1. Stress and Immune Response
Conservation of Pro-inflammatory Transcriptomic Responses in Two Salmonid Species Sharing a Recent Whole Genome Duplication
Naseer, S.1, Clark, T.2, Collet, B.2, Gundappa, M.K.3, Laurent, A.2, Macqueen, D.J.3, Boudinot, P.2 and Martin, S.A.M.1
1 Fish Immunology Research Centre, University of Aberdeen, UK.
2 Université Paris-Saclay, INRAE, UVSQ, VIM, Jouy-en-Josas, France.
3 Roslin Institute, University of Edinburgh, UK.
Summary
Infectious diseases represent one of the most pressing threats to modern aquaculture at a global scale. Control of disease outbreaks is essential for maintaining fish health and welfare, which is crucial for sustainable and profitable aquaculture production. The work presented here is part of the EU Horizon 2020 project, AQUA-FAANG, which aims to understanding the relationship between genotype and phenotype by the improvement of genome annotation in European farmed fish species.
The early innate immune response is critical to the outcome of infection and many previous studies have examined the transcriptomic response at this time. Here, we extend such work to profile both responses in vivo and in vitro within head kidney leukocytes. We used a bacterial infection (heat killed vibrio) as the inducer in both Atlantic salmon and rainbow trout, two closely related salmonids that are extensively used in European aquaculture. Our aim was to determine the commonalities and differences in response to vibrio stimulation between these species through gene expression analysis considering gene evolutionary relationships, including with respect to an ancestral salmonid-specific whole genome duplication (WGD) event.
A total of 48 in vivo and in vitro Atlantic salmon and rainbow trout vibrio stimulated, and control libraries were sequenced (~30 M paired end 150 bp reads per sample). The number of differentially expressed genes was measured using DESeq2 (salmon in vivo up = 1609, down = 2013; in vitro up = 3210, down = 4329; trout in vivo up = 1184, down = 1914; in vitro up = 2931, down = 3752) (adjusted p <0.05, Log2Fold change >1 or <−1). Differentially expressed genes common between in vivo and in vitro stimulations were assessed (salmon up = 594, down = 890; trout up = 558, down = 640). Pathway and gene set enrichment analyses using DAVID indicated a strong immune response from all the stimulated groups. We are examining the conservation of expression responses for key pro-inflammatory gene families between Atlantic salmon and rainbow trout, taking into account orthology and paralogy relationships across species. These data will inform our understanding of the evolution of the innate immune response in salmonid species in relation to their lineage-specific WGD.
Acknowledgments: The AQUA-FAANG project has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement 817923.
Genome Editing to Investigate Genetic Resistance to IPNV in Atlantic Salmon
Jin, Y.H., Robledo, D. and Houston, R.D.
The Roslin Institute, The University of Edinburgh, UK.
Summary
Genetic resistance to infectious pancreatic necrosis virus (IPNV) in Atlantic salmon is almost completely explained by a single locus (QTL) on chromosome 26. Marker-assisted selection based on this QTL in breeding programmes resulted in significantly reduced IPNV outbreaks in the farms. Previously, cdh1 was reported as a cellular receptor for the viral entry [1]. On the other hand, NEDD-8-activating enzyme 1 (nae1) gene was pointed as a putative functional candidate underlying the QTL effect by analyses including whole genome sequencing, functional annotation, and differential expression analysis between salmon fry of homozygous-resistant and homozygous-susceptible genotypes challenged with IPNV [2]. Here, the role of nae1 and cdh1 in IPN resistance was further assessed via CRISPR-Cas9 knockout and subsequent IPNV challenge in Atlantic salmon cell lines. In addition, the impact of chemical inhibition of the Nae1 protein and antibody treatment of Cdh1 against IPNV challenge was also evaluated. Both nae1 knockout and its protein inhibition resulted in a highly significant reduction in IPNV replication. In contrast, cdh1 knockout and its antibody treatment did not have an impact on IPNV replication. Together, these results suggest nae1 as the causative gene underlying the major QTL affecting resistance to IPNV in salmon, and follow-up in vivo studies are currently in progress. Nonetheless, further investigation is necessary to elucidate the role of neddylation in this host–pathogen interaction. The current study is a good example of the potential of combining high-throughput genomics approaches with targeted genome editing to understand the genetic basis of disease resistance in aquaculture species.
References
Moen, T.; Torgersen, J.; Santi, N.; Davidson, W.S.; Baranski, M.; Ødegård, J.; Kjøglum, S.; Velle, B.; Kent, M.; Lubieniecki, K.P.; et al. Epithelial Cadherin Determines Resistance to Infectious Pancreatic Necrosis Virus in Atlantic Salmon.
Genetics 2015,
200, 1313–1326.
https://doi.org/10.1534/genetics.115.175406.
Pavelin, J.; Jin, Y.H.; Gratacap, R.L.; Taggart, J.B.; Hamilton, A.; Verner-Jeffreys, D.W.; Paley, R.K.; Rubin, C.-J.; Bishop, S.C.; Bron, J.E.; et al. The nedd-8 activating enzyme gene underlies genetic resistance to infectious pancreatic necrosis virus in Atlantic salmon.
Genomics 2021,
113, 3842–3850.
https://doi.org/10.1016/j.ygeno.2021.09.012.
Symmetry of Ohnolog Gene Expression in the Interferon Stimulated Pathways of the Tetraploid Common Carp
Blasweiler, A.
Aquaculture and Fisheries, Wageningen University, De Elst 1 Building, 122 6708 WD Wageningen (The Netherlands).
Summary
The common carp (Cyprinus carpio) is a species that came into existence due to a hybridization of two different cyprinid species about 12 MYA ago. The common carp, therefore, contains two subgenomes (A and B) of different ancestral origin, referred to as allo-tetraploidy. Recent studies have shown that the common carp has retained a considerable part of its duplicated genes within its genome, but questions remain as to which degree these duplicated genes have remained active in both subgenomes over time. Genes located on subgenome B appear to have higher expression levels than their counterparts (ohnologs) on subgenome A, suggesting a degree of subgenome dominance. These differences in expression are small; however, rigorous interpretation of subgenome expression requires a good genome assembly and annotation. To this end, we assembled a chromosome-level assembly of the common carp. The new carp genome (WagV4.0; Ensembl ID GCA_905221575.1) facilitated the zooming in on subgenome expression levels by studying, in particular, interferon-stimulated genes (ISGs) as a collective and well-characterized group of genes commonly expressed in response to interferon type-1 following viral infection. Specifically, we looked at 89 ISGs with ancestral origins before the hybridization event, therefore maximizing the chance that both ancestors of the common carp would have had a complete set of these genes present in their genome. Using gene expression analysis, we show that both subgenomes express an almost complete set of ancestral ISGs. We also demonstrate that the ISG homologs present on both subgenomes show diverse degrees of inducibility. Based on our analysis of an ancestral functional pathway, part of complex immune responses to viruses, single subgenome dominance does not appear to be most evident in allo-tetraploid common carp. Rather, both subgenomes present in common carp, and in most cases both gene copies, appear to be of more or less equal importance for coordinating complex physiological responses.
Acknowledgements: This study was supported by the European Union’s Horizon 2020 research and innovation programme under grant agreement No 81792 (AQUA-FAANG).
Characterisation of Functional Mechanisms Underlying Genetic Resistance to Infectious Salmon Anaemia in Atlantic Salmon
Gervais, O.1, Barria, A.1, Papadopoulou, A.1, Gratacap, R.1, Jin, Y.1, Hillestad, B.2, Tinch, A.E.2, Martin, S.A.M.3, Orosa-Puente, B.4, Hassan, M.1, Houston, R.D.1 and Robledo, D.1
1 The Roslin Institute, University of Edinburgh, UK.
2 Benchmark Genetics Norway AS Sandviksboder 3A, N-5035 Bergen, Norway.
3 Institute of Biological and Environmental Sciences, University of Aberdeen, Aberdeen, UK.
4 Institute of Molecular Plant Sciences, University of Edinburgh, UK.
Summary
Infectious Salmon Anaemia Virus (ISAV) causes a notifiable disease in Atlantic salmon and represents a major problem for salmon breeders and producers worldwide. Current prevention and therapeutic methods are not fully effective, and therefore selective breeding to produce ISAV-resistant strains of Atlantic salmon (Salmo salar) is a high priority for the industry. Genomic selection and potentially genome editing can be applied to enhance host resistance. Both approaches can benefit from increased knowledge of the genetic and functional mechanisms of host response and genetic resistance to ISAV. Here, we have combined bulk and single-cell RNA sequencing in in vitro and in vivo models to study Atlantic salmon immune response against ISAV.
First, the transcriptomic changes in response to ISAV were studied in four different tissues (heart, gills, head kidney and spleen) at three timepoints (pre-challenge, 7 and 14 dpi), with a clear but unique response to ISAV in each tissue. Comparisons between four resistant and four susceptible fish per timepoint, selected based on their genomic breeding values estimated from a disease challenge in 1353 fish from the same population, revealed a large number of differentially expressed genes in the head kidney, with a notably smaller number in the other tissues.
To better understand the host–response interaction at the cellular level and evaluate the importance of potential candidate resistance genes, the host response to ISAV was investigated at the single-cell level using an in vitro model (SHK-1 cells, derived from Atlantic salmon head kidney). Cells were challenged with ISAV and single-cell sequenced at 0, 24, 48 and 92 h post-infection. The transcriptome of the challenged Atlantic salmon cells was similar to controls at 24 h, but there was a clear response observed in the 48 h and 96 h samples. This response included several genes related to ubiquitination, and to investigate this process further immunoprecipitation of ubiquitinated proteins was performed in ISAV-challenged SHK-1 cells. An increase in ubiquitination was observed at 24 and 48 h after ISAV infection. Mass spectrometry of the ubiquitin-proteome will be performed to gain insight into the immune mechanisms regulated by ubiquitination in response to ISAV. These results improve our understanding of host responses to ISAV in Atlantic salmon and highlight potential target genes to improve host resistance.
Early Life Microbial Exposures Shape the Crassostrea gigas Immune System for Lifelong and Intergenerational Disease Protection
Fallet, M.1§, Montagnani, C.1§, Petton, B.2, de Lorgeril, J.1,3, Chaparro, C.1, Toulza, E.1, Boitard, S.4, Escoubas, J.M.1, Bulla, I.1, Gueguen, Y.1,5, Vidal-Dupiol, J.1, Grunau, C.1, Mitta, G.1,6* and Cosseau, C.1*
1 IHPE, Univ. Montpellier, CNRS, Ifremer, Univ. Perpignan Via Domitia, Perpignan France.
2 Ifremer, UBO CNRS IRD, LEMAR UMR 6539 Argenton, France.
3 Ifremer, IRD, Univ Nouvelle-Calédonie, Univ La Réunion, ENTROPIE, F-98800 Nouméa, Nouvelle-Calédonie, France.
4 CBGP, Université de Montpellier, CIRAD, INRAE, Institut Agro, IRD, Montpellier, France.
5 MARBEC, Univ Montpellier, CNRS, Ifremer, IRD, Sète, France.
6 Ifremer, UMR 241 Écosystèmes Insulaires Océaniens, Labex Corail, Centre Ifremer du Pacifique, BP 49, 98725 Tahiti, Polynésie française.
§ Both authors contributed equally to this work.
Summary
The interaction of organisms with their surrounding microbial communities influences many biological processes, a notable example of which is the shaping of the immune system in early life. In the Pacific oyster, Crassostrea gigas, the role of the environmental microbial community in immune system maturation—and, importantly, protection from infectious disease—is still an open question. Here, we demonstrate that early life microbial exposure durably improves oyster survival when challenged with the pathogen causing Pacific Oyster Mortality Syndrome (POMS), both in the exposed generation and in the subsequent one. Combining microbiota, transcriptomic, genetic, and epigenetic analyses, we show that the microbial exposure induced changes in epigenetic marks and a reprogramming of immune gene expression leading to long-term and intergenerational immune protection against POMS. We anticipate that this protection likely extends to additional pathogens and may prove to be an important new strategy for safeguarding oyster aquaculture efforts from infectious disease.
Identifying Genes Coding for Semiochemicals in Atlantic Salmon (Salmo salar)
Gulliksen, S.1,2, Burgerhout, E.1, Knutsdatter-Østbye, T.K.1, Krasnov, A.1, Helge Tveiten, H.2 and Robinson, N.1,3
1 Nofima, PO Box 6122, 9292 Tromsø, Norway.
2 UiT Arctic University of Norway, 9019 Tromsø, Norway.
3 Sustainable Aquaculture Laboratory- Temperate and Tropical (SALTT), School of BioSciences, The University of Melbourne, Parkville 3010, Australia.
Summary
Parasitic infestations by sea lice (L. salmonis) are one of the largest challenges in Atlantic salmon aquaculture. They dramatically impact fish health and welfare, and thereby the quality and economy. Some species of salmon (such as Atlantic and chum) are more susceptible to sea lice infestation than others (e.g., pink and coho). For sea lice, semiochemicals, released by salmonids, play a key role in host detection. Studies have shown that they are attracted not only by salmon but also by salmon-conditioned water (SCW). No attraction was seen to water that was conditioned with non-salmonid marine fish or depleted water, where the solid phase was extracted. In our broader project, we are investigating whether coho and pink salmon are less attractive to sea lice than Atlantic and chum and how the semiochemical profile differs between these species. Blocking semiochemical production using CRISPR/Cas9 methodology could potentially prevent infestation with lice.
In a search for target genes that could be involved in the semiochemical production, we found that cytochrome P450 enzymes (CYPs) especially vary in the genomes of salmonids and other marine species. While some types are present in salmonids, they are absent or highly diverged in other marine species. CYPs are involved in pathways producing secondary metabolites and are characterized with high divergence and rapid evolution, so likely to be highly diverged even between salmon species. The expression levels of CYPs were analyzed with qPCR in liver, gill, and skin of Atlantic, chum, coho, and pink salmon samples, which were taken before and after smoltification. Preliminary results show clear differences in expression between salmonid species and tissues. Genes showing high expression in the gill and skin of Atlantic salmon will be preferred as candidate genes and targeted for knock-out using gene editing in Atlantic salmon. The down-stream effects of gene silencing will be investigated by analyses of gene expression and chemical composition of cells and media. Knowledge generated from this study will contribute to our broader understanding of the genetic mechanisms affecting host resistance to sea lice and help formulate future strategies for boosting Atlantic salmon host resistance to reduce the impact of this disease.
Using snRNA-Seq to Elucidate Comparative Resistance to Sea Lice in Salmonids
Ruiz Daniels, R.1, Salisbury, S.1, Gervais, O.1, Villamayor, P.R.1, Monaghan, S.J.2, Bron, J.E.2, Sveen, L.3, Fast, M.D.4, Houston, R.1, Robledo, D.1 and Robinson, N.3,5
1 The Roslin Institute and Royal (Dick) School of Veterinary Studies, University of Edinburgh, Edinburgh, UK.
2 Institute of Aquaculture, Faculty of Natural Sciences, University of Stirling, Stirling, UK.
3 Nofima, PO Box 6122, 9292 Tromsø, Norway.
4 Atlantic Veterinary College—University of Prince Edward Island, Department of Pathology and Microbiology, Charlottetown PEI, Canada.
5 Sustainable Aquaculture Laboratory- Temperate and Tropical (SALTT), School of BioSciences, The University of Melbourne, Parkville 3010, Australia.
Summary
The parasitic sea louse Lepeophtheirus salmonis is a critical threat to the farming of Atlantic salmon (Salmo salar), with losses in Norway alone totalling up to USD 436 million annually. Sea louse infection leads to secondary infections, increases mortality and reduces growth rate and body condition of their host, Atlantic salmon, in both farmed and wild populations. Management through treatment of outbreaks on salmon farms has proven intractable due to lice rapidly evolving resistance to previously effective veterinary medicines.
The susceptibility of salmonid species to sea louse infestation varies significantly. In contrast to Atlantic salmon, it is known that coho salmon (Oncorhynchus kisutch) and pink salmon (O. gorbuscha) are both highly resistant to infection. The objective of our study is to take a comparative approach to elucidate the cellular and genetic mechanisms by which these two species exhibit significant resistance, in contrast to the susceptible species Atlantic salmon and chum salmon (O. keta). With this aim, we conducted a controlled infection experiment over the course of 168 h, generating a time course of infected skin and fin tissues from these four species. Using these samples, we are generating atlases of RNA expression at the level of individual nuclei over the course of sea louse infection in all four species.
We present preliminary results consisting of a snRNA-seq atlas of fins from Atlantic salmon. The cell atlas exhibits significant heterogeneity of cell types, including keratinocytes, mesenchymal cells and immune cells, and we further show that the transcriptomic profiles of these populations allow for the identification of a range of subtypes in each lineage. This atlas will be used as the basis for the comparative analysis of cellular transcriptomics in infected tissues from the other three salmonid species.
Genomic Selection for Disease Resistance to the Copepod Lernanthropus kroveri in European Seabass Using the MEDFIH SNP-Array and Selected Low-Density SNP Panels
Oikonomou, S.1,2, Kazlari, Z.1, Papanna, K.3, Manousaki, T.2, Loukovitis, D.1,4, Dimitroglou, A.3, Kottaras, L.3, Gourzioti, E.3, Pagonis, C.3, Kostandis, A.3, Papaharisis, L.3, Tsigenopoulos, C.S.2 and Chatziplis, D.1
1 Lab. of Agrobiotechnology and Inspection of Agricultural Products, Dept. of Agriculture, International Hellenic University, Greece.
2 Institute of Marine Biology, Biotechnology and Aquaculture (IMBBC), Hellenic Centre for Marine Research (HCMR), Greece.
3 Nireus Aquaculture SA, Greece.
4 Research Institute of Animal Science, ELGO, Greece.
Summary
The copepod Lernanthropus kroyeri is a host-specific parasite affecting the gills of the European seabass. Based on a multi-trait pedigree analysis with growth traits, the heritability of parasite count of the fish was previously estimated to be 0.28 (n = 2425, [1]), while when a selected sub-sample of infected fish (985) was genotyped using the MedFish array [2], the univariate heritability of the parasite count was estimated to be 0.75 using a Pedigree Relationship Matrix (PRM) and 0.71 using a Genomic Relationship Matrix (GRM). A GWAS was performed for parasite count and growth and based on the results, we constructed two low-density panels (SNP–panel 1 and SNP–panel 2) from selected numbers of SNPs, i.e., 1960 and 2907 using as criteria the p-value from the GWAS analysis (p-value < 0.01 and <0.05, respectively). Estimated Breeding Values (EBVs) were estimated using BLUP and Genomic Estimated Breeding Values (GEBVs) were calculated using GBLUP for the three SNP panels (MedFish array, SNP—panel 1 and 2), with BLUPF90. Each time, 20% of the population was selected randomly and its phenotypes were masked; thus, the breeding values (EBVs and GEBVs) were estimated using the information from 80% of the total fish. This process was performed 20 times and the correlation between the predicted values and phenotypes was estimated. A one-way ANOVA with repeated measurements was performed among the four groups showing a significant difference (p-value < 0.01) among the four genetic evaluation procedures. Finally, the use of pedigree showed the lowest correlation, while the use of SNP–panel 2 provided the highest prediction followed by the SNP–panel 1. Additive genetic variance estimates are higher in selected samples, independent of the estimation method (PRM or GRM). The results indicate that small and carefully chosen SNP panels could potentially be utilized in cases of multi-trait genomic evaluation with alternative genotype-by-sequencing methodologies to reduce genotyping cost.
References
Papapetrou, M.; Kazlari, Z.; Papanna, K.; Papaharisis, L.; Oikonomou, S.; Manousaki, T.; Loukovitis, D.; Kottaras, L.; Dimitroglou, A.; Gourzioti, E.; et al. On the trail of detecting genetic (co)variation between resistance to parasite infections (
Diplectanum aequans and
Lernanthropus kroyeri) and growth in European seabass (
Dicentrarchus labrax).
Aquac. Rep. 2021,
20, 100767.
https://doi.org/10.1016/j.aqrep.2021.100767.
Peñaloza, C.; Manousaki, T.; Franch, R.; Tsakogiannis, A.; Sonesson, A.K.; Aslam, M.L.; Allal, F.; Bargelloni, L.; Houston, R.D.; Tsigenopoulos, C.S. Development and testing of a combined species SNP array for the European seabass (Dicentrarchus labrax) and gilthead seabream (Sparus aurata). Genomics 2021, 113, 2096–2107.
Whole Genome Sequencing to Refine the Detection of QTL for Viral Nervous Necrosis in European Sea Bass (Dicentrarchus labrax)
Delpuech, E.1,2, Vandeputte, M.2,3, Phocas, F.3, Bestin, A.1, Besson, M.1, Bajek, A.4, Brunier, J.4, Cherbuin, A.5, Imarazene, B.5, Sourdioux, M.1, Haffray, P.1, Morvezen, R.1 and Allal, F.2
1 SYSAAF, Station LPGP/INRAE, Campus de Beaulieu, 35042 Rennes, France.
2 MARBEC, Univ. Montpellier, Ifremer, CNRS, IRD, INRAE, 34250 Palavas-les-Flots, France.
3 Université Paris-Saclay, INRAE, AgroParisTech, GABI, 78350 Jouy-en-Josas, France.
4 Ecloserie Marine de Gravelines-Ichtus, 59273 Gravelines, France.
5 Ferme Marine Du Douhet, 17840 La Brée Les Bains, France.
Summary
Viral nervous necrosis (VNN) is considered the most impacting disease for the European sea bass industry, leading to mortality up to 90%. Selective breeding is a promising strategy to reduce the frequency and severity of the outbreaks. Several genomic regions for VNN resistance have been identified with SNP arrays [1–3]. This work presents a next level of the genome-wide association studies (GWAS) using whole genome sequencing and imputation in order to refine the genetic architecture of VNN resistance in European seabass.
Four full-sib backcross families were produced by mating four resistant x susceptible hybrid sires from different geographic backgrounds (western, north-eastern and south-eastern Mediterranean Sea) to four susceptible females from the western Mediterranean Sea. All sires and females used in these backcrosses were sequenced on a NovaSeq sequencer. Moreover, offspring were challenged to nervous necrosis virus and genotyped on the ThermoFisher 57K DlabCHIP SNP array [2]. We analysed sequences with a homemade pipeline. In a first step, the variant calling was processed according to the DeepVariant best practice and 2,390,971 SNPs were identified. In a second step, FImpute v2.2 software was used to obtain an imputed sequence for each of the 1334 offspring. Then, GWAS were performed to detect association between 2.4 million imputed SNPs and the VNN resistance trait, using GEMMA software.
A high association was detected on LG12 in three of the four backcross families. In two of them, an additional QTL was located on LGx. We thus validate the strong effect QTL has on LG12 and refine its position compared to previous studies with lower density genotypes.
References
Palaiokostas, C.; Cariou, S.; Bestin, A.; Bruant, J.-S.; Haffray, P.; Morin, T.; Cabon, J.; Allal, F.; Vandeputte, M.; Houston, R.D. Genome-wide association and genomic prediction of resistance to viral nervous necrosis in European sea bass (
Dicentrarchus labrax) using RAD sequencing.
Genet. Sel. Evol.
2018,
50, 30.
https://doi.org/10.1186/s12711-018-0401-2.
Griot, R.; Allal, F.; Phocas, F.; Brard-Fudulea, S.; Morvezen, R.; Bestin, A.; Haffray, P.; François, Y.; Morin, T.; Poncet, C.; et al. Genome-wide association studies for resistance to viral nervous necrosis in three populations of European sea bass (
Dicentrarchus labrax) using a novel 57k SNP array DlabChip.
Aquaculture 2021,
530, 735930.
https://doi.org/10.1016/j.aquaculture.2020.735930.
Faggion, S.; Bertotto, D.; Bonfatti, V.; Freguglia, M.; Bargelloni, L.; Carnier, P. Genomic Predictions of Phenotypes and Pseudo-Phenotypes for Viral Nervous Necrosis Resistance, Cortisol Concentration, Antibody Titer and Body Weight in European Sea Bass.
Animals 2022,
12, 367.
https://doi.org/10.3390/ani12030367.
