
Therapeutic Potential of Mitotic Kinases’ Inhibitors in Cancers of the Gastrointestinal System

 ,
,  ,
, 
Abstract
1. Introduction
2. Gastric Cancer
2.1. Standard-of-Care Treatment of Gastric Cancer
2.2. General Overview of Mitotic Kinases in Gastric Cancer
2.2.1. Cyclin-Dependent Kinases (CDKs)
2.2.2. Aurora Kinases
2.2.3. Polo-Like Kinase 1
2.2.4. Wee1-Like Protein Kinase (WEE1)
2.3. Recent Mitotic Kinase Inhibitors as Therapeutic Interventions for Gastric Cancer
2.3.1. Aurora Kinases
2.3.2. Polo-Like Kinase
2.3.3. Wee1-Like Protein Kinase (WEE1)
2.3.4. Cyclin-Dependent Kinases (CDKs)
3. Liver Cancer
3.1. Standard-of-Care Treatment of Liver Cancer
3.2. General Overview of Mitotic Kinases in Liver Cancer
3.2.1. CDK1
3.2.2. Aurora Kinases
3.2.3. PLK1
3.3. Recent Mitotic Kinase Inhibitors as Therapeutic Interventions for Liver Cancer
3.3.1. CDK1
3.3.2. Aurora Kinase
3.3.3. PLK1
4. Pancreatic Cancer
4.1. Standard-of-Care Treatment of Pancreatic Cancer
4.2. General Overview of Mitotic Kinases in Pancreatic Cancer
4.2.1. Aurora Kinases
4.2.2. Cyclin-Dependent Kinases (CDKs)
4.2.3. Wee1
4.3. Recent Mitotic Kinase Inhibitors as Therapeutic Interventions for Pancreatic Cancer
4.3.1. Aurora A
4.3.2. Cyclin-Dependent Kinases (CDKs)
4.3.3. WEE1
4.3.4. TTK
5. Colorectal Cancer
5.1. Standard-of-Care Treatment of Colorectal Cancer
5.2. General Overview of Mitotic Kinases Inhibitors in Colorectal Cancer
5.2.1. CDKs
5.2.2. Aurora A
5.2.3. Mps1
5.3. Recent Mitotic Kinase Inhibitors as Therapeutic Interventions for Colorectal Cancer
5.3.1. PLK1
5.3.2. Aurora Kinases
5.3.3. CDKs
5.3.4. Mps1
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kapoor, T.M.; Compton, D.A. Searching for the middle ground: Mechanisms of chromosome alignment during mitosis. J. Cell Biol. 2002, 157, 551–556. [Google Scholar] [CrossRef]
- Pines, J. Mitosis: A matter of getting rid of the right protein at the right time. Trends Cell Biol. 2006, 16, 55–63. [Google Scholar] [CrossRef]
- Walczak, C.E.; Cai, S.; Khodjakov, A. Mechanisms of chromosome behaviour during mitosis. Nat. Rev. Mol. Cell Biol. 2010, 11, 91–102. [Google Scholar] [CrossRef]
- Vermeulen, K.; Van Bockstaele, D.R.; Berneman, Z.N. The cell cycle: A review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003, 36, 131–149. [Google Scholar] [CrossRef]
- Ovejero, S.; Bueno, A.; Sacristán, M.P. Working on Genomic Stability: From the S-Phase to Mitosis. Genes 2020, 11, 225. [Google Scholar] [CrossRef]
- Manchado, E.; Guillamot, M.; Malumbres, M. Killing cells by targeting mitosis. Cell Death Differ. 2012, 19, 369–377. [Google Scholar] [CrossRef]
- Chan, K.-S.; Koh, C.-G.; Li, H.-Y. Mitosis-targeted anti-cancer therapies: Where they stand. Cell Death Dis. 2012, 3, e411. [Google Scholar] [CrossRef] [PubMed]
- De Wolf, B.; Kops, G.J.P.L.; Kops, G.J.P.L. Cell Division Machinery and Disease. In Advances in Experimental Medicine and Biology; Springer: Berlin, Germany, 2017. [Google Scholar]
- Goga, A.; Yang, D.; Tward, A.D.; Morgan, D.O.; Bishop, J.M. Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nat. Med. 2007, 13, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Ying, X.; Che, X.; Wang, J.; Zou, G.; Yu, Q.; Zhang, X. CDK1 serves as a novel therapeutic target for endometrioid endometrial cancer. J. Cancer 2021, 12, 2206–2215. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Jiang, Q.; Zhang, C. The role of mitotic kinases in coupling the centrosome cycle with the assembly of the mitotic spindle. J. Cell Sci. 2014, 127, 4111–4122. [Google Scholar] [CrossRef]
- D’Assoro, A.B.; Ehaddad, T.; Egalanis, E. Aurora-A Kinase as a Promising Therapeutic Target in Cancer. Front. Oncol. 2016, 5, 295. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J. The Aurora kinase inhibitors in cancer research and therapy. J. Cancer Res. Clin. Oncol. 2016, 142, 1995–2012. [Google Scholar] [CrossRef] [PubMed]
- Lens, S.M.A.; Medema, R.H. Cytokinesis defects and cancer. Nat. Cancer 2018, 19, 32–45. [Google Scholar] [CrossRef]
- Ma, H.T.; Poon, R.Y. Aurora kinases and DNA damage response. Mutat. Res. Mol. Mech. Mutagen. 2020, 821, 111716. [Google Scholar] [CrossRef]
- Passaro, C.; Chieffi, P. Aurora B: A New Prognostic Marker and Therapeutic Target in Cancer. Curr. Med. Chem. 2011, 18, 482–496. [Google Scholar] [CrossRef]
- Honma, K.; Nakanishi, R.; Nakanoko, T.; Ando, K.; Saeki, H.; Oki, E.; Iimori, M.; Kitao, H.; Kakeji, Y.; Maehara, Y. Contribution of Aurora-A and -B expression to DNA aneuploidy in gastric cancers. Surg. Today 2013, 44, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Sun, Q.; Wang, X. PLK1, A Potential Target for Cancer Therapy. Transl. Oncol. 2016, 10, 22–32. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, G.; Kong, C. FOXM1 participates in PLK1-regulated cell cycle progression in renal cell cancer cells. Oncol. Lett. 2016, 11, 2685–2691. [Google Scholar] [CrossRef]
- Jung, Y.; Kraikivski, P.; Shafiekhani, S.; Terhune, S.S.; Dash, R.K. Crosstalk between Plk1, p53, cell cycle, and G2/M DNA damage checkpoint regulation in cancer: Computational modeling and analysis. NPJ Syst. Biol. Appl. 2021, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.-R.; Shin, S.-B.; Kim, C.-H.; Won, J.-Y.; Xu, R.; Kim, D.-E.; Yim, H. PLK1/vimentin signaling facilitates immune escape by recruiting Smad2/3 to PD-L1 promoter in metastatic lung adenocarcinoma. Cell Death Differ. 2021, 28, 2745–2764. [Google Scholar] [CrossRef] [PubMed]
- Pei, G.; Luo, M.; Ni, X.; Wu, J.; Wang, S.; Ma, Y.; Yu, J. Autophagy Facilitates Metadherin-Induced Chemotherapy Resistance Through the AMPK/ATG5 Pathway in Gastric Cancer. Cell. Physiol. Biochem. 2018, 46, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.S.; Özgüroğlu, M.; Bang, Y.-J.; Di Bartolomeo, M.; Mandalà, M.; Ryu, M.-H.; Fornaro, L.; Olesinski, T.; Caglevic, C.; Chung, H.C.; et al. Pembrolizumab versus paclitaxel for previously treated patients with PD-L1–positive advanced gastric or gastroesophageal junction cancer (GC): Update from the phase III KEYNOTE-061 trial. J. Clin. Oncol. 2020, 38 (Suppl. 15), 4503. [Google Scholar] [CrossRef]
- Parisi, A.; Reim, D.; Borghi, F.; Nguyen, N.T.; Qi, F.; Coratti, A.; Cianchi, F.; Cesari, M. Minimally invasive surgery for gastric cancer: A comparison between robotic, laparoscopic and open surgery. World J. Gastroenterol. 2017, 23, 2376–2384. [Google Scholar] [CrossRef]
