
The Effect of Cation Exchange on the Pore Geometry of Zeolite L

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis and Ion Exchange of K-L
2.2. Powder X-ray Diffraction (PXRD)
2.3. Rietveld Refinements
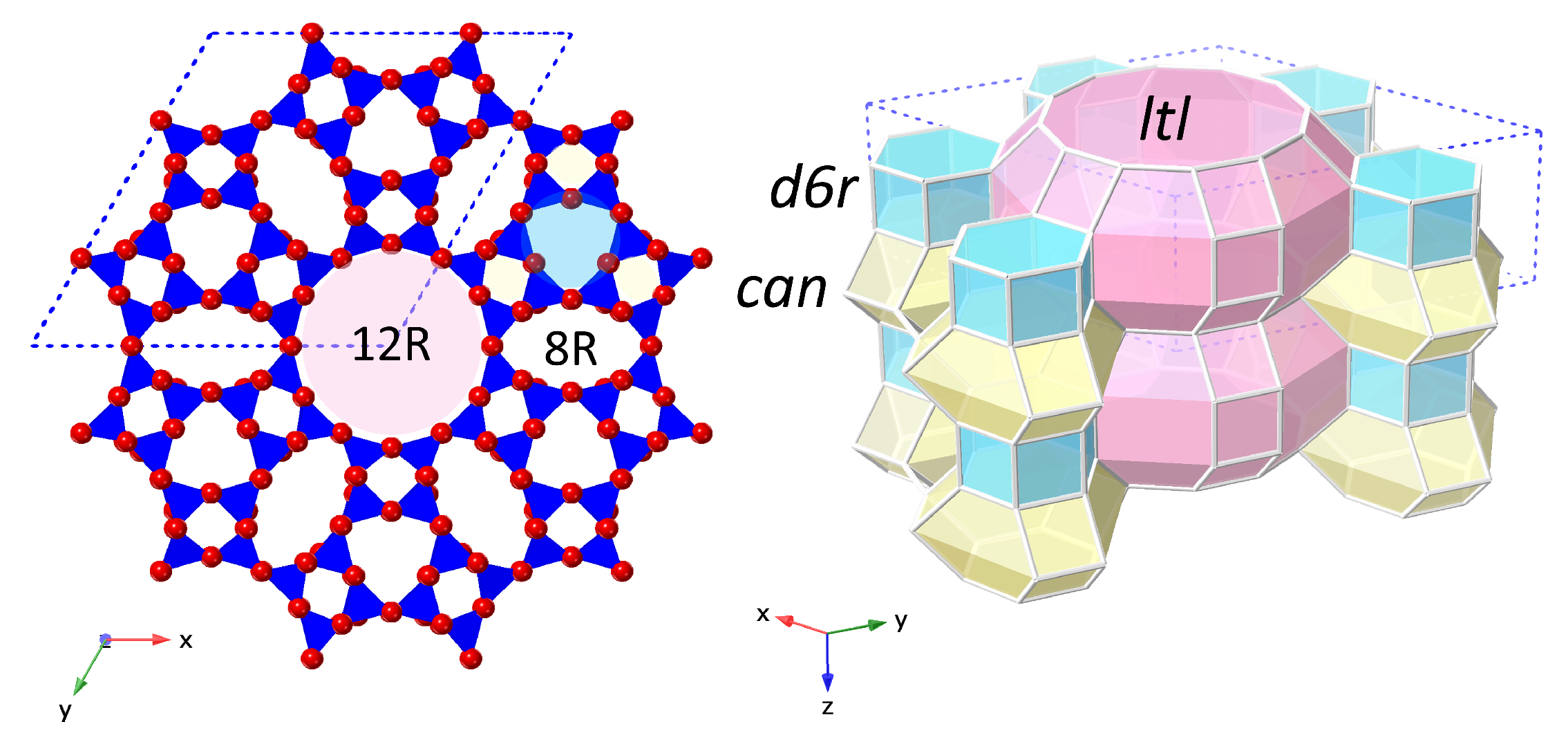
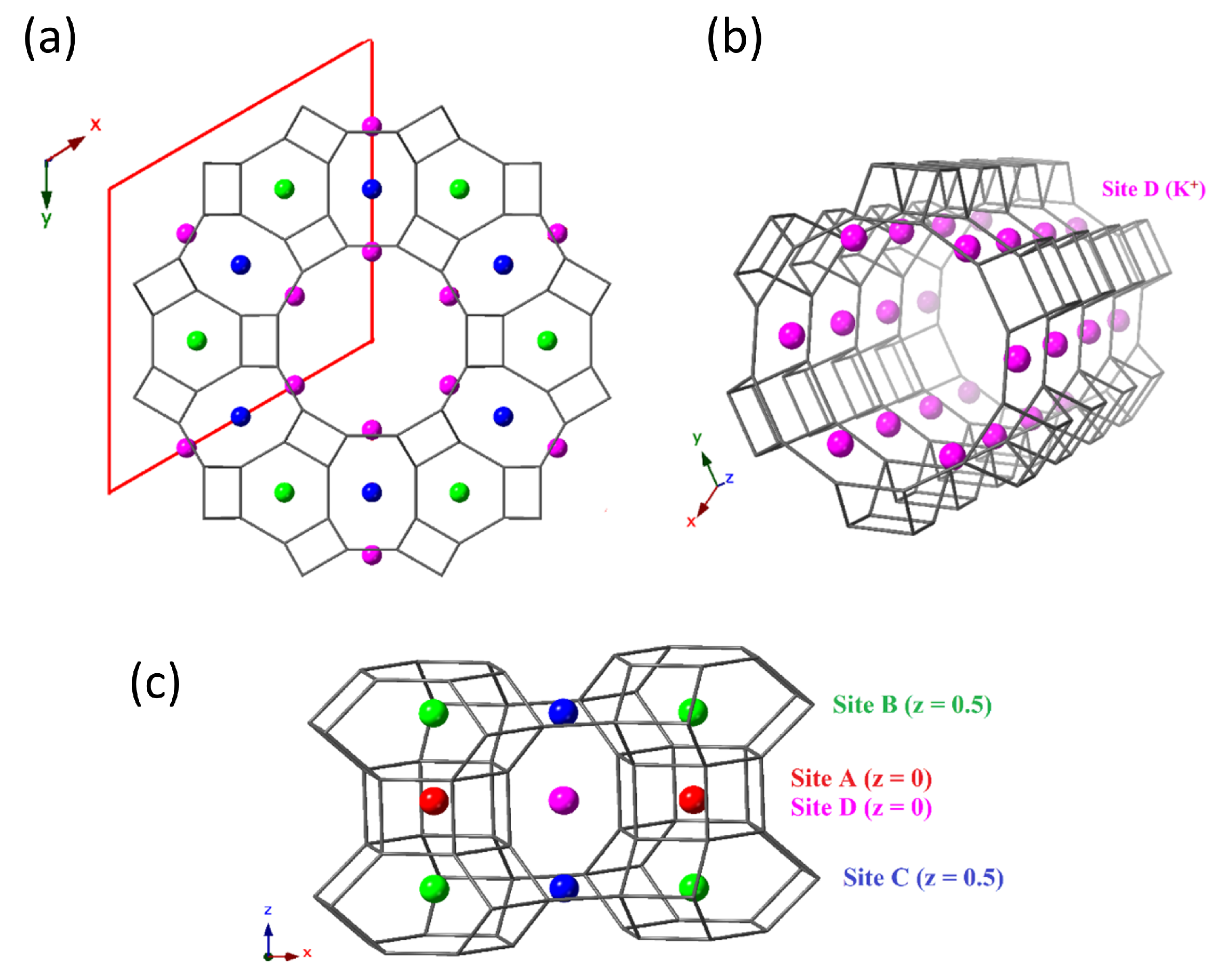
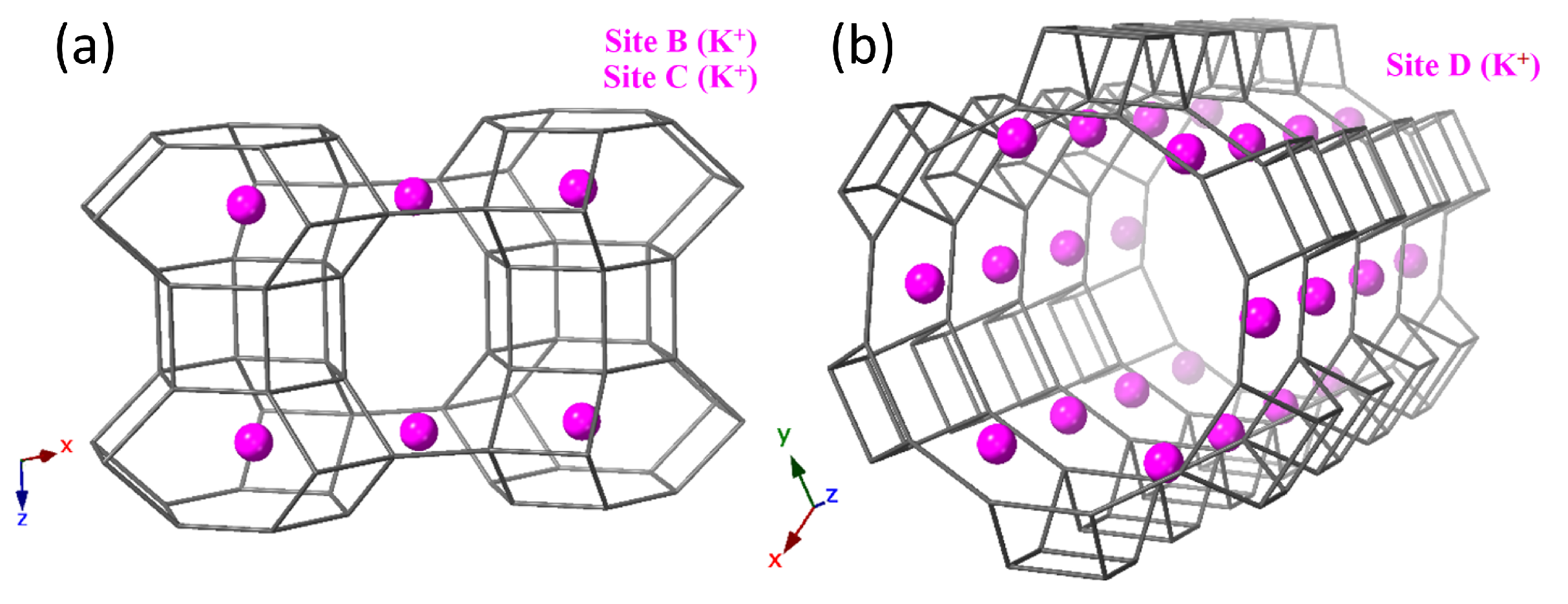
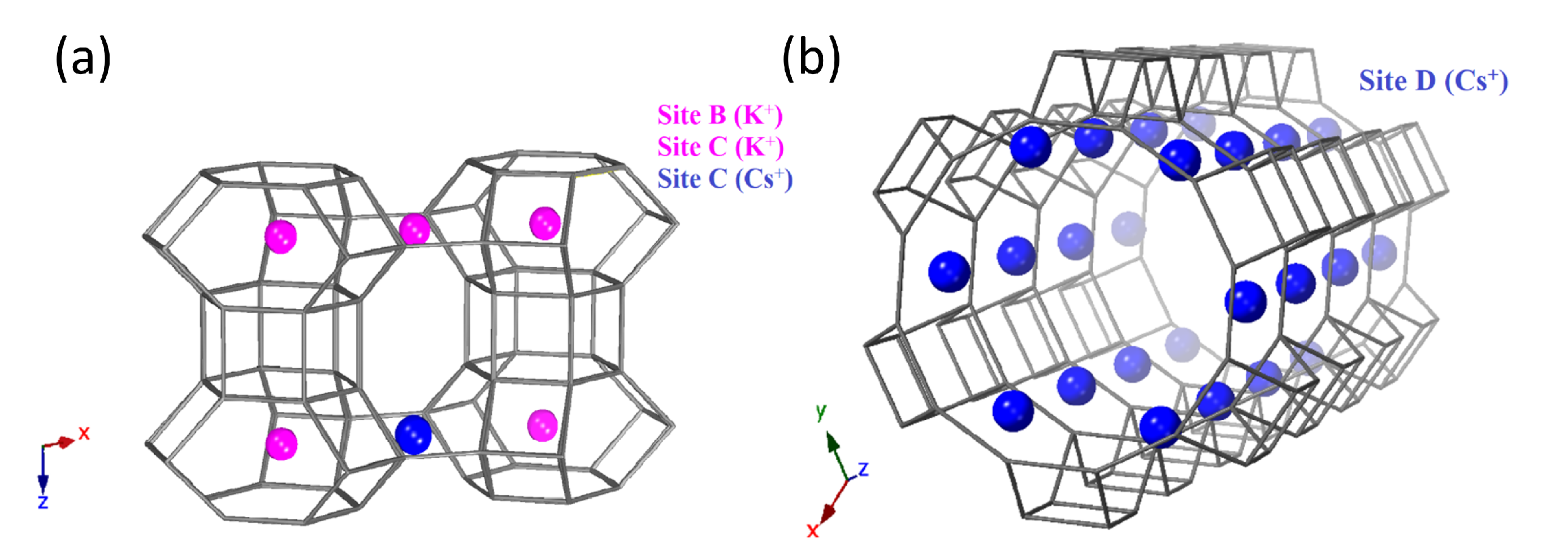
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 1D | One-dimensional |
| 12R | 12-ring |
| 8R | 8-ring |
| can | cancrinite cage |
| d6r | double 6-ring |
Appendix A. Rietveld Refinement Data

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a/b (Å) | c (Å) | () | () |
|---|---|---|---|
| 18.388(3) | 7.529(2) | 90 | 120 |
| Atom | x | y | z | Uiso | Occ. |
|---|---|---|---|---|---|
| K (Site D) | 0 | 0.2918 | 0 | 0.051273 | 0.63 |
| K (Site B) | 0.3333 | 0.6667 | 0.5 | 0.051273 | 1 |
| K (Site C) | 0 | 0.5 | 0.5 | 0.051273 | 1 |
| Si (1) | 0.0946 | 0.3595 | 0.5 | 0.012316 | 0.75 |