Ancestral Physical Stress and Later Immune Gene Family Expansions Shaped Bivalve Mollusc Evolution
Regan, T.1, Stevens, L.2, Peñaloza, C.1, Houston, R.D.1, Robledo, D.1 and Bean, T.P.1
1 The Roslin Institute and Royal (Dick) School of Veterinary Studies, University of Edinburgh, Edinburgh, UK.
2 Tree of Life Programme, Wellcome Sanger Institute, Wellcome Genome Campus, Hinxton, Cambridge, UK.
Summary
Bivalves play vital roles in ocean conservation and food security by acting as ecosystem engineers and underlying >20% of global aquaculture production. Aquaculture production facilities and wild bivalve populations face increasing threats from ocean acidification, emerging disease and global warming; however, our understanding of bivalve biology and evolution is limited. High levels of heterozygosity and repeated regions limited bivalve genome assembly until advances in long read sequencing technology. This has led to a great increase in the number of bivalve assemblies in recent years. By analysing the genomes of 32 species representing each molluscan class, we identified gene families that have undergone expansion during bivalve evolution. Expansions in redox, chaperone and protein recycling gene families were shared across all of Bivalvia. These conserved responses to physical stress mirrors adaptation strategies of other sessile organisms such as plants. Conversely, we discovered that expansions in innate immune response gene families were less conserved across Bivalvia and tended to be species specific or clade specific. This reflects the high level of tolerance bivalves require during constant pathogen exposure. The increasing availability of accurate genome assemblies will provide greater resolution to these analyses. This allows future studies to investigate further points of evolutionary pressure in other understudied taxa and potentially different populations of a single species.
The Impact of Piscirickettsia salmonis Infection on Genome-Wide DNA Methylation Profile in Atlantic Salmon
Mukiibi, R.1, Peñaloza, C.1, Gutierrez, A.1,2, Yáñez, J.M.3,4, Houston, R.D.1 and Robledo, D.1
1 The Roslin Institute and Royal (Dick) School of Veterinary Sciences, The University of Edinburgh, Edinburgh, UK.
2 Institute of Aquaculture, Faculty of Natural Sciences, University of Stirling, Stirling FK9 4LA, UK.
3 Facultad de Ciencias Veterinarias y Pecuarias, Universidad de Chile, University of Chile, Santiago, Chile.
4 Center for Research and Innovation in Aquaculture (CRIA), Universidad de Chile, Santiago, Chile.
Summary
Salmon rickettsial septicaemia (SRS) caused by the intracellular bacteria Piscirickettsia Salmonis generates significant mortalities to farmed Atlantic salmon, particularly in Chile. Due to its economic importance, a wealth of research has focused on the biological mechanisms underlying pathogenicity of P. salmonis, the host response, and genetic variation in host resistance. DNA methylation is a fundamental epigenetic mechanism modulating the response of an organism to internal and external stimuli and plays a key role in host–pathogen interactions via the regulation of gene expression.
In the current study, the role of head kidney and liver DNA methylation in the response to P. salmonis infection was investigated in a commercial Atlantic salmon population. The global DNA methylation profile of 66 juvenile fish was obtained using reduced representation bisulphite sequencing (RRBS). For both head kidney and liver, methylomes of infected animals (3- and 9-days post-infection) and uninfected control animals were compared. Groups of fish with divergent (high and low) breeding values for resistance to P. salmonis infection were also compared to examine the relationship between DNA methylation and genetic resistance.
Head kidney and liver showed organ-specific global methylation patterns, but with similar distribution of methylated sites across gene features. Integration of methylation with RNA sequencing data from the same samples revealed that methylation levels predominantly showed a negative correlation with gene expression; nonetheless, a considerable proportion of positive correlations were also observed. Methylation within the first exon showed the strongest negative correlation with gene expression. A total of 911 and 813 differentially methylated CpG sites were identified between infected and control samples in the head kidney at 3 and 9 days, respectively, whereas only 30 and 44 sites were differentially methylated in the liver. Differential methylation in the head kidney was notably associated with immunological processes such as actin cytoskeleton regulation, phagocytosis, endocytosis and pathogen-associated pattern receptor signaling. Comparison between resistant and susceptible fish identified 113 and 48 differentially methylated sites in the head kidney and liver, respectively. These results contribute to the growing basic understanding of the role of methylation in the regulation of gene expression and response to infectious diseases, and in particular reveal key immunological functions regulated by methylation in Atlantic salmon in response to P. salmonis.
Integration of Host–Pathogen Functional Genomics Data into a Chromosome-Level Turbot (Scophthalmus maximus) Genome Assembly
Aramburu, O., Blanco, A., Bouza, C. and Martínez, P.
University of Santiago de Compostela, Spain.
Summary
Disease resilience is of utmost relevance for turbot aquaculture. Several infectious diseases covering a broad spectrum, from viruses, bacteria to different parasites, have been identified by industry. Since they increase mortality rates, reduce feed conversion ratios and slow down growth, genetic breeding programs for increasing disease resilience are a recognized useful alternative for controlling pathologies. For this, knowledge of the genetic basis underlying resilience using genomic tools is essential to develop the best effective breeding strategies.
In the present study, we compiled the existing genomic information generated in the last decade to construct an integrated atlas of candidate genes and genomic regions involved in pathogen resistance against the main turbot pathogens targeted by industry (Aeromonas salmonicida, Philasterides dicentrarchi, Enteromyxum scophthalmi and the VHS virus) within the chromosome-level turbot genome assembly recently released. Information comprehends reannotated differentially expressed genes (DEG) in different tissues along temporal series, QTL markers associated with important productive traits (disease resistance and growth) and signatures of domestic or wild selection, represented by runs of homozygosity (ROHi) islands and outlier loci for divergent selection.
Most genetic features could be successfully relocated in the turbot assembly including 70–92% of the total DEGs, plus all QTL markers, ROHi and outlier loci. The updated annotation of DEGs for resistance to each pathology demonstrated important changes. Whereas 50–70% of the DEGs’ new annotation was coherent with the original, roughly 15–30% showed imprecise annotations in both assembly versions; ~5% lost their original annotation and ~15% that could not be annotated before now were. Functional profiling and enrichment of these newly annotated genes revealed some key functions in the response to infective diseases, such as chemotaxis, apoptosis regulation, leukocyte differentiation, cell adhesion, iron homeostasis and vascular permeability. Some DEGs, such as celsr1a (cadherin EGF LAG seen-pass G-type receptor 1), fgg (fibrinogen gamma chain) and c1qtnf9 (C1q and TNF related 9) were found near pathogen-associated QTL markers. Additionally, some shared DEGs for resistance to all pathogens were positioned near QTL markers or ROHi, such as hamp (hepcidin-1), plg (plasminogen) and a fibrinogen alpha chain-like gene. Overall, our results provide a global, integrative insight into the genetic architecture of the host–pathogen interaction that could prove useful for future genomic studies to benefit aquaculture breeding programs.
A Single Genomic Region Involving a Putative Chromosome Rearragement in Flat Oyster (Ostrea edulis) Is Associated with Divergent Selection to the Parasite Bonamia ostreae
Sambade, I.M.1, Casanova, A.1, Blanco, A.1, Gundappa, M.2, Bean, T.P.2, Macqueen, D.J.2, Houston, R.D.2, Villalba, A.3, Vera, M.1, Kamermans, P.4 and Martínez, P.1
1 Department of Zoology, Genetics and Physical Anthropology, Faculty of Veterinary. Universidade de Santiago de Compostela. 27002 Lugo, Spain.
2 The Roslin Institute and Royal (Dick) School of Veterinary Studies, Easter Bush, Midlothian, EH25 9RG, UK.
3 Centro de Investigacións Mariñas (CIMA), Consellería do Mar, Xunta de Galicia, 36620 Vilanova de Arousa, Spain.
4 Wageningen Marine Research, P.O. Box 77, Yerseke, The Netherlands.
Summary
European flat oyster (Ostrea edulis) is an ecologically and economically important marine bivalve that has been severely affected by the intracellular parasite Bonamia ostreae. In this study, a flat oyster SNP array (~14,000 SNPs) was used to validate previously reported outlier loci for divergent selection associated with Bonamia ostreae exposure in the northeast Atlantic area. A total of 134 wild and hatchery individuals from the North Sea, collected in naïve- (NV) and long-term-affected (LTA) areas, were analysed. Genetic diversity and differentiation were related to the sampling origin (wild vs. hatchery) when using neutral markers, and to bonamiosis status (NV vs. LTA) when using outlier loci for divergent selection. Two genetic clusters appeared intermingled in all sampling locations when using outlier loci and their frequency was associated with bonamiosis status. When both clusters were compared, outlier datasets showed high genetic divergence (FST > 0.25) unlike neutral loci (FST not ≠ 0). Moreover, the cluster associated with LTA samples showed much higher genetic diversity and significant heterozygote excess with outlier loci, but not with neutral data. Most outliers were mapped on chromosome 8 (OE-C8) of the flat oyster genome, supporting the major quantitative trait locus (QTL) previously suggested for resilience to bonamiosis. Furthermore, differentially expressed genes previously reported between NV and LTA strains showed higher mapping density on OE-C8. A range of relevant immune functions were specifically enriched among genes found on OE-C8, providing hypotheses for resilience mechanisms to an intracellular parasite. The results suggest the application of marker-assisted selection to breed strains of O. edulis with improved resilience to bonamiosis and facilitate the management of oyster beds for production and ecosystem service recovery.
2. Reproduction and Breeding
Integrative Transcriptomics Reveals Lipid Homeostasis Mechanisms in Non-endocrine Cells of the Teleost Pituitary Gland
Ager-Wick, E., Maugars, G., Von Krogh, K., Siddique, K., Nourizadeh-Lillabadi, R., Fontaine, R., Weltzien, F.-A. and Henkel, C.V.
Faculty of Veterinary Medicine, Norwegian University of Life Sciences, Ås, Norway.
Summary
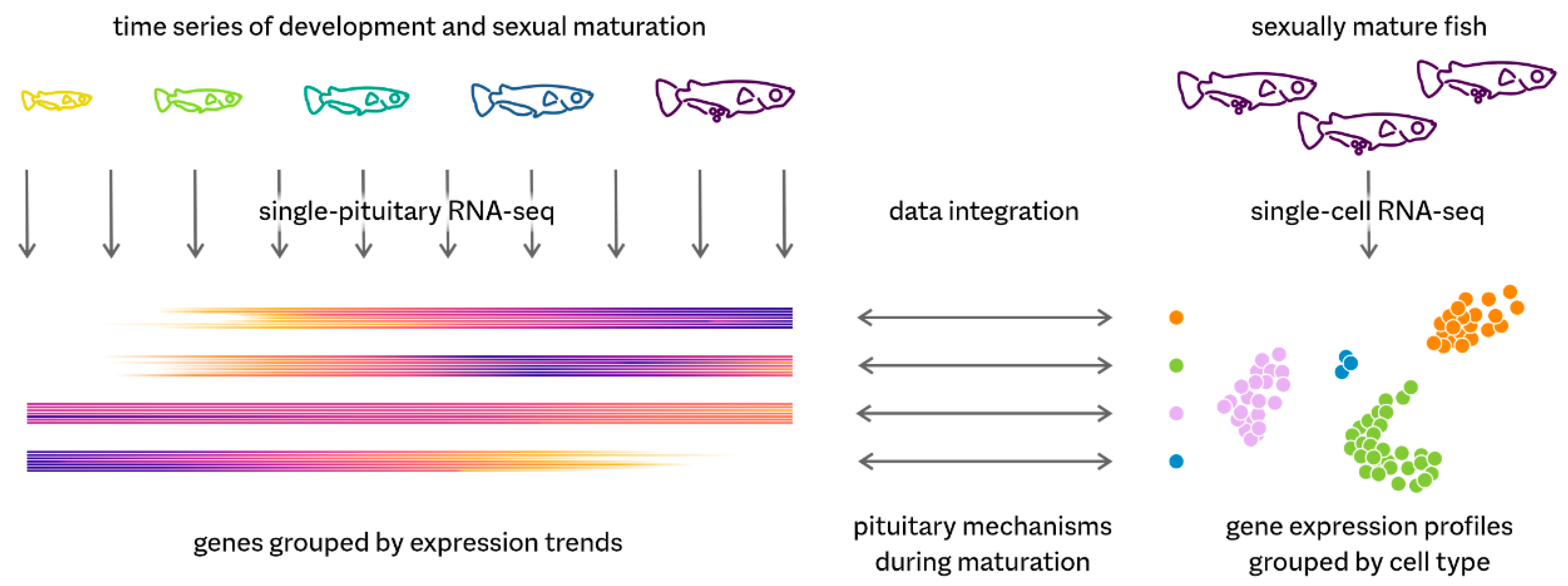
Directing both organismal homeostasis and physiological adaptation, the pituitary is a key endocrine gland in all vertebrates. It communicates the needs of the organism to different organs by secreting hormones into the bloodstream. We have used the model teleost medaka (Oryzias latipes) to investigate pituitary mechanisms using a combination of single-cell transcriptomics (scRNA-seq) and a comprehensive RNA-seq time series (Figure 1).
Figure 1.
Summary of the research presented.
Figure 1.
Summary of the research presented.
Our single-cell data on adult fish reveal nine cell types producing peptide hormones, demonstrating a strict division of labour—each hormone is produced by a dedicated cell type. This contrasts with the tetrapod pituitary, in which for example a single gonadotrope cell can produce both luteinizing hormone and follicle-stimulating hormone. In addition, we identified two distinct populations of prolactin-producing cells. In fish, this hormone is involved in osmoregulation, amongst other functions.
We have complemented this snapshot of the mature pituitary by a developmental RNA-seq time series of single pituitary glands. By linking temporal expression trends to single-cell RNA-seq profiles, we show how the transcriptional activities of all cell types change during sexual maturation. One of the most prominent changes is the decline of the non-endocrine folliculo-stellate cells and especially of a rare subpopulation expressing genes encoding secreted lipid transport proteins. As these genes are typically associated with the liver, this reveals the existence of unexpected connections between endocrine communication, lipid homeostasis, and sexual maturation.
Circulating miRNAs as Biomarkers of Reproduction and Egg Quality
Roza de Abreu, M.1, Cardona, E.1,2, Milhade, L.3, Guyomar, C.4, Skiba-Cassy, S.2 and Bobe, J.1
1 INRAE, LPGP, 35000 Rennes, France
2 INRAE, Univ. Pau & Pays Adour, E2S UPPA, NUMEA, 64310 Saint-Pée-sur-Nivelle, France.
3 IRISA, INRIA, CNRS, Université de Rennes 1, 35000 Rennes, France.
4 INRAE, Sigenae, 31326 Castanet Tolosan, France.
Summary
Circulating miRNAs (c-miRNAs) are found in most, if not all, biological fluids due to their surprising long stability in nuclease-rich fluids. In humans, c-miRNAs have been proposed as non-invasive biomarkers of many pathologies and their relevance to serve as diagnostic or prognostic tools has been established. However, their features in non-pathological contexts and whether their expression profiles reflect normal life history events have received little attention, especially in non-mammalian species. The aim of the present study was to investigate the potential of c-miRNAs to serve as biomarkers of reproduction in fish with special attention for blood plasma and ovarian fluid, in which the eggs are bathed in the body cavity after ovulation.
In a first experiment, the blood plasma c-miRNA repertoire was studied at different reproductive stages using female rainbow trout (Oncorhynchus mykiss) during their second reproductive cycle. In a second experiment, blood plasma and ovarian fluid samples were collected at ovulation and 21 days later, in order to monitor the dramatic decrease in egg quality triggered by post-ovulatory egg ageing in the body cavity. Blood plasma samples were also collected 21 days after stripping to account for reproduction-related physiological changes occurring when eggs are removed from the body cavity. For both experiments, total RNA was extracted from plasma and ovarian fluid and subjected to Illumina small RNA sequencing. Data were analyzed using the Prost! software and the recently characterized rainbow trout miRNAome annotation [1].
Here we show that some c-miRNAs exhibit major changes in their abundance depending on the reproductive stage. This is especially the case for miR-202-5p, for which blood plasma levels were dramatically increased at ovulation, suggesting that this c-miRNA from ovarian origin could be used to predict ovulation. In addition, we observed that miR-202-5p was among the most abundant c-miRNAs in the ovarian fluid at ovulation, while its abundance subsequently decreased after ovulation. Several other c-miRNAs exhibited marked changes in their abundance in ovarian fluid after ovulation. Collectively, these c-miRNAs can serve as non-invasive biomarkers of post-ovulatory egg ageing and can therefore be used to identify and eliminate poor quality eggs when the date of ovulation is not known. Finally, we have identified two c-miRNAs from ovarian origin that are significantly over expressed in the blood plasma 21 days after ovulation when eggs remain in the body cavity. In contrast, these two c-miRNAs exhibit an opposite profile when eggs are stripped. It is known that the presence or removal of the eggs after ovulation triggers differences in FSH and LH plasma levels that have been hypothesized to regulate key oogenetic events in the ovary after ovulation including oogonia proliferation and initiation of the next oogenetic wave. The stripping-dependent regulation of these two ovarian miRNAs after ovulation suggest an important regulatory role. The subsequent identification of their targets will shed new light on the regulation of ovarian cycles in iteroparous fish species.
References
Cardona, E.; Guyomar, C.; Desvignes, T.; Montfort, J.; Guendouz, S.; Postlethwait, J.H.; Skiba-Cassy, S.; Bobe, J. Circulating miRNA repertoire as a biomarker of metabolic and reproductive states in rainbow trout. BMC Biol. 2021, 19, 235.
Transcripts and Proteins Complementary Contributions to Egg Developmental Success in Pikeperch (Sander lucioperca)
Żarski, D.1, Le-Cam, A.2, Frohlich, T.3, Kösters, M.3, Klopp, C.4, Nynca, J.1, Ciesielski, S.5, Sarosiek, B.1, Dryl, K.1, Montfort, J.2, Król, J.6, Fontaine, P.7, Ciereszko, and A:1, Bobe, J.2
1 Department of Gamete and Embryo Biology, Institute of Animal Reproduction and Food Research, Polish Academy of Sciences, Tuwima str. 10, 10-748 Olsztyn, Poland.
2 INRAE, UR1037 Fish Physiology and Genomics, F-35000 Rennes, France.
3 Laboratory for Functional Genome Analysis (LAFUGA), Gene Center, Ludwig Maximilian University of Munich, 81377 Munich, Germany.
4 Sigenae, UR875, INRAE, 31326 Castanet-Tolosan, France.
5 Department of Environmental Biotechnology, University of Warmia and Mazury, Olsztyn, Poland.
6 Department of Salmonid Research, The Stanislaw Sakowicz Inland Fisheries Institute, Olsztyn, Poland.
7 University of Lorraine, INRAE, UR AFPA, F-54000 Nancy, France.
Summary
Molecular profiling of the ova is an excellent approach aiming at understanding biological processes and mechanisms conditioning their developmental competence. Both mRNAs and proteins of maternal origin are important components responsible for embryonic development. However, lack or very weak concordance between protein and transcript levels in vertebrate oocytes and embryos along the early development suggest that maternally derived proteins and mRNAs are playing different roles. Until now, specific roles of the two types of molecules in vertebrates’ unfertilized ova were scarcely considered. Moreover, the integrated transcriptomic–proteomics analysis in relation to developmental competence of the ova is missing. The aim of this study was to compare proteomic and transcriptomic profiles of eggs of pikeperch—ecologically and commercially relevant freshwater fish species—and investigate the involvement of the two molecular profiles in the developmental competence of the eggs.
Our study provides a novel insight into the understanding of the role of maternally derived molecular cargo in the eggs of finfish. The data obtained shed light on the importance of transcriptome in the development of the nervous system suggesting neurogenesis-related mRNAs as a very important, non-genetic inheritance factor. Proteomic analysis highlights the specific role of proteins in the immune response in ovulated eggs. The molecular analysis of egg developmental competence emphasizes post-vitellogenic processes (final oocyte maturation and ovulation) as the ones potentially compromising the transcriptomic profile, with little effect on the proteomic cargo.
The data obtained indicate, for the first time, that transcripts and proteins have complementary functions in the eggs of pikeperch. Our study clearly indicates that the development of the nervous system is very characteristic transcriptomic “maternal legacy” in the ovulated egg whereas defense against pathogens is among the highest priorities of the proteins deposited along the oogenesis. Additionally, the data clearly show that processes directly preceding ovulation have a huge modulatory role on the quality of the ova by the modification of the transcriptomic, but not proteomic, profile. Considering the fact that the mechanisms driving these alterations as well as their consequences are still to be explored, the candidate quality markers, provided for pikeperch for the first time along with the current study, are creating valuable resources for the further exploration of the reproductive capacity in fish.
Acknowledgments: The study was funded by the National Science Center for Poland (project No. 2016/22/M/NZ9/00590).
Vgll3a Alleles Affect the Brain–Pituitary–Gonad Axis in Atlantic Salmon (Salmo salar) via the Regulation of Fshb
Kjærner-Semb, E.1, Crespo, D.1, Vogelsang, P.1, Furmanek, T.1, Fjelldal, P.G.1, Fraser, T.1, Hansen, T.1, Edvardsen, R.B.1, Schulz, R.W.1,2 and Wargelius, A.1
1 Institute of Marine Research, Norway.
2 University of Utrecht, Netherlands.
Summary
Early male maturation represents a major problem in Atlantic salmon farming, both in sea cages and in recirculation aquaculture system facilities. Problems caused by pre-harvest maturation include significantly increased disease susceptibility and osmoregulatory problems, causing higher mortalities, reduced animal welfare, and production losses. We and others have previously reported a strong association between alleles in the vgll3a locus and time of maturation in Atlantic salmon; however, the molecular mechanisms and roles of vgll3a alleles in controlling the time of puberty are largely unknown. Previous studies have indicated a potential role of vgll3a in the salmon gonad, where the expression of vgll3a is regulated in Sertoli cells upon entry into puberty, suggesting a possible connection between vgll3a and the brain–pituitary–gonad (BPG) axis. However, it is unknown which proteins link vgll3a alleles to the BPG axis.
To search for a possible connection between vgll3a alleles and the BPG axis, we used an RNA-seq approach to identify genotype-dependent gene expression in the testis and pituitary of fish stimulated to enter maturation. This was followed by a search for potential endocrine factors with stimulatory or inhibitory effects on the onset of male maturation. We therefore performed ex vivo pituitary incubation experiments with plasma obtained from immature males homozygous for the early (EE) and late (LL) maturation genotypes.
We uncovered several differentially expressed genes and pathways potentially involved in regulating the onset of puberty under the control of vgll3a alleles. Among those, PI3K-Akt signaling was found as the most enriched pathway in the testis. In the pituitary, we found genotype-dependent modulation of genes belonging to the Gnrh signaling pathway, including pituitary follicle-stimulating hormone subunit beta (fshb), the major hormone triggering puberty in vertebrates. Interestingly, the pituitary displayed low levels of vgll3a expression when measured by RNA-seq and qPCR, prompting us to investigate if we could detect the presence of one or more endocrine factors in plasma with stimulatory or inhibitory effects on fshb expression. Interestingly, we observed a significant decrease in fshb expression in pituitaries incubated with plasma from immature LL fish compared to pituitaries incubated with plasma from immature EE fish or negative controls (medium only). Heat denaturation removed the previously observed down-regulation of fshb expression in pituitaries incubated with LL plasma, indicating the presence of one or more (heat-labile) protein/peptide factors in plasma from LL fish with the ability to reduce fshb expression, in turn potentially delaying maturation.
