
Molecular Mechanisms of Oxygen Evolution Reactions for Artificial Photosynthesis


Abstract
:1. Introduction
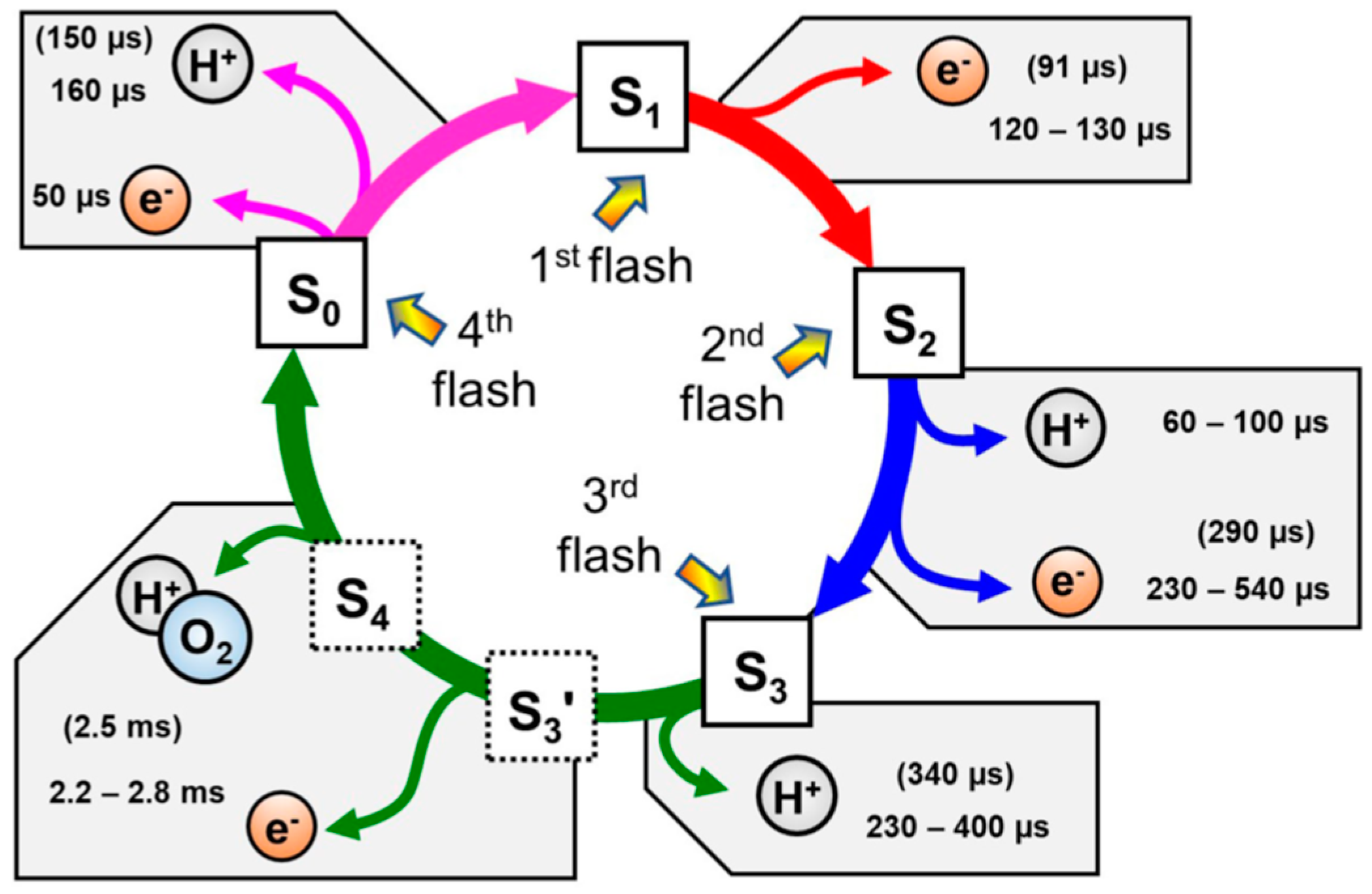
2. Oxygen Evolution Mechanism of Photosystem II (PS II)
3. Devices for Oxygen Evolution
3.1. Water Electrolyzer
3.2. Photoelectrochemical Cell
3.3. Particulate Photocatalysts
4. Methods for Investigating Mechanism
4.1. Electrochemical Analysis
4.2. Fourier Transform InfraRed (FTIR) Spectroscopy
4.3. Raman Spectroscopy
4.4. Electron Paramagnetic Resonance (EPR) Spectroscopy
4.5. Electrochemical Transmission Electron Microscope (EC-TEM)
4.6. X-ray Photoelectron Spectroscopy (XPS)
4.7. X-ray Absorption Spectroscopy (XAS)
4.8. X-ray Emission Spectroscopy (XES)
4.9. Mössbauer Spectroscopy
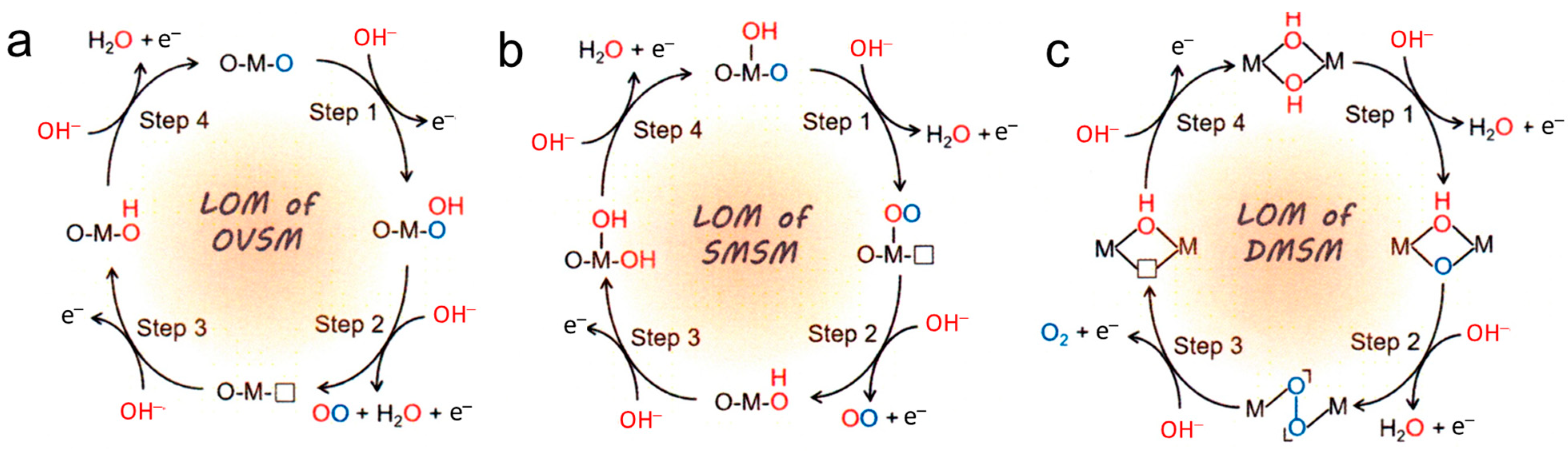
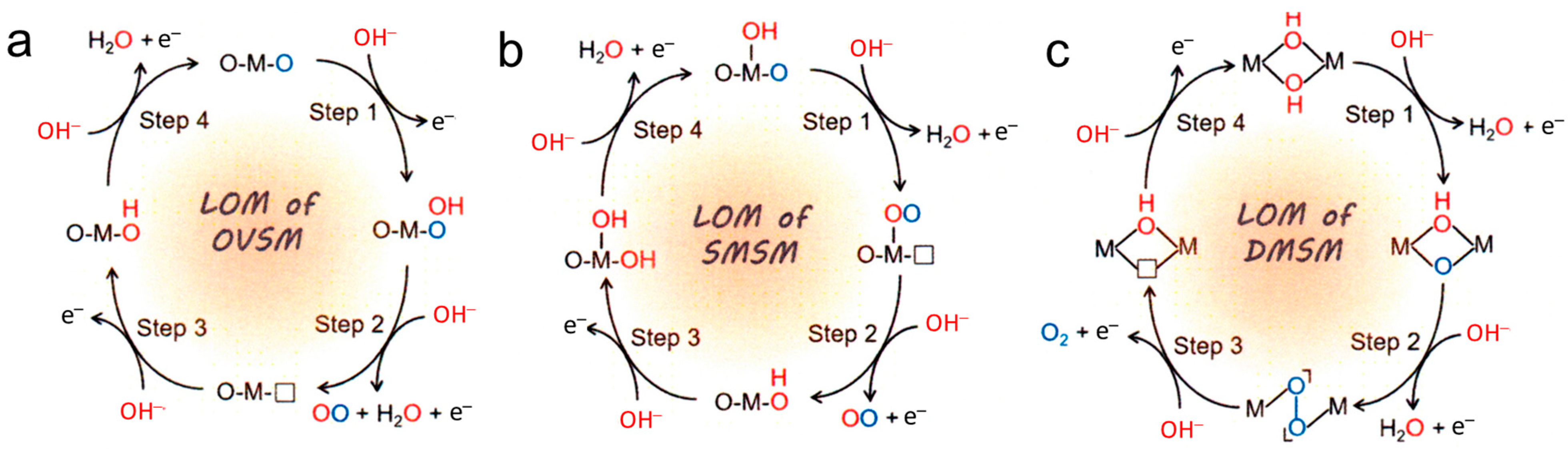
5. Common Mechanisms of OER Catalysis
6. Molecular Mechanism of OER
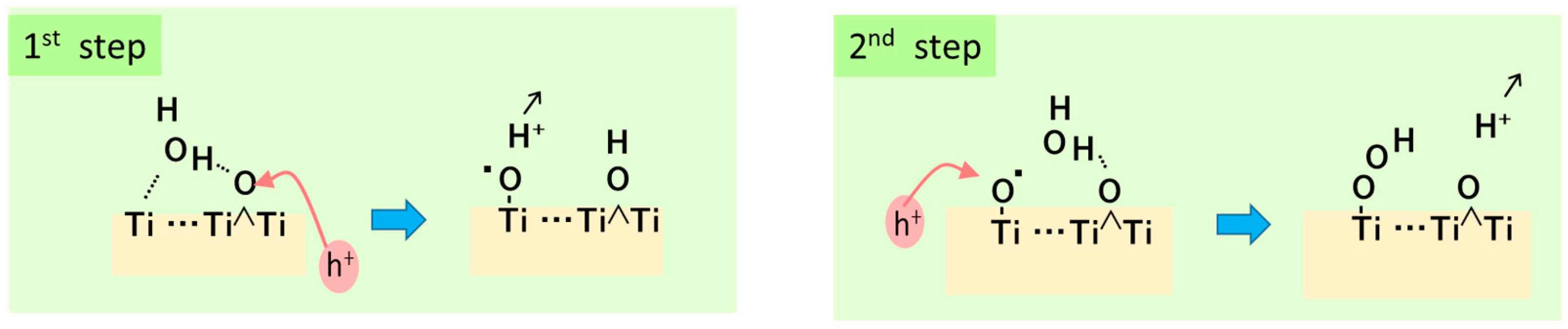
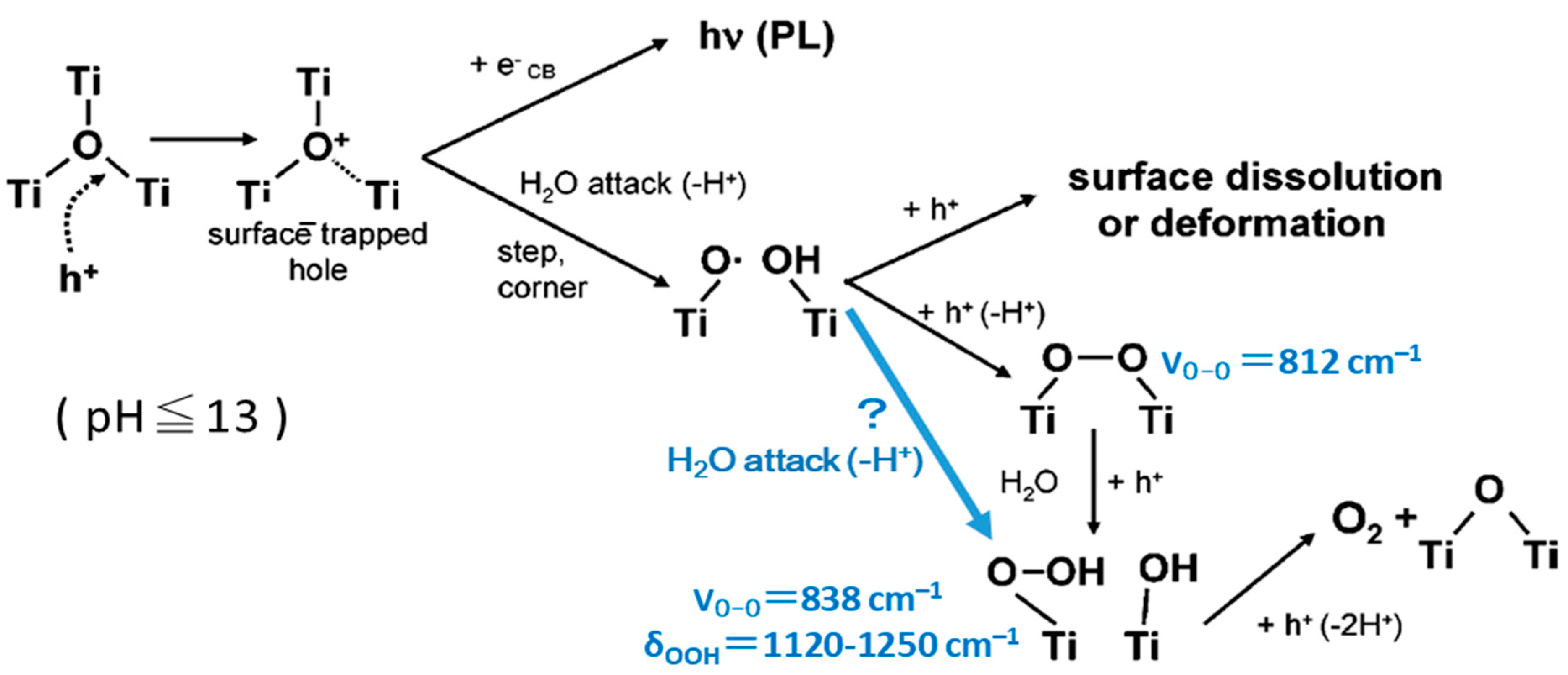
6.1. TiO2
6.2. BiVO4
6.3. SrTiO3
6.4. Ga2O3
6.5. IrO2 and RuO2
6.6. Perovskite as Electrocatalysts
6.7. Transition Metal (TM) Compounds
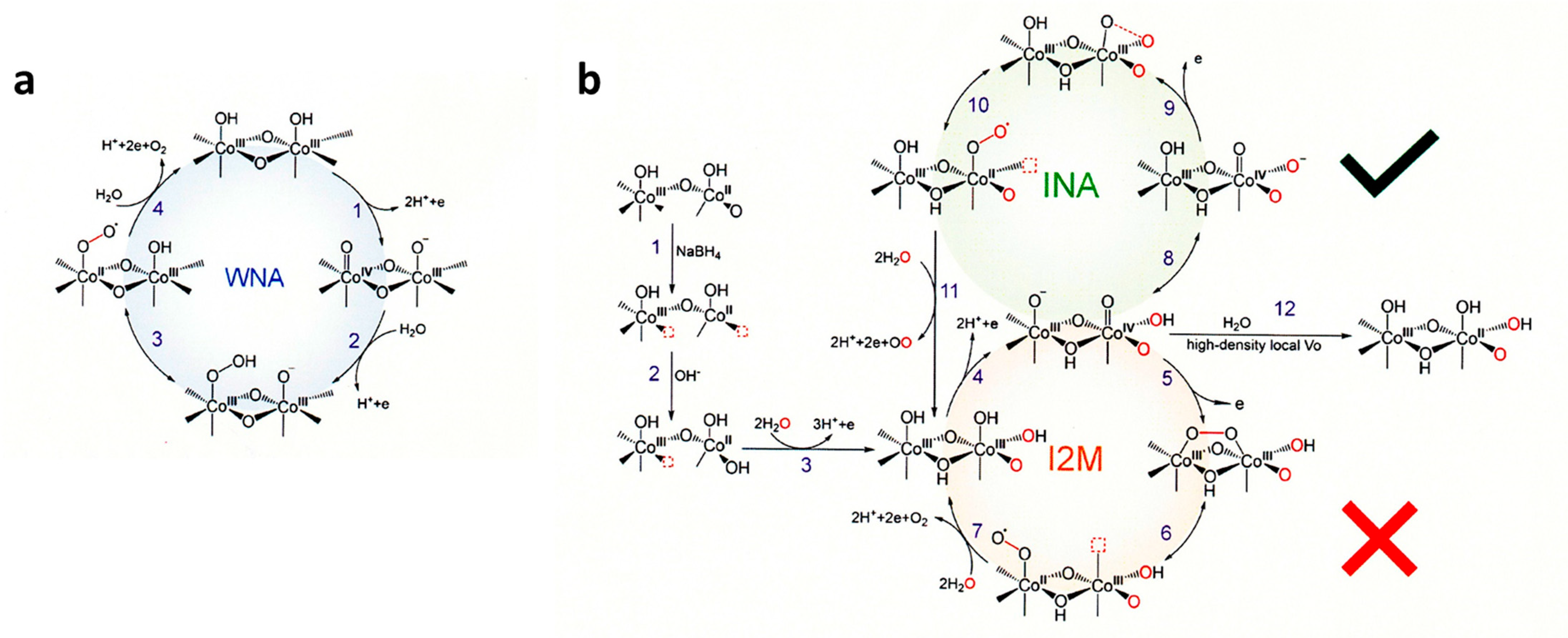
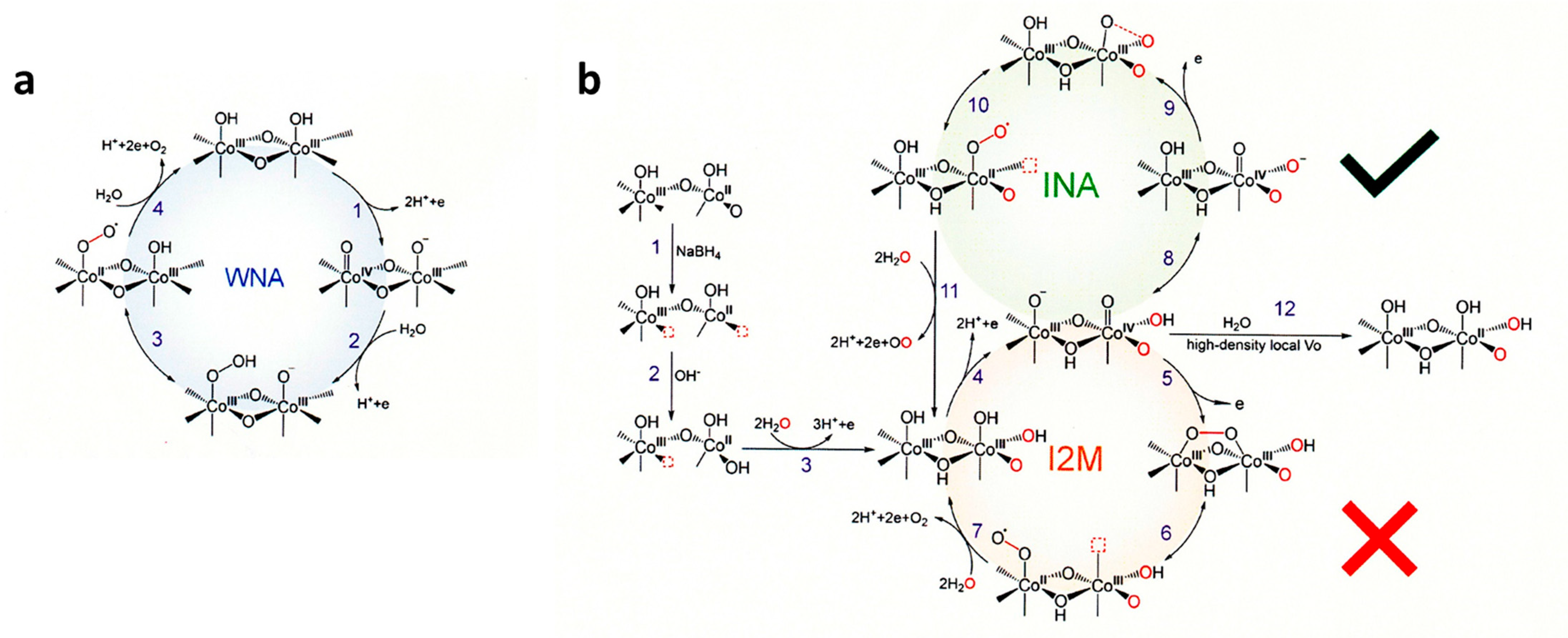
6.7.1. CoOx
6.7.2. NiOx
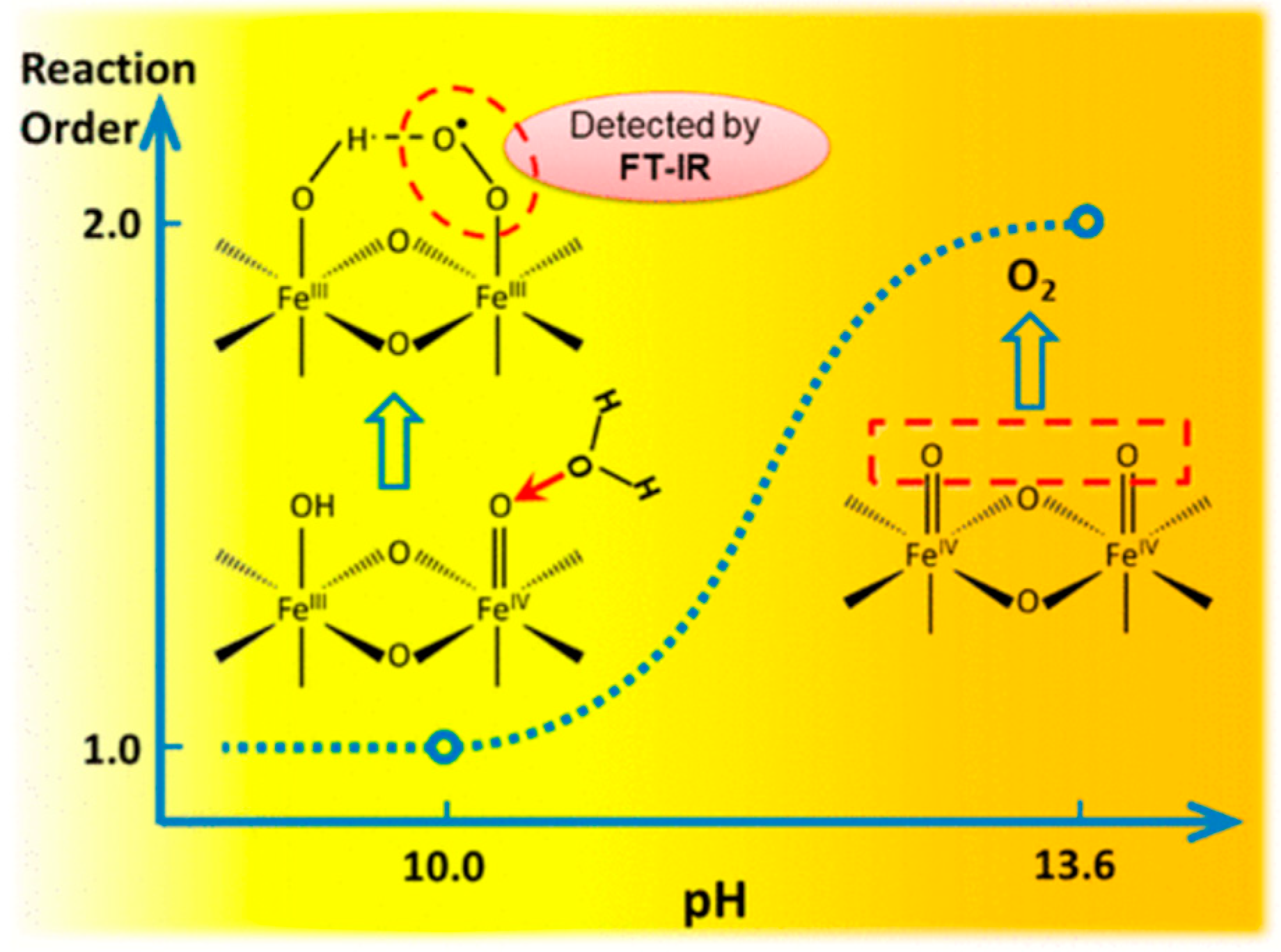
6.7.3. FeOx
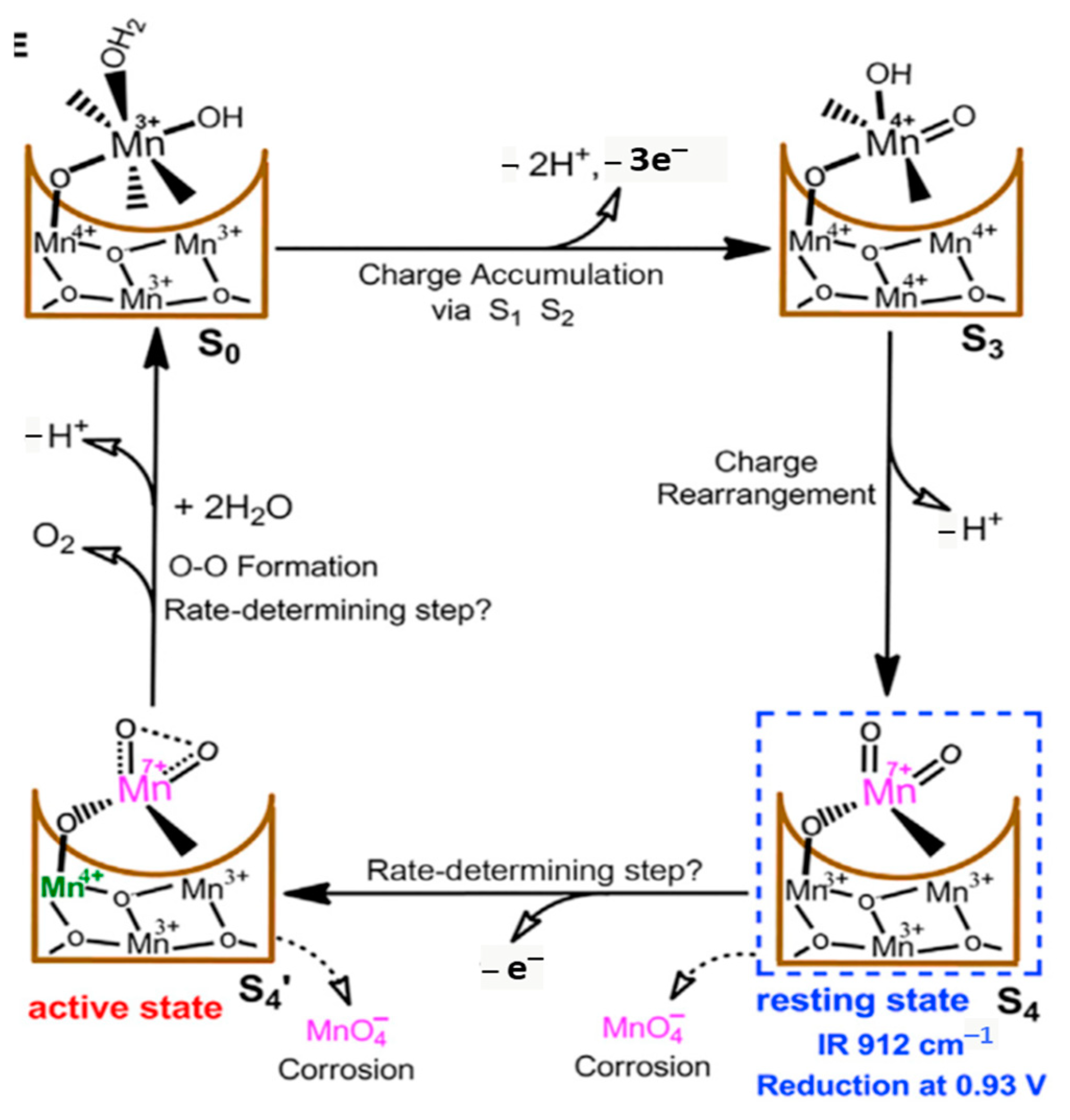
6.7.4. MnOx
6.7.5. Mixed Metal Oxides
6.8. Layered Double Hydroxide (LDH)
6.9. Metal–Organic Framework (MOF)
6.10. Metal Complexes
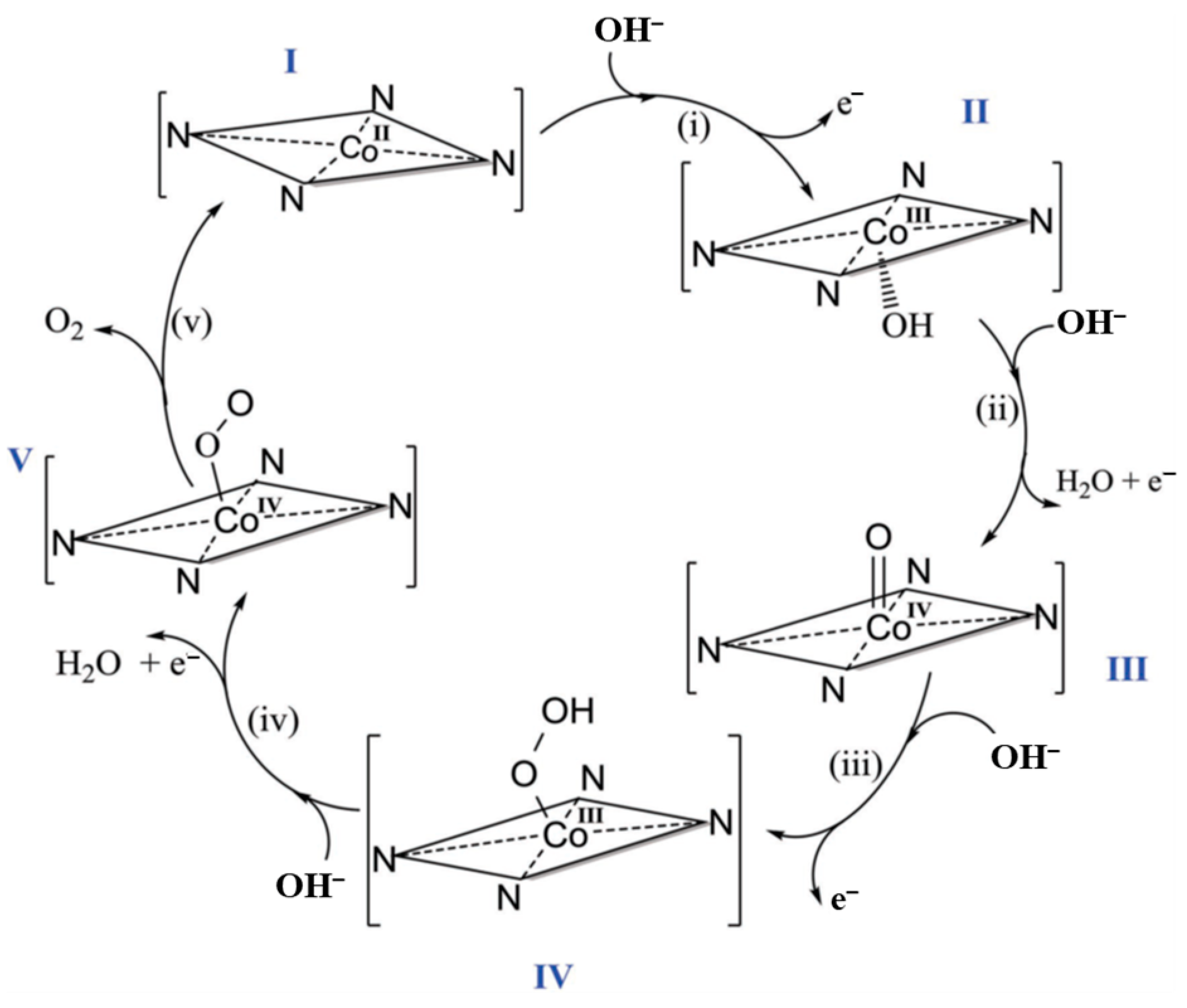
6.10.1. Mn Complexes
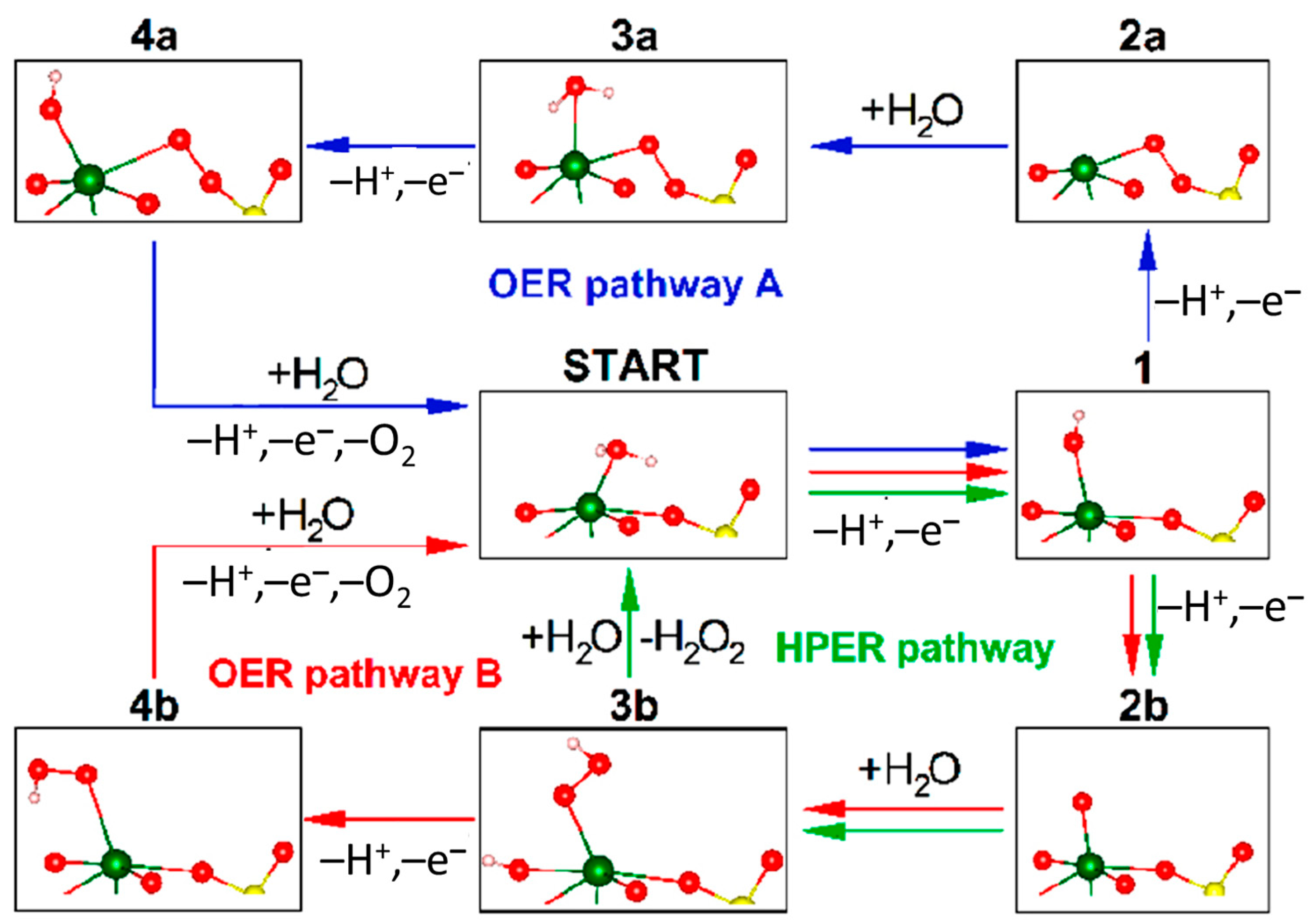
6.10.2. Fe Complexes
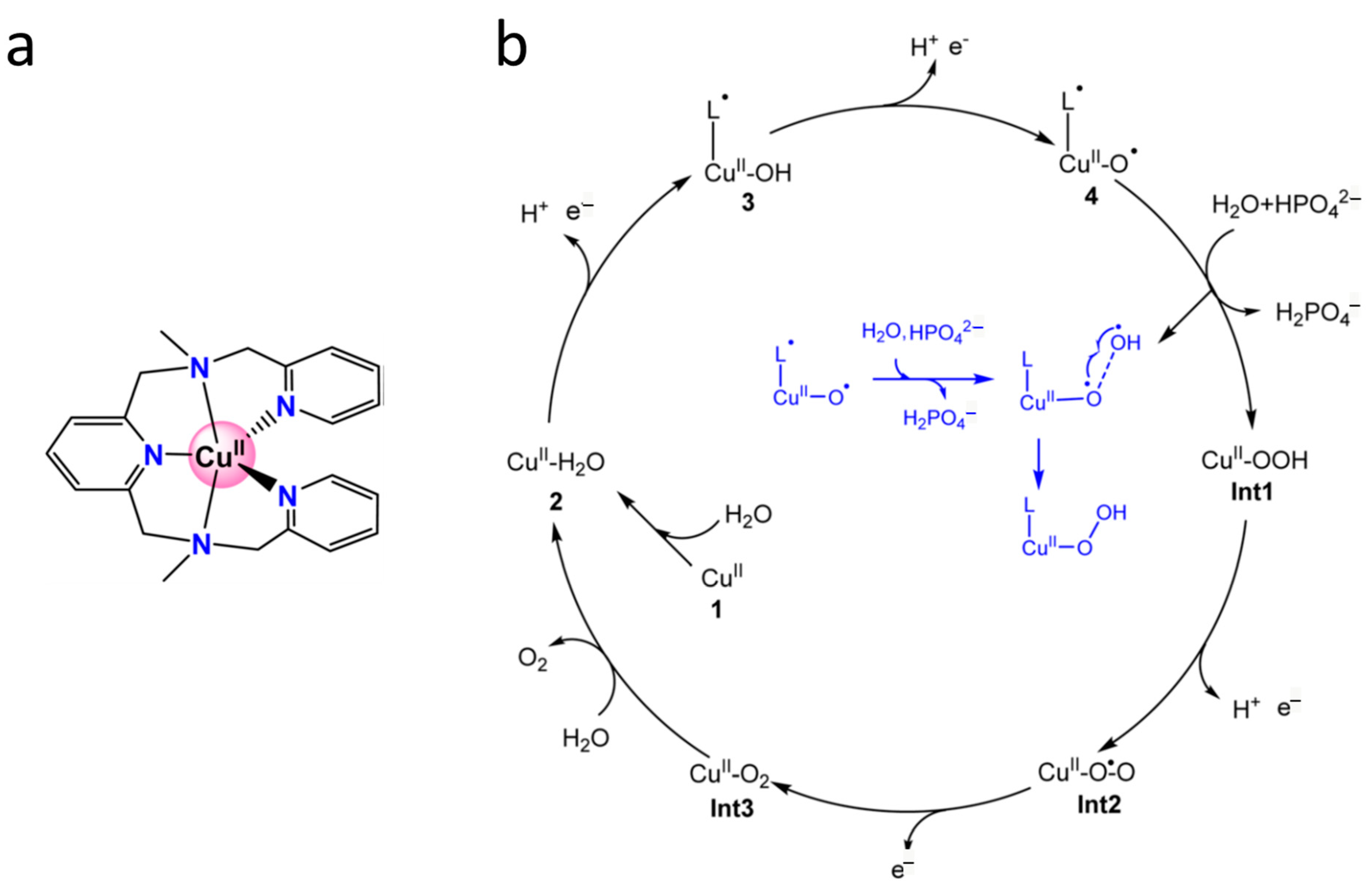
6.10.3. Cu Complexes
6.11. Single-Atom Catalysts (SACs)
6.12. Effect of Surface Functionalization
7. Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hocke, K. Oxygen in the Earth System. Oxygen 2023, 3, 19. [Google Scholar] [CrossRef]
- Nosaka, Y.; Nosaka, A.Y. Generation and detection of reactive oxygen species in photocatalysis. Chem. Rev. 2017, 117, 11302–11336. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, S.; Takayama, T.; Yamaguchi, Y.; Iwase, A.; Kudo, A. CO2 reduction using water as an electron donor over heterogeneous photocatalysts aiming at artificial photosynthesis. Acc. Chem. Res. 2022, 55, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Nosaka, Y.; Nosaka, A. Introduction to Photocatalysis: From Basic Science to Applications; Royal Society of Chemistry: Cambridge, UK, 2016; 272p. [Google Scholar] [CrossRef]
- Oliver, N.; Avramov, A.P.; Nürnberg, D.J.; Dau, H.; Burnap, R.L. From manganese oxidation to water oxidation: Assembly and evolution of the water-splitting complex in photosystem II. Photosynth. Res. 2022, 152, 107–133. [Google Scholar] [CrossRef] [PubMed]
- Najafpour, M.M.; Zaharieva, I.; Zand, Z.; Hosseini, S.M.; Kouzmanova, M.; Hołyńska, M.; Tranca, I.; Larkum, A.W.; Shen, J.-R.; Allakhverdiev, S.I. Water-oxidizing complex in photosystem II: Its structure and relation to manganese-oxide based catalysts. Coord. Chem. Rev. 2020, 409, 213183. [Google Scholar] [CrossRef]
- Yoneda, Y.; Arsenault, E.A.; Yang, S., Jr.; Orcutt, K.; Iwai, M.; Fleming, G.R. The initial charge separation step in oxygenic photosynthesis. Nat. Commun. 2022, 13, 2275. [Google Scholar] [CrossRef]
- Suga, M.; Akita, F.; Sugahara, M.; Kubo, M.; Nakajima, Y.; Nakane, T.; Yamashita, K.; Umena, Y.; Nakabayashi, M.; Yamane, T.; et al. Light-induced structural changes and the site of O=O bond formation in PSII caught by XFEL. Nature 2017, 543, 131–135. [Google Scholar] [CrossRef]
- Suga, M.; Akita, F.; Yamashita, K.; Nakajima, Y.; Ueno, G.; Li, H.; Yamane, T.; Hirata, K.; Umena, Y.; Yonekura, S.; et al. An oxyl/oxo mechanism for oxygen-oxygen coupling in PSII revealed by an X-ray free-electron laser. Science 2019, 366, 334–338. [Google Scholar] [CrossRef]
- Kato, Y.; Noguchi, T. Redox properties and regulatory mechanism of the iron-quinone electron acceptor in photosystem II as revealed by FTIR spectroelectrochemistry. Photosynth. Res. 2022, 152, 135–151. [Google Scholar] [CrossRef]
- Corry, T.A.; O’Malley, P.J. Evidence of O–O Bond formation in the final metastable S3 state of nature’s water oxidizing complex implying a novel mechanism of water oxidation. J. Phys. Chem. Lett. 2018, 9, 6269–6274. [Google Scholar] [CrossRef]
- Rummel, F.; O’Malley, P.J. How nature makes O2: An electronic level mechanism for water oxidation in photosynthesis. J. Phys. Chem. B 2022, 126, 8214–8221. [Google Scholar] [CrossRef]
- Davis, K.M.; Sullivan, B.T.; Palenik, M.C.; Yan, L.; Purohit, V.; Robison, G.; Kosheleva, I.; Henning, R.W.; Seidler, G.T.; Pushkar, Y. Rapid evolution of the photosystem II electronic structure during water splitting. Phys. Rev. X 2018, 8, 041014. [Google Scholar] [CrossRef]
- Simon, P.S.; Makita, H.; Bogacz, I.; Fuller, F.; Bhowmick, A.; Hussein, R.; Ibrahim, M.; Zhang, M.; Chatterjee, R.; Cheah, M.H.; et al. Capturing the sequence of events during the water oxidation reaction in photosynthesis using XFELs. FEBS Lett. 2023, 597, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, A.; Hussein, R.; Bogacz, I.; Simon, R.S.; Ibrahim, M.; Chatterjee, R.; Doyle, M.D.; Cheah, M.H.; Fransson, T.; Chernev, P.; et al. Structural evidence for intermediates during O2 formation in photosystem II. Nature 2023, 617, 629. [Google Scholar] [CrossRef]
- Greife, P.; Schönborn, M.; Capone, M.; Assunção, R.; Narzi, D.; Guidoni, L.; Dau, H. The electron–proton bottleneck of photosynthetic oxygen evolution. Nature 2023, 617, 623. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Zhang, H.; Sibbons, J.; Sun, H.; Wang, H.; Wang, S. Photoelectrochemical water oxidation and longevous photoelectric conversion by a photosystem II electrode. Adv. Energy. Mater. 2021, 11, 2100911. [Google Scholar] [CrossRef]
- de Levie, R. The electrolysis of water. J. Electroanal. Chem. 1999, 476, 92. [Google Scholar] [CrossRef]
- Fujishima, A.; Honda, K.; Kikuchi, S. Photosensitized electrolytic oxidation at TiO2 semiconductor electrodes. J. Chem. Soc. Jan. 1969, 72, 108–113. (In Japanese) [Google Scholar] [CrossRef]
- Longden, T.; Beck, F.J.; Jotzo, F.; Andrews, R.; Prasad, M. ‘Clean’ hydrogen?–Comparing the emissions and costs of fossil fuel versus renewable electricity based hydrogen. Appl. Energy 2022, 306 Pt B, 118145. [Google Scholar] [CrossRef]
- ul Haq, T.; Haik, Y. A roadmap towards sustainable anode design for alkaline water electrolysis. Appl. Catal. B Environ. 2023, 334, 122853. [Google Scholar] [CrossRef]
- An, L.; Wei, C.; Lu, M.; Liu, H.; Chen, Y.; Scherer, G.G.; Fisher, A.C.; Xi, P.; Xu, Z.J.; Yan, C.-H. Recent development of oxygen evolution electrocatalysts in acidic environment. Adv. Mater. 2021, 33, 2006328. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Budiyanto, E.; Tgysgz, H. Principles of water electrolysis and recent progress in cobalt-, nickel-, and iron-based oxides for the oxygen evolution reaction. Angew. Chem. Int. Ed. 2022, 61, e202103824. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Mu, S. Acidic oxygen evolution reaction: Mechanism, catalyst classification, and enhancement strategies. Interdiscip. Mater. 2023, 2, 53–90. [Google Scholar] [CrossRef]
- Vinodh, R.; Kalanur, S.S.; Natarajan, S.K.; Pollet, B.G. Recent advancements of polymeric membranes in anion exchange membrane water electrolyzer (AEMWE): A critical review. Polymers 2023, 15, 2144. [Google Scholar] [CrossRef]
- Lin, Y.; Dong, Y.; Wang, X.; Chen, L. Electrocatalysts for the oxygen evolution reaction in acidic media. Adv. Mater. 2022, 35, 2210565. [Google Scholar] [CrossRef]
- Jia, J.; Seitz, L.C.; Benck, J.D.; Huo, Y.; Chen, Y.; Wei, J.; Ng, D.; Bilir, T.; Harris, J.S.; Jaramillo, T.F. Solar water splitting by photovoltaic-electrolysis with a solar-to-hydrogen efficiency over 30%. Nat. Commun. 2016, 7, 13237. [Google Scholar] [CrossRef]
- Khan, M.A.; Al-Shankiti, I.; Ziani, A.; Wehbe, N.; Idriss, H. A stable integrated photoelectrochemical reactor for H2 production from water attains a solar-to-hydrogen efficiency of 18% at 15 suns and 13% at 207 suns. Angew. Chem. Int. Ed. 2020, 59, 14802–14808. [Google Scholar] [CrossRef]
- Karuturi, S.K.; Shen, H.; Sharma, A.; Beck, F.J.; Varadhan, P.; Duong, T.; Narangari, P.R.; Zhang, D.; Wan, Y.; He, J.-H.; et al. Over 17% efficiency stand-alone solar water splitting enabled by perovskite-silicon tandem absorbers. Adv. Energy Mater. 2020, 10, 2000772. [Google Scholar] [CrossRef]
- Holmes-Gentle, I.; Tembhurne, S.; Suter, C.; Haussener, S. Kilowatt-scale solar hydrogen production system using a concentrated integrated photoelectrochemical device. Nat. Energy 2023, 8, 586–596. [Google Scholar] [CrossRef]
- Anantharaj, S.; Aravindan, V. Developments and perspectives in 3d transition-metal-based electrocatalysts for neutral and near-neutral water electrolysis. Adv. Energy Mater. 2020, 10, 1902666. [Google Scholar] [CrossRef]