- Ychou, M.; Boige, V.; Pignon, J.-P.; Conroy, T.; Bouché, O.; Lebreton, G.; Ducourtieux, M.; Bedenne, L.; Fabre, J.-M.; Saint-Aubert, B.; et al. Perioperative Chemotherapy Compared with Surgery Alone for Resectable Gastroesophageal Adenocarcinoma: An FNCLCC and FFCD Multicenter Phase III Trial. J. Clin. Oncol. 2011, 29, 1715–1721. [Google Scholar] [CrossRef] [PubMed]
- Ende, T.V.D.; ter Veer, E.; Machiels, M.; Mali, R.M.A.; Nijenhuis, F.A.A.; de Waal, L.; Laarman, M.; Gisbertz, S.S.; Hulshof, M.C.C.M.; van Oijen, M.G.H.; et al. The Efficacy and Safety of (Neo)Adjuvant Therapy for Gastric Cancer: A Network Meta-analysis. Cancers 2019, 11, 80. [Google Scholar] [CrossRef]
- Tsai, C.; Mueller, A.; Maubach, J.; Warschkow, R.; Nussbaum, D.P.; Schmied, B.M.; Blazer, D.; Gloor, B.; Worni, M. No Difference in Survival between Neo-Adjuvant Chemotherapy and Neo-Adjuvant Chemoradiation Therapy in Gastric Cardia Cancer Patients: A Contemporary View from the National Cancer Database. Dig. Surg. 2019, 37, 249–257. [Google Scholar] [CrossRef]
- De Vita, F.; Borg, C.; Farina, G.; Geva, R.; Carton, I.; Cuku, H.; Wei, R.; Muro, K. Ramucirumab and paclitaxel in patients with gastric cancer and prior trastuzumab: Subgroup analysis from RAINBOW study. Future Oncol. 2019, 15, 2723–2731. [Google Scholar] [CrossRef]
- Tang, Z.; Li, L.; Tang, Y.; Xie, D.; Wu, K.; Wei, W.; Xiao, Q. CDK 2 positively regulates aerobic glycolysis by suppressing SIRT 5 in gastric cancer. Cancer Sci. 2018, 109, 2590–2598. [Google Scholar] [CrossRef]
- Kamran, M.; Long, Z.-J.; Xu, D.; Lv, S.-S.; Liu, B.; Wang, C.-L.; Xu, J.; Lam, E.; Liu, Q. Aurora kinase A regulates Survivin stability through targeting FBXL7 in gastric cancer drug resistance and prognosis. Oncogenesis 2017, 6, e298. [Google Scholar] [CrossRef]
- Ding, L.; Yang, L.; He, Y.; Zhu, B.; Ren, F.; Fan, X.; Wang, Y.; Li, M.; Li, J.; Kuang, Y.; et al. CREPT/RPRD1B associates with Aurora B to regulate Cyclin B1 expression for accelerating the G2/M transition in gastric cancer. Cell Death Dis. 2018, 9, 1172. [Google Scholar] [CrossRef] [PubMed]
- Wakida, T.; Ikura, M.; Kuriya, K.; Ito, S.; Shiroiwa, Y.; Habu, T.; Kawamoto, T.; Okumura, K.; Ikura, T.; Furuya, K. The CDK-PLK1 axis targets the DNA damage checkpoint sensor protein RAD9 to promote cell proliferation and tolerance to genotoxic stress. eLife 2017, 6, e29953. [Google Scholar] [CrossRef]
- Dang, S.-C.; Wang, F.; Qian, X.-B.; Abdul, M.; Naseer, Q.-A.; Jin, W.; Hu, R.; Gu, Q.; Gu, M. MicroRNA-505 suppresses gastric cancer cell proliferation and invasion by directly targeting Polo-like kinase-1. OncoTargets Ther. 2019, 12, 795–803. [Google Scholar] [CrossRef]
- Dang, S.-C.; Fan, Y.-Y.; Cui, L.; Chen, J.-X.; Qu, J.-G.; Gu, M. PLK1 as a potential prognostic marker of gastric cancer through MEK-ERK pathway on PDTX models. OncoTargets Ther. 2018, 11, 6239–6247. [Google Scholar] [CrossRef]
- Cai, X.P.; Chen, L.D.; Bin Song, H.; Zhang, C.X.; Yuan, Z.W.; Xiang, Z.X. PLK1 promotes epithelial-mesenchymal transition and metastasis of gastric carcinoma cells. Am. J. Transl. Res. 2016, 8, 4172–4183. [Google Scholar]
- Lin, X.; Chen, D.; Zhang, C.; Zhang, X.; Li, Z.; Dong, B.; Gao, J.; Shen, L. Augmented antitumor activity by olaparib plus AZD1775 in gastric cancer through disrupting DNA damage repair pathways and DNA damage checkpoint. J. Exp. Clin. Cancer Res. 2018, 37, 129. [Google Scholar] [CrossRef]
- Kim, H.-Y.; Cho, Y.; Kang, H.; Yim, Y.-S.; Kim, S.-J.; Song, J.; Chun, K.-H. Targeting the WEE1 kinase as a molecular targeted therapy for gastric cancer. Oncotarget 2016, 7, 49902–49916. [Google Scholar] [CrossRef]
- Yuan, C.X.; Zhou, Z.W.; Yang, Y.X.; He, Z.X.; Zhang, X.; Wang, D.; Yang, T.; Pan, S.Y.; Chen, X.W.; Zhou, S.F. Danusertib, a potent pan-Aurora kinase and ABL kinase inhibitor, induces cell cycle arrest and programmed cell death and inhibits epithelial to mesenchymal transition involving the PI3K/Akt/mTOR-mediated signaling pathway in human gastric cancer AGS and NCI-N78 cells. Drug Des. Dev. Ther. 2015, 9, 1293–1318. [Google Scholar] [CrossRef]
- Wang-Bishop, L.; Chen, Z.; Gomaa, A.; Lockhart, A.C.; Salaria, S.; Wang, J.; Lewis, K.B.; Ecsedy, J.; Washington, K.; Beauchamp, R.D.; et al. Inhibition of AURKA Reduces Proliferation and Survival of Gastrointestinal Cancer Cells with Activated KRAS by Preventing Activation of RPS6KB1. Gastroenterology 2019, 156, 662–675.e7. [Google Scholar] [CrossRef]
- Nokihara, H.; Yamada, Y.; Fujiwara, Y.; Yamamoto, N.; Wakui, H.; Nakamichi, S.; Kitazono, S.; Inoue, K.; Harada, A.; Taube, T.; et al. Phase I trial of volasertib, a Polo-like kinase inhibitor, in Japanese patients with advanced solid tumors. Investig. New Drugs 2015, 34, 66–74. [Google Scholar] [CrossRef]
- Liu, H.; Shin, S.H.; Chen, H.; Liu, T.; Li, Z.; Hu, Y.; Liu, F.; Zhang, C.; Kim, D.J.; Liu, K.; et al. CDK12 and PAK2 as novel therapeutic targets for human gastric cancer. Theranostics 2020, 10, 6201–6215. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2016, 2, 16018. [Google Scholar] [CrossRef]
- Caines, A.; Selim, R.; Salgia, R. The Changing Global Epidemiology of Hepatocellular Carcinoma. Clin. Liver Dis. 2020, 24, 535–547. [Google Scholar] [CrossRef]
- Marcon, P.D.S.; Tovo, C.V.; Kliemann, D.A.; Fisch, P.; De Mattos, A.A. Incidence of hepatocellular carcinoma in patients with chronic liver disease due to hepatitis B or C and coinfected with the human immunodeficiency virus: A retrospective cohort study. World J. Gastroenterol. 2018, 24, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, Y.; Kawamura, Y.; Kobayashi, M.; Shindoh, J.; Kobayashi, Y.; Okubo, S.; Muraishi, N.; Kajiwara, A.; Iritani, S.; Fujiyama, S.; et al. Effects of alcohol consumption on multiple hepatocarcinogenesis in patients with fatty liver disease. Hepatol. Res. 2020, 51, 62–68. [Google Scholar] [CrossRef]
- Ayuso, C.; Rimola, J.; Vilana, R.; Burrel, M.; Darnell, A.; García-Criado, Á.; Bianchi, L.; Belmonte, E.; Caparroz, C.; Barrufet, M.; et al. Diagnosis and staging of hepatocellular carcinoma (HCC): Current guidelines. Eur. J. Radiol. 2018, 101, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Cervantes, A.; Chau, I.; Daniele, B.; Llovet, J.M.; Meyer, T.; Nault, J.-C.; Neumann, U.; Ricke, J.; Sangro, B.; et al. Hepatocellular carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv238–iv255. [Google Scholar] [CrossRef]
- Bruix, J.; Chan, S.L.; Galle, P.R.; Rimassa, L.; Sangro, B. Systemic treatment of hepatocellular carcinoma: An EASL position paper. J. Hepatol. 2021, 75, 960–974. [Google Scholar] [CrossRef]