Combined Genomic Approaches to Unravel Sex Determination in the European Seabass
Allal, F.1, Besson, M.2,3, Sánchez-Baizán, N.4, Clota, F.1,3, Goikoetxea, A.1, Sadoul, B.1,5, Ruelle, F.1, Blanc, M.-O.1, Parrinello, H.6, Hermet, S.1, Blondeau-Bidet, E.1, Pratlong, M.6, Piferrer, F.4, Vandeputte, M.1,3 and Geffroy, B.1
1 MARBEC Univ Montpellier, CNRS, Ifremer, IRD, INRAE, Palavas-Les-Flots, France.
2 SYSAAF, Station LPGP/INRAE, Campus de Beaulieu, 35042 Rennes, France.
3 Université Paris-Saclay, INRAE, AgroParisTech, GABI, 78350 Jouy-en-Josas, France.
4 Institut de Ciències del Mar, Spanish National Research Council, Barcelona, Spain.
5 ESE, Ecology and Ecosystem Health, Institut Agro, INRAE, Rennes, France.
6 MGX, BCM, Univ Montpellier, CNRS, INSERM, Montpellier, France.
Summary
Fish sex determination is often considered to be governed by either genetic or environmental factors, but the European seabass (Dicentrarchus labrax) defies this theory. In this species, a polygenic threshold sex determination system was demonstrated [1], where the genetic sex tendency is influenced by larval rearing temperature to determine the phenotypic sex [2]. In this study, we applied two thermal treatments during early larval stage, a low temperature protocol (16 °C, LT), known to favor balanced sex ratios and a high temperature masculinizing protocol (21 °C, HT). We combined various “-omics” approaches to characterize this temperature-dependent polygenic sex determination of European seabass. We produced eight families by mating eight males with the same female. The progenies were reared in common garden under two thermal treatments (LT, HT) in triplicate. Fish at four different key developmental stages encompassing the temperature-sensitive period were sampled. We predicted the genetic sex tendency (eGST) of the animals using a genomic relationship matrix derived from 57K SNPs from the DLabCHIP array [3] with a threshold animal model. This was completed by a transcriptomic approach, whole-body energy measurements. The proportion of females was 53.4% at LT and 25.3% at HT, showing a marked masculinization at high temperature. We found that the eGST accurately predicted the future phenotypic sex. We provided evidence that energetic pathways, concerning the regulation of lipids and glucose, are involved in sex determination and could explain why females tend to exhibit higher energy levels and improved growth compared to males. Overall, we describe for the first time a sex determination system resulting from continuous genetic and environmental influences in an animal, which provides significant progress in our understanding of the mechanisms underlying temperature-induced masculinization in fish.
Acknowledgments: The study was supported by the European Maritime and Fisheries Fund (3S, Seabass Sex and Stress, grant number 4320175237) and from the European Union’s Horizon 2020 Grant Agreement 652831 (AQUAEXCEL2020, Transnational Access project “Transsexbass”).
References
Vandeputte, M.; Dupont-Nivet, M.; Chavanne, H.; Chatain B. A Polygenic Hypothesis for Sex Determination in the European Sea Bass Dicentrarchus labrax. Genetics 2007, 176, 1049–1057.
Piferrer, F.; Blázquez, M.; Navarro, L.; González, A. Genetic, endocrine, and environmental components of sex determination and differentiation in the European sea bass (Dicentrarchus labrax L.). Gen. Comp. Endocrinol. 2005, 142, 102–110.
Griot, R.; Allal, F.; Brard-Fudulea, S.; Morvezen, R.; Haffray, P.; Phocas, F.; Vandeputte, M. APIS: An auto-adaptive parentage inference software that tolerates missing parents. Mol. Ecol. Resour. 2020, 20, 579–590.
Geffroy, B.; Besson, M.; Sánchez-Baizán, N.; Clota, F.; Goikoetxea, A.; Sadoul, B.; Ruelle, F.; Blanc, M.-O.; Parrinello, H.; Hermet, S.; et al. Unraveling the genotype by environment interaction in a thermosensitive fish with a polygenic sex determination system. Proc. Natl. Acad. Sci. USA 2021, 118, e2112660118.
Development of Epigenetic Biomarkers for the European Sea Bass (Dicentrarchus labrax)
Sánchez-Baizán, N.1, Denkena, J.2,3, Allal, F.4, Colomé-Tatché, M.2,3, Vandeputte, M.4,5 and Piferrer, F.1
1 Institut de Ciències del Mar (ICM-CSIC), Barcelona, Spain.
2 Institute of Computational Biology, Helmholtz Zentrum München, German Research Center for Environmental Health, Neuherberg, Germany.
3 Biomedical Center (BMC), Physiological Chemistry, Faculty of Medicine, LMU Munich, Planegg-Martinsried, Germany.
4 MARBEC, Univ. Montpellier, Ifremer CNRS-IRD, Palavas-les-Flots, France.
5 GABI, INRA, AgroParisTech, Université Paris-Saclay, Jouy-en-Josas, France.
Summary
The European sea bass is a gonochoristic species that exhibits sexual size dimorphism in favor of females, the preferred sex in aquaculture, and has a polygenic system of sex determination, where both genetic factors and temperature contribute to population sex ratios. Thus, when reared at temperatures >17 °C, a proportion of the fish which would develop as females under cooler temperature become masculinized (neomales). Epigenetic biomarkers such as DNA methylation changes at specific loci (CpG), which are susceptible to environmental change, hold promise to complement genetic selection of broodstock and could help select fish with desired phenotypic traits resulting from the interplay between genetics and environment. Here, we aim to understand the nature of the epigenetic inheritance and the contribution of the genotype and of the environment to DNA methylation profiles. To this end, we created eight families by crossing one female with eight males which had been reared at different temperature treatments when young: two at low temperature (LT), three at high temperature (HT) and 3 HT neomales (NM). The offspring were also exposed to LT and HT during the thermosensitive period, thus creating a total of 32 groups for analysis (eight parents of three types, two temperatures and two sexes) and gonadal tissues were sampled when fish were over one year old. Based on genotypic (sex tendency) and phenotypic data of the offspring, we selected fish representative of each group by sex, temperature and sire type. In total, we produced 130 libraries by reduced representation bisulfite sequencing to analyze the DNA methylome of grandparents’ gametes (F0, n = 2), sires and dam gametes (F1, n = 9) and offspring juvenile gonad (F2, n = 119). The obtained dataset generated a matrix of high dimensionality (130 samples x ~1,400,000 CpGs per sample). To make it more manageable, we contemplated several approaches to retain the most informative CpG positions. One included the definition of regions that would encompass the methylation data of close CpGs. The length of such regions was determined based on the correlation values of the methylation data between neighboring CpGs as the distance between them increased. We applied unsupervised machine learning algorithms to reduce the dimensionality of these DNA methylation data and identify the factors that contribute the most to the observed variance, in order to reveal underappreciated axes of variation. The purpose is to first identify biomarkers that are linked to sex, parent type and temperature with the ultimate goal to reveal those that are heritable and capable of identifying broodstock that will produce females that will not be masculinized even at high temperatures.
Acknowledgments: Research funded by the EU H2020 Grant 652831 (AQUAEXCEL2020, Transnational Access project “Transsexbass”) and Spanish Ministry of Science and Innovation ‘Epipure’ (PID2019-108888RB-I00) grant to FP.
Early Puberty in the European Sea Bass Females, Dicentrarchus labrax: Changes in Hormone Production and Ovarian Gene Expression
Sempere, L.1, Fernández, C.2, Ibáñez, S.1, Marín, C.3, Molés, G.4, Bouza, C.2, Martínez, P.2 and Felip, A.1
1 Group of Fish Reproductive Physiology, Institute of Aquaculture Torre de la Sal, CSIC, Castellón, Spain.
2 Departamento de Xenética Universidade de Santiago de Compostela, Facultade de Veterinaria, Lugo, Spain.
3 Husbandry Service, Institute of Aquaculture Torre de la Sal, CSIC, Castellón, Spain
4 Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria, Department of Environment, CSIC, Madrid, Spain.
Summary
The European Sea bass is a teleost fish species of high interest in aquaculture that presents sexual dimorphism affecting growth. Under culture conditions, a substantial proportion of fish develop as males that mature one year earlier during their first year of age. The incidence of precocious males brings economic problems for producers and thus the production of highly female-biased stocks is considered as an opportunity to benefit aquaculture. Females grow faster than males and mature later, around 3 years old, reaching a marketable size after a 2-year production cycle. However, the occurrence of early puberty in females is less well known and its determination is necessary for a better accommodation of fish farming. The first objective of this work is to determine the growth history and changes in plasma hormone levels of prepubertal female sea bass based on the ovarian development over the second year of age. Secondly, a gonadal transcriptome under different sexual maturation status is addressed to investigate gene expression profiles influencing oocyte growth in this species. Results show the existence of early differential growth within the female sea bass. Accordingly, slower (small-sized) and faster (large-sized) growing females were observed from several sea bass families with different genetic backgrounds. Our results demonstrated that those large-sized fish usually exhibited an advanced ovarian stage with late vitellogenic oocytes over the second sexual cycle. Thus, early puberty may affect up to 20% of prepubertal female sea bass at 2 years of age. It is worth noting that those females with advanced gonadal growth were usually as much as 17% heavier than females with immature oocytes, although they were not able to release eggs. It bears noting that we observed some fast growth females that exhibited immature ovaries, while some small-sized ones had advanced vitellogenic oocytes. In addition, some factors, including body weight and plasma levels of reproductive hormones contributed to explain the total variance between immature and advanced sea bass females during the fall season, one year before spawning. The gene expression profiles analysed in immature and advanced prepubertal females support the concept that ovary growth is a dynamic physiological process controlled by a complex regulatory network outside and within the follicle.
Acknowledgments: Project funded by the MICINN (AGL2016-75400, PID2019-109548RB-I00). CESGA (
http://www.cesga.es) provided computing facilities. L.S. supported by a FPI fellowship from MICINN (Spain).
Genome Editing to Produce Monosex and Sterile Fish for Aquaculture
Buchanan, J.T, Umazume, T., Tinch, A. and Lauth, X.C.
Center for Aquaculture Technologies 8445 Camino Santa Fe Suite 104, San Diego, California, 92121.
Summary
The ability to produce sterile progeny from broodstock for aquaculture has significant benefits to culture productivity and environmental sustainability. We describe the development of strategies to generate, breed and mass-produce infertile fish. Our solutions rely on precise genetic modifications to create broodstock lines that can be incorporated into breeding programs. These approaches were validated in tilapia but are transferrable to multiple species of fish. We expect that the adoption of these technologies will result in broad economic and environmental benefits for aquaculture.
Our strategy for mass-producing sterile fish is designed to produce monosex, sterile populations in culture. In addition to the benefit of sterile fish, this allows the benefit of sexually dimorphic performance traits in culture. We first investigated gene mutations in two evolutionarily conserved pathways, one governing sex differentiation and the other sexual competency. We created edits in genes necessary for spermiogenesis and steroid hormone synthesis causing male sterility and masculinization, respectively. Double gene edit combinations for these genes produced all-male sterile populations. Likewise, we created variants in genes whose inactivation caused females to develop atrophic ovaries arrested at a previtellogenic stage or string-like ovaries lacking oocytes. We further disrupted genes causing genetic males to sex reverse into females. Double gene edit combinations for these genes produced all-female, sterile populations.
Propagation of the double KO broodstock lines was achieved via germ cell transplantation from a juvenile-edited donor into a germ cell free wild-type recipient embryo. In the resulting recipients, the induced edits had no effect as the genes targeted are not expressed in germ cells. With this approach, we generated fertile broodstock that successfully mass-produced sterile, monosex populations.
Mapping for Males: Sustainable Sex Control in Nile tilapia
D’Cotta, H.1,2, Triay, C.1,2, Sissao, R.3, Bezault, E.5, Conte, M., Courcelle, M., Caminade, P., Toguyeni, A.3, Kocher, T.D.4 and Baroiller, J.F.1,2
1 UMR-ISEM, CNRS, Univ. Montpellier, IRD, EPHE, Montpellier, France.
2 UMR-ISEM, CIRAD, Campus Int. Baillarguet, 34398 Montpellier cedex 5, France.
3 UR ABAQ, Université of N Boni, Bobo-Dioulasso, Burkina Faso.
4 Department of Biology, University of Maryland, College Park, MD 20742, USA.
5 UMR BOREA, CNRS-7208/MNHN/UPMC/IRD-207/UCN/UA, Université des Antilles, 97159 Guadeloupe, France.
Summary
Sexual dimorphism of aquaculture traits is common for farmed fish. The Nile tilapia is the second most important farmed species with a production 6 million tons in 2020. Intensive farming relies on the production of all-males due to males’ higher growth rate, and as a way to avoid uncontrolled reproductions. Currently the large majority of the all-male productions are obtained through androgen treatments. We aim to use more sustainable procedures to produce all-males such as the use of YY males. Until now, the use of YY males has not been reliable. This is because sex determination in Nile tilapia is complex and controlled by several factors. Although sex determinism follows an XX/XY system, the linkage group (LG) carrying the major sex determinant gene has been assigned to either LG1 or LG23, depending on the domesticated strain. Minor parental factors can also be implicated and in addition, high temperatures can override the genetic determinism. It is not clear to what extent these differences in sex determination are due to natural diversity in the mechanisms of sex determination or due to processes of domestication. It is therefore necessary to better understand the genetic basis of sex determinism in order to use this approach to generate all-males. For this, we decided to work on wild populations in Africa that have not suffered domestic manipulations. We underwent a study of sex determination in several wild populations from West (Lake Volta, Lake Kou) and East Africa (Lake Koka and Lake Hora). We used complementary genomic approaches of ddRAD, whole genome sequencing and long Nanopore reads. We were able to determine that the amh region present on LG23 is the major sex-determining region in most of these populations. Nevertheless, our results also show that there is high polymorphism in this SD region. Furthermore, there are populations that lack the male-specific amh duplication on LG23. Hence, there are no universal Y markers for Nile tilapia. It is necessary to work at the population level to identify and validate sex markers, in order to allow the local production of YY males.
A Chromosome-Level Genome Assembly Enables the Identification of the Follicle-Stimulating Hormone Receptor as the Master Sex-Determining Gene in Solea senegalensis
De la Herrán, R.1, Hermida, M.2, Rubiolo, J.2, Gómez-Garrido, J.3, Cruz, F.3, Robles, F.1, Navajas-Pérez, R.1, Blanco, A.2, Rodríguez-Villamayor, P.2, Torres, D.2, Sánchez-Quinteiro, P.4, Ramirez, D.5, Rodríguez, M.E.5, Arias, A.5, Cross, I.5, Duncan, N.6, Martínez-Peña, T.7, Riaza, A.7, Millán, A.8, Gut, M.3, Bouza, C.2, Robledo, D.9, Rebordinos, L.5, Alioto, T.3, Ruíz-Rejón, C.1 and Martínez, P.2
1 Departamento de Genética, Universidad de Granada, Granada, Spain.
2 Departamento de Zoología, Genética y Antropología Física, Universidade de Santiago de Compostela. Lugo, Spain.
3 Centre Nacional d’Anàlisi Genòmica, Barcelona, Spain.
4 Departamento de Anatomía, Producción Animal y Ciencias Clínicas Veterinarias, Universidade de Santiago de Compostela. Lugo, Spain.
5 Departamento de Biomedicina, Biotecnología y Salud Pública, Universidad de Cádiz, Cádiz, Spain.
6 IRTA Sant Carles de la Rapita, Sant Carles de la Rapita, Tarragona, Spain.
7 Stolt Sea Farm SA, Departamento I+D, A Coruña, Spain.
8 Geneaqua SL, Lugo, Spain.
9 The Roslin Institute and Royal (Dick) School of Veterinary Studies, University of Edinburgh, Midlothian, UK.
Summary
Sex determination shows huge variation among fish and a high evolutionary rate. Pleuronectiformes is an emblematic fish group characterized by its adaptation to demersal life and by its compact genomes. Here, we assembled the Senegalese sole genome, a promising European aquaculture species, by combining long- and short-read sequencing (82 contigs, 613 Mb), and further scaffolding using a highly dense genetic map (28,838 markers) constituted of 21 linkage groups (total scaffolding: 607 Mb, 99% of the assembly). Further, we established the correspondence between the new assembly and the 21 chromosomes of its karyotype by using fluorescence in situ hybridization with BAC probes (BAC-FISH). Orthology within Pleuronectiformes was assessed, taking Danio rerio as the outgroup, by using the chromosome-level genomes and annotated proteomes of six important commercial flatfish covering the phylogenetic spectrum of the order: Scophthalmus maximus (Scophthalmidae), Paralychthys olivaceous (Paralychthydae), Cynoglossus semilaevis (Cynoglossidae), Solea senegalensis (Soleidae), Hippoglossus hippoglossus and H. stelopis (Pleuronectidae). We identified 7936 single-gene orthologues shared by the six species, 5879 including D. rerio, that were used to reconstruct their phylogeny and to identify syntenic in the order that were further used to explore chromosome patterns in the evolutionary history of the Senegalese sole. Whole genome resequencing of six males and six females enabled the identification of 41 fixed allelic variants in the follicle-stimulating hormone receptor (fhsr) gene, homozygous in females and heterozygous in males, according to an XX / XY pattern. The observed association was also proved at the species level in a broad sample, which allowed the tuning up of a molecular sexing tool. Fhsr demonstrated differential gene expression between male and female gonads for 86 days post-fertilization, which was still an undifferentiated primordium, even before other testis and ovary marker genes, such as amh and cyp19a1a genes, respectively. Interestingly, the Y-linked allele expressed to a higher level at all stages when detected, suggesting a molecular mechanism for hampering the action of the follicle-stimulating hormone, driving the undifferentiated gonad toward the testis.
Acknowledgments: This study was supported by the Spanish Ministry of Economy and Competitiveness, Grants: AGL2014-57065-R, RTI2018-097110-B-C21 and RTI2018-096847-B-C22, and the European Union’s Horizon 2020 research and innovation programme under grant agreement No 81792 (AQUA-FAANG). We thank Geneaqua SL for their participation and financial support on sequencing.
A Technology for Producing All-Female Progenies of the Flathead Grey Mullet by Using Sex-Reversed Males
David, L.1, Agiv, D.1, Oz, I.1, Marcos-Hadad, E.1, Bennet-Perlberg, A.2 and Ginzbourg, B.3
1 Dept. of Animal Sciences, The Hebrew University of Jerusalem, Israel.
2 Dor research station, Israel Ministry of Agriculture and Rural Development, Israel.
3 Dagon Fish Hatchery, Kibbutz Maagan-Michael, Israel.
Summary
The flathead grey mullet (Mugil cephalus) is a cosmopolitan marine fish food fish, the availability of which depends mainly on capture fishery in seas, but also from increased production in aquaculture on land. Aquaculture production relies heavily on capturing wild fry in estuaries and acclimating them to grow in freshwater/brackish water ponds. Capturing wild fry puts pressure on wild populations, but also hampers further the development of mullet aquaculture since wild fry availability fluctuates over the years. The mullet is a desired aquaculture species, targeted by the EU as a priority species to develop aquaculture in Mediterranean countries. Mullet females grow faster than males and mature females are used also by the roe industry to produce Botarga/Karasumi. Recently, the life cycle of the mullet in captivity was terminated allowing the production of fry in hatcheries. Having carried that out, now is the right time to breed for improved brood stocks. Accordingly, the goal of this research was to establish a technology for producing all-female progenies for aquaculture. The technology is based on producing sex-reversed males, i.e., milt-producing males with a female sex genotype, which are then crossed to normal females for producing all-female progenies. The production of sex-reversed males required developing hormonal sex-reversal protocol and genetic markers to determine the genetic sex. Hormonal sex-reversal was carried out by feeding methyltestosterone-treated food to batches of fry, while experimenting to calibrate the hormonal dose, timing and duration of application. Control and treatment groups were grown for 18 months to maturation, when fish were sampled and sacrificed to visually determine their gonad type. While all control groups had a 1:1 ratio between females and males, in three treatment groups an excess of 63%, 74% and 84% males were identified, indicating that some males were sex-reversed. Identification of genetic sex requires the understanding of the sex-determination system and mapping of the genomic regions affecting sex. Control groups were screened using genotyping by sequencing to identify several thousands of SNP markers used to construct a genetic linkage map. About 280 markers were significantly associated with sex and their mapping identified enrichment on a single linkage group, suggesting a monogenic sex-determination system. Mullet has no karyotypic difference between sexes, but other results suggested an XY/XX system, where the male determines the sex of progeny. Next, markers tightly linked to sex were developed into genotyping assays. These markers had very good accuracy (over 98%) in correctly identifying the sex in some families but lower to none in others, suggesting genetic variation among families in the sex-determining region. Therefore, the use of multiple markers was needed for identifying genetic sex across multiple families. Importantly, marker genotyping of fish from sex-reversed groups and their sib control groups identified which genotypes were of females and which were of males, allowing us to identify males with female genotypes, i.e., sex-reversed males. Such live sex-reversed males were identified by markers and selected as broodfish, which were crossed with normal females to produce, for the first time, an all-female progeny group. We expect that incorporating this technology into the routine production of commercial hatchery fry will push forward mullet aquaculture and profitability of this industry.
Around the Black Pearls: Multi-Omic Approaches in Breeding Programs towards the Improvement of Pearl Quality of the Pacific Pearl Oyster
Dorant, Y.1,2, Ky, C.L.1,2 and Le Luyer, J.1
1 IFREMER, EIO UMR 241, Labex CORAIL, Unité RMPF, Centre Océanologique du Pacifique, Vairao – BP 49 Vairao, Tahiti, Polynésie française.
2 IHPE, Univ Montpellier, CNRS, IFREMER, Univ Perpignan, Montpellier, France.
Summary
The black-lip pearl oyster, Pinctada margaritifera, is an important aquaculture species extensively farmed in French Polynesia to produce the famous “Pacific black pearl”. In recent years, the black pearl industry has deteriorated partly due to the low-quality pearls flooding the market. While the hatchery production of pearl oysters is still developing, in French Polynesia this practice opens a window for selective breeding programs to improve pearl quality. However, the pearl phenotype is the result of a complex process involving a “2-genome” system (graft and receiver) and the effect of environmental drivers. Hence, despite the economic importance of this species, our understanding of the genetic basis and molecular functions controlling pearl quality is limited. Here, we introduce a multi-omic approach to investigate major biological functions underlying pearl quality. First, we used extensive high-throughput transcriptome profiling to identify the genes related to pearl quality and animal growth-related traits. Our results contribute to the refinement of the key genes regulating pearl formation and quality and also provide new insights into biomineralization and growth processes in Mollusca. Second, a first genome draft together with a 70K SNP array was designed in support for testing genomic selection programs in P. margaritifera. Our preliminary results demonstrate that our SNP array can be used to clearly distinguish between families (sibs and parental groups) based on identity-by-state (IBS) clustering parental assignment. This novel SNP array for the black-lip pearl oyster will be applied for genome-wide association and evolutionary genetic studies as well as for genomic selection in oyster breeding programs.
Influence of Genotype-By Environment and Breeding Program Strategy on Genetic Gains Using Genomic Prediction in Barramundi (Lates calcarifer)
Jerry, D.R.1, Jones, D.B.1, Lillehammer, M.2, Massault, C.1, Loughnan, S., Cate, H.S.3, Harrison, P.J.3, Strugnell, J.M.1, Zenger, K.R.1 and Robinson, N.A.2,4
1 College of Science and Engineering, James Cook University, Townsville, Queensland, Australia.
2 Breeding and Genetics, Nofima, Ås, Norway.
3 Mainstream Aquaculture Group Pty Ltd., Werribee, Victoria, Australia.
4 School of BioSciences, University of Melbourne, Parkville, Australia.
Summary
Barramundi (Lates calcarifer) is a tropical, euryhaline fish species that is growing in popularity for aquaculture due to the species’ fast growth rate, hardiness and ability to be farmed in diverse production systems (outdoor ponds to RAS) and environments (freshwater to marine salinities). Recent studies have shown that genotype by environment (GxE) may influence genetic gains and thus GxE needs to be factored into breeding programs. There is also of interest in genomic prediction as an approach to achieve higher genetic gains in commercial breeding programs.
This study utilized a custom 70k Axiom myDesignTM genome-wide SNP array (ThermoFisher ScientificTM) to estimate and compare growth-related genetic parameters (heritability and genetic correlations), genotype by environment and breeding values for barramundi reared in commercial freshwater RAS (VIC) and brackish pond environments (QLD). Genetic gains and accuracy were compared using pedigree (PBLUP)- and genomic (GBLUP)-based approaches at harvest for the body traits of whole fish weight (WFW), standard fish length (SL), body depth (BD), Fulton’s condition factor (K) and body shape index (BS) under three different breeding program scenarios; MULTIPLE, separate breeding programs at each farm site; GENERAL, a single breeding program using the general effects from the interaction model; and SINGLE, a single breeding program based on the performance of individuals within one main farm site.
Moderate to high heritabilities were estimated in barramundi from both farm environments for WFW (PBLUP: 0.35–0.38; GBLUP 0.33–0.35), SL (PBLUP: 0.26–0.32: GBLUP 0.27–0.35), BD (PBLUP 0.38–0.39; GBLUP 0.29–0.30) and K (PBLUP 0.17–0.21; GBLUP 0.14–0.21). Genotype-by-environment interactions were also detected to be present.