- Mayer, M.T. Photovoltage at semiconductor–electrolyte junctions. Curr. Opinion Electrochem. 2017, 2, 104–110. [Google Scholar] [CrossRef]
- Fujishima, A.; Honda, K. Electrochemical photolysis of water at a semiconductor electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef]
- Mesa, C.A.; Francàs, L.; Yang, K.R.; Garrido-Barros, P.; Pastor, E.; Ma, Y.; Kafizas, A.; Rosser, T.E.; Mayer, M.T.; Reisner, E.; et al. Multihole water oxidation catalysis on hematite photoanodes revealed by operando spectroelectrochemistry and DFT. Nat. Chem. 2020, 12, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Rajan, A.G.; Martirez, J.M.P.; Carter, E.A. Why do we use the materials and operating conditions we use for heterogeneous (photo)electrochemical water splitting? ACS Catal. 2020, 10, 11177–11234. [Google Scholar] [CrossRef]
- Tang, R.; Zhou, S.; Zhang, Z.; Zheng, R.; Huang, J. Engineering nanostructure–interface of photoanode materials toward photoelectrochemical water oxidation. Adv. Mater. 2021, 33, 2005389. [Google Scholar] [CrossRef] [PubMed]
- Cendula, P.; Bedoya-Lora, F.E.; Prabhakar, R.R. Semiconductor catalysts for oxygen and hydrogen evolution reactions. ACS Appl. Energy Mater. 2022, 5, 14593–14604. [Google Scholar] [CrossRef]
- Ertem, M.Z.; Kharche, N.; Batista, V.S.; Hybertsen, M.S.; Tully, J.C.; Muckerman, J.T. Photoinduced water oxidation at the aqueous GaN (10) interface: Deprotonation kinetics of the first proton-coupled electron-transfer step. ACS Catal. 2015, 5, 2317–2323. [Google Scholar] [CrossRef]
- Higashi, T.; Nishiyama, H.; Suzuki, Y.; Sasaki, Y.; Hisatomi, T.; Katayama, M.; Minegishi, T.; Seki, K.; Yamada, T.; Domen, K. Transparent Ta3N5 photoanodes for efficient oxygen evolution toward the development of tandem cells. Angew. Chem. Int. Ed. 2019, 58, 2300–2304. [Google Scholar] [CrossRef]
- Pihosh, Y.; Minegishi, T.; Nandal, V.; Higashi, T.; Katayama, M.; Yamada, T.; Sasaki, Y.; Seki, K.; Suzuki, Y.; Nakabayashi, M.; et al. Ta3N5-nanorods enabling highly efficient water oxidation via advantageous light harvesting and charge collection. Energy Environ. Sci. 2020, 13, 1519–1530. [Google Scholar] [CrossRef]
- Ma, Z.; Piętak, K.; Piątek, J.; DeMoulpied, J.R.; Rokicińska, A.; Kuśtrowski, P.; Dronskowski, R.; Zlotnik, S.; Coridan, R.H.; Slabon, A. Semi-transparent quaternary oxynitride photoanodes on GaN underlayers. Chem. Commun. 2020, 56, 13193–13196. [Google Scholar] [CrossRef]
- Huang, D.; Wang, K.; Li, L.; Feng, K.; An, N.; Ikeda, S.; Kuang, Y.; Ng, Y.; Jiang, F. 3.17% efficient Cu2ZnSnS4–BiVO4 integrated tandem cell for standalone overall solar water splitting. Energy Environ. Sci. 2021, 14, 1480–1489. [Google Scholar] [CrossRef]
- Pan, Z.; Chen, S.; Katayama, K. Roles of surface states in cocatalyst-loaded hematite photoanodes for water oxidation. J. Phys. Chem. C 2023, 127, 3904–3909. [Google Scholar] [CrossRef]
- Jeon, T.H.; Han, S.; Kim, B.; Park, C.; Kim, W.; Park, H.; Choi, W. High-valent iron redox-mediated photoelectrochemical water oxidation. ACS Energy Lett. 2022, 7, 59–66. [Google Scholar] [CrossRef]
- Morikawa, T.; Sato, S.; Sekizawa, K.; Suzuki, T.M.; Arai, T. Solar-driven CO2 reduction using a semiconductor/molecule hybrid photosystem: From photocatalysts to a monolithic artificial leaf. Acc. Chem. Res. 2022, 55, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Andrei, V.; Jagt, R.A.; Rahaman; Lari, M.L.; Lazarov, V.K.; MacManus-Driscoll, J.L.; Hoye, R.L.Z.; Reisner, E. Long-term solar water and CO2 splitting with photoelectrochemical BiOI–BiVO4 tandems. Nat. Mater. 2022, 21, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Takanabe, K. Photocatalytic water splitting: Quantitative approaches toward photocatalyst by design. ACS Catal. 2017, 7, 8006–8022. [Google Scholar] [CrossRef]
- Nosaka, Y.; Nosaka, A.Y. Intrinsic nature of photocatalysis by comparing with electrochemistry. Phys. Chem. Chem. Phys. 2020, 22, 7146–7154. [Google Scholar] [CrossRef]
- Wang, Q.; Domen, K. Particulate photocatalysts for light-driven water splitting: Mechanisms, challenges, and design strategies. Chem. Rev. 2020, 120, 919–985. [Google Scholar] [CrossRef]
- Tao, X.; Zhao, Y.; Wang, S.; Li, C.; Li, R. Recent advances and perspectives for solar-driven water splitting using particulate photocatalysts. Chem. Soc. Rev. 2022, 51, 3561–3608. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, H.; Yamada, T.; Nakabayashi, M.; Maehara, Y.; Yamaguchi, M.; Kuromiya, Y.; Nagatsuma, Y.; Tokudome, H.; Akiyama, S.; Watanabe, T.; et al. Photocatalytic solar hydrogen production from water on a 100-m2 scale. Nature 2021, 598, 304–307. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, K.; Xiang, Y.; Wang, J.; Lin, W.; Guo, M.; Ma, G. Facet-oriented assembly of Mo:BiVO4 and Rh:SrTiO3 particles: Integration of p–n conjugated photo-electrochemical system in a particle applied to photocatalytic overall water splitting. ACS Catal. 2022, 12, 2415–2425. [Google Scholar] [CrossRef]
- Zhou, P.; Navid, I.A.; Ma, Y.; Xiao, Y.; Wang, P.; Ye, Z.; Zhou, B.; Sun, K.; Mi, Z. Solar-to-hydrogen efficiency of more than 9% in photocatalytic water splitting. Nature 2023, 613, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, Y.; Li, C.M.; Wang, D. Mechanisms of water oxidation on heterogeneous catalyst surfaces. Nano Res. 2021, 14, 3446–3457. [Google Scholar] [CrossRef]
- Zhao, Y.; Saseendran, D.P.A.; Huang, C.; Triana, C.A.; Marks, W.R.; Chen, H.; Zhao, H.; Patzke, G.R. Oxygen evolution/reduction reaction catalysts: From in situ monitoring and reaction mechanisms to rational design. Chem. Rev. 2023, 123, 6257–6358. [Google Scholar] [CrossRef]
- Shinagawa, T.; Garcia-Esparza, A.T.; Takanabe, K. Insight on Tafel slopes from a microkinetic analysis of aqueous electrocatalysis for energy conversion. Sci. Rep. 2015, 5, 13801. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tao, H.B.; Kuang, M.; Yang, H.B.; Cai, W.; Yan, Q.; Mao, Q.; Liu, B. Advances in thermodynamic-kinetic model for analyzing the oxygen evolution reaction. ACS Catal. 2020, 10, 8597–8610. [Google Scholar] [CrossRef]
- Corby, S.; Rao, R.R.; Steier, L.; Durran, J.R. The kinetics of metal oxide photoanodes from charge generation to catalysis. Nat. Rev. Mater. 2021, 6, 1136–1155. [Google Scholar] [CrossRef]
- Francàs, F.; Corby, S.; Selim, S.; Lee, D.; Mesa, C.A.; Godin, R.; Pastor, E.; Stephens, I.E.L.; Choi, K.-S.; Durrant, J.R. Spectroelectrochemical study of water oxidation on nickel and iron oxyhydroxide electrocatalysts. Nat. Commun. 2019, 10, 5208. [Google Scholar] [CrossRef]
- Franc`as, L.; Selim, S.; Corby, S.; Lee, D.; Mesa, C.A.; Pastor, E.; Choi, K.-S.; Durrant, J.R. Water oxidation kinetics of nanoporous BiVO4 photoanodes functionalised with nickel/iron oxyhydroxide electrocatalysts. Chem. Sci. 2021, 12, 7442. [Google Scholar] [CrossRef]
- Zahran, Z.N.; Mohamed, E.A.; Naruta, Y. Kinetic and mechanism of heterogeneous water oxidation by α-Mn2O3 sintered on FTO electrode. ACS Catal. 2016, 6, 4470–4476. [Google Scholar] [CrossRef]
- Xu, Z.; Liang, Z.; Guo, W.; Zou, R. In situ/operando vibrational spectroscopy for the investigation of advanced nanostructured electrocatalysts. Coord. Chem. Rev. 2021, 436, 213824. [Google Scholar] [CrossRef]
- Li, N.; Cai, L.; Gao, G.; Lin, Y.; Wang, C.; Liu, H.; Liu, Y.; Duan, H.; Ji, Q.; Hu, W.; et al. Operando direct observation of stable water-oxidation intermediates on Ca2−XIrO4 nanocrystals for efficient acidic oxygen evolution. Nano Lett. 2022, 22, 6988–6996. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; de Respinis, M.; Frei, H. Time-resolved observations of water oxidation intermediates on a cobalt oxide nanoparticle catalyst. Nat. Chem. 2014, 6, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Zhou, W.; Zhang, H.; Zhou, W.; Zhao, X.; Li, Y.; Liu, M.; Cheng, W.; Liu, Q. Dynamic evolution of solid–liquid electrochemical interfaces over single-atom active sites. J. Am. Chem. Soc. 2020, 142, 12306–12313. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, X.; Li, Y.; Xu, Y.; Su, H.; Che, W.; He, J.; Zhang, H.; Liu, M.; Zhou, W.; et al. In situ construction of flexible V-Ni redox centers over Ni-based MOF nanosheet arrays for electrochemical water oxidation. Small Methods 2021, 5, 2100573. [Google Scholar] [CrossRef]
- Chu, H.-A.; Hillier, W.; Law, N.A.; Babcock, G.T. Vibrational spectroscopy of the oxygen-evolving complex and of manganese model compounds. Biochim. Biophys. Acta 2001, 1503, 69–82. [Google Scholar] [CrossRef]
- Zhang, B.; Daniel, Q.; Fan, L.; Liu, T.; Meng, Q.; Sun, L. Identifying MnVII-oxo species during electrochemical water oxidation by manganese oxide. iScience 2018, 4, 144–152. [Google Scholar] [CrossRef]
- Zandi, O.; Hamann, T. Determination of photoelectrochemical water oxidation intermediates on haematite electrode surfaces using operando infrared spectroscopy. Nat. Chem. 2016, 8, 778–783. [Google Scholar] [CrossRef]