- Lee, G.C.; Ferrone, C.R.; Vagefi, P.A.; Uppot, R.N.; Tanabe, K.K.; Lillemoe, K.D.; Blaszkowsky, L.S.; Qadan, M. Surgical resection versus ablation for early-stage hepatocellular carcinoma: A retrospective cohort analysis. Am. J. Surg. 2019, 218, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Dean, D.C.; Yu, Z.; Duan, Z. Role of cyclin-dependent kinases (CDKs) in hepatocellular carcinoma: Therapeutic potential of targeting the CDK signaling pathway. Hepatol. Res. 2019, 49, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Li, K.K.W.; Ng, I.O.-L.; Fan, S.T.; Albrecht, J.H.; Yamashita, K.; Poon, R.Y.C. Activation of cyclin-dependent kinases CDC2 and CDK2 in hepatocellular carcinoma. Liver Int. 2002, 22, 259–268. [Google Scholar] [CrossRef]
- Zhao, J.; Han, S.-X.; Ma, J.-L.; Ying, X.; Liu, P.; Li, J.; Wang, L.; Zhang, Y.; Ma, J.; Zhang, L.; et al. The role of CDK1 in apoptin-induced apoptosis in hepatocellular carcinoma cells. Oncol. Rep. 2013, 30, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.B.; Kotturi, H.; Waris, G.; Mohammed, A.; Chandrakesan, P.; May, R.; Sureban, S.; Weygant, N.; Qu, D.; Rao, C.V.; et al. (Z)-3,5,4′-Trimethoxystilbene Limits Hepatitis C and Cancer Pathophysiology by Blocking Microtubule Dynamics and Cell-Cycle Progression. Cancer Res. 2016, 76, 4887–4896. [Google Scholar] [CrossRef]
- He, B.; Yin, J.; Gong, S.; Gu, J.; Xiao, J.; Shi, W.; Ding, W.; He, Y. Bioinformatics analysis of key genes and pathways for hepatocellular carcinoma transformed from cirrhosis. Medicine 2017, 96, e6938. [Google Scholar] [CrossRef]
- Jung, E.U.; Yoon, J.-H.; Lee, Y.-J.; Lee, J.-H.; Kim, B.H.; Yu, S.J.; Myung, S.J.; Kim, Y.J.; Lee, H.-S. Hypoxia and retinoic acid-inducible NDRG1 expression is responsible for doxorubicin and retinoic acid resistance in hepatocellular carcinoma cells. Cancer Lett. 2010, 298, 9–15. [Google Scholar] [CrossRef]
- Shah, M.A.; Kortmansky, J.; Motwani, M.; Drobnjak, M.; Gonen, M.; Yi, S.; Weyerbacher, A.; Cordon-Cardo, C.; Lefkowitz, R.; Brenner, B.; et al. A Phase I Clinical Trial of the Sequential Combination of Irinotecan Followed by Flavopiridol. Clin. Cancer Res. 2005, 11, 3836–3845. [Google Scholar] [CrossRef] [PubMed]
- Richard, C.; Matthews, D.; Duivenvoorden, W.; Yau, J.; Wright, P.S.; Th’Ng, J.P. Flavopiridol Sensitivity of Cancer Cells Isolated from Ascites and Pleural Fluids. Clin. Cancer Res. 2005, 11, 3523–3529. [Google Scholar] [CrossRef][Green Version]
- Zou, Y.; Ruan, S.; Jin, L.; Chen, Z.; Han, H.; Zhang, Y.; Jian, Z.; Lin, Y.; Shi, N.; Jin, H. CDK1, CCNB1, and CCNB2 are Prognostic Biomarkers and Correlated with Immune Infiltration in Hepatocellular Carcinoma. Med. Sci. Monit. 2020, 26, e925289-1–e925289-14. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.-X.; Pan, Y.-Y.; You, C.-G. CDK1, CCNB1, CDC20, BUB1, MAD2L1, MCM3, BUB1B, MCM2, and RFC4 May Be Potential Therapeutic Targets for Hepatocellular Carcinoma Using Integrated Bioinformatic Analysis. Biomed Res. Int. 2019, 2019, 1245072. [Google Scholar] [CrossRef]
- Li, L.; Huang, K.; Zhao, H.; Chen, B.; Ye, Q.; Yue, J. CDK1-PLK1/SGOL2/ANLN pathway mediating abnormal cell division in cell cycle may be a critical process in hepatocellular carcinoma. Cell Cycle 2020, 19, 1236–1252. [Google Scholar] [CrossRef]
- Sun, S.; Wang, W.; Luo, X.; Li, Y.; Liu, B.; Li, X.; Zhang, B.; Han, S.; Li, X. Circular RNA circ-ADD3 inhibits hepatocellular carcinoma metastasis through facilitating EZH2 degradation via CDK1-mediated ubiquitination. Am. J. Cancer Res. 2019, 9, 1695–1707. Available online: http://www.ncbi.nlm.nih.gov/pubmed/31497351%0Ahttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC6726993 (accessed on 20 March 2022).
- Jin, J.; Xu, H.; Li, W.; Xu, X.; Liu, H.; Wei, F. LINC00346 Acts as a Competing Endogenous RNA Regulating Development of Hepatocellular Carcinoma via Modulating CDK1/CCNB1 Axis. Front. Bioeng. Biotechnol. 2020, 8, 54. [Google Scholar] [CrossRef]
- Nikonova, A.S.; Astsaturov, I.; Serebriiskii, I.G.; Dunbrack, R.L., Jr.; Golemis, E.A. Aurora A kinase (AURKA) in normal and pathological cell division. Cell. Mol. Life Sci. 2013, 70, 661–687. [Google Scholar] [CrossRef]
- Chen, C.; Song, G.; Xiang, J.; Zhang, H.; Zhao, S.; Zhan, Y. AURKA promotes cancer metastasis by regulating epithelial-mesenchymal transition and cancer stem cell properties in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2017, 486, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.L.; Yu, H.; Mu, S.-M.; Dong, Y.D.; Li, D.Y. MiR-26a-5p Inhibits Cell Proliferation and Enhances Doxorubicin Sensitivity in HCC Cells via Targeting AURKA. Technol. Cancer Res. Treat. 2019, 18, 1533033819851833. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Hsu, C.-J.; Chou, C.-H.; Lee, H.-L.; Chiang, W.-L.; Su, C.-M.; Tsai, H.-C.; Yang, S.-F.; Tang, C.-H. Variations in the AURKA Gene: Biomarkers for the Development and Progression of Hepatocellular Carcinoma. Int. J. Med. Sci. 2018, 15, 170–175. [Google Scholar] [CrossRef]
- Wu, M.; Zhou, Y.; Fei, C.; Chen, T.; Yin, X.; Zhang, L.; Ren, Z. ID1 overexpression promotes HCC progression by amplifying the AURKA/Myc signaling pathway. Int. J. Oncol. 2020, 57, 845–857. [Google Scholar] [CrossRef]
- Lu, L.; Han, H.; Tian, Y.; Li, W.; Zhang, J.; Feng, M.; Li, Y. Aurora kinase A mediates c-Myc’s oncogenic effects in hepatocellular carcinoma. Mol. Carcinog. 2014, 54, 1467–1479. [Google Scholar] [CrossRef]
- Lin, H.; Huang, Y.-S.; Fustin, J.-M.; Doi, M.; Chen, H.; Lai, H.-H.; Lin, S.-H.; Lee, Y.-L.; King, P.-C.; Hou, H.-S.; et al. Hyperpolyploidization of hepatocyte initiates preneoplastic lesion formation in the liver. Nat. Commun. 2021, 12, 645. [Google Scholar] [CrossRef]
- Yasen, M.; Mizushima, H.; Mogushi, K.; Obulhasim, G.; Miyaguchi, K.; Inoue, K.; Nakahara, I.; Ohta, T.; Aihara, A.; Tanaka, S.; et al. Expression of Aurora B and alternative variant forms in hepatocellular carcinoma and adjacent tissue. Cancer Sci. 2009, 100, 472–480. [Google Scholar] [CrossRef]
- He, Z.-L.; Zheng, H.; Lin, H.; Miao, X.-Y.; Zhong, D.-W. Overexpression of polo-like kinase1 predicts a poor prognosis in hepatocellular carcinoma patients. World J. Gastroenterol. 2009, 15, 4177–4182. [Google Scholar] [CrossRef]