| Si (2) | 0.1662 | 0.4989 | 0.2137 | 0.012316 | 0.75 |
| Al (1) | 0.0946 | 0.3595 | 0.5 | 0.012316 | 0.25 |
| Al (2) | 0.1662 | 0.4989 | 0.2137 | 0.012318 | 0.25 |
| O (1) | 0 | 0.267247 | 0.5 | 0.029498 | 1 |
| O (2) | 0.164702 | 0.329401 | 0.5 | 0.029498 | 1 |
| O (3) | 0.272687 | 0.545376 | 0.261401 | 0.029498 | 1 |
| O (4) | 0.096766 | 0.412575 | 0.323641 | 0.029498 | 1 |
| O (5) | 0.433088 | 0.866174 | 0.269393 | 0.029498 | 1 |
| O (6) | 0.126437 | 0.453851 | 0 | 0.029498 | 1 |
| H2O (1) | 0.069974 | 0.13995 | 0.5 | 0.237862 | 0.39518 |
| H2O (2) | 0 | 0.117857 | 0.5 | 0.237862 | 0.26205 |
| H2O (3) | 0 | 0.151173 | 0.222767 | 0.237862 | 0.500229 |
| H2O (4) | 0 | 0 | 0.078289 | 0.23786 | 0.681591 |
| H2O (5) | 135078 | 0.270158 | 0 | 0.237862 | 0.691272 |
| H2O (6) | 0 | 0.187275 | 0 | 0.237862 | 0.325574 |
| a/b (Å) | c (Å) | () | () |
|---|---|---|---|
| 18.453(5) | 7.558(3) | 90 | 120 |
| Atom | x | y | z | Uiso | Occ. |
|---|---|---|---|---|---|