- Herlihy, D.; Waegele, M.; Chen, X.; Pemmaraju, C.D.; Prendergast, D.; Cuk, T. Detecting the oxyl radical of photocatalytic water oxidation at an n-SrTiO3/aqueous interface through its subsurface vibration. Nat. Chem. 2016, 8, 549–555. [Google Scholar] [CrossRef]
- Su, H.; Zhou, W.; Zhou, W.; Li, Y.; Zheng, L.; Zhang, H.; Liu, M.; Zhang, X.; Sun, X.; Xu, Y.; et al. In-situ spectroscopic observation of dynamic coupling oxygen on atomically dispersed iridium electrocatalyst for acidic water oxidation. Nat. Commun. 2021, 12, 6118. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, X.-P.; Guo, M.; Lv, B.; Guo, K.; Jin, X.; Zhang, W.; Lee, Y.-M.; Fukuzum, S.; Nam, W.; et al. Identifying intermediates in electrocatalytic water oxidation with a manganese corrole complex. J. Am. Chem. Soc. 2021, 143, 14613–14621. [Google Scholar] [CrossRef] [PubMed]
- Vivek, J.P.; Berry, N.G.; Zou, J.; Nichols, R.J.; Hardwick, L.J. In situ surface-enhanced infrared spectroscopy to identify oxygen reduction products in nonaqueous metal–oxygen batteries. J. Phys. Chem. C 2017, 121, 19657–19667. [Google Scholar] [CrossRef]
- Lin, W.; Frei, H. Photochemical and FT-IR probing of the active site of hydrogen peroxide in Ti silicalite sieve. J. Am. Chem. Soc. 2002, 124, 9292–9298. [Google Scholar] [CrossRef]
- Nakamura, R.; Imanishi, A.; Murakoshi, K.; Nakato, Y. In situ FTIR studies of primary intermediates of photocatalytic reactions on nanocrystalline TiO2 films in contact with aqueous solutions. J. Am. Chem. Soc. 2003, 125, 7443–7450. [Google Scholar] [CrossRef]
- Zhou, Z.; Kong, Y.; Tan, H.; Huang, Q.; Wang, C.; Pei, Z.; Wang, H.; Liu, Y.; Wang, Y.; Li, S.; et al. Cation-vacancy-enriched nickel phosphide for efficient electrosynthesis of hydrogen peroxides. Adv. Mater. 2022, 34, 2106541. [Google Scholar] [CrossRef]
- Cheng, W.; Zhao, X.; Su, H.; Tang, F.; Che, W.; Zhang, H.; Liu, Q. Lattice-strained metal–organic-framework arrays for bifunctional oxygen electrocatalysis. Nat. Energy 2019, 4, 115–122. [Google Scholar] [CrossRef]
- Lee, S.; Banjac, K.; Lingenfelder, M.; Hu, X. Oxygen isotope labeling experiments reveal different reaction sites for the oxygen evolution reaction on nickel and nickel iron oxides. Angew. Chem. Int. Ed. 2019, 58, 10295–10299. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Ghatak, A.; Ro, Y.; Guillot, R.; Halime, Z.; Aukauloo, A.; Dey, A. Ligand radical mediated water oxidation by a family of copper o-phenylene bis-oxamidate complexes. Inorg. Chem. 2021, 60, 9442–9455. [Google Scholar] [CrossRef]
- Lin, C.; Li, J.; Li, X.; Yang, S.; Luo, W.; Zhang, Y.; Kim, S.-H.; Kim, D.-H.; Shinde, S.S.; Li, Y.-F.; et al. In-situ reconstructed Ru atom array on α-MnO with enhanced performance for acidic water oxidation. Nat Catal. 2021, 4, 1012–1023. [Google Scholar] [CrossRef]
- Chen, T.; Ding, Q.; Wang, X.; Feng, Z.; Li, C. Mechanistic studies on photocatalytic overall water splitting over Ga2O3-based photocatalysts by operando MS-FTIR spectroscopy. J. Phys. Chem. Lett. 2021, 12, 6029–6033. [Google Scholar] [CrossRef]
- Sivasankar, N.; Weare, W.W.; Frei, H. Direct observation of a hydroperoxide surface intermediate upon visible light-driven water oxidation at an Ir oxide nanocluster catalyst by rapid-scan FT-IR spectroscopy. J. Am. Chem. Soc. 2011, 133, 12976–12979. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, Y.; Mu, X.; Wu, Z.; Jin, X.; Li, J.; Xu, Y.; Yang, L.; Xi, X.; Jang, H.; et al. Spinel-anchored iridium single atoms enable efficient acidic water oxidation via intermediate stabilization effect. ACS Catal. 2023, 13, 3757–3767. [Google Scholar] [CrossRef]
- Shao, M.H.; Adzic, R.R. Spectroscopic identification of the reaction intermediates in oxygen reduction on gold in alkaline solutions. J. Phys. Chem. B 2005, 109, 16563–16566. [Google Scholar] [CrossRef]
- Liu, H.; Frei, H. Observation of O-O bond forming step of molecular Co4O4 cubane catalyst for water oxidation by rapid-scan FT-IR spectroscopy. ACS Catal. 2020, 10, 2138–2147. [Google Scholar] [CrossRef]
- Hunter, B.M.; Thompson, N.B.; Müller, A.M.; Rossman, G.R.; Hill, M.G.; Winkler, J.R.; Gray, H.B. Trapping an iron(VI) water-splitting intermediate in nonaqueous media. Joule 2018, 2, 747–763. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, H.; Liu, A.; Chen, C.; Song, W.; Zhao, J. Rate-limiting O–O bond formation pathways for water oxidation on hematite photoanode. J. Am. Chem. Soc. 2018, 140, 3264–3269. [Google Scholar] [CrossRef]
- Shao, M.-H.; Liu, P.; Adzic, R.R. Superoxide anion is the intermediate in the oxygen reduction reaction on platinum electrodes. J. Am. Chem. Soc. 2006, 128, 7408–7409. [Google Scholar] [CrossRef]
- Li, W.; Fu, H.; Cao, Y.; Wang, H.; Hao Yu, H.; Qiao, Z.; Liang, H.; Peng, F. Mn3O4@C nanoparticles supported on porous carbon as bifunctional oxygen electrodes and their electrocatalytic mechanism. ChemElectroChem 2019, 6, 359–368. [Google Scholar] [CrossRef]
- Hess, C. New advances in using Raman spectroscopy for the characterization of catalysts and catalytic reactions. Chem. Soc. Rev. 2021, 50, 3519–3564. [Google Scholar] [CrossRef]
- Huang, J.; Sheng, H.; Ross, R.D.; Han, J.; Wang, X.; Song, B.; Jin, S. Modifying redox properties and local bonding of Co3O4 by CeO2 enhances oxygen evolution catalysis in acid. Nat. Commun. 2021, 12, 3036. [Google Scholar] [CrossRef]
- Moysiadou, A.; Lee, S.; Hsu, C.-S.; Chen, H.M.; Hu, X. Mechanism of oxygen evolution catalyzed by cobalt oxyhydroxide: Cobalt superoxide species as a key intermediate and dioxygen release as a rate-determining step. J. Am. Chem. Soc. 2020, 142, 11901–11914. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Hu, Y.; Fan, C.; Yang, L.; Zhang, Y.; Li, H.; Xie, W. Surface-enhanced Raman spectroscopic evidence of key intermediate species and role of NiFe dual-catalytic center in water oxidation. Angew. Chem. Int. Ed. 2021, 60, 19774–19778. [Google Scholar] [CrossRef] [PubMed]
- Brückner, A. In situ electron paramagnetic resonance: A unique tool for analyzing structure–reactivity relationships in heterogeneous catalysis. Chem. Soc. Rev. 2010, 39, 4673–4684. [Google Scholar] [CrossRef] [PubMed]
- Brückner, A. In situ EPR spectroscopy in heterogeneous catalysis: Stepchild or ray of hope? Chem. Ingenieur Technik 2014, 86, 1871–1882. [Google Scholar] [CrossRef]
- Chiesa, M.; Giamello, E. On the role and applications of electron magnetic resonance techniques in surface chemistry and heterogeneous catalysis. Catal. Lett. 2021, 151, 3417–3436. [Google Scholar] [CrossRef]
- Balaghi, S.E.; Mehrabani, S.; Mousazade, Y.; Bagheri, R.; Sologubenko, A.S.; Song, Z.; Patzke, G.R.; Najafpour, M.M. Mechanistic understanding of water oxidation in the presence of a copper complex by in situ electrochemical liquid transmission electron microscopy. ACS Appl. Mater. Interfaces 2021, 13, 19927–19937. [Google Scholar] [CrossRef]
- Peña, N.O.; Ihiawakrim, D.; Han, M.; Lassalle-Kaiser, B.; Carenco, S.; Sanchez, C.; Laberty-Robert, C.; Portehault, D.; Ersen, O. Morphological and structural evolution of Co3O4 nanoparticles revealed by in situ electrochemical transmission electron microscopy during electrocatalytic water oxidation. ACS Nano 2019, 13, 11372–11381. [Google Scholar] [CrossRef]
- Zhao, G.; Yao, Y.; Lu, W.; Liu, G.; Guo, X.; Tricoli, A.; Zhu, Y. Direct observation of oxygen evolution and surface restructuring on Mn2O3 nano catalysts using in situ and ex situ transmission electron microscopy. Nano Lett. 2021, 21, 7012–7020. [Google Scholar] [CrossRef]
- Mom, R.; Frevel, L.; Velasco-Vélez, J.-J.; Plodinec, M.; Knop-Gericke, A.; Schlögl, R. The oxidation of platinum under wet conditions observed by electrochemical X-ray photoelectron spectroscopy. J. Am. Chem. Soc. 2019, 141, 6537–6544. [Google Scholar] [CrossRef]
- van Oversteeg, C.H.M.; Doan, H.Q.; de Groot, F.M.F.; Cuk, T. In situ X-ray absorption spectroscopy of transition metal based water oxidation catalysts. Chem. Soc. Rev. 2017, 46, 102–125. [Google Scholar] [CrossRef]
- Beaumont, S.K. Soft XAS as an in situ technique for the study of heterogeneous catalysts. Phys. Chem. Chem. Phys. 2020, 22, 18747–18756. [Google Scholar] [CrossRef] [PubMed]
- Cutsail III, G.E.; DeBeer, S. Challenges and opportunities for applications of advanced X-ray spectroscopy in catalysis research. ACS Catal. 2022, 12, 5864–5886. [Google Scholar] [CrossRef]
- Park, J.; Cho, J. Advances in understanding mechanisms of perovskites and pyrochlores as electrocatalysts using in-situ X-ray absorption spectroscopy. Angew. Chem. Int. Ed. 2020, 59, 15314–15324. [Google Scholar] [CrossRef]
- Chang, C.-J.; Zhu, Y.; Wang, J.; Chen, H.-C.; Tung, C.-W.; Chu, Y.-C.; Chen, H.M. In situ X-ray diffraction and X-ray absorption spectroscopy of electrocatalysts for energy conversion reactions. J. Mater. Chem. A 2020, 8, 19079–19112. [Google Scholar] [CrossRef]