- Pellegrino, R.; Calvisi, D.F.; Ladu, S.; Ehemann, V.; Staniscia, T.; Evert, M.; Dombrowski, F.; Schirmacher, P.; Longerich, T. Oncogenic and tumor suppressive roles of polo-like kinases in human hepatocellular carcinoma. Hepatology 2009, 51, 857–868. [Google Scholar] [CrossRef]
- Sun, W.; Su, Q.; Cao, X.; Shang, B.; Chen, A.; Yin, H.; Liu, B. High Expression of Polo-Like Kinase 1 Is Associated with Early Development of Hepatocellular Carcinoma. J. Genom. 2014, 2014, 312130. [Google Scholar] [CrossRef]
- Bouhlal, H.; Ouled-Haddou, H.; DeBuysscher, V.; Singh, A.R.; Ossart, C.; Reignier, A.; Hocini, H.; Fouquet, G.; Al Baghami, M.; Eugenio, M.S.; et al. RB/PLK1-dependent induced pathway by SLAMF3 expression inhibits mitosis and control hepatocarcinoma cell proliferation. Oncotarget 2016, 7, 9832–9843. [Google Scholar] [CrossRef][Green Version]
- Zhang, C.; Wang, X.; Fang, D.; Xu, P.; Mo, X.; Hu, C.; Abdelatty, A.; Wang, M.; Xu, H.; Sun, Q.; et al. STK39 is a novel kinase contributing to the progression of hepatocellular carcinoma by the PLK1/ERK signaling pathway. Theranostics 2021, 11, 2108–2122. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.-T.; Yu, H.-Q.; Fang, L.; Tan, Y.; Liu, Z.-Y.; Wu, D.; Zhang, J.; Xiong, H.-J.; Xie, C.-M. Elevated FBXO45 promotes liver tumorigenesis through enhancing IGF2BP1 ubiquitination and subsequent PLK1 upregulation. eLife 2021, 10, e70715. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Deng, W.; Jiang, B.; Liu, S.; Tang, M.; Liu, Y.; Zhang, J. Hsa-let-7b inhibits cell proliferation by targeting PLK1 in HCC. Gene 2018, 673, 46–55. [Google Scholar] [CrossRef]
- Zhang, H.; Diab, A.; Fan, H.; Mani, S.K.; Hullinger, R.; Merle, P.; Andrisani, O. PLK1 and HOTAIR Accelerate Proteasomal Degradation of SUZ12 and ZNF198 during Hepatitis B Virus–Induced Liver Carcinogenesis. Cancer Res. 2015, 75, 2363–2374. [Google Scholar] [CrossRef]
- Wu, C.X.; Wang, X.Q.; Chok, S.H.; Man, K.; Tsang, S.H.Y.; Chan, A.C.Y.; Ma, K.W.; Xia, W.; Cheung, T.T. Blocking CDK1/PDK1/β-Catenin signaling by CDK1 inhibitor RO3306 increased the efficacy of sorafenib treatment by targeting cancer stem cells in a preclinical model of hepatocellular carcinoma. Theranostics 2018, 8, 3737–3750. [Google Scholar] [CrossRef]
- Zhou, J.; Han, S.; Qian, W.; Gu, Y.; Li, X.; Yang, K. Metformin induces miR-378 to downregulate the CDK1, leading to suppression of cell proliferation in hepatocellular carcinoma. OncoTargets Ther. 2018, ume 11, 4451–4459. [Google Scholar] [CrossRef]
- Yin, S.; Yang, S.; Luo, Y.; Lu, J.; Hu, G.; Wang, K.; Shao, Y.; Zhou, S.; Koo, S.; Qiu, Y.; et al. Cyclin-dependent kinase 1 as a potential target for lycorine against hepatocellular carcinoma. Biochem. Pharmacol. 2021, 193, 114806. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; Li, S.; Peng, Q.; Guo, Y.; Ji, J.; Zhang, Z.; Xue, Y.; Liu, Y.; Shi, X. Anti-malarial drug dihydroartemisinin downregulates the expression levels of CDK1 and CCNB1 in liver cancer. Oncol. Lett. 2021, 22, 653. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, T.; Zhou, H.; Feng, B.; Chen, Y.; Zhi, Y.; Wang, R. A Novel Aurora-A Inhibitor (MLN8237) Synergistically Enhances the Antitumor Activity of Sorafenib in Hepatocellular Carcinoma. Mol. Ther.-Nucleic Acids 2018, 13, 176–188. [Google Scholar] [CrossRef]
- Liu, F.; Wang, G.; Wang, X.; Che, Z.; Dong, W.; Guo, X.; Wang, Z.; Chen, P.; Hou, D.; Zhang, Q.; et al. Targeting high Aurora kinases expression as an innovative therapy for hepatocellular carcinoma. Oncotarget 2017, 8, 27953–27965. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, M.; Yu, X.; Liu, T.; Li, T.; Zhou, L.; Liu, W.; Li, W.; Gao, F. Butein suppresses hepatocellular carcinoma growth via modulating Aurora B kinase activity. Int. J. Biol. Sci. 2018, 14, 1521–1534. [Google Scholar] [CrossRef]
- Zhu, Q.; Yu, X.; Zhou, Z.-W.; Luo, M.; Zhou, C.; He, Z.-X.; Chen, Y.; Zhou, S.-F. A quantitative proteomic response of hepatocellular carcinoma Hep3B cells to danusertib, a pan-Aurora kinase inhibitor. J. Cancer 2018, 9, 2061–2071. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Yu, X.; Zhou, Z.-W.; Zhou, C.; Chen, X.-W.; Zhou, S.-F. Inhibition of Aurora A Kinase by Alisertib Induces Autophagy and Cell Cycle Arrest and Increases Chemosensitivity in Human Hepatocellular Carcinoma HepG2 Cells. Curr. Cancer Drug Targets 2017, 17, 386–401. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, Z.-G.; Luo, Y.; Cui, H.; Wang, H.-Y.; Jia, Y.-F.; Gao, Y.-T. Dual targeting of Polo-like kinase 1 and baculoviral inhibitor of apoptosis repeat-containing 5 in TP53-mutated hepatocellular carcinoma. World J. Gastroenterol. 2020, 26, 4786–4801. [Google Scholar] [CrossRef]
- Dietrich, P.; Freese, K.; Mahli, A.; Thasler, W.E.; Hellerbrand, C.; Bosserhoff, A.K. Combined effects of PLK1 and RAS in hepatocellular carcinoma reveal rigosertib as promising novel therapeutic “dual-hit” option. Oncotarget 2017, 9, 3605–3618. [Google Scholar] [CrossRef]
- Xu, L.; Zhu, Y.; Shao, J.; Chen, M.; Yan, H.; Li, G.; Zhu, Y.; Xu, Z.; Yang, B.; Luo, P.; et al. Dasatinib synergises with irinotecan to suppress hepatocellular carcinoma via inhibiting the protein synthesis of PLK1. Br. J. Cancer 2017, 116, 1027–1036. [Google Scholar] [CrossRef]
- Fawzy, F.; Canada, A.; Fawzy, N. Cancer Treatment. Encycl. Stress 2007, 384–387. [Google Scholar] [CrossRef]
- Milella, M.; Bassi, C.; Boggi, U.; Brunetti, O.; Cavaliere, A.; Crippa, S.; De Vita, F.; Falconi, M.; Frassineti, G.L.; Giommoni, E.; et al. Evolving pancreatic cancer treatment: From diagnosis to healthcare management. Crit. Rev. Oncol. 2021, 169, 103571. [Google Scholar] [CrossRef] [PubMed]
- Cancer Today. (n.d.). Available online: https://gco.iarc.fr/today/online-analysis-pie?v=2020&mode=cancer&mode_population=continents&population=900&populations=900&key=total&sex=0&cancer=39&type=1&statistic=5&prevalence=0&population_group=0&ages_group%5B%5D=0&ages_group%5B%5D=17&nb_items=7&group_cancer=1&include_nmsc=1&include_nmsc_other=1&half_pie=0&donut=0 (accessed on 31 March 2022).
- Chandana, S.; Babiker, H.M.; Mahadevan, D. Therapeutic trends in pancreatic ductal adenocarcinoma (PDAC). Expert Opin. Investig. Drugs 2018, 28, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer. JAMA J. Am. Med. Assoc. 2021, 326, 851–862. [Google Scholar] [CrossRef] [PubMed]
- PDQ Adult Treatment Editorial Board. Pancreatic Cancer Treatment (Adult) (PDQ®). PDQ Cancer Information Summaries. 2022. Available online: https://www-ncbi-nlm-nih-gov.sire.ub.edu/books/NBK65957/ (accessed on 31 March 2022).