| K (Site B) | 0.3333 | 0.6667 | 0.5 | 0.0298 | 1 |
| K (Site C) | 0 | 0.5 | 0.5 | 0.0298 | 0.3751 |
| Cs (Site C) | 0 | 0.5 | 0.5 | 0.0298 | 0.446 |
| Cs (Site D) | 0 | 0.275554 | 0 | 0.0298 | 0.7238 |
| Si (1) | 0.104606 | 0.364589 | 0.5 | 0.0152 | 0.75 |
| Si (2) | 0.170499 | 0.500876 | 0.207083 | 0.0152 | 0.75 |
| Al (1) | 0.0946 | 0.3595 | 0.5 | 0.0152 | 0.25 |
| Al (2) | 0.1662 | 0.4989 | 0.2137 | 0.0152 | 0.25 |
| O (1) | 0 | 0.289743 | 0.5 | 0.0341 | 1 |
| O (2) | 0.161538 | 0.32308 | 0.5 | 0.0341 | 1 |
| O (3) | 0.274509 | 0.549017 | 0.276285 | 0.0341 | 1 |
| O (4) | 0.11304 | 0.433529 | 0.309766 | 0.0341 | 1 |
| O (5) | 0.427542 | 0.855089 | 0.255541 | 0.0341 | 1 |
| O (6) | 0.114231 | 0.437655 | 0 | 0.0341 | 1 |
| H2O (1) | 0 | 0.127013 | 0.334108 | 0.2514 | 0.6092 |
| H2O (2) | 0.101592 | 0.203184 | 0 | 0.2514 | 0.3631 |
| a/b (Å) | c (Å) | () | () |
|---|---|---|---|
| 18.386(5) | 7.529(3) | 90 | 120 |
| Atom | x | y | z | Uiso | Occ. |
|---|---|---|---|---|---|