- Zeng, Y.; Li, X.; Wang, J.; Sougrati, M.T.; Huang, Y.; Zhang, T.; Liu, B. In situ/operando Mössbauer spectroscopy for probing heterogeneous catalysis. Chem Catal. 2021, 1, 1215–1233. [Google Scholar] [CrossRef]
- Scherthan, L.; Schmidt, S.F.M.; Auerbach, H.; Hochdörffer, T.; Wolny, J.A.; Bi, W.; Zhao, J.; Hu, M.Y.; Toellner, T.; Alp, E.E.; et al. 161Dy time-domain synchrotron Mössbauer spectroscopy for investigating single-molecule magnets incorporating Dy ions. Angew. Chem. Int. Ed. 2019, 58, 3444–3449. [Google Scholar] [CrossRef]
- Kunze, S.; Grosse, P.; Lopez, M.B.; Sinev, I.; Zegkinoglou, I.; Mistry, H.; Timoshenko, J.; Hu, M.Y.; Zhao, J.; Alp, E.E.; et al. Operando NRIXS and XAFS investigation of segregation phenomena in Fe-Cu and Fe-Ag nanoparticle catalysts during CO2 electroreduction. Angew. Chem. Int. Ed. 2020, 59, 22667–22674. [Google Scholar] [CrossRef]
- Craig, M.J.; Coulter, G.; Dolan, E.; Soriano-López, J.; Mates-Torres, E.; Schmitt, W.; García-Melchor, M. Universal scaling relations for the rational design of molecular water oxidation catalysts with near-zero overpotential. Nat. Commun. 2019, 10, 4993. [Google Scholar] [CrossRef]
- Siahrostami, S.; Li, G.-L.; Viswanathan, V.; Nørskov, J.K. One- or two-electron water oxidation, hydroxyl radical, or H2O2 evolution. J. Phys. Chem. Lett. 2017, 8, 1157–1160. [Google Scholar] [CrossRef]
- Man, I.C.; Su, H.-Y.; Calle-Vallejo, F.; Hansen, H.A.; Martínez, J.I.; Inoglu, N.G.; Kitchin, J.; Jaramillo, T.F.; Nørskov, J.K.; Rossmeisl, J. Universality in oxygen evolution electrocatalysis on oxide surfaces. ChemCatChem 2011, 3, 1159–1165. [Google Scholar] [CrossRef]
- Zhang, N.; Chai, Y. Lattice oxygen redox chemistry in solid-state electrocatalysts for water oxidation. Energy Environ. Sci. 2021, 14, 4647–4671. [Google Scholar] [CrossRef]
- Nosaka, Y. Water photo-oxidation over TiO2—History and reaction mechanism. Catalysts 2022, 12, 1557. [Google Scholar] [CrossRef]
- Wang, D.; Sheng, T.; Chen, J.; Wang, H.-F.; Hu, P. Identifying the key obstacle in photocatalytic oxygen evolution on rutile TiO2. Nat. Catal. 2018, 1, 291–299. [Google Scholar] [CrossRef]
- Li, B.; Wu, S.; Gao, X. Theoretical calculation of a TiO2-based photocatalyst in the field of water splitting: A review. Nanotechnol. Rev. 2020, 9, 85. [Google Scholar] [CrossRef]
- Malik, A.S.; Fredin, L.A. Unraveling the water oxidation mechanism on a stoichiometric and reduced rutile TiO2(100) surface using first-principles calculations. J. Phys. Chem. C 2023, 127, 3444–3451. [Google Scholar] [CrossRef]
- Kakuma, Y.; Nosaka, A.Y.; Nosaka, Y. Difference of TiO2 photocatalytic mechanism between rutile and anatase studied by the detection of active oxygen and surface species in water. Phys. Chem. Chem. Phys. 2015, 17, 18691–18698. [Google Scholar] [CrossRef]
- Li, F.; Chen, J.-F.; Gong, X.-Q.; Hu, P.; Wang, D. Subtle structure matters: The vicinity of surface Ti5C cations alters the photooxidation behaviors of anatase and rutile TiO2 under aqueous environments. ACS Catal. 2022, 12, 8242–8251. [Google Scholar] [CrossRef]
- Nakamura, R.; Nakato, Y. Primary intermediates of oxygen photoevolution reaction on TiO2 (rutile) particles, revealed by in situ FTIR absorption and photoluminescence measurements. J. Am. Chem. Soc. 2004, 126, 1290–1298. [Google Scholar] [CrossRef]
- Nakamura, R.; Okamura, T.; Ohashi, N.; Imanishi, A.; Nakato, Y. Molecular mechanisms of photoinduced oxygen evolution, PL emission, and surface roughening at atomically smooth (110) and (100) n-TiO2 (rutile) surfaces in aqueous acidic solutions. J. Am. Chem. Soc. 2005, 127, 12975–12983. [Google Scholar] [CrossRef]
- Zhuang, Y.-B.; Cheng, J. Deciphering the anomalous acidic tendency of terminal water at rutile(110)–water interfaces. J. Phys. Chem. C 2023, 127, 10532–10540. [Google Scholar] [CrossRef]
- Kudo, A.; Miseki, Y. Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 2009, 38, 253–278. [Google Scholar] [CrossRef]
- Wang, Q.; Hisatomi, T.; Jia, Q.; Tokudome, H.; Zhong, M.; Wang, C.; Pan, Z.; Takata, T.; Nakabayashi, M.; Shibata, N.; et al. Scalable water splitting on particulate photocatalyst sheets with a solar-to-hydrogen energy conversion efficiency exceeding 1. Nat. Mater 2016, 15, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Rather, R.A.; Mehta, A.; Lu, Y.; Valant, M.; Fang, M.; Liu, W. Influence of exposed facets, morphology and hetero-interfaces of BiVO4 on photocatalytic water oxidation: A review. Int. J. Hydrogen Energy 2021, 46, 21866–21888. [Google Scholar] [CrossRef]
- Ma, Y.; Pendlebury, S.R.; Reynal, A.; Formal, F.L.; Durrant, J.R. Dynamics of photogenerated holes in undoped BiVO4 photoanodes for solar water oxidation. Chem. Sci. 2014, 5, 2964–2973. [Google Scholar] [CrossRef]
- Abdellaoui, I.; Islam, M.M.; Remeika, M.; Higuchi, Y.; Kawaguchi, T.; Harada, T.; Budich, C.; Maeda, T.; Wada, T.; Ikeda, S.; et al. Photocarrier recombination dynamics in BiVO4 for visible light-driven water oxidation. J. Phys. Chem. C 2020, 124, 3962–3972. [Google Scholar] [CrossRef]
- Tran-Phu, T.; Fusco, Z.; Bernardo, I.D.; Lipton-Duffin, J.; Toe, C.Y.; Daiyan, R.; Gengenbach, T.; Lin, C.-H.; Bo, R.; Nguyen, H.T.; et al. Understanding the role of vanadium vacancies in BiVO4 for efficient photoelectrochemical water oxidation. Chem. Mater. 2021, 33, 3553–3565. [Google Scholar] [CrossRef]
- Walsh, A.; Yan, Y.; Huda, M.N.; Al-Jassim, M.M.; Wei, S.-H. Band edge electronic structure of BiVO4: Elucidating the role of the Bi s and V d orbitals. Chem. Mater. 2009, 21, 547–551. [Google Scholar] [CrossRef]
- Hu, J.; Zhao, X.; Chen, W.; Su, H.; Chen, Z. Theoretical insight into the mechanism of photoelectrochemical oxygen evolution reaction on BiVO4 anode with oxygen vacancy. J. Phys. Chem. C 2017, 121, 18702–18709. [Google Scholar] [CrossRef]
- Liu, G.; Li, F.; Zhu, Y.; Lia, J.L.; Sun, L. Cobalt doped BiVO4 with rich oxygen vacancies for efficient photoelectrochemical water oxidation. RSC Adv. 2020, 10, 28523–28526. [Google Scholar] [CrossRef]
- Liu, T.; Liu, R.; Li, Q.; Yang, J. Theoretical insight into the role of defects and facets in the selectivity of products in water oxidation over bismuth vanadate (BiVO4). ACS Sustain. Chem. Eng. 2020, 8, 1980–1988. [Google Scholar] [CrossRef]
- Wang, W.; Strohbeen, P.J.; Lee, D.; Zhou, C.; Kawasaki, J.K.; Choi, K.-S.; Liu, M.; Galli, G. The role of surface oxygen vacancies in BiVO4. Chem. Mater. 2020, 32, 2899–2909. [Google Scholar] [CrossRef]
- Steinitz-Eliyahu, R.; Hernangómez-Pérez, D.; Hegner, F.S.; Nikačević, P.; López, N.; Refaely-Abramson, S. Mixed excitonic nature in water-oxidized BiVO4 surfaces with defects. Phys. Rev. Mater. 2022, 6, 065402. [Google Scholar] [CrossRef]
- Huang, M.; He, W.; Xu, Z.; Zhu, H. Enhanced catalytic mechanism of twin-structured BiVO4. J. Phys. Chem. Lett. 2021, 12, 10610–10615. [Google Scholar] [CrossRef] [PubMed]
- Nikačević, P.; Hegner, F.S.; Galán-Mascarós, J.R.; López, N. Influence of oxygen vacancies and surface facets on water oxidation selectivity toward oxygen or hydrogen peroxide with BiVO4. ACS Catal. 2021, 11, 13416–13422. [Google Scholar] [CrossRef]
- Park, H.S.; Leonard, K.C.; Bard, A.J. Surface interrogation scanning electrochemical microscopy (SISECM) of photoelectrochemistry at a W/Mo-BiVO4 semiconductor electrode. J. Phys. Chem. C 2013, 117, 12093–12102. [Google Scholar] [CrossRef]
- Nakabayashi, Y.; Nishikawa, M.; Saito, N.; Terashima, C.; Fujishima, A. Significance of hydroxyl radical in photoinduced oxygen evolution in water on monoclinic bismuth vanadate. J. Phys. Chem. C 2017, 121, 25624–25631. [Google Scholar] [CrossRef]
- Nakabayashi, Y.; Suzuki, N.; Terashima, C.; Fujishima, A. In situ infrared analysis for the process of water photo-oxidation on monoclinic bismuth vanadate. J. Phys. Chem. C 2021, 125, 18579–18587. [Google Scholar] [CrossRef]
- Sun, Q.; Ren, K.; Qi, L. Boosting the performance of BiVO4 photoanodes by the simultaneous introduction of oxygen vacancies and cocatalyst via photoelectrodeposition. ACS Appl. Mater. Interfaces 2022, 14, 37833–37842. [Google Scholar] [CrossRef]
- Pan, L.; Wu, J.; Xu, X.; Lv, F.; Chen, Y.; Guo, L. Photoelectrochemical performance of bismuth vanadate photoanode for water splitting under concentrated light irradiation. Int. J. Hydrogen Energy 2023, 48, 13479–13488. [Google Scholar] [CrossRef]
- Majumder, S.; Su, X.; Kim, K.H. Effective strategy of incorporating Co3O4 as a co-catalyst onto an innovative BiVO4/Fe2TiO5 core-shell heterojunction for effective photoelectrochemical water-splitting application. Surf. Interfaces. 2023, 39, 102936. [Google Scholar] [CrossRef]
- Avcıoǧlu, C.; Avcıoǧlu, S.; Bekheet, M.F.; Gurlo, A. Photocatalytic overall water splitting by SrTiO3: Progress report and design strategies. ACS Appl. Energy Mater. 2023, 6, 1134–1154. [Google Scholar] [CrossRef]