- Gugenheim, J.; Crovetto, A.; Petrucciani, N. Neoadjuvant therapy for pancreatic cancer. Update Surg. 2021, 74, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Tischer, J.; Gergely, F. Anti-mitotic therapies in cancer. J. Cell Biol. 2018, 218, 10–11. [Google Scholar] [CrossRef]
- Bayraktar, S.; Lima, C.M.R. Emerging cell-cycle inhibitors for pancreatic cancer therapy. Expert Opin. Emerg. Drugs 2012, 17, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Filho, S.M.; dos Santos, E.O.; Bertoldi, E.R.M.; Scalabrini, L.C.; Heidrich, V.; Dazzani, B.; Levantini, E.; Reis, E.M.; Bassères, D.S. Aurora A kinase and its activator TPX2 are potential therapeutic targets in KRAS-induced pancreatic cancer. Cell. Oncol. 2020, 43, 445–460. [Google Scholar] [CrossRef]
- Bavetsias, V.; Linardopoulos, S. Aurora Kinase Inhibitors: Current Status and Outlook. Front. Oncol. 2015, 5, 278. [Google Scholar] [CrossRef]
- García-Reyes, B.; Kretz, A.-L.; Ruff, J.-P.; von Karstedt, S.; Hillenbrand, A.; Knippschild, U.; Henne-Bruns, D.; Lemke, J. The Emerging Role of Cyclin-Dependent Kinases (CDKs) in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2018, 19, 3219. [Google Scholar] [CrossRef]
- Piao, J.; Zhu, L.; Sun, J.; Li, N.; Dong, B.; Yang, Y.; Chen, L. High expression of CDK1 and BUB1 predicts poor prognosis of pancreatic ductal adenocarcinoma. Gene 2019, 701, 15–22. [Google Scholar] [CrossRef]
- Dong, S.; Huang, F.; Zhang, H.; Chen, Q. Overexpression of BUB1B, CCNA2, CDC20, and CDK1 in tumor tissues predicts poor survival in pancreatic ductal adenocarcinoma. Biosci. Rep. 2019, 39, BSR20182306. [Google Scholar] [CrossRef] [PubMed]
- Wijnen, R.; Pecoraro, C.; Carbone, D.; Fiuji, H.; Avan, A.; Peters, G.J.; Giovannetti, E.; Diana, P. Cyclin Dependent Kinase-1 (CDK-1) Inhibition as a Novel Therapeutic Strategy against Pancreatic Ductal Adenocarcinoma (PDAC). Cancers 2021, 13, 4389. [Google Scholar] [CrossRef]
- di Rorà, A.G.L.; Cerchione, C.; Martinelli, G.; Simonetti, G. A WEE1 family business: Regulation of mitosis, cancer progression, and therapeutic target. J. Hematol. Oncol. 2020, 13, 126. [Google Scholar] [CrossRef]
- Guo, S.; Fesler, A.; Huang, W.; Wang, Y.; Yang, J.; Wang, X.; Zheng, Y.; Hwang, G.-R.; Ju, J. Functional Significance and Therapeutic Potential of miR-15a Mimic in Pancreatic Ductal Adenocarcinoma. Mol. Ther.-Nucleic Acids 2019, 19, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhu, S.; Zhong, M.; Yang, M.; Sun, X.; Liu, J.; Kroemer, G.; Lotze, M.; Zeh, H.J.; Kang, R.; et al. Inhibition of Aurora Kinase A Induces Necroptosis in Pancreatic Carcinoma. Gastroenterology 2017, 153, 1429–1443.e5. [Google Scholar] [CrossRef]
- Ozcimen, A.; Kirbiyik, I. The effect of danusertib, an Aurora kinase inhibitor, onto the cytotoxicity, cell cycle and apoptosis in pancreatic ductal adenocarcinoma cells. J. Cancer Res. Ther. 2021, 17, 1419. [Google Scholar] [CrossRef] [PubMed]
- Mathison, A.; Salmonson, A.; Missfeldt, M.; Bintz, J.; Williams, M.; Kossak, S.; Nair, A.; de Assuncao, T.M.; Christensen, T.; Buttar, N.; et al. Combined AURKA and H3K9 Methyltransferase Targeting Inhibits Cell Growth by Inducing Mitotic Catastrophe. Mol. Cancer Res. 2017, 15, 984–997. [Google Scholar] [CrossRef]
- Kim, E.J.-H.; Semrad, T.J.; Gandara, D.R.; Riess, J.; Li, T.; Yu, A.; Matsukuma, K.; Mack, P.C.; Kelly, K. Phase I study of the combination of alisertib (MLN8237) and gemcitabine in advanced solid tumors. J. Clin. Oncol. 2015, 33, 2526. [Google Scholar] [CrossRef]
- Uboha, N.V.; Milhem, M.M.; Kovacs, C.; Amin, A.; Magley, A.; Das Purkayastha, D.; Piha-Paul, S.A. Phase II study of spartalizumab (PDR001) and LAG525 in advanced solid tumors and hematologic malignancies. J. Clin. Oncol. 2019, 37, 2553. [Google Scholar] [CrossRef]
- Pecoraro, C.; Parrino, B.; Cascioferro, S.; Puerta, A.; Avan, A.; Peters, G.J.; Diana, P.; Giovannetti, E.; Carbone, D. A New Oxadiazole-Based Topsentin Derivative Modulates Cyclin-Dependent Kinase 1 Expression and Exerts Cytotoxic Effects on Pancreatic Cancer Cells. Molecules 2021, 27, 19. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Chen, P.; Liu, K.; Liu, J.; Zhou, B.; Wu, R.; Peng, Q.; Liu, Z.-X.; Li, C.; Kroemer, G.; et al. CDK1/2/5 inhibition overcomes IFNG-mediated adaptive immune resistance in pancreatic cancer. Gut 2020, 70, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.G.; Zahurak, M.; Shah, M.; Weekes, C.D.; Hansen, A.; Siu, L.L.; Spreafico, A.; LoConte, N.; Anders, N.M.; Miles, T.; et al. A Phase I Study of Dinaciclib in Combination with MK-2206 in Patients with Advanced Pancreatic Cancer. Clin. Transl. Sci. 2020, 13, 1178–1188. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Geng, J.; Zhang, L.; Wang, Y.; Niu, N.; Fang, Y.; Liu, F.; Shi, J.; Zhang, Z.-G.; Sun, Y.-W.; et al. THZ1 reveals CDK7-dependent transcriptional addictions in pancreatic cancer. Oncogene 2019, 38, 3932–3945. [Google Scholar] [CrossRef]
- Hartman, S.J.; Bagby, S.M.; Yacob, B.W.; Simmons, D.M.; MacBeth, M.; Lieu, C.H.; Davis, S.L.; Leal, A.D.; Tentler, J.J.; Diamond, J.R.; et al. WEE1 Inhibition in Combination with Targeted Agents and Standard Chemotherapy in Preclinical Models of Pancreatic Ductal Adenocarcinoma. Front. Oncol. 2021, 11, 642328. [Google Scholar] [CrossRef]