| K (Site B) | 0.3333 | 0.6667 | 0.5 | 0.0848 | 1 |
| K (Site C) | 0 | 0.5 | 0.5 | 0.0848 | 1 |
| K (Site D) | 0 | 0.293855 | 0 | 0.0848 | 0.5696 |
| Li (Site D) | 0 | 0.357731 | 0 | 0.0848 | 0.1119 |
| Si (1) | 0.0946 | 0.3595 | 0.5 | 0.0152 | 0.75 |
| Si (2) | 0.1662 | 0.4989 | 0.2137 | 0.0152 | 0.75 |
| Al (1) | 0.0946 | 0.3595 | 0.5 | 0.0152 | 0.25 |
| Al (2) | 0.1662 | 0.4989 | 0.2137 | 0.0152 | 0.25 |
| O (1) | 0 | 0.264863 | 0.5 | 0.0431 | 1 |
| O (2) | 0.165997 | 0.331993 | 0.5 | 0.0431 | 1 |
| O (3) | 0.272915 | 0.545829 | 0.258433 | 0.0431 | 1 |
| O (4) | 0.099272 | 0.419473 | 0.320544 | 0.0431 | 1 |
| O (5) | 0.430069 | 0.860139 | 0.270699 | 0.0431 | 1 |
| O (6) | 0.131235 | 0.459158 | 0 | 0.0431 | 1 |
| H2O (1) | 0.108684 | 0.217367 | 0.5 | 0.2196 | 0.2751 |
| H2O (2) | 0 | 0.145016 | 0.275609 | 0.2196 | 0.4133 |
| H2O (3) | 0.142988 | 0.28598 | 0 | 0.2196 | 0.5295 |
| H2O (4) | 0 | 0.178042 | 0 | 0.2196 | 0.3054 |
References
- Barrer, R.M. Hydrothermal Chemistry of Zeolites. In Department of Chemistry Imperial College of Science and Technology London; Academic Press Inc.: Cambridge, MA, USA, 1982; p. 360. [Google Scholar]
- Abdo, F.S.; Wilson, T.S. Zeolites in Industrial Catalysis. In Zeolites in Catalysis: Properties and Applications; The Royal Society of Chemistry: London, UK, 2017; Section 9; pp. 310–350. [Google Scholar]
- Sartbaeva, A.; Rees, N.H.; Edwards, P.P.; Ramirez-Cuesta, A.J.; Barney, E. Local probes show that framework modification in zeolites occurs on ammonium exchange without calcination. J. Mater. Chem. A 2013, 1, 7415–7421. [Google Scholar] [CrossRef]
- Price, L.; Leung, M.K.; Sartbaeva, A. Local and Average Structural Changes in Zeolite A upon Ion Exchange. Magnetochemistry 2017, 3, 42. [Google Scholar] [CrossRef] [Green Version]
- Baerlocher, C.; McCusker, L. Database of Zeolite Structures. Available online: http://www.iza-structure.org/databases/ (accessed on 20 April 2022).