GBLUP breeding value estimation accuracy and prediction of genetic gain were higher than PBLUP for all traits in the three breeding scenarios evaluated. Genomic prediction under the MULTIPLE breeding strategy was shown to return the highest genetic gains (28–45% in VIC and 33–40% in QLD), in comparison to PBLUP. In the SINGLE scenario, genetic gains were the same for each individual farm site as MULTIPLE, but achieved only 48–66% in the alternative farm site where records were not collected. GENERAL, which operates at half the cost of MULTIPLE, achieved 72–97% of the expected gains of MULTIPLE across the farm sites, suggesting that it would be the most cost-effective scenario for genomic selection in the production environments evaluated.
Acknowledgments: This project was supported by CRC project funding from the Department of Industry, Innovation and Science, Commonwealth of Australia.
Using Genome-Wide Ancestry Pattern in European Sea Bass to Assess Phenotypic Variation and Ensure Sustainable Aquaculture
Leitwein, M.1, Durif, G.2, Gagnaire, P.-A.3, Vergnet, A.1, Clota, F.1,4, Duranton, M.1, Vandeputte, M.1,4 and Allal, F.1
1 MARBEC, Univ. Montpellier, Ifremer, CNRS, IRD, INRAE 34250 Palavas-les-Flots, France.
2 Institut Montpelliérain Alexander Grothendieck, Université de Montpellier, France
3 ISEM, Univ Montpellier, CNRS, EPHE, IRD, Montpellier, France.
4 Université Paris-Saclay, INRAE, AgroParisTech, GABI, 78350 Jouy-en-Josas, France.
Summary
The European sea bass (Dicentrarchus labrax) is one of the most socio-economically important species in Mediterranean aquaculture and has undergone domestication and selective breeding during the last two decades. The European sea bass is subdivided in three main populations (Atlantic (ATL), West Mediterranean (WM) and East Mediterranean (EM)) displaying significant phenotypic differences in growth, disease resistance, sex ratio and fat content, which is of interest for aquaculture [1–3]. Consequently, understanding the genomic specificity of each lineage and the evolutionary adaptive or maladaptive consequences of admixture between them became of great importance for their conservation and exploitation [4]. More specifically, unravelling how those populations are adapted to their environment, their adaptive potential and their genomic and phenotypic diversity is of interest to improve efficient breeding programs.
By using genome-wide genotyping data (57K SNPs chips) of over 900 individuals from the EM, WM and ATL populations, we were able to (i) develop a high-density linkage map for both the Atlantic and the Mediterranean lineage, (ii) assess ancestry and characterize admixture pattern among individuals and (iii) identify genomic regions of interest for aquaculture by coupling genome-wide pattern of haplotype introgression and phenotypic data (growth rate, weight, phenotypic sex).
We demonstrate that assessing ancestry among individuals reveals a mosaic of admixture shaped by evolutionary mechanisms such as genomic incompatibilities and local heterosis. We also show that individual phenotypic response is associated with the genome-wide ancestry pattern. Consequently, taking into consideration the evolutionary mechanisms at play is necessary to better understand the genetic basis of traits and ensure sustainable aquaculture production.
References
Doan Q, K.; Vandeputte, M.; Chatain, B.; Haffray, P.; Vergnet, A.; Breuil, G.; Allal, F. Genetic variation of resistance to Viral Nervous Necrosis and genetic correlations with production traits in wild populations of the European sea bass (Dicentrarchus labrax). Aquaculture 2017, 478, 1–8.
Guinand, B.; Vandeputte, M.; Dupont-Nivet, M.; Vergnet, A.; Haffray, P.; Chavanne, H.; Chatain, B. Metapopulation patterns of additive and nonadditive genetic variance in the sea bass (Dicentrarchus labrax). Ecol. Evol. 2017, 7, 2777–2790.
Faggion, S.; Vandeputte, M.; Chatain, B.; Gagnaire, P.A.; Allal, F. Population-specific variations of the genetic architecture of sex determination in wild European sea bass Dicentrarchus labrax L. Heredity 2019, 122, 612–621.
Duranton, M.; Allal, F.; Fraïsse, C.; Bierne, N.; Bonhomme, F.; Gagnaire, P.-A. The origin and remolding of genomic islands of differentiation in the European sea bass. Nat. Commun. 2018, 9, 2518.
A Comparison of Genomic Coancestry Matrices for Maintaining Genetic Variability Using Simulations and Turbot Data
Morales-González, E.1, Fernández, J.1, Saura, M.1, Fernández, A.1, Pong-Wong, R.2, Toro, M.A.3, Cabaleiro, S.4, Martínez, P.5 and Villanueva, B.1
1 INIA-CSIC, Dpto. Mejora Genética Animal, Crta. de la Coruña, km 7.5, 28040 Madrid, Spain.
2 The Roslin Institute and R(D)SVS of the University of Edinburgh, Midlothian EH25 9RG, Roslin, UK.
3 Universidad Politécnica de Madrid, Dpto. Producción Agraria, Ciudad Universitaria, 28040 Madrid, Spain.
4 CETGA, Cluster de Acuicultura de Galicia, Punta do Couso, s/n. 15695, Aguino-Ribeira, A Coruna, Spain.
5 Universidade de Santiago de Compostela, Dpto. de Xenética, Campus de Lugo, 27002 Lugo, Spain.
Summary
Genetic diversity and inbreeding are fundamental parameters in the genetic management of populations. The application of the optimal contributions method is the most efficient strategy to control the loss of genetic diversity and the increase in inbreeding. This method operates by optimizing the contributions of candidates as to minimize the weighted global coancestry. This leads to the highest levels of genetic diversity and, under random mating, to the lowest inbreeding levels in the next generation. The required coancestry matrix has been traditionally computed from pedigree data but following the development of genomic tools, genomic coancestry matrices can be used instead. Two commonly used measures of genomic coancestry include those based on the i) proportion of shared alleles by two individuals and ii) VanRaden’s methodology to compute the realized genomic relationship matrices. Using different matrices in the optimal contributions method may lead to different levels of diversity maintained but also to different trajectories of the change in allele frequencies. Maintaining allele frequencies may be important to preserve the uniqueness of a particular population. In this study, through computer simulations we evaluate the genetic variability, the change in allele frequencies and the number of loci fixed when different genomic matrices are used in the optimization. The genetic variability was measured as the expected heterozygosity and the change in allelic frequencies was measured using the Kullback–Leibler divergence criterion. Additionally, the optimization was performed using real genomic data from a domesticated population of turbot. Simulation and turbot results showed that coancestry coefficients based on the proportion of shared alleles retained higher variability than those based on realized genomic relationship matrices. However, coefficients based on the proportion of shared alleles also led to stronger changes in allelic frequencies and to the fixation of a larger number of loci. Thus, the choice of the genomic measure of coancestry will depend on the specific objective, i.e., to maintain genetic variability or the genetic composition of the population.
Optimising Genomic Selection for Aquaculture Breeding Programmes in Small-Scale Operations and Developing Countries
Kriaridou, C.1, Tsairidou, S.2, Fraslin, C.1, Looseley, M.3, Johnston, I.A.3, Houston, R.1 and Robledo, D.1
1 The Roslin Institute, University of Edinburgh, UK
2 Global Academy of Agriculture and Food Security, University of Edinburgh, UK
3 Xelect Ltd., UK
Summary
Genomic selection results in improved rates of genetic gain for production traits compared to traditional selection. While the cost of sequencing and genotyping is decreasing, it remains expensive and hinders the large-scale application of genomic selection, especially in small- and medium-sized aquaculture farms. Genotype imputation can predict untyped single nucleotide polymorphisms (SNPs) in populations genotyped at low densities (LDs) from a reference population genotyped at a higher density (HD) and has the potential to lower the cost of genotyping in aquaculture breeding programmes. This project investigates best practices for the broader application of genotype imputation, by designing and evaluating cost-effective genotyping strategies.
In this study, eight LD SNP panels (ranging from 300 to 6000 SNPs) were generated in silico for four aquaculture species, Atlantic salmon, turbot, Pacific oyster and common carp. SNPs were selected 1) proportionally to chromosome length and evenly distributed according to physical position or 2) based on linkage disequilibrium. The LD SNP panels were then imputed to HD (8103 SNPs for carp to 78035 SNPs for Atlantic salmon) with three different software (AlphaImpute v.2, FImpute v.3 and findhap v.4). Correlation between the true and the imputed genotypes was used to evaluate the accuracy of genotype imputation.
The results of this study revealed that FImpute v.3 was faster and more accurate for most of the densities that were tested in all the species (Figure 1). Imputation accuracy increased with increasing density of SNP panel, reaching values above 0.95 in three species and 0.80 in Pacific oyster for the LD panel with 6000 SNPs. Furthermore, we found that SNP selection based on linkage disequilibrium slightly increased the accuracy of imputation for the lowest LD panels. The different scenarios will be used to test the accuracy of prediction of genomic breeding values for traits of interest in the four species. Our results are the first step towards a more affordable genomic selection that combines genotyping of an LD panel and imputation.
Figure 1.
Comparison of genotype imputation accuracy between the three imputation software for Atlantic salmon, turbot, Pacific oyster and carp when selecting SNPs for the LD panel with method 1.
Figure 1.
Comparison of genotype imputation accuracy between the three imputation software for Atlantic salmon, turbot, Pacific oyster and carp when selecting SNPs for the LD panel with method 1.
Plant-Based Diet Supplemented with Hermetia illucens in Combination with Poultry By-Product Meal—A Step Closer to Sustainable Aquafeeds for European Sea Bass
Bušelić, I.1, Lepen-Pleić, I.1, Šegvić-Bubić, T.1, Kaitetzidou, E.2, Tibaldi, E.3, Grubišić, L.1 and Sarropoulou, E.2
1 Laboratory of Aquaculture, Institute of Oceanography and Fisheries, Croatia
2 Institute of Marine Biology, Biotechnology and Aquaculture, Hellenic Centre for Marine Research Crete, Greece
3 Department of Agri-Food, Environmental and Animal Science, University of Udine, Italy
Summary
Within the ongoing Interreg AdriAquaNet project, new feeds were designed and tested at a laboratory scale on sub-adult European sea bass, aiming to enhance the innovation and sustainability of Adriatic aquaculture. For the present study, three tested diet formulations were selected for a comprehensive analysis of transcriptomic profiles of sub-adult European sea bass intestines after performed feeding trial. Selected formulations included two control diets, one rich in fish-derived ingredients (CF) and one rich in plant-derived ingredients (CV). The test diet (VH10P30) contained the same vegetable: fish lipid ratio as the CV diet, replacing crude proteins from the plant-based sources with 10 % of crude proteins from a commercial defatted Hermetia illucens pupae meal and 30 % of poultry by-product meal. Based on quality, 24 samples of total RNA were selected for cDNA library preparation, comprising 4 biological replicates per feeding treatment of two selected intestinal parts, pyloric caeca, and distal intestine (pyloric caeca and distal intestine were paired as subsamples). Single-end 3’UTR sequencing was performed using NextSeq 500 System. Two analyses were performed for differential expression analysis, comparing the VH10P30 test treatment to CF treatment as the positive control, and comparing the VH10P30 test treatment to CV treatment as the negative control. In total, 1963 (915 up and 1048 down) differentially expressed (DE) genes were found in the distal intestine of European sea bass in the VH10P30 treatment vs. CF treatment. Using CV treatment as the negative control, there were 711 (439 up and 272 down) DE genes in the distal intestine of European sea bass in the VH10P30 treatment. In contrast to the distal intestine, no clear differences were detected between the diet treatments in the pyloric caeca of the experimentally fed European sea bass. Possible explanation for these results could be the occurrence of functional specialization along the intestinal tract of European sea bass [1]. Taking into account growth performance and overall fitness of the European sea bass in the VH10P30 treatment, H. illucens larval meal and poultry by-product meal demonstrated great potential as alternative protein sources for European sea bass aquaculture.
References
Calduch-Giner, J.A.; Sitjà-Bobadilla, A.; Pérez-Sánchez, J. Gene Expression Profiling Reveals Functional Specialization along the Intestinal Tract of a Carnivorous Teleostean Fish (Dicentrarchus labrax). Front. Physiol. 2016, 7, 359.
OsHV-1 Replication in Pacific Oyster Tissue Explants
Potts, R.W.A., Regan, T., Houston, R.D. and Bean, T.P.
The Roslin Institute, University of Edinburgh, UK.
Summary
Pacific oysters (Crassostrea gigas) are one of the most important aquaculture species globally. Sustainable production of oysters is hampered by outbreaks of mass mortality caused by Oyster Herpes Virus (OsHV-1). Better understanding of the genetics underlying resistance to OsHV-1 is essential to preventing future outbreaks, as vaccination or treatments are not feasible in the field. Studies of OsHV-1 often rely on natural outbreaks or are influenced by confounding factors in the marine environment, which is a limited approach compared to infection studies in other taxa.
The aim of this study was to use tissue explants challenged with OsHV-1 as a model infection system that can be used in a controlled environment, overcoming issues of biological contamination, environmental stability and more. The development of a system for maintaining whole tissues from Pacific oysters is an exciting development that has been investigated for the first time [1]. This system facilitates investigation into the response to infection in oysters from different backgrounds of disease resistance [2]. Furthermore, tissue-specific responses will be valuable for better understanding OsHV-1 virology. Key questions to address are the main site of OsHV-1 entry into the oyster and potential sites for virus latency or senescence.
The OsHV-1 lifecycle in the laboratory has been achieved, which is a crucial addition to the OsHV-1 toolkit. Quantitative PCR of viral DNA has revealed that oyster gills seem to be essential for viral infection. Furthermore, the resistance phenotype of the donor oyster seems to be reflected in the disease response of the tissue explant, suggesting the model is representative of the whole oyster responses. The development of this model system has the potential to inform selective breeding and gene editing approaches to enhancing disease resistance.
References
Potts, R.W.; Gutierrez, A.P.; Cortés-Araya, Y.; Houston, R.D.; Bean, T.P. Developments in marine invertebrate primary culture reveal novel cell morphologies in the model bivalve Crassostrea gigas. PeerJ 2020, 8, e9180.
Dégremont, L.; Morga, B.; Maurouard, E.; Travers, M. Susceptibility variation to the main pathogens of Crassostrea gigas at the larval, spat and juvenile stages using unselected and selected oysters to OsHV-1 and/or V. aestuarianus. J. Invertebr. Pathol. 2021, 183, 107601.
Understanding Early Life History on the Fish Gut Microbiome Development: Lessons from Nile tilapia
Kokou, F., Deng, Y. and Verdegem, M.
Wageningen University and Research, Aquaculture and Fisheries Group, Wageningen, Netherlands.
Summary
From birth onwards all organisms are colonized by microbes. Initial colonization during developmental windows such as birth, appears to shape the gut microbial community in adults with lifelong effects on, for example, health and metabolism. While microbes can have profound effects on their hosts, we are only beginning to understand how variation in history of the initial colonization either through feed or the rearing environment may shape host performance throughout life. The rearing environment and the feed are important factors shaping the initial colonization, while their long-term effects on fish performance are not well-understood.
We used next-generation sequencing in series of experiments to describe changes in the microbial community composition in Nile tilapia Oreochromis niloticus post-larvae. In these different experiments, fish were exposed to different rearing environments (RAS, Biofloc, Flow-through) and different diets (high versus low fish meal, and prebiotic addition: b-glucans). The microbial communities of the water and gut were characterized by the sequencing of the 16S rRNA gene.
The results show that early life environment and diet can affect the gut microbiome composition. Although the developmental stage has a strong impact on the gut microbiome composition, we observed a long-term effect of the rearing environment (Biofloc vs Flow-through) on the gut microbial interactions. With regard to the dietary effects, low fishmeal diets resulted in a lower overall performance compared to the high fishmeal diets. The addition of the prebiotic b-glucan on low fishmeal diets did not show a positive effect on performance nor on the gut microbiome. These findings suggest that the rearing environment plays a major role in modulating the fish gut microbiome during early stages. Further studies are required to understand to effect of different dietary stimuli during early life on fish performance. The interpretation of such results is highly crucial as a better understanding of the fish gut–microbe interactions is a great challenge that can allow the production of quality aquafeeds but also rearing protocols for hatcheries that will ensure optimal animal performance.
The Genetic Background Drives the Reshaping of the Gut Microbiome by Feed Additives in Farmed Gilthead Sea Bream (Sparus aurata)
Naya-Català, F.1, Piazzon, M.C.1, Torrecillas, S.4, Fontanillas, R.2, Hostins, B.3, Calduch-Giner, J.1, Sitjà-Bobadilla, A.1, Montero, D.4, Pérez-Sánchez, J.1
1 Nutrigenomics and Fish Growth Endocrinology and Fish Pathology Groups, Institute of Aquaculture Torre de la Sal (IATS-CSIC), Castellón, Spain.
2 Skretting Aquaculture Research Centre, Sjohagen, Norway.
3 INVE Technologies NV, Dendermonde, Belgium.
4 Grupo de Investigación en Acuicultura (GIA), Instituto Universitario ECOAQUA, Universidad de Las Palmas de Gran Canaria, Las Palmas, Canary Islands, Spain.
Summary
The use of feed additives has expanded rapidly as an alternative for antibiotics and chemotherapeutics, with also the capacity to modify the composition of gut microbiota. In farmed fish, and in gilthead sea bream in particular, there is evidence that the host genetic background has a major impact on gut microbiota. Thus, families selected for fast growth have a more flexible microbiota capable of exerting a wider nutritionally mediated response with less microbial community changes. However, nutrition and genetic interactions remain poorly explored in fish, and the aim of this study was to unravel how the microbiota of fish selected (GS) and unselected (NGS) for growth are differentially regulated by oil-coated feed additives. The basal diet (CTRL, no feed additives) was a low fishmeal/fish-containing algae oil, poultry by-products, and plant ingredients. Experimental diets were oil-coated with the additives: organic acids (OA), Bacillus-species probiotics (PROB), or natural plant extracts (PHYTO). Fish were fed to visual satiety with the CTRL diet for two weeks. After this adaptation period, the different supplemented diets were used with a high additive dose (7.5–10 g/kg) for 2 weeks, decreasing thereafter to 2–5 g/kg until the end of the trial (97 days). Then, adherent microbiota were obtained from the anterior intestine. Illumina sequencing of microbiota yielded a mean of 62,594 reads per sample, which were assigned to 1156 OTUs at 97% identity threshold. A significant lower richness and diversity were found in the GS fish, which was mainly evidenced by a higher abundance of Actinobacteria in GS-PROB. To study in more detail the observed differences on gut microbial populations, supervised partial least-squares discriminant analyses (PLS-DA) were used. When all populations were analysed as a whole (GS vs NGS), dispersal was markedly lower in GS fish. This pattern was further evidenced for fish fed the CTRL, PROB and OA diets but not for the PHYTO additive, explaining the statistically validated PLS-DA models more than 80% of the total variance. This genetically guided group differentiation was driven by a total of 104 OTUs. Regarding diet and genetic interactions: i) no effect was detected with the PHYTO additive, ii) the OA reshaped the gut microbiota in NGS with a decrease in Photobacterium damselae sp. and an increase in Paracoccus and Acinetobacter genera, and iii) the PROB diet modified the gut microbiota of both GS and NGS fish, with a higher abundance of Kocuria and Bacillus genera, which reflected the establishment of the probiotic bacteria in the mucosal adherent surface, favouring long-term health-promoting probiotic effects.
Acknowledgments: AquaIMPACT (H2020 #818367), RYC2018-024049-I & ESF.
1-Carbon Nutrients Impact Histone Modification Enrichment in the Atlantic Salmon Genome
Putman, A.1, Catrin-Adam, A.2, Takaya Saito, T.2, Espe, M.2, Skjaerven, K.2 and Miska, E.1
1 Gurdon Institute, University of Cambridge.
2 Norwegian Institute of Marine Research.
Summary
Histone post-translational modifications are dynamic regulators of the genome. Despite their crucial role in genome regulation, the impact of dietary nutrients upon histone modification landscapes remains poorly understood, particularly in fish models. In this project, by supplying Atlantic salmon (Salmo salar) with a surplus nutrient package, we measure the impact of altered one-carbon (1C) metabolism on the enrichment of two histone modifications associated with activation: H3K27ac and H3K4me3. Crucial to cell function, one-carbon (1C) metabolism provisions methyl groups for a range of biological processes. These particularly include the regulation of gene expression via DNA methylation and histone post-translational modifications. The activity of the 1C metabolism is dependent upon the availability of select nutrients, particularly folic acid, pyridoxine (B6), cobalamin (B12), and methionine. To measure the impact of an increased intake of 1C nutrients upon growth, epigenetic state, and transcriptome, we raised groups of Atlantic salmon (Salmo salar) on two plant-based diets with varying levels of 1C nutrients: one control (control 1C) using nutrients at recommended levels and one moderate+ diet (1C+) with increased levels of 1C nutrients [1]. Respectively, the control and 1C+ diets contained 2.6 mg/kg and 4.8 mg/kg folate, 6.75 mg/kg and 9.31 mg/kg B6, 0.15 mg/kg and 0.18 mg/kg B12, and 6.7 g/kg and 9.5 g/kg methionine [2,3]. Muscle tissues from the control and 1C+ treatment groups were collected for various sequencing applications during both freshwater and saltwater life stages. To characterize the changes in chromatin landscape resulting from the limitation of 1C nutrients, we mapped the enrichment of two histone post-translational marks associated with activation: H3K4me3 (a marker of active promoters) and H3K27ac (marking active enhancers and promoters). We performed Cut&Run of both marks in single muscle samples from the control and 1C+ treatment groups (n = 2 per tank, 3 tanks/treatment). Sequencing was performed for both freshwater and saltwater life stages. Levels of gene expression (RNA-seq) and DNA methylation (RRBS) were measured in these same muscle samples, allowing for multi-omic comparisons across data types [2,4]. In this presentation, we characterize the impact of the altered 1C metabolism on two major histone modifications in a teleost model, contributing to the growing area of investigation concerning the impact of nutrition on the chromatin architecture across the genome.
References
National Research Council.
Nutrient Requirements of Fish and Shrimp; The National Academies Press: Washington, DC, USA, 2011; p. 392.
https://doi.org/10.17226/13039.
Adam, A.C.; Saito, T.; Espe, M.; Whatmore, P.; Fernandes, J.M.D.O.; Vikeså, V.; Skjærven, K.H. Metabolic and molecular signatures of improved growth in Atlantic salmon (Salmo salar) fed surplus levels of methionine, folic acid, vitamin B6 and B12 throughout smoltification. Br. J. Nutr. 2021, 127, 1–14.
Espe, M.; Vikeså, V.; Helgøy Thomsen, T.; Adam, A.C.; Saito, T.; Skjærven, K.H. Atlantic salmon fed a nutrient package of surplus methionine, vitamin B12, folic acid and vitamin B6 improved growth and reduced the relative liver size, but when in excess growth reduced. Aquac. Nutr. 2020, 26, 477–489.
Saito, T.; Whatmore, P.; Taylor, J.F.; Fernandes, J.M.; Adam, A.C.; Tocher, D.R.; Espe, M.; Skjærven, K.H. Micronutrient supplementation affects transcriptional and epigenetic regulation of lipid metabolism in a dose-dependent manner. Epigenetics 2021, 16, 1217–1234.
Nutrient-Sensitive Epigenetic Regulation in Salmon Muscle
Adam, A.C.1, Saito, T.1, Putman, A.L.K.2,3, Espe, M.1, Fernandes, J.M.O.4, Miska, E.A.2,3 and Skjærven, K.H.1
1 Institute of Marine Research (Norway).
2 The Gurdon Institute, Wellcome Trust/Cancer Research UK, Cambridge (United Kingdom).
3 Department of Genetics, University of Cambridge, Cambridge (United Kingdom).
4 Faculty of Biosciences and Aquaculture, Nord University (Norway).
Summary
Methionine along with folic acid, vitamin B6 (pyridoxine) and vitamin B12 (cobalamin) are important micronutrients in the one-carbon (1C) metabolism. Recent research* points to their importance in improved Atlantic salmon (Salmo salar) growth through smoltification [1,2]. The availability of those 1C nutrients can affect histone tail modifications and DNA methylation that together regulate mRNA expression and thereby control metabolism, which is one underlying explanation for nutritional programming. Epigenetic changes during early life stages such as early impact in pre-smolts can program life-long consequences on physiology, robustness and growth. Understanding how growth is controlled by those non-genetic mechanisms becomes important for a rapidly growing aquaculture industry whose concerns are to optimize production, sustainability and quality.