- Diehl, J.N.; Klomp, J.E.; Snare, K.R.; Hibshman, P.S.; Blake, D.R.; Kaiser, Z.D.; Gilbert, T.S.; Baldelli, E.; Pierobon, M.; Papke, B.; et al. The KRAS-regulated kinome identifies WEE1 and ERK coinhibition as a potential therapeutic strategy in KRAS-mutant pancreatic cancer. J. Biol. Chem. 2021, 297, 101335. [Google Scholar] [CrossRef] [PubMed]
- Cuneo, K.C.; Morgan, M.A.; Sahai, V.; Schipper, M.J.; Parsels, L.A.; Parsels, J.D.; Devasia, T.; Al-Hawaray, M.; Cho, C.S.; Nathan, H.; et al. Dose Escalation Trial of the Wee1 Inhibitor Adavosertib (AZD1775) in Combination with Gemcitabine and Radiation for Patients with Locally Advanced Pancreatic Cancer. J. Clin. Oncol. 2019, 37, 2643–2650. [Google Scholar] [CrossRef] [PubMed]
- Stratford, J.K.; Yan, F.; Hill, R.A.; Major, M.; Graves, L.M.; Der, C.; Yeh, J.J. Genetic and pharmacological inhibition of TTK impairs pancreatic cancer cell line growth by inducing lethal chromosomal instability. PLoS ONE 2017, 12, e0174863. [Google Scholar] [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries and 25 major cancers in 2018. Eur. J. Cancer 2018, 103, 356–387. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Bender, U.; Rho, Y.; Barrera, I.; Aghajanyan, S.; Acoba, J.; Kavan, P. Adjuvant Therapy for Stages II and III Colon Cancer: Risk Stratification, Treatment Duration, and Future Directions. Curr. Oncol. 2019, 26, 43–52. [Google Scholar] [CrossRef]
- Mielcarska, S.; Kula, A.; Dawidowicz, M.; Kiczmer, P.; Chrabańska, M.; Rynkiewicz, M.; Wziątek-Kuczmik, D.; Świętochowska, E.; Waniczek, D. Assessment of the RANTES Level Correlation and Selected Inflammatory and Pro-Angiogenic Molecules Evaluation of Their Influence on CRC Clinical Features: A Preliminary Observational Study. Medicina 2022, 58, 203. [Google Scholar] [CrossRef] [PubMed]
- Bin Wang, H.; Yao, H.; Li, C.S.; Liang, L.X.; Zhang, Y.; Chen, Y.X.; Fang, J.-Y.; Xu, J. Rise of PD-L1 expression during metastasis of colorectal cancer: Implications for immunotherapy. J. Dig. Dis. 2017, 18, 574–581. [Google Scholar] [CrossRef]
- Venook, A.P.; Niedzwiecki, D.; Lenz, H.-J.; Innocenti, F.; Fruth, B.; Meyerhardt, J.A.; Schrag, D.; Greene, C.; O’Neil, B.H.; Atkins, J.N.; et al. Effect of First-Line Chemotherapy Combined with Cetuximab or Bevacizumab on Overall Survival in Patients with KRAS Wild-Type Advanced or Metastatic Colorectal Cancer: A randomized clinical trial. JAMA 2017, 317, 2392–2401. [Google Scholar] [CrossRef]
- Zhao, Q.; Ouyang, X.; Wan, X.; Gajiwala, K.S.; Kath, J.C.; Jones, L.H.; Burlingame, A.L.; Taunton, J. Broad-Spectrum Kinase Profiling in Live Cells with Lysine-Targeted Sulfonyl Fluoride Probes. J. Am. Chem. Soc. 2017, 139, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Baur, F.; Nietzer, S.L.; Kunz, M.; Saal, F.; Jeromin, J.; Matschos, S.; Linnebacher, M.; Walles, H.; Dandekar, T.; Dandekar, G. Connecting Cancer Pathways to Tumor Engines: A Stratification Tool for Colorectal Cancer Combining Human in Vitro Tissue Models with Boolean in Silico Models. Cancers 2019, 12, 28. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, M.H.; Lam, F.; Zhong, L.; Teo, T.; Adams, J.; Yu, M.; Milne, R.W.; Pepper, C.; Lokman, N.A.; Ricciardelli, C.; et al. Targeting CDK9 for treatment of colorectal cancer. Mol. Oncol. 2019, 13, 2178–2193. [Google Scholar] [CrossRef]
- Tanaka, H.; Wada, M.; Park, J. HASPIN kinase inhibitor CHR-6494 suppresses intestinal polyp development, cachexia, and hypogonadism in Apc min/+ mice. Eur. J. Cancer Prev. 2019, 29, 481–485. [Google Scholar] [CrossRef]
- Schmit, T.L.; Ahmad, N. Regulation of mitosis via mitotic kinases: New opportunities for cancer management. Mol. Cancer Ther. 2007, 6, 1920–1931. [Google Scholar] [CrossRef]
- Fadaka, A.O.; Sibuyi, N.R.S.; Bakare, O.O.; Klein, A.; Madiehe, A.M.; Meyer, M. Expression of cyclin-dependent kinases and their clinical significance with immune infiltrates could predict prognosis in colorectal cancer. Biotechnol. Rep. 2021, 29, e00602. [Google Scholar] [CrossRef]
- Koh, H.M.; Jang, B.G.; Hyun, C.L.; Kim, Y.S.; Hyun, J.W.; Chang, W.Y.; Maeng, Y.H. Aurora Kinase A Is a Prognostic Marker in Colorectal Adenocarcinoma. J. Pathol. Transl. Med. 2017, 51, 32–39. [Google Scholar] [CrossRef]
- Zhang, L.; Jiang, B.; Zhu, N.; Tao, M.; Jun, Y.; Chen, X.; Wang, Q.; Luo, C. Mitotic checkpoint kinase Mps1/TTK predicts prognosis of colon cancer patients and regulates tumor proliferation and differentiation via PKCα/ERK1/2 and PI3K/Akt pathway. Med. Oncol. 2019, 37, 5. [Google Scholar] [CrossRef]
- Welburn, J.P.I.; Jeyaprakash, A.A. Mechanisms of Mitotic Kinase Regulation: A Structural Perspective. Front. Cell Dev. Biol. 2018, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Lu, T.; Wang, L.; Wu, C.; Chen, M.; Kuo, C.; Shyu, R.; Tsai, F. Tazarotene-induced gene 1 interacts with Polo-like kinase 2 and inhibits cell proliferation in HCT116 colorectal cancer cells. Cell Biol. Int. 2021, 45, 2347–2356. [Google Scholar] [CrossRef]