- Newsam, J.M. Structures of dehydrated potassium zeolite L at 298 and 78 K and at 78 K containing sorbed perdeuteriobenzene. J. Phys. Chem. 1989, 93, 7689–7694. [Google Scholar] [CrossRef]
- Newsam, J.M. Structural characterization of dehydrated gallium zeolite L. Mater. Res. Bull. 1986, 21, 661–672. [Google Scholar] [CrossRef]
- Calzaferri, G.; Huber, S.; Maas, H.; Minkowski, C. Host–guest antenna materials. Angew. Chem. Int. Ed. 2003, 42, 3732–3758. [Google Scholar] [CrossRef]
- Vohra, V.; Bolognesi, A.; Calzaferri, G.; Botta, C. Multilevel organization in hybrid thin films for optoelectronic applications. Langmuir 2009, 25, 12019–12023. [Google Scholar] [CrossRef]
- Calzaferri, G.; Meallet-Renault, R.; Bruhwiler, D.; Pansu, R.; Dolamic, I.; Dienel, T.; Adler, P.; Li, H.R.; Kunzmann, A. Designing Dye-Nanochannel Antenna Hybrid Materials for Light Harvesting, Transport and Trapping. ChemPhysChem 2011, 12, 580–594. [Google Scholar] [CrossRef]
- Insuwan, W.; Jungsuttiwong, S.; Rangsriwatananon, K. Host–guest composite materials of dyes loaded zeolite LTL for antenna applications. J. Lumin. 2015, 161, 31–36. [Google Scholar] [CrossRef]
- Insuwan, W.; Rangsriwatananon, K.; Meeprasert, J.; Namuangruk, S.; Surakhot, Y.; Kungwan, N.; Jungsuttiwong, S. Combined experimental and theoretical investigation on fluorescence resonance energy transfer of dye loaded on LTL zeolite. Microporous Mesoporous Mater. 2017, 241, 372–382. [Google Scholar] [CrossRef]
- Meeprasert, J.; Kungwan, N.; Jungsuttiwong, S.; Namuangruk, S. Location and reactivity of extra-framework cation in the alkali exchanged LTL zeolites: A periodic density functional study. Microporous Mesoporous Mater. 2014, 195, 227–239. [Google Scholar] [CrossRef]
- Fois, E.; Tabacchi, G.; Calzaferri, G. Interactions, Behavior, And Stability of Fluorenone inside Zeolite Nanochannels. J. Phys. Chem. C 2010, 114, 10572–10579. [Google Scholar] [CrossRef] [Green Version]
- Jentoft, R.E.; Tsapatsis, M.; Davis, M.E.; Gates, B.C. Platinum Clusters Supported in Zeolite LTL: Influence of Catalyst Morphology on Performance in n-Hexane Reforming. J. Catal. 1998, 179, 565–580. [Google Scholar] [CrossRef]
- Bertucci, A.; Lülf, H.; Septiadi, D.; Manicardi, A.; Corradini, R.; De Cola, L. Intracellular Delivery of Peptide Nucleic Acid and Organic Molecules Using Zeolite-L Nanocrystals. Adv. Healthc. Mater. 2014, 3, 1812–1817. [Google Scholar] [CrossRef] [PubMed]
- Besoukhanova, C.; Guidot, J.; Barthomeuf, D.; Breysse, M.; Bernard, J.R. Platinum–zeolite interactions in alkaline L zeolites. Correlations between catalytic activity and platinum state. J. Chem. Soc. Faraday Trans. Phys. Chem. Condens. Phases 1981, 77, 1595–1604. [Google Scholar] [CrossRef]
- Ramezani Shabolaghi, K.; Irani, M. Ethanol adsorption in cation-exchanged linde type L zeolite, studied by molecular simulations. Comput. Theor. Chem. 2022, 1207, 113498. [Google Scholar] [CrossRef]