Two experimental diets provided by Skretting ARC were fed to Atlantic salmon 6 weeks prior to smoltification until 3 months after saltwater transfer in triplicate tanks at Skretting’s research station. A control diet included 1C nutrients on requirement and recommended levels, whereas a 1C+ diet contained a moderate surplus package to support maximal performance [1]. Pre- and post-smolt muscle from both groups were taken for global metabolic profiling (n = 3), RNA-sequencing (n = 9), DNA methylation analysis (RRBS, n = 9) and analysis of histone tail methylation (H3K4me3) and acetylation (H3K27ac) marks (CUT&RUN, n = 6). Both metabolic and gene expression signatures revealed significant 1C nutrient-dependent changes already at pre-smolt, but differences intensified when analyzing post-smolt muscle [2]. DNA methylation and histone tail modification data are under analysis and key results from all sequencing approaches will be presented.
The 1C+ feed fed over the challenging smoltification period resulted in the best growth performance in the saltwater period [1]. The overall metabolic profile in the muscle of salmon fed the control diet suggests a lower amino acid utilization for protein synthesis and increased methionine metabolization in polyamine and redox homeostasis, whereas gene expression profiles are indicative of compensatory growth regulation at local muscle tissue level. Integration of metabolic, transcriptomic with DNA methylation and histone tail modification profiles will reveal possible epigenetic mechanisms involved in improved growth that highlight the employment of nutritional programming strategies on healthy and robust growth. * The Norwegian Research Council (NRC): 295118 (EpiFishGrowth) and 267787 (NutrEpi).
References
Espe, M.; Vikeså, V.; Thomsen, T.H.; Adam, A.; Saito, T.; Skjærven, K.H. Atlantic salmon fed a nutrient package of surplus methionine, vitamin B12, folic acid and vitamin B6 improved growth and reduced the relative liver size, but when in excess growth reduced. Aquac. Nutr. 2019, 26, 477–489.
Adam, A.C.; Saito, T.; Espe, M.; Whatmore, P.; Fernandes, J.M.D.O.; Vikeså, V.; Skjærven, K.H. Metabolic and molecular signatures of improved growth in Atlantic salmon (
Salmo salar) fed surplus levels of methionine, folic acid, vitamin B
6 and B
12 throughout smoltification.
Br. J. Nutr. 2021,
127, 1–14.
https://doi.org/10.6084/m9.figshare.14484489.
Seasonal Broodstock Management Influences the Epigenetic Gene Regulation in Atlantic Salmon Progeny
Skjærven, K.H.1, Saito, T.1, Fernandes, J.M.O.2, Mommens, M.3, Adam, A.C.1 and Espe, M.1
1 Institute of Marine Research (IMR), Bergen, Norway.
2 Nord University, Bodø, Norway.
3 AquaGen AS, Trondheim, Norway.
Summary
In the salmon aquaculture industry, the spawning season adjustment of female broodstock is a common practice to make the products available throughout the year. The methods of manipulating spawning seasons have been well established and optimized by controlling multiple environmental factors, such as water temperature, light cycle, and feeding schedules. This practice has also attracted research interest in the health and well-being of the broodstock as well as their offspring when spawning seasons are adjusted. Here, the aim of this study is to investigate how two manipulated spawning seasons—early season (September) and late season (January)—have influenced the nutritional status and the profiles of gene expression and DNA methylation compared to the regular spawning season (November).
As several studies indicated that the one-carbon (1C) metabolism is one of the key biological pathways to affect the nutritional status of both broodstock and offspring inter-generationally, we measured 1C nutrients (vitamin B6, vitamin B12, folate, and methionine) along with free amino acids and lipid classes in the muscle and liver tissues of broodstock, newly fertilized eggs, and first-feeding larvae. The nutritional analysis revealed that the manipulated spawning seasons significantly altered the levels of nutrition in both broodstock and offspring. Specifically, broodstock from the early season appeared to incorporate less nutrients into the eggs, whereas broodstock from the late season indicated hunger.
To further investigate the effects of the seasonal changes, we analyzed gene expression and DNA methylation in the liver of first-feeding larvae. The over-representation analysis on the Kyoto Encyclopedia of Genes and Genomes (KEGG) database based on gene expression differences revealed that several metabolic and cell regulatory pathways were significantly enriched for the manipulated spawning seasons. Among them, mitotic cell cycle progression (Cell cycle, KEGG ID: sasa04110) showed a clear differential expression pattern as most associated genes were significantly down-regulated. The seasonal changes also affected DNA methylation profiles with having over 3000 differentially methylated CpG sites identified. Specifically, multiple CpG sites were hyper- or hypo-methylated in genes related to cell cycle regulation, developmental outcomes, and metabolism.
This study suggests that the assessment of optimal nutritional requirements for Atlantic salmon broodstock could be more effective if different spawning seasons are taken into consideration. It has also revealed potential biological pathways and epigenetic regulations in offspring affected by altered spawning seasons of broodstock.
Transcriptomics of Vertebral Anomalies in Senegalese sole (Solea senegalensis)
Bouza, C.1, Fernández, J.C.1, Losada, A.P.2, de Azevedo, A.M.2, Álvarez-Dios, J.A.3, Barreiro, A.2, Quiroga, M.I.2, Vázquez, S.2 and Martínez, P.1
1 Zoology, Genetics and Physical Anthropology Department, Faculty of Veterinary, Universidade de Santiago de Compostela, Lugo, Spain.
2 Veterinary Clinical Sciences Department, Faculty of Veterinary, Universidade Santiago de Compostela, Lugo, Spain.
3 Department of Applied Mathematics, Faculty of Mathematics, Universidade Santiago de Compostela, Santiago de Compostela, Spain.
Summary
Skeletal anomalies are frequent pathologies in fish aquaculture, affecting animal quality and welfare, as well as production, with special impact in certain flatfish species, such as the Senegalese sole (Solea senegalensis). In this context, the present multidisciplinary research aimed at deepening the knowledge of the genetic basis and molecular mechanisms underlying osteogenesis processes and the development of vertebral anomalies in S. senegalensis. The experimental trial was carried out in the Marine Research Center ECIMAT (Vigo, Spain) starting from a commercial batch of fertilized eggs under two thermal incubation conditions (18 °C and 22 °C), to evaluate the incidence of vertebral anomalies and growth. Stereomicroscopy, radiographic and image assessment methods were optimized for monitoring the vertebral status at larval and juvenile stages, to classify the main types of vertebral anomalies and support sampling collections for further transcriptomic analyses. Vertebral bone tissues from unaffected and anomalous vertebral phenotypes were collected for RNA extraction at 30 and 130 days after hatching. Muscle and fin tissues were also sampled in the latter juvenile stage. An RNA-seq analysis including three replicates per condition was performed after quality-filtered reads aligned against the Senegalese sole genome. A reference transcriptome of the vertebral body bone of Senegalese sole was obtained as a basis for further identification of gene expression profiles and pathways related to the development of skeletal anomalies in the conditions tested. Sets of differentially expressed genes were detected between control and malformed vertebral phenotypes across tissues and life-stages under different incubation temperatures, providing insights into the functional mechanisms regulating vertebral development and growth in Senegalese sole. These results may be useful to search for biomarkers of fish quality and welfare in aquaculture and for comparative analysis with other farmed and model teleosts and vertebrates, with production and biomedicine purposes.
Acknowledgments: Spanish Ministry of Science, Innovation and Universities & European Regional Development Fund (RTI2018-097110-B-C21). AM de Azevedo received a postdoctoral contract from Consellería de Cultura, Educación e Ordenación Universitaria, Xunta de Galicia, Spain.
4. Structural Genomics and Population Genetics
Chromosome Level Genome Assembly for the Meagre, Argyrosomus regius
Papadogiannis, V.1, Tsakogiannis, A.1, Kristoffersen, J.B.1, Nousias, O.1,2, Manousaki, T.1 and Tsigenopoulos, C.S.1
1 Evolutionary Genomics and Bioinformatics Lab (GenomeNerds), Hellenic Centre for Marine Research (HCMR), Institute of Marine Biology Biotechnology and Aquaculture (IMBBC).
2 Department of Biology, University of Crete, Greece.
Summary
The meagre, Argyrosomus regius, has recently become a species of increasing economic interest for the Mediterranean aquaculture and there is extensive ongoing work to boost the breeding efficiency. Access to the complete genomic sequence would provide an important resource for studying quantitative trait-associated loci and explore the genetic diversity of different aquaculture stocks and wild populations. Here, we present the first complete nuclear genome for A. regius, produced through a combination of long- and short-read technologies and an efficient in-house developed pipeline for assembly and polishing. Scaffolding using linkage map data allowed us to reconstruct a chromosome level assembly with high completeness, complemented with careful gene annotation and repeat masking. We use this new resource to study the evolution of the meagre genome as well as of other Scianidae family species, via a comparative analysis of more than thirty high-quality teleost genomes. Following phylogenomic reconstruction, we show highly conserved synteny within the group. Duplication analysis identifies immune-related gene family expansions in the meagre and duplications in Scianidae that could be connected to their unique muscular and sensory adaptations. Multiple genome alignment and base-wise study of evolutionary rate across the meagre genome reveal additional candidate loci related to growth, developmental and immune adaptations. We consider that this genomic dataset will add an important new tool for aquaculture studies and greatly facilitate culture and breeding work in the species.
Acknowledgments: The study has received funding from the Greek Republic through the “MeagreGen” project under the call “Special Actions AQUACULTURE” in the Operational Program “Competitiveness Entrepreneurship and Innovation 2014–2020. This research was further supported through computational resources provided by IMBBC of the HCMR Zorbas HPC infrastructure.
Chromosome Level Reference Genome for a British European Flat Oyster (Ostrea edulis)
Gundappa, M.K.1, Huang, J.1, Penaloza, C.1, Regan, T.1, Tanguy, A.2, Houston, R.D.1, Bean, T.P.1 and Macqueen, D.J.1
1 The Roslin Institute and Royal (Dick) School of Veterinary Studies, University of Edinburgh, Easter Bush Campus.
2 Station Biologique de Roscoff, Laboratoire Adaptation et Diversité en Milieu Marin (UMR 7144 AD2M CNRS-Sorbonne Université), Place Georges Tessier, 29680 Roscoff, France.
Summary
The European flat oyster (Ostrea edulis) is a bivalve mollusks species naturally distributed across Europe. This keystone species was an integral part of human diet in Europe for centuries until anthropogenic activities and disease outbreaks severely reduced wild populations. Large-scale restoration activities and aquaculture over the last few decades have led to an increase in the natural populations and commercial production of this species. However, despite a growing interest in the use of genetic tools to support population management and breeding goals in flat oyster, a reference genome assembly has been lacking to support such research. Here we report a high-quality chromosome-level genome assembly and annotation for European flat oyster generated using a combination of high-coverage Nanopore long-read data, high-accuracy short-read data and Omni-C scaffolding. An initial contig assembly (N50: 2.38Mb) was scaffolded into 10 pseudo-chromosomes using Omni-C data and further verified for scaffolding accuracy using a newly generated linkage map. The finished assembly had a total length of 935.13 Mb with scaffold-N50 of 95.56 Mb. Annotation of the genome using multi-tissue RNA sequence data, protein evidence and ab initio gene prediction resulted in the identification of ~35,000 protein-coding genes in the assembly. The genome exhibited extensive chromosome-level synteny with other high-quality bivalve molluscan genome assemblies, including a flat oyster genome for a French population generated independently by a collaborating team based at Station Biologique de Roscoff. We investigated the structural variation (SV) landscape in the flat oyster genome using resequencing data generated using long-read (6 individuals) and short-read (24 individuals) technologies. These variants were overlapped against the genome annotation to identify SVs with potential major impacts on gene function. A comparative genomics approach was taken to characterize gene family evolution in Ostrea, including loss and copy number expansion, carried out in a phylogenetic context capturing other bivalve molluscans. The current study, in addition to enhancing genetic resources available for flat oyster research in support of conservation and aquaculture, improves our understanding of bivalve genome evolution.
ddRAD-Seq Reveals the Genetic Structure and Detects Signals of Selection in Italian Brown Trout
D’Agaro, E.1^, Magris, G.1,2^, Marroni, F.1,2^, Vischi, M.1, Chiabà, C.1, Scaglione, D.3, Kijas, J.4, Messina, M.1, Tibaldi, E.1 and Morgante, M.1,2
1 Department of Agricultural, Food, Environmental and Animal Sciences (Di4A), University of Udine, Italy.
2 Istituto di Genomica Applicata, Udine, Italy.
3 IGA technology services, s.r.l., Udine, Italy.
4 CSIRO, St Lucia QLD 4067, Australia.
^ These authors contributed equally to this work.
Summary
Brown trout is one of the most widespread freshwater fish species in Europe. The evolutionary history of and phylogenetic relationships between brown trout populations are complex, and this is especially true for Italian populations which are heavily influenced in different ways by stocking practices. The characterization of the genetic structure of Italian brown trout populations may give information on the risk of losing endemic Italian populations due to lack of genetic diversity or to admixture with stocking populations.
The identification of signatures of selection and the information deriving from dense genotyping data will help genotype-informed breeding programs. We used a ddRAD-seq approach to obtain more than 100,000 single nucleotide polymorphisms (SNPs) and to characterize the population structure and signatures of selection in 90 brown trout samples. Italian brown trout populations are genetically differentiated, although the stocking practices have introduced strong admixture in endemic Italian trout, especially with the Atlantic lineage. Most of the analyzed populations showed high levels of kinship and inbreeding.
We detected putative signatures of selection using different approaches and investigated if the regions were enriched for functional categories. Several regions putatively under selection and characterized by a reduction in heterozygosity across all the studied populations are enriched for genes involved in the response to viral infections.
Our results, which show evidence of admixture with the Atlantic lineage (commonly used for stocking), confirm the need for controlling stocking practices, in order to avoid the erosion of the endemic gene pool; given the apparently high levels of kinship and inbreeding in local populations, our results also show the need to take action for increasing gene diversity.
In addition, we used the genetically distinct lineages to detect signatures of selection and we identified putative signatures of selection in several regions associated with resistance to infectious diseases. These constitute candidate regions for the study of resistance to infections in wild and farmed trout.
Genome-Wide Detection of Positive Selection in Rainbow Trout Populations
Paul, K., Restoux, G. and Phocas, F.
Université Paris-Saclay, INRAE, AgroParisTech, GABI, 78350 Jouy-en-Josas, France.
Summary
Rainbow trout (Oncorhynchus mykiss) is native to North America and its domestication began in the 1870s. Then introduced in other continents, rainbow trout has been selected in various breeding programs since the 1970s [1]. Domestication and selection have led to captive phenotypes different from the wild due to genetic changes (adaptation, inbreeding and genetic drift). To detect signatures of recent selection among domesticated lines, we analyzed the genotypes of 177 fish from 4 populations:;20 sequenced fish from a wild American population (WA) coming from 4 rivers in the northwest of USA [1]; 14 fish from the INRAE synthetic line (SY) created by intercrossing several domesticated lines in the 1980s to produce a population with a large genetic diversity; and 143 fish coming from two French commercial lines (LB and LC) selected since the 1990s mainly for improved growth performance. The three domesticated lines were genotyped using a new HD Axiom Trout Genotyping Array with 665K SNPs designed in the HypoTemp project and funded by the European Maritime and Fisheries Fund (n° PFEA470019FA1000016). After quality control, 546,903 SNPs remained for the analysis with all SNP polymorphic in at least one population. Differentiation among the three domesticated lines was medium as already reported [2] with Fst values ranging from 0.10 to 0.12, while the Fst values between the French lines and WA population were large (0.27–0.28). A Cross-Population Extended Haplotype Homozygosity method (XP-EHH) was used to detect selected alleles that have risen to high frequency or fixation in one but not all populations [3]. Figure 1 presents the associated p-values when comparing EHH between WA and SY lines. For all comparisons between WA and any of the three domesticated lines, genome-wide significant signatures of selection were detected on chromosome 17 (Omy17) in a region containing 69 genes spanning from 18 to 21 Mb on the GCF_013265735.2 reference genome, and on Omy26 in a large region spanning from 39 to 48 Mb. Some other highly significant signatures were only observed when comparing WA with only two domesticated lines: on Omy13 from 59 to 62 Mb, on Omy18 from 11 to 13 Mb and on Omy21 from 13 to 17 Mb. Two-by-two comparisons among the domesticated lines only gave chromosome-wide significant p-values with consistent signals on chromosomes 12, 16, 21 and 32. Further work will characterize the main gene functions beyond these regions under positive selection.
Figure 1.
Manhattan plot of the log transformed XP-EHH test values for WA versus SY line. The horizontal lines represent the 5% genome-wide and chromosome-wide significant thresholds after Bonferroni correction.
Figure 1.
Manhattan plot of the log transformed XP-EHH test values for WA versus SY line. The horizontal lines represent the 5% genome-wide and chromosome-wide significant thresholds after Bonferroni correction.
References
Gao, G.; Nome, T.; Pearse, D.E.; Moen, T.; Naish, K.A.; Thorgaard, G.H.; Lien, S.; Palti, Y. A new single nucleotide polymorphism database for rainbow trout generated through whole genome resequencing. Front. Genet. 2018, 9, 147.
D’ambrosio, J.; Phocas, F.; Haffray, P.; Bestin, A.; Brard-Fudulea, S.; Poncet, C.; Quillet, E.; Dechamp, N.; Fraslin, C.; Charles, M.; et al. Genome-wide estimates of genetic diversity, inbreeding and effective size of experimental and commercial rainbow trout lines undergoing selective breeding. Genet. Sel. Evol. 2019, 51, 1–15.
Sabeti, P.C.; Varilly, P.; Fry, B.; Lohmueller, J.; Hostetter, E.; Cotsapas, C.; Xie, X.; Byrne, E.H.; McCarroll, S.A.; Gaudet, R.; et al. Genome-wide detection and characterization of positive selection in human populations. Nature 2007, 449, 913–918.
Exploiting Intraspecific Variation in Wild Populations to Improve Arctic Charr (Salvelinus alpinus) Aquaculture Selective Breeding Practices
Salisbury, S.J.1, McCracken, G.R.1, Perry, R.2, Keefe, D.3, Layton, K.K.S.4,5, Kess, T.4, Nugent, C.M.6, Leong, J.S.7, Bradbury, I.R.1,4,5, Koop, B.F.7,8, Ferguson, M.M.6 and Ruzzante, D.E.1
1 Department of Biology, Dalhousie University, Halifax, NS, Canada
2 Department of Environment, Fish and Wildlife Division, Government of Yukon, Whitehorse, YT, Canada.
3 Department of Fisheries, Forestry, and Agriculture, Government of Newfoundland and Labrador, Corner Brook, NL, Canada.
4 Department of Fisheries and Oceans, Northwest Atlantic Fisheries Centre, St. John’s, NL, Canada.
5 Department of Ocean Sciences, Memorial University of Newfoundland, St. John’s, NL, Canada.
6 Department of Integrative Biology, University of Guelph, Guelph, ON, Canada.
7 Department of Biology, University of Victoria, Victoria, BC, Canada.
8 Centre for Biomedical Research, University of Victoria, Victoria, BC, Canada.
Current Address for KKS Layton: School of Biological Sciences, University of Aberdeen, Aberdeen, United Kingdom.
Summary
Early maturation to the detriment of growth, maximum size, and flesh quality, is a pressing problem in the aquaculture production of Arctic charr (Salvelinus alpinus) and other salmonid species. Historically, early maturation was believed to be a plastic trait in charr, driving intensive research into optimizing environmental conditions in aquaculture practices. However, the observation of sympatric, genetically differentiated morphs that differ in the size at which they mature (small vs. large) in wild charr populations suggests the potential for a genetic underpinning to maturation timing in this species. Previous work had identified such morphs in Quebec and Labrador, Canada in the form of sympatric small and big morphs in landlocked lakes (without access to the sea) as well as sympatric small resident and large anadromous morphs. These populations are particularly relevant to informing aquaculture practices as they are within close proximity to the wild Fraser River population, the source of an important aquaculture strain of this species. We therefore identified five locations in Labrador, Canada with sympatric small and large morphs of wild charr and used an 87K Axiom SNP chip to investigate for candidate genes consistently differentiating sympatric morphs. Strong genome-wide genetic differentiation supported a genetic basis to morph differentiation. We found several SNPs, genes, and paralogs which repeatedly differentiated morphs across locations. This included pappalysin-2, a gene previously associated with growth in mice. While no single locus consistently differed between morphs in all locations (potentially because multiple genetic pathways may underpin maturation timing), we reveal a number of potential candidate loci which could be investigated further in an experimental setting and may improve Arctic charr selective breeding practices in future. Our results further demonstrate the utility of investigating natural variation in wild populations of aquaculture strains to inform selective breeding practices.
5. Physiology and Development
A Pipeline for Analysis of Allele Specific Expression from RNA Seq Data Reveals Salinity Dependent Response in Nile tilapia
Campo, A., Gershoni, M., Doron-Faigenboim, A. and Cnaani, A.
Department of Poultry and Aquaculture, Institute of Animal Science, Agricultural Research Organization, Rishon LeTsiyon, Israel.
Summary
Allelic imbalance (AI) is a phenomenon that depicts cis- and trans-effects on gene expression regulation, can illustrate imprinting events, and is an arduous effort to retrieve due to the multiple sources of bias present in a pipeline for RNA-seq and variant calling tasks. Major advances in software packages and tools for variant calling and allelic imbalance detection have successfully achieved individual variability. However, very little of these tools spread outside the medical research to be applied into the aquaculture field. In the present study, we introduce a pipeline to detect allelic imbalance reducing the bias in critical steps such as genome mapping. The identification of significant allelic imbalance is resolved by binomial test after the removal of monoallelic gene expression (MAE). We employed the pipeline on transcriptome data of Nile tilapia (Oreochromis niloticus) grown in fresh and salty water. We then validated the pipeline SNP calling from the transcriptome with whole genome sequencing of the same individuals. Our results indicate that specific imbalanced expression of alleles is critical in the sampled tissues. Calculation of allele frequencies confirmed these AI differences in tissues and revealed further differences in allele expression related to water salinity. Overall, AI analysis adds an additional layer of information that can be obtained from transcriptome sequences and enables a wider view of genetic basis underlying phenotypic variation.
Oxytocinergic Regulation of the Homeostatic Response to Cold Stress in Nile tilapia
Biran J.1, Segev-Hadar A.1, Krispin S.1,2, Olthof A.M.3, Hyatt K.C.3, Haller L.1, Barki A.1, Nitzan T.1, Levkowitz G.4, Kanadia R.N.3 and Cnaani A.1
1 Department of Poultry and Aquaculture, Institute of Animal Science, Agricultural Research Organization, Rishon LeTsiyon, Israel.
2 Faculty of Life Sciences, Bar-Ilan University, Ramat-Gan, Israel.
3 Physiology and Neurobiology Department, University of Connecticut, Storrs, CT 06269, USA.
4 Department of Molecular Cell Biology, Weizmann Institute of Science, PO Box 26, Rehovot, 7610001, Israel.
Summary
Similar to poikilotherms, cultured fish exposed to the environmental extremes of cold temperatures experience a stressful metabolic challenge, which elicits physiological responses required to maintain cellular homeostasis. These responses include modified glucose or lipid metabolism, altered gene expression and alternative splicing, and endocrine and immune system activity. Additionally, heat seeking may not resolve the homeostatic needs of tropical poikilotherms under unpredictable extreme cold events, which occur frequently due to global climate change and aquaculture conditions. In spite of the physiological responses that occur in poikilothermic vertebrates, the prevailing notion is that these reactions are passive. Here, we explored molecular hypothalamic and physiological responses to cold stress in Nile tilapia (Oreochromis niloticus). We show that cold exposed tilapia exhibit complex homeostatic responses, including plasma glucose and cortisol concomitant with reduced plasma lactate and metabolic rate. Hypothalamic transcriptome analysis revealed increased oxytocin expression. Using CRISPR knockout of oxytocin and its receptors, and pharmacological blockage of oxytocin signaling, further affected temperature-dependent metabolic rate in two cold-exposed fish species. This indicates that oxytocin a known thermoregulator in homeotherms actively regulates temperature-related homeostasis in fish. Overall, our findings show that the fish brain actively responds to cold temperatures by regulating metabolic physiology. Moreover, we identify oxytocin signaling as an adaptive and evolutionarily conserved metabolic regulator of temperature-related homeostasis.