- Klauck, P.J.; Bagby, S.M.; Capasso, A.; Bradshaw-Pierce, E.L.; Selby, H.M.; Spreafico, A.; Tentler, J.J.; Tan, A.C.; Kim, J.; Arcaroli, J.J.; et al. Antitumor activity of the polo-like kinase inhibitor, TAK-960, against preclinical models of colorectal cancer. BMC Cancer 2018, 18, 136. [Google Scholar] [CrossRef] [PubMed]
- Capasso, A.; Pitts, T.M.; Klauck, P.J.; Bagby, S.M.; Westbrook, L.; Kaplan, J.; Soleimani, M.; Spreafico, A.; Tentler, J.J.; Diamond, J.R.; et al. Dual compartmental targeting of cell cycle and angiogenic kinases in colorectal cancer models. Anti-Cancer Drugs 2018, 29, 827–838. [Google Scholar] [CrossRef]
- Robb, C.M.; Kour, S.; Contreras, J.I.; Agarwal, E.; Barger, C.J.; Rana, S.; Sonawane, Y.; Neilsen, B.K.; Taylor, M.; Kizhake, S.; et al. Characterization of CDK(5) inhibitor, 20-223 (aka CP668863) for colorectal cancer therapy. Oncotarget 2017, 9, 5216–5232. [Google Scholar] [CrossRef]
- Faisal, A.; Mak, G.W.Y.; Gurden, M.D.; Xavier, C.P.R.; Anderhub, S.J.; Innocenti, P.; Westwood, I.M.; Naud, S.; Hayes, A.; Box, G.; et al. Characterisation of CCT271850, a selective, oral and potent MPS1 inhibitor, used to directly measure in vivo MPS1 inhibition vs therapeutic efficacy. Br. J. Cancer 2017, 116, 1166–1176. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Inhibitor | Model | Main Findings | Reference |
|---|---|---|---|---|
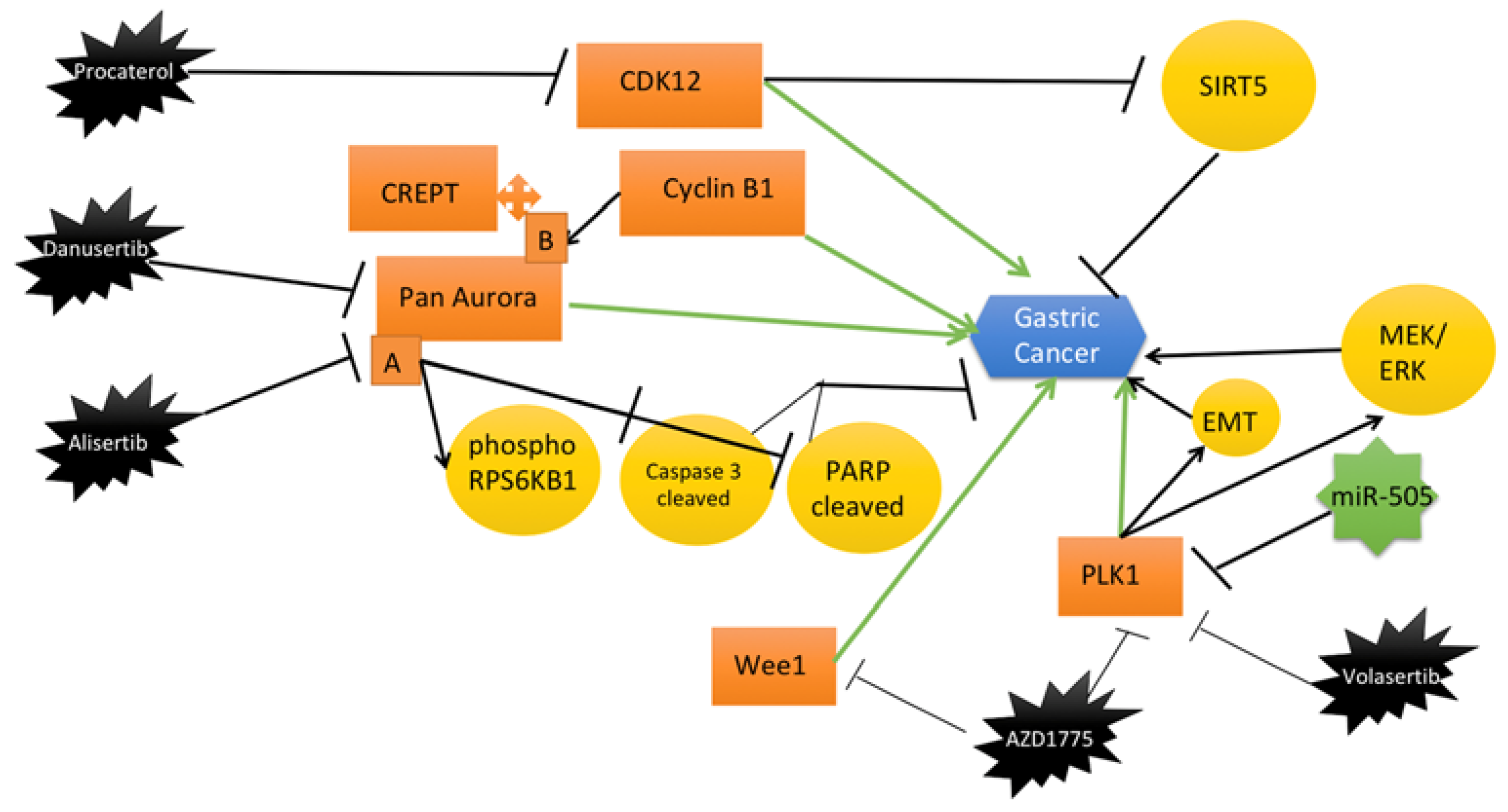
| Aurora and CDK2 | Danusertib | NCI-N78 and AGS human GC cell line. | Danusertib inhibits Aurora and CDK2 expression in GC cell lines. The proportion of GC cells in the G1 phase is reduced. | [38] |
| Aurora A | Alisertib | Mice with xenograft tumors tissues. | Alisertib reduces tumor volume. Downregulation of Aurora A by alisertib reduces the level of phosphorylated RPS6KB1. | [39] |
| Wee1 | AZD1775 | The GC cell lines (AGS, YCC-2, MKN28, KATO III, SNU-216, SNU-601, SNU-638, SNU-668, and SNU-719. Orthotopic mouse model for GC. | AZD1775 induces apoptosis and cell cycle arrest. Tumor size and weight were reduced in mice treated with AZD1775 compared to control mice. | [37] |
| CDK12 | Procaterol | Cell xenograft NU/NU mouse models (CDXs) and patient-derived xenograft NOD/SCID mouse models (PDXs) were conducted to study. | Procaterol bounds to and inhibits CDK12 kinase activity. | [41] |
| PLK1 | Volasertib | Japanese patients with advanced solid tumors. Methods In this phase I, open-label, dose-escalation trial, sequential patient cohorts. | Volasertib has been investigated as a selective Polo-like kinase inhibitor that resulted in mitotic arrest and apoptosis in Japanese patients with gastric cancer. | [40] |
| WEE1/(PLK1) | AZD1775 | Human GC cell lines MKN45 and AGS. female NOD/SCID mice. | Administration of AZD1775-olaparib amed at WEE1/PLK1 and the expression of PLK1 reduced in AGS and MKN45 cell lines, which exerted stronger antigrowth efficacy than olaparib alone. The combination of AZD1775 and olaparib has led to higher inhibition in gastric tumor growth than their monotherapy in mice. | [36] |
| Target | Inhibitor | Model | Main Findings | Reference |
|---|---|---|---|---|
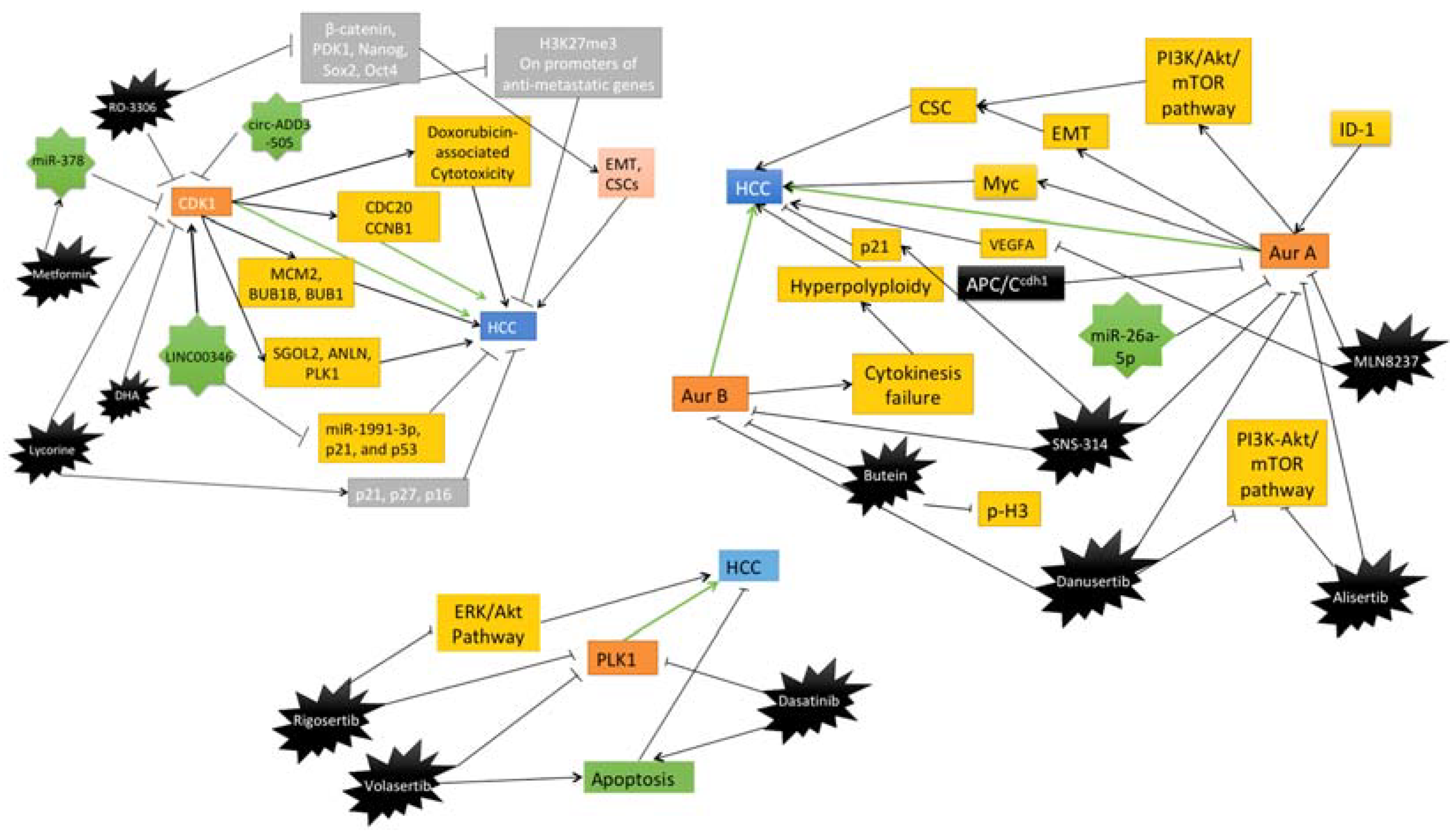
| CDK1 | RO-3306 | HCC patient-derived xenograft (PDX) tumor models treated with sorafenib alone or with RO-3306 | Downregulation of PK1-beta-Catenin pathway, reduced CSC growth, decrease in EMT profile | [79] |
| CDK1 | Metformin | Human HCC cell lines HepG2 and Hep3B, and nude xenograft mice models | Induction of miR-378 which inhibits CDK1 mRNA and causes reduction in HCC cell proliferation | [80] |
| CDK1 | Lycorine | HCC cells | Inhibition of CDK1 expression directly and induces expression of p21, p16 and p21 | [81] |
| CDK1 | Dihydroartemisinin (DHA) | HCC cells | Inhibition of the cyclin B1-CDK1 pathway and reduces the HCC cell proliferation | [82] |
| Aurora A | MLN8237 | HepG2 and SMMC-7721 cell lines, HepG2 xenograft mice models | Inhibition of p-Akt, p-MAPK, CDK4, cyclin D1, and ultimately progression of HCC. | [83] |
| Pan-Aurora kinases | SNS-314 | HepG2, SMMC-7721 and HCC-LM6 Liver cancer cell lines | Reduction of YAP and accumulates p21 to induce polyploidy and apoptosis in HCC cells. | [84] |
| Aurora B | Butein | HepG2 and Hep3B cells, xenograft models | Induction of apoptosis by decreasing p-H3 and Aurora B, marked by reduced Ki67 | [85] |
| Pan-Aurora kinases | Danusertib | Hep3B cell line | Induction of autophagy and mitochondria-dependent apoptosis by changing the PI3K/Akt/mTOR signaling pathway | [86] |
| Aurora A | Alisertib | HepG2 cell line | Regulation of PI3K/Akt/mTOR pathway, induces autophagy, and increase sensitivity to doxorubicin and cisplatin | [87] |
| PLK1 | Volasertib | P53 mutated HCC cells, Xenograft model | Induction of apoptosis selectively in p53 mutated HCC cells | [88] |
| PLK1 | Rigosertib | HCC cell lines | Inhibition of ERK and Akt signaling, reduces proliferation and causes cell cycle arrest | [89] |
| PLK1 | Dasatinib | HCC cell lines and xenograft models | Inhibition of protein synthesis of PLK1 and increases the apoptosis rate of HCC cells | [90] |
| Target | Inhibitor | Model | Main Findings | Reference |
|---|---|---|---|---|
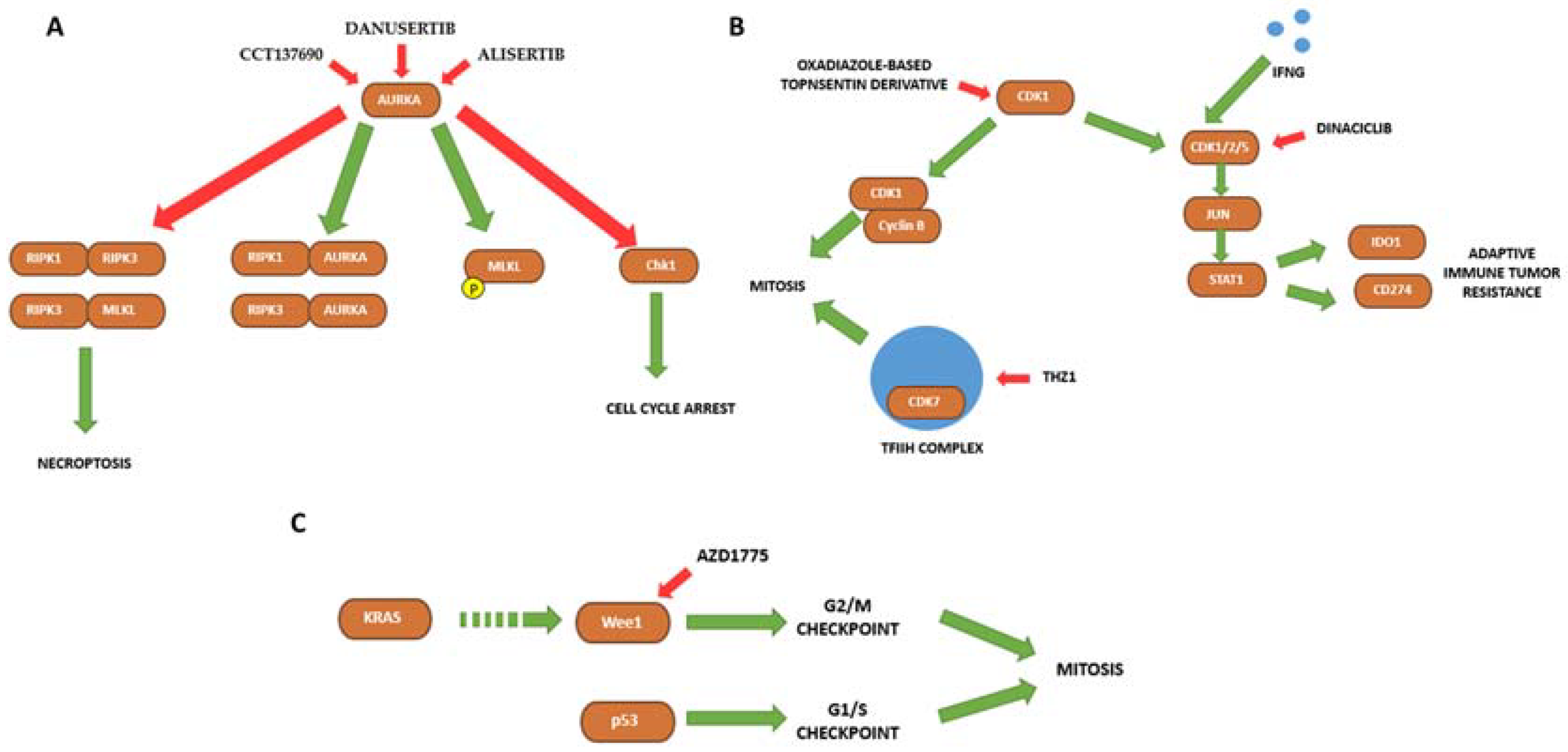