- Barrer, R.M.; Villiger, H. The crystal structure of the synthetic zeolite L. Z. Kristallogr. Cryst. Mater. 1969, 128, 352–370. [Google Scholar] [CrossRef]
- Bhat, S.D.; Niphadkar, P.S.; Gaydhankar, T.R.; Awate, S.V.; Belhekar, A.A.; Joshi, P.N. High temperature hydrothermal crystallization, morphology and yield control of zeolite type K-LTL. Microporous Mesoporous Mater. 2004, 76, 81–89. [Google Scholar] [CrossRef]
- Larson, A.C.; VonDreele, R.B. GSAS General Structure Analysis System Operation Manual; Los Alamos National Laboratory: Santa Fe, NM, USA, 2000; Volume 86, pp. 1–179. [Google Scholar]
- Gigli, L.; Arletti, R.; Fois, E.; Tabacchi, G.; Quartieri, S.; Dmitriev, V.; Vezzalini, G. Unravelling the high-pressure behaviour of dye-zeolite L hybrid materials. Crystals 2018, 8, 79. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Kao, C.C.; Kim, S.J.; Lee, H.-H.; Lee, D.R.; Shin, T.J.; Choi, J.-Y. Water nanostructures confined inside the quasi-one-dimensional channels of ltl zeolite. Chem. Mater. 2007, 19, 6252–6257. [Google Scholar] [CrossRef]
- Newell, P.A.; Rees, L.V.C. Ion-exchange and cation site locations in zeolite L. Zeolites 1983, 3, 22–27. [Google Scholar] [CrossRef]
- Seoung, D.; Lee, Y.; Kim, S.J.; Lee, H.-H.; Ahn, D.; Shin, N.-S.; Vogt, T.; Lee, Y. Pressure-induced hydration and cation migration in a Cs+ exchanged gallosilicate zeolite LTL: Synchrotron X-ray powder diffraction study at ambient and high pressures. Microporous Mesoporous Mater. 2010, 136, 75–82. [Google Scholar] [CrossRef]



| Zeolite | Channel HO/ Unit-Cell | Cation Cluster HO/ Unit-Cell | Channel Aperture (Å) | |
|---|---|---|---|---|
| O1-O1 | O2-O2 | |||
| Li-L | 11.62 | 9.79 | 9.739 | 10.572 |
| K-L | 17.41 | 13.47 | 9.825 | 10.491 |
| Cs-L | 9.49 | 2.18 | 10.694 | 10.326 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Price, L.A.; Jones, Z.; Nearchou, A.; Stenning, G.; Nye, D.; Sartbaeva, A. The Effect of Cation Exchange on the Pore Geometry of Zeolite L. AppliedChem 2022, 2, 149-159. https://doi.org/10.3390/appliedchem2030011
Price LA, Jones Z, Nearchou A, Stenning G, Nye D, Sartbaeva A. The Effect of Cation Exchange on the Pore Geometry of Zeolite L. AppliedChem. 2022; 2(3):149-159. https://doi.org/10.3390/appliedchem2030011
Chicago/Turabian StylePrice, Lisa A., Zöe Jones, Antony Nearchou, Gavin Stenning, Daniel Nye, and Asel Sartbaeva. 2022. "The Effect of Cation Exchange on the Pore Geometry of Zeolite L" AppliedChem 2, no. 3: 149-159. https://doi.org/10.3390/appliedchem2030011
APA StylePrice, L. A., Jones, Z., Nearchou, A., Stenning, G., Nye, D., & Sartbaeva, A. (2022). The Effect of Cation Exchange on the Pore Geometry of Zeolite L. AppliedChem, 2(3), 149-159. https://doi.org/10.3390/appliedchem2030011