Neural Regulation of Bone Mineral Homeostasis in Fish: Functional and Transcriptional Characterization of pth4 Neurons
Méndez-Martínez, L.1, Guerrero-Peña, L.1, Suarez-Bregua, P.1, Naranjo, S.2, Tena, J.J.2 and Rotllant, J.1
1 Acuabiotec Lab, Department of Biotechnology & Aquaculture. Institute of Marine Research, (IIM-CSIC), 36208 Vigo, Spain.
2 Centro Andaluz de Biología del Desarrollo (CABD), Consejo Superior de Investigaciones Científicas/Universidad Pablo de Olavide, Sevilla, Spain.
Summary
Understanding the mechanism of bone mineralization and skeletal development is key to prevent skeletal deformities, which still represent one of the major problems in aquaculture. Our previous studies revealed that the novel Pth4 peptide was involved in bone metabolism through phosphate regulation. The pth4 gene is specifically expressed in two bilateral groups of neurons in the hypothalamic area of the zebrafish brain. To characterize the pth4 neurons and their role in bone metabolism, we performed a targeted cell ablation using a stable transgenic pth4 promoter-driven EGFP (pth4:EGFP) zebrafish line. This resulted in impaired mineralization and altered expression of phosphate regulation and osteoblast differentiation genes. To understand the basic biological properties of Pth4 neurons, we examined the transcriptome of adult zebrafish Pth4 neurons. EGFP-labeled Pth4 neurons were isolated from the brain of pth4:EGFP transgenic zebrafish. Four hundred Pth4 neurons (from 10 brains) were collected for each biological replicate using the FACS technique. RNA sequencing of two biological replicates was performed and analyzed. The pth4 neurons showed a transcriptional profile similar to that from other hypothalamic dopaminergic neurons. Our dataset provides an extensive resource for understanding the molecular mechanisms underlying function of adult zebrafish Pth4 neurons. It also establishes a framework for the future characterization of genes expressed in Pth4 neurons and the study of Pth4 neuron evolution.
Acknowledgments: This research was funded by the MCIN/AEI/10.13039/501100011033 grant numbers AGL2014-52473R and AGL2017-89648P to JR and by “ERDF A way of making Europe”.
P1—Protective Effect of Bacteria Isolated from the Natural Microbiota of the Pacific Oyster Crassostrea gigas against OsHV-1 µVar and Vibrio aestuarianus Infections
Dantan, L.1, Toulza, E.1, Gueguen, Y.2, Degrémont, L.3, Morga, B.3, Vidal Dupiol, J.1, Petton, B.4, Fleury, Y.5, Mege, M.3, Maurouard, E.3, Alienne, J.-F.1, Courtay, G.1, Romatif, O.1, Carcassonne, P.1 and Cosseau, C.1
1 IHPE, Univ. Montpellier, CNRS, Ifremer, Univ. Perpignan Via Domitia, Perpignan France.
2 MARBEC, Univ Montpellier, CNRS, Ifremer, IRD, Sète, France.
3 Ifremer, SG2M, LPGMM, La Tremblade, France.
4 Ifremer, UBO CNRS IRD, LEMAR UMR 6539 Argenton, France.
5 Univ Bretagne Sud, Univ Bretagne Occidentale, Lab Biotechnol & Chim Marine, EA3884, F-29334 Quimper, France.
Summary
Recently, marine diseases have increased in frequency and severity in association with global changes and mollusks of economic interest are particularly concerned. One of the most striking examples of devastating diseases is the Pacific Oyster Mortality Syndrome (POMS) caused by the ostreid herpes virus 1 µVar (OsHV-1 µVar) that emerged in 2008 and which heavily impacts Crassostrea gigas production worldwide by affecting juvenile oysters. Adult oysters are also regularly affected by infectious diseases, especially infections by bacterial species of the genius Vibrio such as V. aestuarianus. The present work aims to find sustainable strategies to help fight against these infectious diseases. We propose to take profit of the beneficial effect of the natural oyster microflora to develop probiotics. It has been reported that some bacterial species are preferentially associated with oysters with better survival capacity. In addition, previous work has shown that the immune defenses of oysters can be stimulated by exposing them to microorganisms from the natural environment during their larval development. Following these encouraging results, we have generated a collection of bacterial species from disease resistant C. gigas and we have characterized their effect on the oysters either by exposure during larval development or by direct contact at the juvenile stage. We have shown that it is possible to enhance oyster survival capacity using potential probiotic bacteria by two mechanisms: (1) Adding a cocktail of probiotic during larval stages leads to an improvement in survival against OsHV-1 µVar, a possible immunomodulatory mechanism currently being investigated. (2) We have identified antimicrobial-producing bacterial strains that display a protective effect towards oyster pathogens in vitro and in vivo during oyster exposure at the juvenile stage, leading to an increase in their survival during an infection with OsHV-1 µVar or Vibrio aestuarianus. These findings open new avenues for the development of microbiome-targeted prophylactic approaches to mitigate diseases in oyster farming.
P3—β-Glucans as an Immunostimulant in a GALT Leucocyte Primary Cell Model
Porter, D.1, Peggs, D.2, McGurk, C.2 and Martin, S.A.M.1
1 Scottish Fish Immunology Research Centre, School of Biological Sciences, University of Aberdeen, Aberdeen, AB24 2TZ.
2 Skretting ARC, Sjøhagen 15, 4016 Stavanger, Norway.
Summary
The development of functional feeds for farmed fish are considered a significant factor in advancing fish health and performance. Despite recent advances in this area many of the mechanisms by which the immune response is affected are not fully understood. β-glucans are a common feed additive due to their main method of action as a prebiotic but have also demonstrated some immunostimulatory actions causing the upregulation of key pro-inflammatory genes during feeding trials. Cell lines and primary cell cultures are used as an alternative method to identify immune responses which also addresses 3Rs in reducing whole animal experiments. In this study, cell culture models were used to act as the nutrition health interface with a variety of cell culture (RTS11 and RTgutGC) and primary culture models used (GALT leucocytes). Two β-glucans and several pathogen-associated molecular patterns (PAMPS) were used to elucidate key pro-inflammatory and viral markers which may be altered due to functional ingredients.
Our results suggest that primary cultures of GALT leucocytes can respond to PAMPs and immunostimulants, with similar but not the same response as permanent cell lines, with differences thought to be due to the greater diversity of cell types in the primary cultures. The results demonstrate the viability of GALT leucocytes to act as a valuable precursor to feeding trials. The results from the GALT leucocyte stimulations show that stimulation with the PAMPs, Poly I:C, PHA and recombinant IL-1β, show significant responses to target genes. Stimulation with β-glucans with responses causing an upregulation in il-1β, il-8, il-4 and tnfα in all cell types is in agreement with published data [1]. This study suggests further research into the direct mechanisms of actions of both β-glucans will help define the direct immunological effects of these functional feed components, enabling focused future designs of diets. This work was funded by a PhD studentship from University of Aberdeen and Co funded by Skretting ARC.
References
Douxfils, J.; Fierro-Castro, C.; Mandiki, S.; Emile, W.; Tort, L.; Kestemont, P. Dietary β-glucans differentially modulate immune and stress-related gene expression in lymphoid organs from healthy and Aeromonas hydrophila-infected rainbow trout (Oncorhynchus mykiss). Fish Shellfish Immunol. 2017, 63, 285–296.
P4—Exploring New Insights into the Relationship between Growth and Sexual Maturation in Sea Bass
Sempere, L.1, Ibáñez, S.1, Marín, C.2, Molés, G.3, Calduch-Giner, J.4, Pérez-Sánchez, J.4 and Felip, A.1
1 Group of Fish Reproductive Physiology, Institute of Aquaculture Torre de la Sal, CSIC, Castellón, Spain.
2 Husbandry Service, Institute of Aquaculture Torre de la Sal, CSIC, Castellón, Spain.
3 Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria, Department of Environment, CSIC, Madrid, Spain.
4 Nutrigenomics and Fish Growth Endocrinology Group, Institute of Aquaculture Torre de la Sal, CSIC, Castellón, Spain.
Summary
Growth is one of the most desirable traits in an aquaculture species, which is in turn closely related to early maturity, a complex biological process that reduces the final yield of fish farming operations. Thus, while the selection of rapid-growth fish is highly attractive for the aquaculture sector, it is also common to select against early maturing fish. The correlation between body size and age at maturation is still not entirely understood in fish and several studies have reported different associations between the two traits. In this study, biometric and physiological parameters are being evaluated in order to determine the relationship of small- and large-sized female sea bass and its ovarian growth. Fish were individually tagged and growth and gonadal development were evaluated during the second and third year of age. Fish have been sacrificed at the end of each sexual cycle and performance indicators are being restored to a specific point in time based on the oocyte development. Temporal hormonal profiles, including Fsh, sex steroids, vitellogenin and Igf-1 plasma levels are being determined. Furthermore, genes with a putative role in the relationship between both growth and reproductive systems will be analysed. Results show the existence of early differential growth within the female sea bass.
Thus, slow-growing (small-sized) and fast-growing (large-sized) female sea bass have been identified and their relationship to gonadal maturity has been considered. We have identified four female phenotypes as follows: (a) small-sized and immature gonads, (b) small-sized and maturing gonads, (c) large-sized and immature gonads and (d) large-sized and maturing gonads, over the second and third sexual cycles of this species. Accordingly, physiological aspects and also molecular factors are being considered to elucidate key events contributing to the relationship between these traits. A comprehensive understanding of the relationship between growth and maturation in sea bass will contribute to the attainment of new insights for improving breeding conditions and production of this teleost species.
Acknowledgments: Project funded from the Ministry of Science and Innovation (AGL2016-75400, PID2019-109548RB-I00). L.S. supported by a FPI fellowship from MICINN (Spain).
P5—Genetic Variation Associated with Divergent Selection to Marteilia cochillia in the Common Cockle (Cerastoderma edule)
Pampín, M.1, Casanova, A.1, Cao, A.2, Fernández, C.1, Iglesias, D.2, Carballal, M.J.2, Pardo, B.G.1, Vera, M.1, Villalba, A.2 and Martínez, P.1
1 Department of Zoology, Genetics and Physical Antropology, Faculty of Veterinary, University of Santiago de Compostela, Spain.
2 Centro de Investigacións Mariñas, Consellería do Mar, Xunta de Galicia, Spain.
Summary
The common cockle (Cerastoderma edule) is fundamental for marine coastal ecosystems and a valuable socioeconomic resource for the communities where it is harvested. In 2012, the cockle beds of the Arousa Ría (Galicia), northwest of Spain, were seriously decimated due to a disease caused by the protozoan Marteilia cochillia responsible for marteiliosis. Marked-assisted selection programmes could be instrumental in developing parasite-resilient strains for restoring affected seabeds. Our study aimed to identify single nucleotide polymorphisms (SNP) markers across the cockle’s genome, potentially associated with resilience to marteiliosis. For this purpose, two complementary approaches were followed: (i) a population genomics analysis in samples collected in 2018 from a long-term-affected shellfish bed in the Arousa estuary, Lombos de Ulla, before and after the annual outbreak of marteiliosis, taken as reference naïve samples in 2012 in the same estuary, before the first report of marteliosis in Galicia. Samples were genotyped using 2b-RAD on a panel of 9.154 SNPs mapped on the cockle’s genome on a sample of 77 individuals—38 from 2012 and 39 from 2018. Low but significant genetic differentiation (p < 0.05) was detected between samples exposed and non-exposed to the parasite (average FST = 0.061). A total of 170 outliers for divergent selection were detected, which were able to discriminate individuals infected vs non-infected or naïve. Genomic windows with consistent signals of divergent selection were identified and gene mined yielding 90 genes enriched in binding molecular functions (ion, organic cyclic compound, and heterocyclic compound binding), catalytic activities and biologic and binding; (ii) a second approach, using a cockle’s digestive gland transcriptomic study was addressed to identify SNPs associated with the 767 differentially expressed genes (DEGs) detected in the aforementioned marteiliosis outbreak by comparing non-infected vs infected cockles across a temporal series (four points in 2018 and 2019). Among the ~45,000 SNPs detected, two were selected per DEG for comparing samples with different infection status. A total of 123 SNPs with significant differentiation between infected and non-infected samples (FST p < 0.05) were identified, 41 of them showing a gradual increase or decrease in allelic frequencies related to the cockle’s infection level. Finally, a panel of 60 candidate SNPs, combining both approaches, were selected to assess their association with resilience against the parasite M. cochillia in an ongoing common garden field experiment.
Acknowledgments: This study was funded by the European Union through the project COCKLES within the INTERREG-AA programme (EAPA_458/2016).
P6—Eggs from Different Rainbow Trout Females Show Different Developmental Competence for Induced Gynogenesis—A Transcriptome Perspective
Ocalewicz, K.1, Gurgul, A.2, Polonis, M.1 and Dobosz, S.3
1 Faculty of Oceanography and Geography, University of Gdansk, Poland.
2 Centre for Experimental and Innovative Medicine, University of Agriculture in Kraków, Poland.
3 Department of Salmonid Research, Inland Fisheries Institute in Olsztyn, Poland.
Summary
In the present research, eggs from four rainbow trout females were gynogenetically activated and subjected to High Hydrostatic Pressure (HHP) shock to provide four groups of Doubled Haploids (DHs). The quality of eggs from different clutches was comparable as rainbow trout from the control groups had similar survival rates during embryogenesis and after hatching. However, interclutch differences were observed in gynogenetic variants of the experiment and the survival of DH specimens from different groups varied from 3% to 57% during embryogenesis. Transcriptome analysis of eggs from different females exhibited inter-individual differences in maternal genes’ expression. Eggs originating from females whose gynogenetic offspring had the highest survival showed increased expression of 46 genes when compared to eggs from three other females. Eggs with the highest survival of gynogenetic embryos showed the up-regulation of genes that are associated with cell survival, migration and differentiation (tyrosine-protein kinase receptor TYRO3-like gene), triglyceride metabolism (carnitine O-palmitoyltransferase 1 gene), biosynthesis of polyunsaturated fat (3-oxoacyl-acyl-carrier-protein reductase gene), early embryogenic development (protein argonaute-3 gene, leucine-rich repeat-containing protein 3-like gene), 5S RNA binding (ribosome biogenesis regulatory protein homolog), embryonic neurogenesis and tissue modelling during development (transcription factor SOX-11-like gene) and senescence and aging (telomerase reverse transcriptase, TERT gene), among others. These genes may be considered as candidate genes whose expression enables gynogenesis in rainbow trout. Special attention has been dedicated to the TERT gene that maintains the length of the telomeric DNA and its high expression in rainbow trout eggs with high developmental capacity for gynogenesis suggests telomerase may also be involved in processes related to egg quality, early ontogeny and gynogenetic development in fish.
P7—Integrating NGS Techniques in Bone Analysis of a Miniaturized Fish
de Azevedo, A.M.1, Losada, A.P.1, Bouza, C.2,3, Pardo, B.G.2,3, Blanco, A.3, Vázquez, S.1,3 and Quiroga, M.I.1,3
1 Department of Anatomy, Animal Production and Clinical Veterinary Sciences, Faculty of Veterinary Science, Universidade de Santiago de Compostela, Spain.
2 Department of Zoology, Genetics and Physical Anthropology, Faculty of Veterinary Science, Universidade de Santiago de Compostela, Spain.
3 Aquaculture Institute, Campus Vida, Santiago de Compostela, Spain.
Summary
Zebrafish (Danio rerio) continues to gain more importance in the scientific community as a model to investigate skeletal disorders both in humans and farmed fish species. In this species, there are few studies on the molecular events that occur in small specific regions of the skeleton, for which transcriptome sequencing techniques can increase the knowledge obtained by conventional microscopy. The objective of this work was to design a practical procedure to study the cellular and molecular mechanisms that occur during the development of zebrafish caudal complex vertebrae. A total of 71 juvenile zebrafish between 6 and 12 mm in total length were used. The fish were individually stored in RNA stabilization reagent and in 4% paraformaldehyde to perform transcriptomic, stereomicroscopic and microscopic techniques, respectively. For transcriptomic analyses, fish were pooled into two homogeneous groups of 20 samples each, from which the caudal complex preurals and urals were dissected. RNA extraction and sequencing were carried out. Individuals fixed in paraformaldehyde were processed for bone staining with alizarin red S, and for histopathologic study by means of glycol methacrylate embedding. Specific genes and functions associated with transcriptome profiles of fish bone were detected in the vertebral samples. Stereomicroscopic and histological techniques complemented the transcriptomic analysis allowing the detailed observation of bone development and mineralization of the caudal complex and facilitated the sampling of the small vertebral elements. This approach could be applied to multidisciplinary studies of other bone structures or specific skeletal anomalies in other fish species used in biomedical and aquaculture research.
Acknowledgments: The authors would like to thank Profs. P.E. Witten and A. Huysseune (Biology Department, Ghent University, Belgium) for their constructive comments. This work was performed within the framework of a project funded by the BioReDes Strategic Group (ED431E 2018/09). AM de Azevedo received a postdoctoral contract from Consellería de Cultura, Educación e Ordenación Universitaria, Xunta de Galicia, Spain.
P8—Comprehensive Analysis of the Olfactory Organ Transcriptome in Senegalese sole (Solea senegalensis) and Its Potential Implications in Fish Reproduction
Villamayor, P.R.1,2†, Torres, D.1,2†, Blanco, A.1, Riaza, A.3, Sánchez-Quinteiro, P.2, Robledo, D.4 and Martínez, P.1
1 Department of Genetics, Veterinary Faculty, Universidade de Santiago de Compostela (USC), Spain.
2 Department of Anatomy, Animal Production and Veterinary Science, Veterinary Faculty (USC), Spain.
3 Acuiculture Stolt Sea Farm, S.A., A Coruña, Spain.
4 The Roslin Institute, University of Edinburgh, UK.
† Both authors contributed equally to this work.
Summary
Olfaction is critical for fish survival, detecting environmental cues essential for food detection, predator avoidance or mating and leading to behavioral changes. Solea senegalensis is a flatfish that can reproduce in captivity when captured from the wild; however, males reared in captivity (F1) fail to reproduce despite normal gonad development. This is currently the main concern of the aquaculture of this species. We hypothesize that exposure to different life histories in attractive or aversive chemical/olfactory cues (wild vs. farm) might underlie the low male reproductive performance in F1 individuals. In fish, the sense of smell is controlled by large multigenic families of olfactory receptors (OR, TAAR, VR). Olfactory receptors are expressed individually in olfactory sensory neurons (OSNs), following the rule one receptor-one neuron, complicating their study. OSNs are found within the olfactory epithelium, which is integrated into a multilamellar and rosette-shaped structure located in the rostral part of the head, called olfactory rosette. Despite its potential importance in fish reproduction, the olfactory rosette remains understudied. As a first step towards understanding the role of the fish olfactory system in fish reproduction, we characterized the olfactory rosette transcriptome and the sole olfactory gene repertoire.
To do so, we dissected the olfactory rosettes of 12 individuals (10-month-old juveniles and 27-month-old adults, three males and three females per group) and performed RNA extraction (RIN > 9). Samples were then pooled into a single sample, from which library construction and long-read Nanopore sequencing technology was performed. In total, 71 olfactory/odorant receptors and a single VR (type 2) are found in the sole genome. The genomic localization of these receptors in the sole genome and the comparison with other flatfish genomes suggest that the sole olfactory receptor repertoire originated via tandem gene duplications during the adaptation of the order Pleuronectiformes to a demersal lifestyle. The sole olfactory transcriptome contains 13,000 expressed genes and 9535 show homology to known proteins. In total, 55 olfactory/odorant receptors were present in the sole olfactory organ transcriptome. The only sole VR was also found, showing 10 times more expression than the average OR. We also detected 438 enriched functions in the Senegalese sole olfactory organ transcriptome, such as receptor ligand activity, G protein receptor activity, synaptic transmission and androgen and steroid hormone receptor activity. Future studies are necessary to decipher the function of the 3465 unannotated genes and their conservation across Pleuronectiformes and teleosts. This work sets the basis to study the role of the fish olfactory epithelium in reproduction, potentially leading to an improvement in the reproductive performance of Senegal sole and other fish species in captivity, a key issue for aquaculture.
P9—The Physiological Impact of Total Fish Meal Replacement with Soybean Meal in Rainbow Trout (Oncorhynchus mykiss) Diets: New Clues from Circulating miRNAs
Fernández, I.1,2, Toledo-Solís, F.J.2,3, Larrán, A.M.2, Ortiz-Delgado, J.B.4, Sarasquete, C.4, Dias, J.5 and Morais, S.6
1 Centro Oceanográfico de Vigo (IEO-CSIC), 36390 Vigo, Spain.
2 Aquaculture Research Center (ITACyL), 40196 Zamarramala, Segovia, Spain.
3 Consejo Nacional de Ciencia y Tecnología (CONACYT), 03940 Mexico City, Mexico.
4 Instituto de Ciencias Marinas de Andalucía (CSIC), 11510, Puerto Real, Cádiz, Spain.
5 SPAROS Ltd., 8700-221 Olhão, Portugal.
6 Lucta S.A., Bellaterra, Spain.
Summary
Aquaculture long-term sustainability depends on the successful replacement of fish meal (FM) by alternative raw materials. Until now, FM replacement without compromising growth and/or wellbeing has been generally limited to 50–75% in most carnivorous fish species. FM replacement is still majorly based on vegetable protein sources, particularly on soybean meal (SBM). High FM replacement with SBM is known to decrease growth, feed intake, energy and fat digestibility, and inducing an inflammatory response in the distal intestine (Klinger and Naylor, 2016). The specific mechanisms are not fully understood and are probably multifactorial. Recently, the use of high-throughput tools in fish species has shown that nutritional interventions induce an altered profile of particular circulating miRNAs in blood plasma, which are considered a new class of integrative biomarkers (Toledo-Solís et al., 2021).
Here, the effects of feeding rainbow trout (Oncorhynchus mykiss) juveniles (23.2 ± 0.2 g mean weight) with diets containing 12% of FM and 8% of SBM (Control diet) or 0% of FM and 26.2% of SBM (SBM diet) for 63 days were characterized in triplicates. Fish growth performance, feed apparent digestibility, activity of digestive enzymes, and histopathological analysis were performed. The identification of differentially expressed circulating miRNAs from blood plasma was conducted using small RNA-Seq analysis.
Fish fed SBM and control diets did not shown differences in growth, SGR, FCR, fillet’s proximal composition, and hepatosomatic index. However, control fish showed a significantly lower viscerosomatic index (13.8 vs 18.1%). Additionally, control fish had higher amylase (51.7 vs 30.8 u/mg) but lower aminopeptidase (21.6 vs 32.0 u/mg) enzymatic activities than fish from SBM group. Histological analysis revealed that fish fed with the SBM diet had a higher number of microvilli fusions in proximal intestine, and lower brush border integrity and submucosa layer width in distal intestine. Analysis of miRNAs from the blood plasma of 10 specimens (five from each group; with 15–22 million reads) identified five particular miRNAs differentially expressed. Present results allowed us to identify new biomarkers of total FM replacement by SBM. Furthermore, the bioinformatic prediction of targeted mRNAs by these five miRNAs has enable us to identify new molecular pathways explaining the physiological impact of FM replacement by SBM.
Acknowledgments: Partially funded by Lucta S.A. and the MET2VI project (Ref. RTI2018-099029-A-I00) from ERDF and MICIU. I.F. acknowledges RYC2018-025337-I contract.