- Chen, X.; Choing, S.N.; Aschaffenburg, D.J.; Pemmaraju, C.D.; Prendergast, D.; Cuk, T. The formation time of Ti–O• and Ti–O•–Ti radicals at the n-SrTiO3/aqueous interface during photocatalytic water oxidation. J. Am. Chem. Soc. 2017, 139, 1830–1841. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Aschaffenburg, D.J.; Cuk, T. Selecting between two transition states by which water oxidation intermediates decay on an oxide surface. Nat. Catal. 2019, 2, 820–827. [Google Scholar] [CrossRef]
- Vinogradov, I.; Singh, S.; Lyle, H.; Paolino, M.; Mandal, A.; Rossmeisl, J.; Cuk, T. Free energy difference to create the M-OH• intermediate of the oxygen evolution reaction by time-resolved optical spectroscopy. Nat. Mater. 2022, 21, 88–94. [Google Scholar] [CrossRef]
- Lyle, H.; Singh, S.; Magnano, E.; Nappini, S.; Bondino, F.; Yazdi, S.; Cuk, T. Assessing and quantifying thermodynamically concomitant degradation during oxygen evolution from water on SrTiO3. ACS Catal. 2023, 13, 8206–8218. [Google Scholar] [CrossRef]
- Zhou, X.; Hensen, E.J.M.; van Santen, R.A.; Li, C. DFT simulations of water adsorption and activation on low-index α-Ga2O3 surfaces. Chem. Eur. J. 2014, 20, 6915–6926. [Google Scholar] [CrossRef]
- Wang, Q.; Nakabayashi, M.; Hisatomi, T.; Sun, S.; Akiyama, S.; Wang, Z.; Pan, Z.; Xiao, X.; Watanabe, T.; Yamada, T.; et al. Oxysulfide photocatalyst for visible-light-driven overall water splitting. Nat. Mater. 2019, 18, 827–832. [Google Scholar] [CrossRef]
- Yu, H.; Ge, J. Recent advances in Ru-based electrocatalysts toward acid electrochemical water oxidation. Curr. Opinion Electrochem. 2023, 39, 101296. [Google Scholar] [CrossRef]
- Raman, A.S.; Vojvodic, A. Providing atomistic insights into the dissolution of rutile oxides in electrocatalytic water splitting. J. Phys. Chem. C 2022, 126, 922–932. [Google Scholar] [CrossRef]
- Liu, S.; Chang, Y.; He, N.; Zhu, S.; Wang, L.; Liu, X. Competition between lattice oxygen and adsorbate evolving mechanisms in rutile Ru-based oxide for the oxygen evolution reaction. ACS Appl. Mater. Interf. 2023, 15, 20563–20570. [Google Scholar] [CrossRef]
- Pavlovic, Z.; Ranjan, C.; Gao, Q.; van Gastel, M.; Schlögl, R. Probing the structure of a water-oxidizing anodic iridium oxide catalyst using Raman spectroscopy. ACS Catal. 2016, 6, 8098–8105. [Google Scholar] [CrossRef]
- Ooka, H.; Yamaguchi, A.; Takashima, T.; Hashimoto, K.; Nakamura, R. Efficiency of oxygen evolution on iridium oxide determined from the pH dependence of charge accumulation. J. Phys. Chem. C 2017, 121, 17873–17881. [Google Scholar] [CrossRef]
- Bozal-Ginesta, C.; Rao, R.R.; Mesa, C.A.; Liu, X.; Hillman, S.A.J.; Stephens, I.E.L.; Durrant, J.R. Redox-state kinetics in water-oxidation IrOX electrocatalysts measured by operando spectroelectrochemistry. ACS Catal. 2021, 11, 15013–15025. [Google Scholar] [CrossRef]
- Czioska, S.; Boubnov, A.; Escalera-López, D.; Geppert, J.; Zagalskaya, A.; Röse, P.; Saraçi, E.; Alexandrov, V.; Krewer, U.; Cherevko, S.; et al. Increased Ir−Ir interaction in iridium oxide during the oxygen evolution reaction at high potentials probed by operando spectroscopy. ACS Catal. 2021, 11, 10043–10057. [Google Scholar] [CrossRef]
- Ping, Y.; Nielsen, R.J.; Goddard III, W.A. The reaction mechanism with free energy barriers at constant potentials for the oxygen evolution reaction at the IrO2(110) surface. J. Am. Chem. Soc. 2017, 139, 149–155. [Google Scholar] [CrossRef]
- Binninger, T.; Doublet, M.-L. The Ir–OOOO–Ir transition state and the mechanism of the oxygen evolution reaction on IrO2(110). Energy Environ. Sci. 2022, 15, 2519–2528. [Google Scholar] [CrossRef]
- Liao, F.; Yin, K.; Ji, Y.; Zhu, W.; Fan, Z.; Li, Y.; Zhong, J.; Shao, M.; Kang, Z.; Shao, Q. Iridium oxide nanoribbons with metastable monoclinic phase for highly efficient electrocatalytic oxygen evolution. Nat. Commun. 2023, 14, 1248. [Google Scholar] [CrossRef]
- Xie, Y.; Chang, C.; Luo, F.; Yang, Z. Modulation in the d band of Ir by core–shell construction for robust water splitting electrocatalysts in acid. ACS Appl. Mater. Interfaces. 2023, 15, 20081–20088. [Google Scholar] [CrossRef]
- Reksten, A.H.; Thuv, H.; Seland, F.; Sunde, S. The oxygen evolution reaction mechanism at IrXRu1−XO2 powders produced by hydrolysis synthesis. J. Electroanal. Chem. 2018, 819, 547–561. [Google Scholar] [CrossRef]
- Rao, R.R.; Kolb, M.J.; Halck, N.B.; Pedersen, A.F.; Mehta, A.; You, H.; Stoerzinger, K.A.; Feng, Z.; Hansen, H.A.; Zhou, H.; et al. Towards identifying the active sites on RuO2(110) in catalyzing oxygen evolution. Energy Environ. Sci. 2017, 10, 2626–2637. [Google Scholar] [CrossRef]
- Liang, Q.; Bieberle-Hütter, A.; Brocks, G. Anti-ferromagnetic RuO2: A stable and robust OER catalyst over a large range of surface terminations. J. Phys. Chem. C 2022, 126, 1337–1345. [Google Scholar] [CrossRef]
- Stoerzinger, K.A.; Diaz-Morales, O.; Kolb, M.; Rao, R.R.; Frydendal, R.; Qiao, L.; Wang, X.R.; Halck, N.B.; Rossmeisl, J.; Hansen, H.A.; et al. Orientation-dependent oxygen evolution on RuO2 without lattice exchange. ACS Energy Lett. 2017, 2, 876–881. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, R.; Ding, Y.; Zhang, B.; Li, H.; Bai, B.; Li, M.; Cui, Y.; Xiao, J.; Wu, Z.-S. Unraveling oxygen vacancy site mechanism of Rh-doped RuO2 catalyst for long-lasting acidic water oxidation. Nat. Commun. 2023, 14, 1412. [Google Scholar] [CrossRef]
- Zagalskaya, A.; Alexandrov, V. Role of defects in the interplay between adsorbate evolving and lattice oxygen mechanisms of the oxygen evolution reaction in RuO2 and IrO2. ACS Catal. 2020, 10, 3650–3657. [Google Scholar] [CrossRef]
- Wang, X.; Wan, X.; Qin, X.; Chen, C.; Qian, X.; Guo, Y.; Xu, Q.; Cai, W.-B.; Yang, H.; Jiang, K. Electronic structure modulation of RuO2 by TiO2 enriched with oxygen vacancies to boost acidic O2 evolution. ACS Catal. 2022, 12, 9437–9445. [Google Scholar] [CrossRef]
- Godínez-Salomón, J.F.; Ospina-Acevedo, F.; Albiter, L.A.; Bailey, K.O.; Naymik, Z.G.; Mendoza-Cruz, R.; Balbuena, P.B.; Rhodes, C.P. Titanium substitution effects on the structure, activity, and stability of nanoscale ruthenium oxide oxygen evolution electrocatalysts: Experimental and computational study. ACS Appl. Nano Mater. 2022, 5, 11752–11775. [Google Scholar] [CrossRef]
- Shi, Z.; Li, J.; Wang, Y.; Liu, S.; Zhu, J.; Yang, J.; Wang, X.; Ni, J.; Jiang, Z.; Zhang, L.; et al. Customized reaction route for ruthenium oxide towards stabilized water oxidation in high-performance PEM electrolyzers. Nat. Commun. 2023, 14, 843. [Google Scholar] [CrossRef]
- Xue, Y.; Fang, J.; Wang, X.; Xu, Z.; Zhang, Y.; Lv, Q.; Liu, M.; Zhu, W.; Zhuang, Z. Sulfate-functionalized RuFeOx as highly efficient oxygen evolution reaction electrocatalyst in acid. Adv. Funct. Mater. 2021, 31, 2101405. [Google Scholar] [CrossRef]
- Shang, C.; Xiao, X.; Xu, Q. Coordination chemistry in modulating electronic structures of perovskite-type oxide nanocrystals for oxygen evolution catalysis. Coord. Chem. Rev. 2023, 485, 215109. [Google Scholar] [CrossRef]
- Hong, W.T.; Stoerzinger, K.A.; Lee, Y.-L.; Giordano, L.; Grimaud, A.; Johnson, A.M.; Hwang, J.; Crumlin, E.J.; Yange, W.; Shao-Horn, Y. Charge-transfer-energy-dependent oxygen evolution reaction mechanisms for perovskite oxides. Energy Environ. Sci. 2017, 10, 2190–2200. [Google Scholar] [CrossRef]
- Shi, Z.; Wang, X.; Ge, J.; Liu, C.; Xing, W. Fundamental understanding of the acidic oxygen evolution reaction: Mechanism study and state-of-the-art catalysts. Nanoscale 2020, 12, 13249–13275. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Zheng, Y.; Hu, Y.; Huang, B.; Ji, D.; Sun, M.; Li, J.; Peng, Y.; Si, R.; Xi, P.; et al. Artificially steering electrocatalytic oxygen evolution reaction mechanism by regulating oxygen defect contents in perovskites. Sci. Adv. 2022, 8, eabq3563. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Huang, J.; Zhu, Y.; Zhou, H.; Hu, Z.; Liao, Y.-K.; Lai, Y.-H.; Chen, C.-T.; Chu, Y.-H.; Zhang, K.H.L.; et al. Structural anisotropy determining the oxygen evolution mechanism of strongly correlated perovskite nickelate electrocatalyst. ACS Sustain. Chem. Eng. 2021, 9, 4262–4270. [Google Scholar] [CrossRef]
- Sun, Y.; Wu, C.-R.; Ding, T.-Y.; Gu, J.; Yan, J.-W.; Cheng, J.; Zhang, K.H.L. Direct observation of the dynamic reconstructed active phase of perovskite LaNiO3 for the oxygen evolution reaction. Chem. Sci. 2023, 14, 5906–5911. [Google Scholar] [CrossRef]
- Lee, S.; Kishore, M.R.A.; Kim, D.; Kang, H.; Chun, J.; Oh, L.S.; Park, J.H.; Kim, H.J.; Yoo, J.S.; Lim, E. Direct O–O coupling promoted the oxygen evolution reaction by dual active sites from Ag/LaNiO3 interfaces. ACS Appl. Energy Mater. 2022, 5, 14658–14668. [Google Scholar] [CrossRef]
- Chen, H.; Shi, L.; Sun, K.; Zhang, K.; Liu, Q.; Ge, J.; Liang, X.; Tian, B.; Huang, Y.; Shi, Z.; et al. Protonated iridate nanosheets with a highly active and stable layered perovskite framework for acidic oxygen evolution. ACS Catal. 2022, 12, 8658–8666. [Google Scholar] [CrossRef]
