In Vitro Immunity: An Overview of Immunocompetent Organ-on-Chip Models. Front. Immunol. 2024, 15, 1373186. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.O.; Rodriguez-Romera, A.; Reyat, J.S.; Olijnik, A.-A.; Colombo, M.; Wang, G.; Wen, W.X.; Sousos, N.; Murphy, L.C.; Grygielska, B.; et al. Human Bone Marrow Organoids for Disease Modeling, Discovery, and Validation of Therapeutic Targets in Hematologic Malignancies. Cancer Discov. 2023, 13, 364–385. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.; Xu, R.; Yi, G.; Li, Z.; Zhang, H.; Qi, S.; Huang, G. Patient-Derived Organoids in Human Cancer: A Platform for Fundamental Research and Precision Medicine. Mol. Biomed. 2024, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Magré, L.; Verstegen, M.M.A.; Buschow, S.; van der Laan, L.J.W.; Peppelenbosch, M.; Desai, J. Emerging Organoid-Immune Co-Culture Models for Cancer Research: From Oncoimmunology to Personalized Immunotherapies. J. Immunother. Cancer 2023, 11, e006290. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Company | Platform | Model Description | Ref. |
|---|---|---|---|
| TissUse | HUMIMIC Chip2 | Human lymph node-on-chip model with integrated lymphatic vasculature and recirculating human T- and dendritic cells, enabling the study of lymphatic–endothelial interactions, immune cell trafficking, and antigen-specific immune responses. | [45] |
| Mimetas | Organoplate | Platform using 64 perfused HUVEC tubules to model endothelial inflammation under exposure to cytokines and immune cells, capturing barrier disruption and morphological change for studying vascular inflammation. | [46] |
| Hesperos | 3-Organ system | Human multi-organ immune system-on-a-chip with recirculating monocytic cells simulating both targeted and systemic immune responses, enabling assessment of tissue-specific infiltration, cytokine profiles, and immune-mediated toxicity. | [47] |
| Cherry Biotech | Cubix | Multi-well microfluidic adaptor integrated with reconstructed human skin and flowing monocyte-like immune cells, enabling dynamic, skin-on-chip culture with control over gas and flow conditions, supporting immune activation studies. | [48] |
| Alveolix | AXLung-on-Chip System | Lung-on-chip model combining human pulmonary endothelial cells, circulating peripheral blood mononuclear cells, and mechanical breathing motion reveals that immune–endothelial interactions under lipopolysaccharide challenge significantly exacerbate inflammation and barrier disruption. | [49] |
| AIM Biotech | idenTx | RCC-on-a-chip platform recreating tumor spheroids in a collagen extracellular matrix, enabling assessment of drug responses and tumor cell migration, incorporating engineered human cytotoxic T lymphocytes to study antigen-specific immune-mediated tumor killing. | [30] |
| Strengths | Current Limitations |
|---|---|
| Physiologically relevant design: Enables simulation of tissue-specific architecture, flow conditions, and mechanical stress, better reflecting the tumor microenvironment [81,82,83]. | Restricted immune complexity: Most models only include one or two immune cell types, lacking full immune repertoire [82]. |
| Immune–tumor co-culture compatibility: Supports direct interaction between tumor cells and immune components (e.g., T cells, CAR-T, macrophages) [82]. | Short experimental lifespan: Co-cultures are typically viable for less than 10 days, limiting long-term studies [84]. |
| Real-time monitoring: Optical clarity and microfluidic control allow for live-cell imaging, migration tracking, and real-time immune response evaluation [83,85]. | Absence of systemic dynamics: Models lack full-body immune circulation or multiorgan crosstalk relevant in metastatic or immunomodulatory settings [82,86]. |
| Ethically favorable: Helps reduce reliance on animal testing, aligning with regulatory efforts toward human-relevant in vitro alternatives [81,83]. | Inconsistency in chip fabrication: Manual handling and variable gel properties can lead to differences in structure and flow patterns [85]. |
| Customizable and modular: Devices can be tailored for specific cancer types, immune interactions, or treatment scenarios [82,83]. | Standardization hurdles: Lack of harmonized protocols complicates cross-platform comparison and clinical translation [85,87]. |
| Useful for immunotherapy evaluation: Some platforms allow testing of immune checkpoint inhibitors, cytokine blockers, or engineered cell therapies [82]. | Immune cell adhesion and blockage: Immune cells can adhere to microchannel surfaces or become blocked in MPSs [86,87]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Große, M.; Burchardt, M.; Pinto, P.C. Next-Generation Cancer Models for Drug Testing: Recent Advances in Immunocompetent Microphysiological Systems. Future Pharmacol. 2025, 5, 36. https://doi.org/10.3390/futurepharmacol5030036
Große M, Burchardt M, Pinto PC. Next-Generation Cancer Models for Drug Testing: Recent Advances in Immunocompetent Microphysiological Systems. Future Pharmacology. 2025; 5(3):36. https://doi.org/10.3390/futurepharmacol5030036
Chicago/Turabian StyleGroße, Marlene, Martin Burchardt, and Pedro Caetano Pinto. 2025. "Next-Generation Cancer Models for Drug Testing: Recent Advances in Immunocompetent Microphysiological Systems" Future Pharmacology 5, no. 3: 36. https://doi.org/10.3390/futurepharmacol5030036
APA StyleGroße, M., Burchardt, M., & Pinto, P. C. (2025). Next-Generation Cancer Models for Drug Testing: Recent Advances in Immunocompetent Microphysiological Systems. Future Pharmacology, 5(3), 36. https://doi.org/10.3390/futurepharmacol5030036