| Aurora Kinase A | CCT137690 | Human pancreatic cancer cell lines (PANC1, PANC2.03, CFPAC1, MiaPaCa2, BxPc-3 and PANC02) and mouse PDAC cell line KPC. Murine subcutaneous tumors (PANC1, PANC02 and KPC injected in athymic nude or B6 mice). Orthotopic tumors (KPC cells in B6 mice). | CCT137690 serves as an Aurora Kinase A inhibitor that induces necrosis-like death in pancreatic cancer cells in vitro and in vivo. | [109] |
| Aurora Kinase A | Danusertib | Human pancreatic cancer cell line CFPAC-1. | Danusertib exerts cytotoxic effects, induces apoptosis, and accumulates cells at S and G2/M phases in CFPAC-1 cell line. | [110] |
| Aurora Kinase A | Alisertib | Human pancreatic cancer cell lines (BxPC-3, Capan-2, L3.6, MiaPaCa-2, PANC-1) and mouse PDAC cell line Pan02. Organoids from Ela-Kran mice. Orthotopic xenografts injecting Pan02 cells in C57BL/6 mice. | Alisertib in combination with H3K9 methyltransferase inhibitors is more effective in reducing pancreatic cancer cell viability in vitro and in vivo via inducing mitotic catastrophe. | [111] |
| CDK1/2/5 | Dinaciclib | Human pancreatic cancer cell line CFPAC-1, and mouse cell lines KPC and CT26. Human clinical samples from pancreatic cancer patients who underwent surgery. Animal models: vaccination murine model, subcutaneous xenografts, orthotopic xenografts, Pdx-1-Cre and KRASG12D/+ transgenic mice. | Dinaciclib treatment triggers caspase-dependent apoptosis and histone-dependent immunogenic cell death in pancreatic cancer cells. Dinaciclib in combination of IGNF exerts anticancer effects both in vitro and in vivo. | [115] |
| CDK7 | THZ1 | Human pancreatic cancer cell lines (BxPC-3, MiaPaCa-2, SW1990, PANC-1, ASPC-1) and human pancreatic epithelial cell line HPDE6-C7. Pancreatic patient-derived tumor cells. Patient-derived xenografts in BALB/c mice. KC and KPC mice. | CDK7 inhibition has cytotoxic effects and induces apoptosis in pancreatic cancer cells both in vitro and in vivo. Susceptibility to THZ1 treatment in pancreatic cancer cells is associated with MYC expression. | [117] |
| Wee1 | AZD1775 | Human pancreatic cancer cell lines (BxPC-3, MiaPaCa-2, PANC-1, and L3.3). Patient-Derived Xenografts. | AZD1775 in combination with irinotecan or capecitabine shows anti-tumor effects in PDAC both in vitro and in vivo. | [118] |
| Wee1 | AZD1775 | Human pancreatic cancer cell lines (AsPC-1, Panc10.05, SW-1990, MiaPaCa-2, PANC-1, HPAC, HPAF-II, Pa01C, Pa02C, Pa14C, and Pa16C) and organoids. | AZD1775 treatment exerts cytotoxic effects in KRAS mutated PDAC. AZD1775 and ERK inhibition causes synergistic growth arrest and apoptosis in vitro. | [119] |
| TTK | AZ3146 | Human pancreatic cancer cell lines (HPDE, HPNE, BxPC-3, Panc02.03, MiaPaCa-2, HPAC, PANC-1, Panc 10.05, Capan2, T3M4, AsPC-1, HPAF-II, SW1990, HuPT3, Capan1, CFPAC). | AZ3146 treatment decreases cell proliferation in PDAC cell lies. | [121] |
| Target | Inhibitor | Model | Main Findings | Reference |
|---|---|---|---|---|
| P-Plk1 | TAK-960 | CRC cell lines (HCT116, WIDR, DLD1 and COLO678) | Colonization of CRC cell lines treated with TAK960 was reduced in a dose-dependent manner. PPlk1 decreased when CRC cell lines were treated with TAK960. | [138] |
| Aurora A/B | ENMD-2076 | Forty-seven CRC cell lines had been tested. patient-derived xenograft (PDX) models. | Exposure to ENMD2076 resulted in G2/M cell cycle arrest, increased aneuploidy, and cell death in responsive cell lines. | [139] |
| CDKs | CP668863 | CRC cell lines included GEO, HCT116 and HT29 their colorectal cancer xenograft model used GEO cells. | CP668863to target CDK2 and CDK5 in CRC cell lines. CP668863-treated tumors had low phospho focal adhesion kinase (pFAK) levels. | [140] |
| Mps1 | CCT271850 | Human colon cancer HCT116 cells. Xenograft mice models using DLD1-GFP-MPS1 cells. | CCT271850 strongly suppressed MPS1 kinase activity in biochemical and cellular assays and in vivo models. Moderate efficacy of CCT271850 as a single agent in a xenograft model of human colorectal cancer. | [141] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Javed, A.; Malagraba, G.; Yarmohammadi, M.; Perelló-Reus, C.M.; Barceló, C.; Rubio-Tomás, T. Therapeutic Potential of Mitotic Kinases’ Inhibitors in Cancers of the Gastrointestinal System. Future Pharmacol. 2022, 2, 214-237. https://doi.org/10.3390/futurepharmacol2030015
Javed A, Malagraba G, Yarmohammadi M, Perelló-Reus CM, Barceló C, Rubio-Tomás T. Therapeutic Potential of Mitotic Kinases’ Inhibitors in Cancers of the Gastrointestinal System. Future Pharmacology. 2022; 2(3):214-237. https://doi.org/10.3390/futurepharmacol2030015
Chicago/Turabian StyleJaved, Aadil, Gianluca Malagraba, Mahdieh Yarmohammadi, Catalina M. Perelló-Reus, Carles Barceló, and Teresa Rubio-Tomás. 2022. "Therapeutic Potential of Mitotic Kinases’ Inhibitors in Cancers of the Gastrointestinal System" Future Pharmacology 2, no. 3: 214-237. https://doi.org/10.3390/futurepharmacol2030015
APA StyleJaved, A., Malagraba, G., Yarmohammadi, M., Perelló-Reus, C. M., Barceló, C., & Rubio-Tomás, T. (2022). Therapeutic Potential of Mitotic Kinases’ Inhibitors in Cancers of the Gastrointestinal System. Future Pharmacology, 2(3), 214-237. https://doi.org/10.3390/futurepharmacol2030015