- Han, J.; Guan, J. Multicomponent transition metal oxides and (oxy)hydroxides for oxygen evolution. Nano Res. 2023, 16, 1913–1966. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, K.H.L.; Hofmann, J.P.; O’Shea, V.A.; Oropeza, F.E. The electronic structure of transition metal oxides for oxygen evolution reaction. J. Mater. Chem. A 2021, 9, 19465–19488. [Google Scholar] [CrossRef]
- Feng, Y.; Yang, H.; Wang, X.; Hu, C.; Jing, H.; Cheng, J. Role of transition metals in catalyst designs for oxygen evolution reaction: A comprehensive review. Int. J. Hydrogen Energy 2022, 47, 17946–17970. [Google Scholar] [CrossRef]
- Guo, T.; Li, L.; Wang, Z. Recent development and future perspectives of amorphous transition metal-based electrocatalysts for oxygen evolution reaction. Adv. Energy Mater. 2022, 12, 2200827. [Google Scholar] [CrossRef]
- Huang, C.-J.; Xu, H.-M.; Shuai, T.-Y.; Zhan, Q.-N.; Zhang, Z.-J.; Li, G.-R. A review of modulation strategies for improving catalytic performance of transition metal phosphides for oxygen evolution reaction. App. Catal. B Environ. 2023, 325, 122313. [Google Scholar] [CrossRef]
- Zhang, W.; Cao, R. Switching the O–O bond formation mechanism by controlling water activity. Chem 2021, 7, 1981–1992. [Google Scholar] [CrossRef]
- Takata, T.; Jiang, J.; Sakata, Y.; Nakabayashi, M.; Shibata, N.; Nandal, V.; Seki, K.; Hisatomi, T.; Domen, K. Photocatalytic water splitting with a quantum efficiency of almost unity. Nature 2020, 581, 411–414. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Deng, R.; Guo, M.; Zhang, Q. Recent progress of advanced Co3O4-based materials for electrocatalytic oxygen evolution reaction in acid: From rational screening to efficient design. Int. J. Hydrogen Energy, 2023; in press. [Google Scholar] [CrossRef]
- Favaro, M.; Yang, J.; Nappini, S.; Magnano, E.; Toma, F.M.; Crumlin, E.J.; Yano, J.; Sharp, I.D. Understanding the oxygen evolution reaction mechanism on CoOX using operando ambient-pressure X-ray photoelectron spectroscopy. J. Am. Chem. Soc. 2017, 139, 8960–8970. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.; Li, J.; Yang, K.R.; Wang, Y.; He, D.; Thorne, J.E.; Croslow, S.; Dong, Q.; Zhao, Y.; Prostko, G.; et al. Observation of a potential-dependent switch of water-oxidation mechanism on Co-oxide-based catalysts. Chem 2021, 7, 2101–2117. [Google Scholar] [CrossRef]
- Wang, S.; Jiang, Q.; Ju, S.; Hsu, C.-S.; Chen, H.M.; Zhang, D.; Song, F. Identifying the geometric catalytic active sites of crystalline cobalt oxyhydroxides for oxygen evolution reaction. Nat. Commun. 2022, 13, 6650. [Google Scholar] [CrossRef]
- Kang, W.; Wei, R.; Yin, H.; Li, D.; Chen, Z.; Huang, Q.; Zhang, P.; Jing, H.; Wang, X.; Li, C. Unraveling sequential oxidation kinetics and determining roles of multi-cobalt active sites on Co3O4 catalyst for water oxidation. J. Am. Chem. Soc. 2023, 145, 3470–3477. [Google Scholar] [CrossRef]
- Lin, Y.; Yu, L.; Tang, L.; Song, F.; Schlögl, R.; Heumann, S. In situ identification and time-resolved observation of the interfacial state and reactive intermediates on a cobalt oxide nanocatalyst for the oxygen evolution reaction. ACS Catal. 2022, 12, 5345–5355. [Google Scholar] [CrossRef]
- Zhou, D.; Li, F.; Zhao, Y.; Wang, L.; Zou, H.; Shan, Y.; Fu, J.; Ding, Y.; Duan, L.; Liu, M.; et al. Mechanistic regulation by oxygen vacancies in structural evolution promoting electrocatalytic water oxidation. ACS Catal. 2023, 13, 4398–4408. [Google Scholar] [CrossRef]
- Ren, X.; Ji, Y.; Zhai, Y.; Yuan, N.; Ding, J.; Li, Y.; Yan, J.; Liu, S.F. Self-assembled CoOOH on TiO2 for enhanced photoelectrochemical water oxidation. J. Energy Chem. 2021, 60, 512–521. [Google Scholar] [CrossRef]
- Yao, N.; Wang, G.; Jia, H.; Yin, J.; Cong, H.; Chen, S.; Luo, W. Intermolecular energy gap-induced formation of high-valent cobalt species in CoOOH surface layer on cobalt sulfides for efficient water oxidation. Angew. Chem. Int. Ed. 2022, 61, e202117178. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Li, H.; Xia, Y.; Wang, L.; Yin, K.; Wei, Y.; Liu, X.; Luo, S. Co3O4 nanocrystals with oxygen vacancy-rich and highly reactive (222) facet on carbon nitride scaffolds for efficient photocatalytic oxygen evolution. ACS Appl. Mater. Interfaces 2020, 12, 44608–44616. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, K.; Auer, A.A. Oxygen evolution reaction electrocatalysis on cobalt(oxy)hydroxide: Role of Fe impurities. J. Phys. Chem. C 2022, 126, 18623–18635. [Google Scholar] [CrossRef]
- Li, L.-F.; Li, Y.-F.; Liu, Z.-P. Oxygen evolution activity on NiOOH catalysts: Four-coordinated Ni cation as the active site and the hydroperoxide mechanism. ACS Catal. 2020, 10, 2581–2590. [Google Scholar] [CrossRef]
- Wang, X.; Xi, S.; Huang, P.; Du, Y.; Zhong, H.; Wang, Q.; Borgna, A.; Zhang, Y.-W.; Wang, Z.; Wang, H.; et al. Pivotal role of reversible NiO6 geometric conversion in oxygen evolution. Nature 2022, 611, 702–708. [Google Scholar] [CrossRef]
- Wang, H.; Lu, S. Light inducing the geometric conversion of NiO6 to trigger a faster oxygen evolution reaction pathway: The coupled oxygen evolution mechanism. Energy Environ. Mater. 2023, 6, e12558. [Google Scholar] [CrossRef]
- Jiang, J.; Sun, F.; Zhou, S.; Hu, W.; Zhang, H.; Dong, J.; Jiang, Z.; Zhao, J.; Li, J.; Yan, W.; et al. Atomic-level insight into super-efficient electrocatalytic oxygen evolution on iron and vanadium co-doped nickel (oxy)hydroxide. Nat. Commun. 2018, 9, 2885. [Google Scholar] [CrossRef]
- Rao, R.R.; Corby, S.; Bucci, A.; García-Tecedor, M.; Mesa, C.A.; Rossmeisl, J.; Giménez, S.; Lloret-Fillol, J.; Stephens, I.E.L.; Durrant, J.R. Spectroelectrochemical analysis of the water oxidation mechanism on doped nickel oxides. J. Am. Chem. Soc. 2022, 144, 7622–7633. [Google Scholar] [CrossRef]
- Li, T.; Zhang, L.; Wang, J.; Zhang, X.; Zhang, L.; Wang, M.; Yan, C.; Tao Qian, T. Facilitating reconstruction of the hetero interface electronic structure by the enriched oxygen vacancy for the oxygen evolution reaction. Inorg. Chem. 2023, 62, 10504–10512. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, J.; Wang, Z.; Yuan, H.; Lu, Z.; Luo, B.; Tian, E.; Waterhouse, G.I.N. Structural and electronic engineering of Ir-doped Ni-(oxy)hydroxide nanosheets for enhanced oxygen evolution activity. ACS Catal. 2021, 11, 5386–5395. [Google Scholar] [CrossRef]
- Tian, B.; Shin, H.; Liu, S.; Fei, M.; Mu, Z.; Liu, C.; Pan, Y.; Sun, Y.; Goddard III, W.A.; Ding, M. Double-exchange-induced in situ conductivity in nickel-based oxyhydroxides: An effective descriptor for electrocatalytic oxygen evolution. Angew. Chem. Int. Ed. 2021, 60, 16448–16456. [Google Scholar] [CrossRef]
- Xiong, Y.; He, P. A review on electrocatalysis for alkaline oxygen evolution reaction (OER) by Fe-based catalysts. J. Mater. Sci. 2023, 58, 2041–2067. [Google Scholar] [CrossRef]
- Righi, G.; Plescher, J.; Schmidt, F.-P.; Campen, R.K.; Fabris, S.; Knop-Gericke, A.; Schlögl, R.; Jones, T.E.; Teschner, D.; Piccinin, S. On the origin of multihole oxygen evolution in haematite photoanodes. Nat. Catal. 2022, 5, 888–899. [Google Scholar] [CrossRef]
- Adak, M.K.; Mallick, L.; Samanta, K.; Chakraborty, B. Slow O–H dissociation in the first-order oxygen evolution reaction kinetics on polycrystalline γ-FeO(OH). J. Phys. Chem. C 2023, 127, 154–168. [Google Scholar] [CrossRef]
- Gao, L.; Tang, C.; Liu, J.; He, L.; Wang, H.; Ke, Z.; Li, W.; Jiang, C.; He, D.; Cheng, L.; et al. Oxygen vacancy-induced electron density tuning of Fe3O4 for enhanced oxygen evolution catalysis. Energy Environ. Mater. 2021, 4, 392–398. [Google Scholar] [CrossRef]
- Ouyang, Q.; Cheng, S.; Yang, C.; Lei, Z. Ni, Co, and Yb cation Co-doping and defect engineering of FeOOH nanorods as an electrocatalyst for the oxygen evolution reaction. Inorg. Chem. 2023, 62, 1719–1727. [Google Scholar] [CrossRef]
- Li, C.; Chen, M.; Xie, Y.; Wang, H.; Jia, L. Boosting photoelectrochemical water splitting of bismuth vanadate photoanode via novel co-catalysts of amorphous manganese oxide with variable valence states. J. Colloid. Interf. Sci. 2023, 636, 103–112. [Google Scholar] [CrossRef]
- Mousazade, Y.; Mohammadi, M.R.; Chernev, P.; Bikas, R.; Song, Z.; Lis, T.; Dau, H.; Najafpour, M.M. Water oxidation by a manganese–potassium cluster: Mn oxide as a kinetically dominant “true” catalyst for water oxidation. Catal. Sci. Technol. 2018, 8, 4390–4398. [Google Scholar] [CrossRef]
- Seo, H.; Park, S.; Cho, K.H.; Choi, S.; Ko, C.; Randriamahazaka, H.; Nam, K.T. Complex impedance analysis on charge accumulation step of Mn3O4 nanoparticles during water oxidation. ACS Omega 2021, 6, 18404–18413. [Google Scholar] [CrossRef]
- Yoon, S.; Seo, H.; Jin, K.; Kim, H.G.; Lee, S.-Y.; Jo, J.; Cho, K.H.; Ryu, J.; Yoon, A.; Kim, Y.-W.; et al. Atomic reconstruction and oxygen evolution reaction of Mn3O4 nanoparticles. J. Phys. Chem. Lett. 2022, 13, 8336–8343. [Google Scholar] [CrossRef]