P10—Molecular Pathways Induced by Recombinant Gonadotropins in Mugil cephalus Were Typical of Natural Oogenesis
Ramos-Júdez, S.1, Danis, T.2, Angelova, N.2, Tsakogiannis, A.2, Giménez, I.3, Tsigenopoulos, C.2, Duncan, N.1* and Manousaki, T.2*
1 IRTA, 43540 Tarragona, Spain.
2 IMBBC- HCMR, 71003 Heraklion, Greece.
3 Rara Avis Biotec, S.L., 46002 Valencia, Spain.
Summary
The flathead grey mullet (Mugil cephalus) remains arrested at early stages of gametogenesis in intensive captivity conditions. Treatment with homologous single-chain recombinant gonadotropins (rGths) produced in CHO cells (rFsh and rLh) induced vitellogenesis to produce viable eggs and larvae. The present study aimed to characterize the molecular pathways and gene expression patterns throughout the rGths-induced vitellogenesis. Repeated ovarian samples were collected through cannulation from the same five females at four stages of ovarian development: (1) initial arrested gonad (previtellogenesis), (2) early–mid vitellogenesis after rFsh application, (3) late vitellogenesis after combined rFsh and rLh treatment, and (4) full-grown oocytes after rLh application. The RNASeq libraries were constructed for all the described stages. The Illumina HiSeq4000 paired-end sequencing generated a total of 614,942,156 raw paired reads of which 506,875,944 were maintained after trimming (Trimmomatic v0.39). The assembled transcriptome (Trinity v2.8.5) consisted of 513,643 transcripts with an average contig length of 919.18 nucleotides (nt) and N50 value of 1561 nt. BUSCO v3.1.0 indicated an 86.4% of transcriptome completeness (against the vertebrate orthologs database). An average of 89.68 % of the reads were back-mapped (Bowtie2). The final assembly was constituted of 287,089 transcripts with an expression value of FPKM ≥ 1 (relative abundances of transcripts by RSEM) which were annotated in the SwissProt, GO and KEGG databases (Trinotate v3.2.1). Differentially expressed genes (DEGs) were identified between stages by DESeq2 v1.26.0 performed in R v3.6.1. and excluding genes with < 30 reads in all samples. Enrichment of pathways analysis was conducted for DEGs.
Throughout vitellogenesis, more genes were significantly (p < 0.05) upregulated than down-regulated. Enriched molecular pathways were typical of the pathways described during natural oogenesis in other fish species. The rFsh application induced the upregulation of genes enriched in pathways related to cholesterol metabolism, ovarian steroidogenesis, ovarian growth and differentiation, lipid accumulation, and cell-to-cell adhesion pathways. The application of rFsh and rLh at early-to-mid vitellogenesis induced the upregulation of genes significantly enriched in pathways related to lysosomes activity, while the application of rLh at late vitellogenesis induced the downregulation of genes enriched in the synthesis of estrogens and in cell-to-cell adhesion pathways, and the upregulated gene enrichment in the synthesis of C-21 steroids; processes linked with the switch from vitellogenesis to oocyte maturation.
Acknowledgments: The study was funded by AQUAEXCEL 2020 from the European Union’s Horizon 2020 (grant No. 652831). Ovary samples were provided by the Project RTI2018-094710-R-100 funded by the MICIU (Spain). Computations were performed at the HPC bioinformatics platform of HCMR. S.R. had a PhD grant from AGAUR co-financed by the European Social Fund.
P11—Regulation of Gene Expression Levels by 3′UTR Modification
Raudstein, M.1, Barvik, M.2, Edvardsen, R.B.1 and Ellingsen, S.2
1 Reproduction and developmental biology, Institute of Marine Research, Norway.
2 Department of Biological Sciences, University of Bergen, Norway.
Summary
Infectious diseases attributable to pathogens such as bacteria and viruses are a major challenge to the Norwegian salmon aquaculture industry. Regulation of gene expression levels can contribute to increased disease resistance in farmed animals [1] and may propose a solution to the disease problem. One way to regulate gene expression is through the modification of the untranslated regions (UTRs) of mRNA. Adenylate-uridylate-rich elements (AU-rich elements, AREs) in the 3′ UTRs may have a destabilizing effect on the mRNA, thereby reducing the amount of protein produced [2]. We have isolated AU-rich 3′ UTR sequences from Atlantic salmon genes to investigate their destabilizing potential on mRNA levels using zebrafish as a model system.
We identified and isolated 3′ UTR AREs from salmon immune genes. In total, 60 bp long oligos containing either the wild-type sequence or a mutated sequence with disrupted AREs were cloned into the 3′ UTR of a GFP expression vector. GFP mRNA was produced from the vector by in vitro transcription and injected into one-cell-staged zebrafish embryos.
We observed that the fish injected with mRNA containing AREs in their 3′UTR had lower GFP fluorescence intensity compared to those injected with mRNA containing mutated AREs. Our results indicate that the salmon AREs we identified have a destabilizing effect on the mRNA. However, when these elements were disrupted, more GFP is produced. The next step is to inject salmon embryos with the same GFP mRNAs to confirm if the AREs have a similar effect in salmon as observed in zebrafish.
Future plans include using genome editing in Atlantic salmon to regulate the expression of key genes with antiviral properties. Developing a method to assess the effect of putative regulatory 3′ UTR elements allows us to more readily decide which elements should be focused on and is an important step towards improved disease resistance in farmed Atlantic salmon.
References
Cooper, C.A.; Tizard, M.L.; Stanborough, T.; Moore, S.C.; Chandry, P.S.; Jenkins, K.A.; Wise, T.G.; O’Neil, T.E.; Layton, D.S.; Morris, K.R.; et al. Overexpressing ovotransferrin and avian β-defensin-3 improves antimicrobial capacity of chickens and poultry products. Transgenic Res. 2018, 28, 51–76.
Vejnar, C.E.; Messih, M.A.; Takacs, C.M.; Yartseva, V.; Oikonomou, P.; Christiano, R.; Stoeckius, M.; Lau, S.; Lee, M.T.; Beaudoin, J.-D.; et al. Genome wide analysis of 3′ UTR sequence elements and proteins regulating mRNA stability during maternal-to-zygotic transition in zebrafish. Genome Res. 2019, 29, 1100–1114.
P12—Is Microbiota Composition Correlated to Changes in Permissiveness in Pacific Oyster Mortality Syndrome?
Duperret, L.1, Kunselman, E.2, Boismorand, L.1, Petton, B.3, Morga, B.4, Faury, N.4, Degremont, L.4, Pernet, F.3, Vidal-Dupiol, J.1, Lagorce, A.1, Toulza, E.1 and Mitta, G.1
1 IHPE, Univ. Montpellier, CNRS, Ifremer, Univ. Perpignan Via Domitia, Perpignan, France.
2 Center for Marine Biotechnology and Biomedicine, Scripps Institution of Oceanography, University of California San Diego, La Jolla, CA, USA.
3 Ifremer, LEMAR UMR 6539, UBO/CNRS/IRD/Ifremer, Argenton-en-Landunvez, France.
4 Ifremer, SG2M, LPGMM, La Tremblade, France.
Summary
Since 2008, Crassostrea gigas (C. gigas) have experienced severe annual mortality due to the emergence of a panzootic disease, referred to as the Pacific Oyster Mortality Syndrome (POMS). The outbreak of this disease coincided with the detection of a microvariant of the Ostreid Herpesvirus called OsHV1-µVar. This viral infection is followed by microbiota dysbiosis, inducing bacteremia due to the colonization of the animal by opportunistic and pathogenic bacteria. POMS permissiveness depends on several factors including temperature, oyster nutrition and age. However, the effect of these different factors on the holobiont (virus, bacteria and host) in permissive and non-permissive conditions remains unknown. To address these questions, two “pathogen-free” susceptible oyster families (full-sib) were exposed to the disease under various temperatures and levels of nutrition. The experimental design used, called cohabitation, was thought to mimic a natural infection. As expected, the mortality rates were higher when the parameters were fitting permissive conditions, meaning temperatures between 16 °C and 24 °C and ad libitum diet. The effect of these factors on the bacterial component of the holobiont is the focal point of this analysis. A metabarcoding approach was used to follow the dynamics of the microbial community through time under different permissive and non-permissive conditions. Results are still in progress, but we expect to see shifts in the microbiome composition over time and a divergence in the microbiota composition under different temperatures and diets.
P13—Temperature-Dependent miRNA Repertoire Adaptation of Greater Amberjack (Seriola dumerili) Developmental Stages
Kaitetzidou, E., Papandroulakis, N., Mitrizakis, N. and Sarropoulou, E.
Institute of Marine Biology, Biotechnology and Aquaculture, Hellenic Centre for Marine Research (HCMR), Heraklion, Crete, Greece.
Summary
Temperature is a strong effector of numerous and diverse physiological processes in fish which has been shown to be mainly regulated by epigenetic mechanisms. Among them, microRNAs (miRNA) hold a leading role in the post-transcriptional regulation of gene expression. Particularly, temperature shifts may result in altered miRNA profiles of teleost embryos and larva influencing key developmental processes.
The present study targets the effects of the rearing temperature of the greater amberjack (Seriola dumerili), a species with high-quality meat, high commercial value, and a significant growth rate, which makes the greater amberjack a strong candidate for the development of the aquaculture sector. Therefore, eggs were collected from sea cages in North Crete and transferred to the HCMR Aqualabs facilities where they were kept at 20 °C or 24 °C. Samples from the first feeding to the mid-metamorphosis stage were collected, and miRNA libraries were constructed and sequenced at a NextSeq500 Illumina platform.
Subsequent analysis revealed miRNA profiles associated with staging and also with the different rearing temperatures, whereas further consideration was given to miRNAs implicated in growth processes. Overall, it is suggested that uncovering regulative molecular mechanisms may assist in overcoming the limits of greater amberjack domestication.
P14—Design of a Targeted Low-Density SNP-Chip for Greek Populations of the European Seabass and the Gilthead Seabream
Papadogiannis, V.1, Manousaki, T.1, Oikonomou, S.2, Kazlari, Z.2, Chatziplis, D.2 and Tsigenopoulos, C.S.1
1 Evolutionary Genomics and Bioinformatics Lab (GenomeNerds), Hellenic Centre for Marine Research (HCMR), Institute of Marine Biology Biotechnology and Aquaculture (IMBBC).
2 Lab. of AgroBiotechnology and Inspection of Agricultural Products, Department of Agriculture, School of Geotechnical Sciences, International Hellenic University (IHU).
Summary
The European seabass (Dicentrarchus labrax) and the gilthead seabream (Sparus aurata) are the two most economically important species for Mediterranean aquaculture and Greece is the biggest producer in the EU, accounting for almost 60% of the production for both species. Initial breeding programs for either species have made significant progress in uncovering the genetics of growth-related traits. The development of a low-density SNP-array (SNP-chip), utilizing selected SNP, for both species would offer a highly sought genomic tool to increase the rate of genetic improvement in traits that are already selected with classical selection methods, but also, in many other traits that are considered to be of great importance in the Greek aquaculture. The mapping of quantitative trait loci (QTL) affecting production traits of economic importance in the genome of both species will be highly valuable for breeding programs through the utilization of marker-assisted selection and/or the application of new high-accuracy methods in the genetic evaluations, such as genomic selection. In this work, we carry out selective SNP discovery using large genomic datasets from different Greek populations of both species. Using SNPs that are prevalent in these populations, we design a novel specific SNP-chip that will allow cost-effective and more targeted studies compared to larger generic arrays.
P15—Genome Functional Annotation of Host Defense Response in Generated Immunomaps Using Unstimulated vs. Stimulated Immune Status in Gilthead Seabream (Sparus aurata)
Papadopoulou, A.1,2, Papadogiannis, V.1 Radojicic, J.1, Manousaki, T.1, Katharios, P.1, Sarropoulou E.1 and Tsigenopoulos C.S.1
1 Institute of Marine Biology, Biotechnology and Aquaculture (IMBBC), Hellenic Centre for Marine Research (HCMR), Thalassocosmos, Gournes Pediados, Crete, Greece.
2 School of Medicine, University of Crete, Greece.
Summary
Understanding the genomic basis of immune function and disease resistance in farmed fish species represents a major economical, as well as welfare task in aquaculture breeding. In the current work, we focused on developing genome-wide functional annotation maps (‘ImmunoMaps’) that represent host defense responses of the immune system in the presence of two distinct classes of disease agents, viruses and bacteria, in gilthead seabream (
Sparus aurata). This experiment was conducted as part of the AquaFAANG project (
www.aqua-faang.eu) that aimed to develop comparative framework protocols for generating biological samples to be used in genome functional annotation of immune function research. With high-quality genome assemblies, this project contributes to in-depth comparative examination of the genomic basis for immune function in six targeted farmed species (turbot, gilthead seabream, European seabass, Atlantic salmon, rainbow trout and common carp). Our experimental work included the standardization of experimental protocols for host immunity activation in the head kidney, the major lymphoid organ that plays a critical role in the generation of pro-inflammatory and anti-microbial responses. Two different pathogen-associated molecular patterns (PAMP) were applied in vivo by injecting fish and in vitro in primary head kidney leukocyte cultures, in order to study their robust innate responses. A total of 24 gilthead seabream individuals were stimulated in vivo and in vitro with mimics of bacterial (neutralized
Vibrio) and polyinosinic:polycytidylic acid (poly I:C) viral infections versus 12 control individuals. Library preparations were performed for transposase-accessible chromatin (ATAC-seq) and transcriptome (RNA-seq) and their analyses using pipelines from the nf-core initiative. The expected outcome of this work is the identification of the regions in the genome that are activated by the stimulation of innate immune mechanisms. Preliminary results indicate success in acquiring high-quality data and novel functional annotation of fish immune phenotypes and provide the basis for immuno-biological comparisons within and between targeted species.
Acknowledgments: The AQUA-FAANG project was funded from the European Union’s Horizon 2020 research and innovation program under grant agreement 817923. Computational analyses were performed at the HPC bioinformatics platform of HCMR.
P16—Examining Genetic Diversity and Chromatin Accessibility in Aquaculture-Relevant Tilapia Species
Mehta, T.K.1, Ciezarek, A.1, Ford, A.2, Turner, G.3, Genner, M.J.4, Penman, D.5, Di Palma, F.6,7 and Haerty, W.1,7
1 Organisms and Ecosystems, Earlham Institute, Norwich, UK.
2 Department of Life Sciences, University of Roehampton, London, UK.
3 School of Natural Sciences, Bangor University, Bangor, UK.
4 School of Biological Sciences, University of Bristol, Bristol, UK.
5 University of Stirling, Stirling, UK.
6 Genome British Columbia, Vancouver, Canada.
7 School of Biological Sciences, University of East Anglia, Norwich, UK.
Summary
Tilapia cichlid fish of the genus Oreochromis, native to Africa and the Middle East, are farmed in over 120 countries/territories and account for ~12% of all global inland finfish aquaculture production. The exponential growth in tilapia aquaculture production is largely due to their suitability for aquaculture systems and unlike most other finfish species, tilapia can grow and reproduce in many culture systems. However, climate change is leading to extreme cold events and competition is decreasing freshwater resources. Therefore, there is a need to develop aquaculture systems based on saline waters with a broad temperature range. By determining the genetic bases responsible for such adaptive traits e.g., growth and salinity acclimation, we are able to genotype desirable traits that can be ultimately bred into farmed strains.
We recently reported striking cases of gene regulatory network (GRN) rewiring for adaptive trait genes and confirmed that discrete transcription factor binding site (TFBS) mutations disrupt regulatory edges in the Nile tilapia (Oreochromis niloticus) genome. To further genotype the observed variation at O. niloticus gene regulatory sites, we used genome-wide sequencing data across 575 individuals from 27 tilapia species from across Tanzania and East Africa, to identify 68.8 million SNPs in the O. niloticus genome. A significant proportion of these SNPs are identified in non-coding regions of the O. niloticus genome. To analyse the gene regulatory impact of these non-coding SNPs, we developed and optimised the assay for transposase-accessible chromatin using sequencing (ATAC-seq) from five O. niloticus fish tissues (brain, eye, liver, gill and testis). This allowed us to characterise and compare tissue-specific activity of regulatory SNPs, including the integration of TFBS and GRN predictions that could be associated with aquaculture-relevant traits.
P17—Telomere Length and Telomerase Activity in Dwarf Androgenetic Rainbow Trout (Oncorhynchus mykiss) and Normally Developed Individuals
Panasiak, L. and Ocalewicz, K.
Faculty of Oceanography and Geography, University of Gdańsk, Poland.
Summary
Telomeres are nucleoprotein complexes composed of tandemly repeated TTAGGG sequences that occur at the end of chromosomes. The main function of this structure is to protect ends of chromosomes from losing genetic information during cell division. Due to DNA replication, about 50–200bp (base pair) of telomeres are lost. This erosion is compensated by telomerase which is a large ribonucleoprotein complex composed by a subunit of reverse transcriptase (TERT) and the RNA component (TR), which provides a template for telomeres synthesis. In contrast to mammals where telomerase will not be active in the vast majority of somatic tissues and telomeres will shorten during ontogenetic development, in fish the enzyme is present in many tissues and telomeres do not simply shorten during aging. Experiments on a variety of fish species have revealed that increased telomerase is linked to telomere length maintenance and somatic growth. In the present study, the length of the telomeric DNA and telomerase activity were assessed in one-year-old dwarf androgenetic individuals (dDH), normally developed androgenetic specimens (nDH) and their normally developed siblings from the control group. Dwarf rainbow trout exhibited significantly lower body length and weight when compared to their normally developed siblings. Telomere-length-related fluorescence (TLF) measured by Q-FISH (Quantitative Fluorescent in situ Hybridization) and telomeric PNA probe in nDH, dDH and control rainbow trout, equaled 12.52 ± 2.78, 13.42 ± 3.60 and 15.29 ± 3.51 (mean ± SD), respectively. Statistical analysis did not show statistically significant differences between telomere-length-related fluorescence in rainbow trout from different examined groups. Telomerase activity in the liver of nDH, dDH and individuals from the control group equaled 2.02 ± 0.55, 2.06 ± 0.36 and 2.52 ± 0.31, respectively. In the muscles, the enzyme activity was 0.78 ± 0.39, 1.01 ± 0.76 and 0.43 ± 0.19 in the nDH, dDH and individuals from the control group, respectively. However, statistical analysis did not show significant differences in liver and muscle telomerase activity among all examined fish groups. The activity of telomerase in the skin examined from nDH, dDH and individuals from the control group were as follows: 0.82 ± 0.26, 0.41 ± 0.19 and 0.37 ± 0.08, respectively. Significantly (p < 0.05) higher activity of telomerase was in nDH skin compared to dDH and the control group. Equal length of telomeres observed in the rainbow trout siblings with normal and disturbed growth is thus explained by the comparable activity of telomerase in such fish that compensate for any loss of the telomeric DNA. The present research indicates that retarded growth in the androgenetic rainbow trout is not associated with restricted telomerase activity.
P18—Reprogramming of Epigenetic Marks during Early Development in Vertebrates, with Emphasis on the European Sea Bass
Beato, S.1, Sánchez-Baizán, N.1, Felip, A.2 and Piferrer, F.1
1 Institut de Ciències del Mar (ICM-CSIC), Barcelona, Spain.
2 Instituto de Acuicultura de Torre de la Sal (IATS-CSIC), Castellón, Spain.
Summary
Epigenetics concerns the mechanisms that alter gene expression and that do not imply changes to the DNA sequence. DNA methylation, one of the major epigenetic mechanisms, is commonly associated with gene expression silencing and occurs mostly in CpG sites. The methylation status of CpGs constitutes epigenetic marks that can not only be replicated during mitotic division but can also be inherited from parents to offspring via germ cells. DNA methylation has important roles in vertebrates and underlies phenomena such as genomic imprinting and reprogramming. Reprogramming concerns changes in maternally and/or paternally derived epigenetic marks during early zygote and embryo development and for germ cell specification, contributing to the development of a new individual. In animals, germ cell specification can take place either by reprogramming differentiated epiblast cells through inductive signals received from neighboring tissues (inductive species), or by inheriting the germ plasm, a small area of cytoplasm within the oocyte, containing all the signals necessary for germ cell specification (performative species). Reprogramming is well studied in mammals (inductive sp.) and thus both human and mice are characterized by two waves of DNA demethylation: one taking place during early stages of embryo development and another during germ cell specification. In anuran amphibians such as Xenopus tropicalis, the genome is heavily methylated at both intergenic and intragenic regions throughout gastrulation, but global demethylation does not occur (performative sp). In fish, in which PGC specification commonly occurs through the inheritance of the germplasm, information is available only for a few species. In zebrafish (Danio rerio), maternal methylome gradually changes to resemble that of the sperm, allowing zygotic genome activation. Given that future PGCs inherit the signals necessary for their specification from the germplasm, demethylation is not required (performative sp.). In medaka (Oryzias latipes), strategies similar to mammals seem to be adopted, since in both early embryo and PGCs, methylation reprogramming occurs but neither transcripts nor protein encoded by the vasa homolog olvas, typical of performative species, was found in cleavage stage embryos, questioning whether or not medaka was indeed a performative species. Finally, in Kryptolebias marmoratus, a self-fertilizing hermaphrodite fish, epigenome reprogramming lasts longer than in other vertebrates and global demethylation is achieved at a late embryonic stage. Taken together, these data suggest that in fish reprogramming may be species-specific. Consequently, our objective is to determine what is the situation during European sea bass (Dicentrarchus labrax; Perciformes) embryo development, firstly because it is phylogenetically distant from medaka (Cyprinodontiformes) and zebrafish (Cypriniformes) and, second, to improve our knowledge about epigenetic inheritance. This could help to devise breeding strategies able to maintain certain favorable traits. Methylation will be quantified in sperm, oocyte and embryo, first by ELISA assay and then by whole genome bisulfite sequencing. Different early developmental stages, from zygote to 90% epiboly, will be tested. In addition, 5mC methylation will be compared with h5mC methylation in order to better define the role of active demethylation during epigenome reprogramming.
Acknowledgments: Research funded by the Spanish Ministry of Science and Innovation ‘Epipure’ (PID2019-108888RB-I00) grant to FP.
P20—Transgenerational Inheritance of Circular RNAs in Fish
Diab, F., Nedoluzhko, A., Rbbani, G. and Fernandes, J.M.O.
Faculty of Biosciences and Aquaculture, Nord University, Bodø, Norway
Summary
Circular RNAs (circRNAs) are non-coding RNAs that play a significant role in various biological processes, including embryonic development [1]. These regulatory molecules can directly/indirectly modulate gene expression and possess a plenitude of special functions, including RNA-binding protein regulation, microRNA sponging and protein translation [2]. It is well known that parentally inherited RNA transcripts participate in embryonic development until maternal-to-zygotic transition [3,4]. Despite significant progress in the study of the role of circRNAs in embryogenesis, our knowledge about their inheritance in fish remains limited.
Our study aims to describe circRNA expression profiles during early embryogenesis in zebrafish. First, to identify parentally inherited circRNAs, we sampled zebrafish gametes (unfertilized egg and sperm) as well as embryos at stages up to the 90% epiboly stage. Total RNA was extracted, treated with RNase R and rRNA depletion kit to ensure the complete removal of ribosomal and linear RNAs. CircRNA multiplexed libraries were prepared and sequenced on the NextSeq 500 (Illumina). CircRNA prediction was performed for each ribosomal RNA depleted RNA-seq library using CIRI2 in silico prediction tool [5].
The identification of circRNAs passed on from parents to their offspring shows that these molecules play an essential role in the transmission and expression of modified phenotypes in the next generation. This knowledge may be applied for the selection of desired traits in aquaculture species through environmental and nutritional manipulation.
Acknowledgments: This study has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no 683210) and from the Research Council of Norway under the Toppforsk programme (grant agreement no 250548/F20).
References
Liu, H.; Hu, Y.; Yin, J.; Yan, X.Y.; Chen, W.J.; Jiang, C.Y.; Hu, X.S.; Wang, X.Y.; Zhu, J.G.; Yu, Z.B.; et al. Profiles analysis reveals circular RNAs involving zebrafish physiological development.
J. Cell. Physiol. 2019,
234, 15922–15933.
https://doi.org/10.1002/jcp.28250.
Nedoluzhko, A.; Sharko, F.; Rbbani, M.G.; Teslyuk, A.; Konstantinidis, I.; Fernandes, J.M. CircParser: a novel streamlined pipeline for circular RNA structure and host gene prediction in non-model organisms.
PeerJ 2020,
8, e8757.
https://doi.org/10.7717/peerj.8757.
Bouleau, A.; Desvignes, T.; Traverso, J.M.; Nguyen, T.; Chesnel, F.; Fauvel, C.; Bobe, J. Maternally Inherited npm2 mRNA Is Crucial for Egg Developmental Competence in Zebrafish1.
Biol. Reprod. 2014,
91, 43.
https://doi.org/10.1095/biolreprod.114.119925.