- Wang, Y.; Sharma, A.; Duong, T.; Arandiyan, H.; Zhao, T.; Zhang, D.; Su, Z.; Garbrecht, M.; Beck, F.J.; Karuturi, S.; et al. Direct solar hydrogen generation at 20% efficiency using low-cost materials. Adv. Energy Mat. 2021, 11, 2101053. [Google Scholar] [CrossRef]
- Zhang, J.; Winkler, J.R.; Gray, H.B.; Hunter, B.M. Mechanism of nickel−iron water oxidation electrocatalysts. Energy Fuels 2021, 35, 19164–19169. [Google Scholar] [CrossRef]
- Yang, Y.; Du, X.; Wang, S.; Zhao, K.; Wang, L.; Qi, Z.; Yang, W.; Hao, J.; Shi, W. Cation transport effect on nickel iron oxyhydroxide electrodes in the oxygen evolution reaction. Ind. Eng. Chem. Res. 2022, 61, 16702–16710. [Google Scholar] [CrossRef]
- Acharya, P.; Manso, R.H.; Hoffman, A.S.; Bakovic, S.I.P.; Kékedy-Nagy, L.; Bare, S.R.; Chen, J.; Greenlee, L.F. Fe coordination environment, Fe-incorporated Ni(OH) phase, and metallic core are key structural components to active and stable nanoparticle catalysts for the oxygen evolution reaction. ACS Catal. 2022, 12, 1992–2008. [Google Scholar] [CrossRef]
- Qian, H.; Wei, J.; Yu, C.; Tang, F.; Jiang, W.; Xia, D.; Gan, L. In situ quantification of the active sites, turnover frequency, and stability of Ni–Fe (oxy)hydroxides for the oxygen evolution reaction. ACS Catal. 2022, 12, 14280–14289. [Google Scholar] [CrossRef]
- Avcı, O.N.; Sementa, L.; Fortunelli, A. Mechanisms of the oxygen evolution reaction on NiFe2O4 and CoFe2O4 inverse-spinel oxides. ACS Catal. 2022, 12, 9058–9073. [Google Scholar] [CrossRef]
- Deng, H.; Jiang, H.; Wang, K.; Wang, Z.; Wang, B.; Zhou, Z.; Li, J. Coupling the vanadium-induced amorphous/crystalline NiFe2O4 with phosphide heterojunction toward active oxygen evolution reaction catalysts. Nanotechnol. Rev. 2022, 11, 3165–3173. [Google Scholar] [CrossRef]
- Schoen, M.A.W.; Calderon, O.; Randell, N.M.; Jimenez-Villegas, S.; Daly, K.M.; Chernikov, R.; Trudel, S. Local structural changes in polyamorphous (Ni,Fe)O electrocatalysts suggest a dual-site oxygen evolution reaction mechanism. J. Mater. Chem. A 2021, 9, 13252–13262. [Google Scholar] [CrossRef]
- Sugawara, Y.; Kamata, K.; Ishikawa, A.; Tateyama, Y.; Yamaguchi, T. Efficient oxygen evolution electrocatalysis on CaFe2O4 and its reaction mechanism. ACS Appl. Energy Mater. 2021, 4, 3057–3066. [Google Scholar] [CrossRef]
- Zheng, J.; Peng, X.; Xu, Z.; Gong, J.; Wang, Z. Cationic defect engineering in spinel NiCo2O4 for enhanced electrocatalytic oxygen evolution. ACS Catal. 2022, 12, 10245–10254. [Google Scholar] [CrossRef]
- Li, A.; Kong, S.; Guo, C.; Ooka, H.; Adachi, K.; Hashizume, D.; Jiang, Q.; Han, H.; Xiao, J.; Nakamura, R. Enhancing the stability of cobalt spinel oxide towards sustainable oxygen evolution in acid. Nat. Catal. 2022, 5, 109–118. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, L.; Cao, Z.; Kozlov, S.M.; de Arquer, F.P.G.; Dinh, C.T.; Li, J.; Wang, Z.; Zheng, X.; Zhang, L.; et al. High-valence metals improve oxygen evolution reaction performance by modulating 3d metal oxidation cycle energetics. Nat. Catal. 2020, 3, 985–992. [Google Scholar] [CrossRef]
- Simondson, D.; Chatti, M.; Bonke, S.A.; Tesch, M.F.; Golnak, R.; Xiao, J.; Hoogeveen, D.A.; Cherepanov, P.V.; Gardiner, J.L.; Tricoli, A.; et al. Stable acidic water oxidation with a cobalt–iron–lead oxide catalyst operating via a cobalt-selective self-healing mechanism. Angew. Chem. Int. Ed. 2021, 60, 15821–15826. [Google Scholar] [CrossRef] [PubMed]
- Karim, A.V.; Hassani, A.; Eghbali, P.; Nidheesh, P.V. Nanostructured modified layered double hydroxides (LDHs)-based catalysts: A review on synthesis, characterization, and applications in water remediation by advanced oxidation processes. Curr. Opinion Solid State and Mater. Sci. 2022, 26, 100965. [Google Scholar] [CrossRef]
- Xu, S.-M.; Pan, T.; Dou, Y.-B.; Yan, H.; Zhang, S.-T.; Ning, F.-Y.; Shi, W.-Y.; Wei, M. Theoretical and experimental study on MIIMIII -layered double hydroxides as efficient photocatalysts toward oxygen evolution from water. J. Phys. Chem. C 2015, 119, 18823–18834. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, L.; Feng, R.; Wang, S.; Hsu, C.-S.; Ashfaq, Y.N.; Zhang, A.C.; Wu, H.; Chen, H.-M.; Zhang, W.; et al. Oxygen vacancies unfold the catalytic potential of NiFe-layered double hydroxides by promoting their electronic transport for oxygen evolution reaction. ACS Catal. 2023, 13, 6000–6012. [Google Scholar] [CrossRef]
- Kang, J.; Qiu, X.; Hu, Q.; Zhong, J.; Gao, X.; Huang, R.; Wan, C.; Liu, L.-M.; Duan, X.; Guo, L. Valence oscillation and dynamic active sites in monolayer NiCo hydroxides for water oxidation. Nat. Catal. 2021, 4, 1050–1058. [Google Scholar] [CrossRef]
- Wang, Z.; Goddard III, W.A.; Xiao, H. Potential-dependent transition of reaction mechanisms for oxygen evolution on layered double hydroxides. Nat. Commun. 2023, 14, 4228. [Google Scholar] [CrossRef]
- Xu, J.; Li, Z.; Chen, D.; Yang, S.; Zheng, K.; Ruan, J.; Wu, Y.; Zhang, H.; Chen, J.; Xie, F.; et al. Active electrocatalyst in the oxygen evolution reaction and flexible zinc–air batteries. ACS Appl. Mater. Interfaces 2021, 13, 48774–48783. [Google Scholar] [CrossRef]
- Sun, Z.; Lin, L.; He, J.; Ding, D.; Wang, T.; Li, J.; Li, M.; Liu, Y.; Li, Y.; Yuan, M.; et al. Regulating the spin state of FeIII enhances the magnetic effect of the molecular catalysis mechanism. J. Am. Chem. Soc. 2022, 144, 8204–8213. [Google Scholar] [CrossRef]
- Qiao, C.; Usman, Z.; Wei, J.; Gan, L.; Hou, J.; Hao, Y.; Zhu, Y.; Zhang, J.; Cao, C. Efficient O–O coupling at catalytic interface to assist kinetics optimization on concerted and sequential proton–electron transfer for water oxidation. ACS Nano 2023, 17, 12278–12289. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Han, X.; Zhou, X.; Chen, J.; Wang, J.; Chen, Y.; Yu, L.; Zhang, N.; Li, J.; Wang, S.; et al. Electronic modulation with Pt-incorporated NiFe layered doublehydroxide for ultrastable overall water splitting at 1000 mA cm−2. App. Catal. B Environ. 2023, 331, 122683. [Google Scholar] [CrossRef]
- Gao, Z.-W.; Liu, J.-Y.; Chen, X.-M.; Zheng, X.-L.; Mao, J.; Liu, H.; Ma, T.; Li, L.; Wang, W.-C.; Du, X.-W. Engineering NiO/NiFe LDH intersection to bypass scaling relationship for oxygen evolution reaction via dynamic tridimensional adsorption of intermediates. Adv. Mater. 2019, 31, 1804769. [Google Scholar] [CrossRef]
- Luo, Y.; Wu, Y.; Wu, D.; Huang, C.; Xiao, D.; Chen, H.; Zheng, S.; Chu, P.K. NiFe-layered double hydroxide synchronously activated by heterojunctions and vacancies for the oxygen evolution reaction. ACS Appl. Mater. Interfaces 2020, 12, 42850–42858. [Google Scholar] [CrossRef]
- Zhai, P.; Wang, C.; Zhao, Y.; Zhang, Y.; Gao, J.; Sun, L.; Hou, J. Regulating electronic states of nitride/hydroxide to accelerate kinetics for oxygen evolution at large current density. Nat. Commun. 2023, 14, 1873. [Google Scholar] [CrossRef]
- Liang, X.; Li, Y.; Fan, H.; Deng, S.; Zhao, X.; Chen, M.; Pan, G.; Xiong, Q.; Xia, X. Bifunctional NiFe layered double hydroxide@Ni3S2 hetero structure as efficient electrocatalyst for overall water splitting. Nanotechnology 2019, 30, 484001. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Liu, X.; Li, H.; Sun, Z.; Cao, M.; Li, Z.; Fang, C.; Zhou, J.; Cao, C.; Dong, J.; et al. A semiconductor-electrocatalyst nano interface constructed for successive photoelectrochemical water oxidation. Nat. Commun. 2023, 14, 2574. [Google Scholar] [CrossRef] [PubMed]
- Guru, S.; Rao, G.R. Review—Strategic design of layered double hydroxides and graphitic carbon nitride heterostructures for photoelectrocatalytic water splitting applications. J. Electrochem. Soc. 2022, 169, 046515. [Google Scholar] [CrossRef]
- Singh, B.; Singh, A.; Yadav, A.; Indra, A. Modulating electronic structure of metal-organic framework derived catalysts for electrochemical water oxidation. Coord. Chem. Rev. 2021, 447, 214144. [Google Scholar] [CrossRef]
- Li, C.; Wang, G.; Li, K.; Liu, Y.; Yuan, B.; Lin, Y. FeNi-based coordination crystal directly serving as efficient oxygen evolution reaction catalyst and its density functional theory insight on the active site change mechanism. ACS Appl. Mater. Interfaces 2019, 11, 20778–20787. [Google Scholar] [CrossRef]
- Gu, M.; Wang, S.-C.; Chen, C.; Xiong, D.; Yi, F.-Y. Iron-based metal–organic framework system as an efficient bifunctional electrocatalyst for oxygen evolution and hydrogen evolution reactions. Inorg. Chem. 2020, 59, 6078–6086. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, S.; Li, Z.; Chu, H.; Zhou, W. Insight into the surface-reconstruction of metal–organic framework-based nanomaterials for the electrocatalytic oxygen evolution reaction. Coord. Chem. Rev. 2023, 484, 215117. [Google Scholar] [CrossRef]
- Zhang, W.; Li, F.; Fu, Z.; Dai, S.; Pan, F.; Li, J.; Zhou, L. Co-MOF nanosheets etched by FeCl2 solution for enhanced electrocatalytic oxygen evolution. Energy Fuels 2022, 36, 4524–4531. [Google Scholar] [CrossRef]
- Sunil, J.; Narayana, C.; Kumari, G.; Jayaramulu, K. Raman spectroscopy. An ideal tool for studying the physical properties and applications of metal–organic frameworks (MOFs). Chem. Soc. Rev. 2023, 52, 3397–3437. [Google Scholar] [CrossRef]
- Yu, R.; Wang, C.; Liu, D.; Wang, X.; Yin, J.; Du, Y. Self-reconstruction of Fe-doped Co-metal–organic frameworks boosted electrocatalytic performance for oxygen evolution reaction. Inorg. Chem. 2023, 62, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.H.; Li, Q.H.; Yu, D.Y.; Tang, Y.H.; Lin, D.Y.; Wang, F.; Zhang, J. Hybrid zeolitic imidazolate frameworks for promoting electrocatalytic oxygen evolution via dual-site relay mechanism. Inorg. Chem. 2021, 60, 3074–3081. [Google Scholar] [CrossRef]
- Jayaramulu, K.; Masa, J.; Morales, D.M.; Tomanec, O.; Ranc, V.; Petr, M.; Wilde, P.; Chen, Y.-T.; Zboril, R.; Schuhmann, W.; et al. Ultrathin 2D cobalt zeolite-imidazole framework nanosheets for electrocatalytic oxygen evolution. Adv. Sci. 2018, 5, 1801029. [Google Scholar] [CrossRef]
- Benkó, T.; Lukács, D.; Li, M.; Pap, J.S. Redox-active ligands in artificial photosynthesis: A review. Environ. Chem. Lett. 2022, 20, 3657–3695. [Google Scholar] [CrossRef]
- Li, M.; Liao, R.-Z. Water oxidation catalyzed by a bioinspired tetranuclear manganese complex: Mechanistic study and prediction. ChemSusChem 2022, 15, e202200187. [Google Scholar] [CrossRef]
- Chen, Q.-F.; Guo, Y.-H.; Yu, Y.-H.; Zhang, M.-T. Bioinspired molecular clusters for water oxidation. Coord. Chem. Rev. 2021, 448, 214164. [Google Scholar] [CrossRef]
- Zhang, Q.; Guan, J. Mono-/multinuclear water oxidation catalysts. ChemSusChem 2019, 12, 3209–3235. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, P. Transition metal based coordination complexes as catalysts for water oxidation. Resonance 2022, 27, 1185–1209. [Google Scholar] [CrossRef]
- Masaya, M.; Kondo, M.; Kuga, R.; Kurashige, Y.; Yanai, T.; Hayami, S.; Praneeth, V.K.K.; Yoshida, M.; Yoneda, K.; Kawata, S.; et al. A pentanuclear iron catalyst designed for water oxidation. Nature 2016, 530, 465–468. [Google Scholar] [CrossRef]
- Chen, G.; Lam, W.W.Y.; Lo, P.K.; Man, W.-L.; Chen, L.; Lau, K.-C.; Lau, T.-C. Mechanism of water oxidation by ferrate(VI) at pH 7–9. Chem. Eur. J. 2018, 24, 18735–18742. [Google Scholar] [CrossRef]
- den Boer, D.; Konovalov, A.I.; Siegler, M.A.; Hetterscheid, D.G.H. Unusual water oxidation mechanism via a redox-active copper polypyridyl complex. Inorg. Chem. 2023, 62, 5303–5314. [Google Scholar] [CrossRef]
- Li, Y.-Y.; Wang, X.-Y.; Li, H.-J.; Chen, J.-Y.; Kou, Y.-H.; Li, X.; Wang, Y. Theoretical study on the mechanism of water oxidation catalyzed by a mononuclear copper complex: Important roles of a redox non-innocent ligand and HPO42− anion. RSC Adv. 2023, 13, 8352–8359. [Google Scholar] [CrossRef]
- Iqbal, S.; Safdar, B.; Hussain, I.; Zhang, K.; Chatzichristodoulou, C. Trends and prospects of bulk and single-atom catalysts for the oxygen evolution reaction. Adv. Energy Mater. 2023, 13, 2203913. [Google Scholar] [CrossRef]
- Kumar, P.; Kannimuthu, K.; Zeraati, A.S.; Roy, S.; Wang, X.; Wang, X.; Samanta, S.; Miller, K.A.; Molina, M.; Trivedi, D.; et al. High-density cobalt single-atom catalysts for enhanced oxygen evolution reaction. J. Am. Chem. Soc. 2023, 145, 8052–8063. [Google Scholar] [CrossRef]
- Cipriano, L.A.; Liberto, G.D.; Pacchioni, G. Superoxo and peroxo complexes on single-atom catalysts: Impact on the oxygen evolution reaction. ACS Catal. 2022, 12, 11682–11691. [Google Scholar] [CrossRef]
- Barlocco, I.; Cipriano, L.A.; Liberto, G.D.; Pacchioni, G. Does the oxygen evolution reaction follow the classical OH*, O*, OOH* path on single atom catalysts? J. Catal. 2023, 417, 351–359. [Google Scholar] [CrossRef]
- He, Y.; Zhou, X.; Jia, Y.; Li, H.; Wang, Y.; Liu, Y.; Tan, Q. Advances in transition-metal-based dual-atom oxygen electrocatalysts. Small 2023, 19, 2206477. [Google Scholar] [CrossRef]
- Fang, C.; Zhou, J.; Zhang, L.; Wan, W.; Ding, Y.; Sun, X. Synergy of dual-atom catalysts deviated from the scaling relationship for oxygen evolution reaction. Nat. Commun. 2023, 14, 4449. [Google Scholar] [CrossRef]
- Sayama, K.; Arakawa, H. Effect of carbonate salt addition on the photocatalytic decomposition of liquid water over Pt–TiO2 catalyst. J. Chem. Soc. Faraday Trans. 1997, 93, 1647–1654. [Google Scholar] [CrossRef]
- Kusama, H.; Kodera, M.; Yamashita, K.; Sayama, K. Insights into the carbonate effect on water oxidation over metal oxide photocatalysts/photoanodes. Phys. Chem. Chem. Phys. 2022, 24, 5894–5902. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Laberty-Robert, C.; Batuk, D.; Cibin, G.; Chadwick, A.V.; Pimenta, V.; Yin, W.; Zhang, L.; Tarascon, J.-M.; Grimaud, A. Phosphate ion functionalization of perovskite surfaces for enhanced oxygen evolution reaction. J. Phys. Chem. Lett. 2017, 8, 3466–3472. [Google Scholar] [CrossRef] [PubMed]
- Tsuneda, T.; Ten-no, S. Water–oxidation mechanism of cobalt phosphate co-catalyst in artificial photosynthesis: A theoretical study. Phys. Chem. Chem. Phys. 2022, 24, 4674–4682. [Google Scholar] [CrossRef]
- Liu, M.; Li, N.; Wang, X.; Zhao, J.; Zhong, D.-C.; Li, W.; Bu, X.-H. Photosystem II inspired NiFe-based electrocatalysts for efficient water oxidation via second coordination sphere effect. Angew. Chem. Int. Ed. 2023, 62, e202300507. [Google Scholar] [CrossRef] [PubMed]
- Gao1, Y.; Wu, C.; Sen, S.; Tan, Y. Hexadecyltrimethylammonium hydroxide promotes electrocatalytic activity for the oxygen evolution reaction. Commun. Chem. 2020, 3, 154. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oxidation Step | /cm−1 | Mode | Sample | [Reference] Year | |
|---|---|---|---|---|---|
| 1 | Co(IV)=O | 840 | νMO | Co3O4 | [64] 2014 |
| Ni-O· | 908 | Ni-NC | [65] 2020 | ||
| Ni-O· | 916 | Ni-Vac | [65] 2020 | ||
| Ni-O· | 1170–1330 | NiV-MOF | [66] 2021 | ||
| Mn(IV)=O | 712–755 | Mn complex | [67] 2001 | ||
| Mn(V)=O | 970–981 1 | Mn complex | [67] 2001 | ||
| Mn(VII)=O | 912 | NiFe-LDH | [68] 2018 | ||
| Fe(IV)=O | 898 | α-Fe2O3 | [69] 2016 | ||
| Ti(IV)-O· | 795 | n-SrTiO3 | [70] 2016 | ||
| Ir-O | 784 | Atomic Ir | [71] 2015 | ||
| Ir(VI)=O | 946 | CaIrO2 | [63] 2022 | ||
| 2 | Mn(IV)=O2 | 810 | Mn complex | [72] 2021 | |
| Li-OOH | 1170 | Li in DMSO | [73] 2017 | ||
| Ti-OOH | 837 | νOO | Ti-SiO2 | [74] 2002 | |
| Ti-OOH | 838 | νOO | TiO2 | [75] 2003 | |
| Ti-OOH | 1120–1250 | δOOH | TiO2 | [75] 2003 | |
| Ni-OOH | 1060 | Ni-Vac | [76] 2022 | ||
| Ni(IV)-OOD | 1048 | NiFe·MOF | [77] 2019 | ||
| Ni-OO- | 850–1200 | NiOOH | [78] 2019 | ||
| Cu-OO- | 1180 | Cu complex | [79] 2021 | ||
| Ru-OOH | 986 | RuO2 | [80] 2021 | ||
| Ga-OOH | 978 | Ga2O3 | [81] 2021 | ||
| Ir (VI)-OO- | 870 | νOO | CaIrO2 | [63] 2022 | |
| Ir(III)-OOH | 830 | νOO | IrO2 | [82] 2011 | |
| Ir-OOH | 1055 | Atomic Ir | [71] 2021 | ||
| Ir-OOH | 1065 | Ir/Co3O4 | [83] 2023 | ||
| Au-OOH | 1268 | Au electrode | [84] 2005 | ||
| Co-OO-Co | 833 | νOO | Co3O4 | [85] 2020 | |
| Li-OO-Li | 780, 830 | Li in DMSO | [73] 2017 | ||
| Ti-OO-Ti | 812 | TiO2 | [75] 2003 | ||
| Fe(VI)(=O)2 | 840, 856 | NiFe-LDH | [86] 3018 | ||
| M-OO-M | 1089 | Ru/MnO2 | [80] 2021 | ||
| 3 | Co(III)-OO·(HO) | 1013 | Co3O4 | [64] 2014 | |
| Fe-OO·(HO) | 1096 | α-Fe2O3 | [87] 2018 | ||
| Li-OO· | 1127–1139 | Li in DMSO | [73] 2017 | ||
| M-OO | 1128 | Ru/MnO2 | [80] 2021 | ||
| Pt-OO· | 1005–1016 | Pt in Alkaline | [88] 2006 | ||
| Mn-OO | 1080 | Mn3O4 | [89] 2019 |
| Mode | Ti-OOH | Ti-O18OH | Ti-18O18OH | Ti-OOD | |
|---|---|---|---|---|---|
| Experimental a | δ(OOH) | 1120–1250 cm−1 | ∆ = −220–−320 cm−1 | ||
| ν(OO) | 838 cm−1 | ∆ = −19 cm−1 | ∆ = −45 cm−1 | ∆ = −67–−97 cm−1 | |
| DFT calculation b | δ(OOH) | 1257 cm−1 | ∆ = −2 cm−1 | ∆ = −8 cm−1 | ∆ = −299 cm−1 |
| ν(OO) | 881 cm−1 | ∆ = −23 cm−1 | ∆ = −49 cm−1 | ∆ = −22 cm−1 |
| Mode | Bi-OOH | Bi-O18OH | Bi-18O18OH | Bi-OOD | |
|---|---|---|---|---|---|
| BiVO4 Experiment a | δ(OOH) | 1233 cm−1 | ∆ = −8 cm−1 | ∆ = −313 cm−1 | |
| ν(OO) | 1035 cm−1 | ∆ = −50 cm−1 | ∆ = −45 cm−1 | ||
| BiOOH DFT calc. b | δ(OOH) | 1257 cm−1 | ∆ = −2 cm−1 | ∆ = −8 cm−1 | ∆ = −299 cm−1 |
| ν(OO) | 881 cm−1 | ∆ = −23 cm−1 | ∆ = −50 cm−1 | ∆ = −22 cm−1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nosaka, Y. Molecular Mechanisms of Oxygen Evolution Reactions for Artificial Photosynthesis. Oxygen 2023, 3, 407-451. https://doi.org/10.3390/oxygen3040027
Nosaka Y. Molecular Mechanisms of Oxygen Evolution Reactions for Artificial Photosynthesis. Oxygen. 2023; 3(4):407-451. https://doi.org/10.3390/oxygen3040027
Chicago/Turabian StyleNosaka, Yoshio. 2023. "Molecular Mechanisms of Oxygen Evolution Reactions for Artificial Photosynthesis" Oxygen 3, no. 4: 407-451. https://doi.org/10.3390/oxygen3040027
APA StyleNosaka, Y. (2023). Molecular Mechanisms of Oxygen Evolution Reactions for Artificial Photosynthesis. Oxygen, 3(4), 407-451. https://doi.org/10.3390/oxygen3040027