P22—Mitochondrial DNA Methylation Profiling in Nile tilapia (Oreochromis niloticus) through Nanopore Sequencing
Tripathy, P.S.1, Nedoluzhko, A.1,2, Skjærven, K.3, Piferrer, F.4 and Fernandes, J.M.O.1
1 Faculty of Biosciences and Aquaculture, Nord University, Bodø, Norway.
2 European University at St. Petersburg, St. Petersburg, Russia.
3 Institute of Marine Research, Bergen, Norway.
4 Institute of Marine Sciences, CSIC, Barcelona, Spain.
Summary
The mitochondrion is a cellular organelle that plays a central role in the regulation of respiration and metabolic processes in the cell. Thus, it is implicated in the growth, metabolism and aging of an organism. The first study on mitochondrial DNA (mtDNA) epigenetics was published at the beginning of the 1970s [1]. The identification of DNA methyltransferase 1 in mammalian mitochondria attracted researchers towards the study of the mitoepigenome [2]. However, data about the methylation patterns of mtDNA in adult fish tissues are still absent, apart from a single report [3]. Mitochondria play a critical role in the production of reactive oxygen species, which in turn is essential for muscle function and growth. Growth is one of the most important traits in the aquaculture sector [4]. In the present study, mtDNA methylation profiles have been analysed in Nile tilapia (Oreochromis niloticus), fast muscle using Nanopore long-read sequencing. O. niloticus fast tissue was sampled and used for genomic DNA isolation and construction of PCR-free libraries. The Nanopore MinION Mk1C sequencing workflow generated 673 Fast5 files, which were used for basecalling and methylation analysis with Megalodon v2.4.2 and Remora v0.1.2. The mean sequencing read length was 3275 bp and the coverage obtained for Nile tilapia mtDNA was 1082X. This methylation data analysis based on Nanopore long-read sequencing constitutes a valuable resource for future research on the mitoepigenome of farmed fishes.
Acknowledgments: This work was supported by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no 683210) and by the Research Council of Norway under the Toppforsk programme (grant agreement no 250548/F20), with additional support from Notur/NorStore (project NN9337K).
References
Nass, M.M. Differential methylation of mitochondrial and nuclear DNA in cultured mouse, hamster and virus-transformed hamster cells In vivo and in vitro methylation.
J. Mol. Biol. 1973,
80, 155–175.
https://doi.org/10.1016/0022-2836(73)90239-8.
Shock, L.S.; Thakkar, P.V.; Peterson, E.J.; Moran, R.G.; Taylor, S.M. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 3630–3635.
Nedoluzhko, A.; Mjelle, R.; Renström, M.; Skjærven, K.H.; Piferrer, F.; Fernandes, J.M.O. The first mitochondrial 5-methylcytosine map in a non-model teleost (Oreochromis niloticus) reveals extensive strand-specific and non-CpG methylation. Genomics 2021, 113, 3050–3057.
Gjedrem, T. Selection and Breeding Programs in Aquaculture; Springer: Berlin/Heidelberg, Germany, 2005.
P23—Circular RNAs as Potential Growth Biomarkers in Nile Tilapia
Rabbani, G.1, Nedoluzhko, A.1, Siriyappagouder, P.1, Sharko, F.2, Galindo-Villegas, J.1, Joshi, R.3, Raeymaekers, J.A.M.1 and Fernandes, J.M.O.1
1 Faculty of Biosciences and Aquaculture, Nord University, 8049 Bodø, Norway.
2 LLC ELGENE, 109029 Moscow, Russia.
3 GenoMar Genetics, 0252 Oslo, Norway.
Summary
Circular RNAs (circRNAs) are non-polyadenylated, non-coding RNAs produced by alternative back-splicing. They are considered as potential biomarkers in different biological processes and efficient in participating in microRNA (miRNA) sponging and epigenetic regulation of gene expression. There are some reports in farm animals (sheep, goat, chicken) that describe the functional roles of circRNAs in muscle growth [1]. However, our knowledge of their expression in muscle and functional mechanisms related to growth is still limited in teleosts. Fast and enhanced fish muscle growth is one of the main traits of interest for the global aquaculture industry, which is currently targeted mainly through diet improvements and selective breeding. In this study, we used fast- and slow-growing Nile tilapia from the third generation of our in-house domestication program, which have been reared in a freshwater recirculating aquaculture system. Nile tilapia having an average total length of 29.28 ± 2.15 cm and 13.8 ± 1.98 cm were considered fast- and slow-growing fish, respectively. We profiled mRNA, miRNA and circRNA expression in muscle tissue to provide insights into their potential function in muscle growth.
The results showed that 1947 mRNAs, 9 microRNAs and 8 circRNAs were differentially expressed between fast- and slow-growing Nile tilapia individuals. Gene ontology and KEGG analysis revealed that the differentially expressed mRNAs are involved in the regulation of myoblast differentiation, striated muscle cell development, mRNA splicing via spliceosome and spliceosomal snRNP complex; the last two processes are import components in circRNA production during pre-mRNA splicing. Bioinformatic target prediction revealed oni-miR-34, oni-miR-130b and oni-miR-202 have complimentary binding sites for cirMef2c and muscle growth-associated mRNAs. A ceRNA network was constructed through integrative analysis of the interplay between cirMef2c, 3 miRNA and 65 mRNAs. Potential interactions were found in circMef2C-oni-miR-34-Igfbp2, circMef2C-oni-miR-130b-5p-myod1 and circMef2C-oni-miR-202-fgf14, which likely play an important role in muscle gain throughout ontogeny. Our data suggest that these circRNAs may prove to be novel growth biomarkers with potential application in the aquaculture sector.
Acknowledgments: This work was supported by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no 683210, 2016) and by the Research Council of Norway under the Toppforsk programme (grant agreement no 250548/F20, 2016).
References
Rbbani, G.; Nedoluzhko, A.; Galindo-Villegas, J.; Fernandes, J. Function of Circular RNAs in Fish and Their Potential Application as Biomarkers.
Int. J. Mol. Sci. 2021,
22, 7119.
https://doi.org/10.3390/ijms22137119.
P25—Novel In Vitro Models Using Primary Cell Cultures and Genome Editing to Study WSSV Resistance in Pacific Whiteleg Shrimp
Florea, A.1, Robledo, D.1, Matthews, J.2, Tait-Burkard, C.1, Houston, R.D.1 and Bean, T.P.1
1 The Roslin Institute, Easter Bush Campus, Midlothian University of Edinburgh, UK.
2 Roslin Technologies, Roslin Innovation Centre, Easter Bush Campus, Edinburgh, UK.
Summary
Shrimp are one of the most important groups of species in global aquaculture. The Pacific whiteleg shrimp (Litopenaeus vannamei) is the most cultured species, accounting for over 50% of the shrimp worldwide production [1]. Unfortunately, the industry’s sustainability is threatened by significant annual losses due to infectious diseases, 60% caused by viral pathogens such as the white spot syndrome virus (WSSV) [2]. The efforts to combat the disease are impeded by the lack of effective treatments available and the absence of an adaptive immune system in crustaceans, meaning there is little potential to develop an efficient vaccine against the disease [3]. Instead, this project aims to employ precision genetics tools such as CRISPR/Cas9 technology in order to help minimize the economic losses among crustaceans caused by WSSV outbreaks.
We aim to improve host resistance to WSSV in whiteleg shrimp by knocking out priority candidate genes in primary cell cultures. We have established successful in vitro models using lymphoid and hemocyte cells from adult shrimp. The next step is to perform high-throughput transcriptomic analysis pre- and post-WSSV infection using single-nuclei RNA-seq. This will allow us to systematically choose the candidate genes for our CRISPR/Cas9 knockouts. If successful, the results could be used as a foundation for breeding new stocks of WSSV-resistant shrimp and set a new base for developing therapeutic strategies in shrimp aquaculture.
References
FAO.
GLOBEFISH Highlights April 2020 Issue, with Annual 2019 Statistics—A Quarterly Update on World Seafood Markets, No. 2–2020; FAO: Rome, Italy, 2020.
https://doi.org/10.4060/ca9528en.
Chandrika, S.K.; Puthiyedathu, S.T. Challenges and prospects of Viral Envelope protein VP28-based control strategies to combat white spot syndrome virus in penaeid shrimps: A review.
Rev. Aquac. 2020, 13, 734–743.
https://doi.org/10.1111/raq.12497.
P26—Repetitive DNA Sequences of Senegalese sole (Solea senegalensis) and Its Implications in Chromosome Structure
Navajas-Pérez, R.1, Robles, F.1, De la Herrán, R.1, Cross, I.2, Hermida, M.3, Rodríguez, M.E., Robledo, D.4, Martínez, P.3 Rebordinos, L.2 and Ruíz-Rejón, C.1
1 Departamento de Genética, Universidad de Granada, Granada, Spain.
2 Departamento de Biomedicina, Biotecnología y Salud Pública, Universidad de Cádiz, Cádiz, Spain.
3 Departamento de Zoología, Genética y Antropología Física, Universidade de Santiago de Compostela. Lugo, Spain.
4 The Roslin Institute and Royal (Dick) School of Veterinary Studies, University of Edinburgh, Midlothian, UK.
Summary
The Senegalese sole (Solea senegalensis) is a promising aquaculture species. In order to identify repetitive DNA sequences and ascertain its role in the chromosome structure, the assembled chromosomes and scaffolds of S. senegalensis were analyzed with WindowMasker (Morgulis et al. 2006). Repetitive DNA makes up to 8% of its genome and was grouped into three main categories: simple repeats (2.81%), low-complexity motifs (0.32%), and transposable elements (TEs) (4.73%). The TE-derived fraction was very similar to that found in the other known flatfish genome (5.8% and 5% in C. semilaevis and S. maximus, respectively). The Senegalese sole genome displayed a higher TE proportion than T. nigroviridis and Fugu rubripes (<3%), but much lower than that observed in other fish such as Danio rerio (>40%).
For a more accurate detection of tandemly arrayed sequences, 1000K raw fastq Illumina paired sequences (500K from each sex) were analyzed using Repeat Explorer. No significant differences between repetitive families from male and female genomes were detected. Main repetitive DNA families included ribosomal DNA (5S and 45S, accounting for 0.38% and 0.25% of the genome, respectively), centromeric DNA (satellite DNA PvuII, 0.83%), and other putative satellite DNAs (1.59%).
The location of the main repetitive clusters was determined by blast (e-value < 1e−10) against the assembled chromosomes. Clusters were considered tandemly arrayed when consensus sequences included at least five consecutive hits of the assembly. Chromosome coordinates of the loci were obtained using Geneious (
https://www.geneious.com) and idiograms were constructed by RIdeogram (Hao et al., 2020). Centromeres were positioned in all chromosomes of Senegalese sole by mapping the PvuII satellite DNA and other pericentromeric clusters. Telomeres were positioned by mapping the vertebrate telomeric motif. Several clusters were found to be accumulated in multiple loci in all chromosomes, while others showed a unique location (as miRNA 430).
Acknowledgments: This study was supported by the Spanish Ministry of Economy and Competitiveness, Grants: RTI2018-097110-B-C21 and RTI2018-096847-B-C22.
P27—Deepening the Knowledge of the Sexual Determination and Differentiation Pathway in Sturgeons
Robles, F., Navajas-Pérez, R., De la Herrán, R., Bakkali, M., García-Zea, A. and Ruiz-Rejón, J.C.
Departamento de Genética. Universidad de Granada. Granada. Spain.
Summary
Sturgeons are a group of fish with high economic value mainly due to the production of caviar. Additionally, they are considered living fossils and are all the members of the group on the red list of endangered species. Increasing our knowledge of their mechanism of sex differentiation and determination would be an important achievement from both the basic and the applied point of view.
In this work, we studied 55 candidate genes to fish sex development (Yue et al., 2015). We used an Illumina database of cDNA libraries from gonads of 2-year-old males (2) and females (2) of Acipenser naccarii (Adriatic sturgeon). The presence of these genes was comparatively studied using sturgeon gonad transcriptomic data from 454-sequencing of a 6-month-old A. naccarii (Vidoto et al., 2013), Illumina sequences from a 3-year-old A. sinensis (Yue et al., 2015), and selective qPCR data of a 3-year-old A. baeri (Berbejillo et al., 2012).
From this set of genes, we have found 42 of them, 2-fold that found by Vidoto et al. 2013 in A. naccarii: dmrt1 and gsdf, both major sex-determining genes of fish; sox3 and sox4 from sox family; one transcription factor, bmp15; six receptors, fshr, gnrhr, igf-1r, pdgfr-b, era, and erb; one hormone gene, fsh; one gene belonging to the double sex and mab-3 (DM) domain, dmrt3; the signaling molecules ctnnb1, igf-1, dhh, pdgf, and wnt4; the recombinase dmc1; and the esteridogenic enzymes fst, srd5A1, srd5A2, and srd5A3. Only one gene, rspo, was not detected in this work despite the fact that it was found previously in transcriptome gonads of 6-month-old A. naccarii. Additionally, this gene was not found in gonad transcriptome from a 3-year-old A. sinensis (Yue et al., 2015).
When A. naccarii and A. sinensis Illumina transcriptomes from gonad are compared, 27 genes out of 55 are present in both species, 13 were only detected in A. naccarii, and seven were exclusive to A. sinensis. Lastly, eight genes (AMHR2, cyp11b, dmrt6, pdgf, RSPO-1, sdY, SRD5A2, and SRD5A3) are not detected in any of these species.
Finally, of the 42 genes detected in A. naccarii in this study, three (aromatase, foxl2, and gnrhr) were only present in females and four (dmrt3, igf1, lhx1, and sox11) were only present in males. These data shed light on our knowledge about the chronology of the expression of genes involved in gonad development of this group of species.
Acknowledgments: This study has been funded by the research project P07-CVI-03296 “Implicaciones y aplicaciones de la genética molecular en los esturiones (Familia Acipenseridae)” of the Consejería de Innovación, Ciencia y Empresa de la Junta de Andalucía.
P28—Cytogenomic Characterization and Chromosome Evolution in Solea senegalensis
Ramírez, D.1, Rodríguez, M.E.1, Cross, I.1, Arias-Pérez, A.1, Merlo M.A.1, Anaya, M.1, Mendizábal, M.1, Sánchez, C.1, Portela-Bens, S.1, Martínez, P.2, de la Herrán R.3, Ruiz-Rejón C.3 and Rebordinos, L.1
1 Departamento de Biomedicina, Biotecnología y Salud Pública, Universidad de Cádiz, Cádiz, Spain.
2 Departamento de Zoología, Genética y Antropología Física, Universidade de Santiago de Compostela. Lugo, Spain.
3 Departamento de Genética, Universidad de Granada, Granada, Spain.
Summary
The Pleuronectiformes order, which includes several commercially important species, has undergone extensive chromosome evolution. One of these species is Solea senegalensis, a flatfish with 2n = 42 chromosomes and a XX/XY sex determination system. In this study, a cytogenomics approach and integration with previous maps was applied to characterize the karyotype of the species.
Using FISH, a total of 126 BAC clones were located in the chromosomes and sequenced by NGS. Of these, 38 BACs contained microsatellite markers. To select microsatellite markers, we initially selected 74 out of the 129 belonging to 27 linkage groups (LGs) described by Molina-Luzón et al. (2015). Each microsatellite was associated with one of the 27 LGs, except LG5, LG17, LG23, and LG26. With this information, we were able to integrate the cytogenetic map of S. senegalensis with genetic and physical maps previously obtained by Molina-Luzón et al. (2015) and Guerrero-Cózar et al. (2021), respectively.
Synteny analysis of S. senegalensis was carried out using two flatfish as reference: Cynoglossus semilaevis and Scophthalmus maximus. Most S. senegalensis chromosomes (or chromosome arms for metacentrics and submetacentrics) showed a one-to-one macrosyntenic pattern with the other two species: i) BACs of the metacentric chromosomes (C) 1, 2, and 4 each had two orthologue chromosomes in C. semilaevis and S. maximus, except C3 and C5, with only one ortholog in the other two reference species; ii) the subtelocentric pairs C6–9 and the twelve acrocentric pairs (C10–21) all showed a one-to-one orthologue chromosome in the other species.
In addition, using Repeat Masker program, we studied how repetitive sequences could have played a role in the evolution of S. senegalensis biarmed (C3, C5–9) and acrocentric (C11, C12, and C16) chromosomes, which showed the highest rearrangements regarding the reference species. A greater abundance of TEs and other repeat elements (coverage and NL/Mb) adjacent to telomeric regions were observed in C3, C7, C9, and C16. This pattern was also previously reported in C1, C2, and C4 of S. senegalensis (Rodríguez et al., 2019, 2021). However, in C11, a greater abundance of DNA transposons were detected in interstitial BACs. This chromosome is syntenic to several chromosomes of the other two flatfish species, which suggests rearrangements during its evolution. A similar situation was also found in C16 (for microsatellites and low complexity sequences), but not for TEs (retroelements and DNA transposons). These differences in the distribution and abundance of repetitive elements in chromosomes which have undergone remodeling processes during the course of evolution also suggest a possible role for simple repeat sequences in rearranged regions.
Acknowledgments: This study was supported by the Spanish Ministry of Economy and Competitiveness, (Grants: RTI2018-097110-B-C21 and RTI2018-096847-B-C22) and Junta de Andalucía-FEDER (P20-00938).
P29—Hypergravity Induces Alterations in Gene Expression and Epigenetics in Zebrafish
Ribas, L.1, Anglada-Escudé, G.2, Joly, S.1 and Salazar, M.1,2,
1 Department of Renewable Marine Resources, Institute of Marine Sciences, (ICM-CSIC), Barcelona, Spain.
2 Institute of Space Sciences, (IEEC-CSIC), Barcelona, Spain.
Summary
All forms of life on our planet have been formed under a common factor, gravity; for which it is easy to deduce that any change in this constant could trigger important alterations in these forms of life in the near future settlements beyond Earth. Although some studies use altered gravity to evaluate different animal and cellular models and systems, including fish models, almost no data regarding molecular mechanisms in fish are found. This work aims to understand the effects of hypergravity on gene expression and epigenetics in zebrafish during the early stages of development. We designed a centrifuge by adding two arms with gondolas to place zebrafish larvae at a force of three gravities for 6 days post-fertilization (dpf). The survival and hatching rate were measured daily, and while no significant changes were found in the hatching rate, the survival of larvae were decreased up to 20%. The gene expression and global DNA methylation were evaluated in larvae at the end of the treatment, and we found over-expression on some evaluated genes, and a significant global DNA hypermethylation in the larvae exposed to hypergravity. Taken together, these findings show how alterations in Earth’s gravity could affect some basic biological responses in fish, and thus are the first steps in exploring molecular events in cultured fish in space-related environments.
P30—MicroRNA Expression Profile in Brackish Water and Freshwater Populations of Two Stickleback Fishes
Shankregowda, A.M.1, Nedoluzhko, A.1,2, Brand, A.F.3, Rbbani, G.1, Siriyappagouder, P.1, Bal, T.P1, Fernandes, J.M.O.1 and Raeymaekers, J.A.M.1
1 Faculty of Biosciences and Aquaculture, Nord University, N-8049 Bodø, Norway.
2 Paleogenomics Laboratory, European University at Saint Petersburg, 191187 St.- Petersburg, Russia.
3 Department of Biology, Norwegian University of Science and Technology, Høgskoleringen 5, 7491, Trondheim, Norway.
Summary
The capacity of species to respond to environmental changes largely depends on their evolutionary potential, which is rooted in the amount and structure of adaptive genetic variation. However, species may compensate for low evolutionary potential through phenotypic or behavioural plasticity. Adaptive plastic responses to natural selection may be modulated by the regulation of gene expression, for instance via epigenetic modifications based on non-coding RNA regulatory molecules (including, microRNAs) and DNA modifications. In this work, we used the stickleback model to identify microRNA (miRNA) regulatory molecules and investigate their role in freshwater adaptation. To do so, we compared brackish water and freshwater populations of two coexisting and closely related fishes, the three-spined (Gasterosteus aculeatus) and nine-spined stickleback (Pungitius pungitius). These two species have been shown to differ markedly in the strength and nature of local adaptation, where the three-spined stickleback shows stronger phenotypic and genomic differentiation among brackish water and freshwater populations than the nine-spined stickleback. Across the same study sites (Belgian–Dutch lowlands), the two species thus seem to cope with landscape-level ecological heterogeneity in fundamentally different ways. First, we will investigate the importance of gene regulation by miRNAs for the set of genes that are putatively targeted by natural selection. This set of genes will be obtained by screening highly differentiated genomic regions between brackish water and freshwater populations. Subsequently, each of the putative genes will be checked for the presence of miRNA targets. Finally, we will quantify and compare the level of differential miRNAs and gene expression between brackish water and freshwater populations among the two species. We expect that miRNA regulatory mechanisms are more important in the nine-spined stickleback, as such mechanisms may compensate for the apparent lack of genomic differentiation across populations from contrasting salinities. Since adaptation to different environmental conditions is important for several economically relevant fishes, our study of the variation in miRNA expression profiles across populations and species is also relevant for aquaculture.
P31—Development of Genomic Markers Associated to Production Traits in Lumpfish (Cyclopterus lumpus)
Gutierrez, A.P.1, Davie, A.1, Migaud, H.1, Palma, P.1, Cavrois-Rogacki, T.2 and Bekaert, M.1
1 Institute of Aquaculture, University of Stirling, Stirling, Scotland.
2 Otter Ferry Seafish Ltd., Tighnabruaich, Argyll, Scotland.
Summary
Biological control of sea lice infection in Atlantic salmon cages has become an important alternative to tackle one of the most important diseases affecting salmon aquaculture. Cleaner fish species have gained great importance in the control of sea lice, among them, the use of lumpfish (Cyclopterus lumpus). Current lumpfish production is reliant on wild caught broodstock to meet the increasing demand. Lumpfish life cycle has been closed and hatchery reproduction is now possible. Genomic resources are called to play a fundamental role in the improvement of selective breeding practices of aquaculture species. The development of these resources has been scarce for emerging species such as lumpfish; therefore, there is an imperative need to obtain genomic information that will support the establishment of effective and sustainable selective breeding programs. Ten lumpfish families from wild origin were produced in the spring of 2019 at the Otter Ferry SeaFish (OFS) facilities. Four of these families were maintained in separate tanks until they reached a mean weight of 3–9g. Fin clips were obtained from all parents and from the four families including the 50 bigger, 50 smaller and 25 random fish from each family. Two ddRAD libraries were prepared, containing all 536 samples and were sequenced on Illumina Novaseq 6000 platform. Genome-wide association analysis was performed using the package R/SNPassoc v1.9-2 and R/qtl2 v0.20, analyzing four traits, including gender, weight, condition factor and standard length. After SNP calling and quality control, 10,630 informative SNPs were identified. Association analyses were able to identify many genomic regions linked to the analyzed traits. The analysis of gender showed the highest association, identifying a single major QTL located in chromosome 13. Markers located in this region were further analyzed and a set of 16 markers was capable of accurately predicting the sex in all samples. Analysis of growth traits showed a polymorphic behavior as expected, identifying significant association in many chromosomes, showing evidence of overlap in the QTL regions identified for weight and length in chromosomes 7, 12, 13, 15, 17 and 21. Of particular importance was the association between growth and gender shown in a region of chromosome 13 which could have implications in selective breeding. Moving forward, markers located in these identified QTL regions are candidates for the development of low-density SNP panels that will provide a low-cost alternative for producers to use genomic information in the development of selective breeding programs and improve production traits.
P32—Fish Mucosa “Smells” Rotten Eggs: Mucosal Immunotoxicity of Hydrogen Sulphide in Atlantic Salmon
Alipio, H.R.1,2, Bergstedt, J.H.3, Albaladejo-Riad, N.4, Carletto, D.5 and Lazado, C.C.1
1 Nofima, Norwegian Institute of Food, Fisheries and Aquaculture Research 1433 Ås, Norway.
2 Aquaculture and Fisheries Group, Wageningen University and Research, 6708 PB Wageningen, Netherlands.
3 Technical University of Denmark, DTU Aqua, Section for Aquaculture, The North Sea Research Centre, PO Box 101, 9850 Hirtshals, Denmark.
4 Immunobiology for Aquaculture Group, Department of Cell Biology and Histology, Faculty of Biology, Campus of International Excellence, Campus Mare Nostrum, University of Murcia, Murcia, Spain.
5 Department of Chemical, Biological, Pharmaceutical and Environmental Sciences, University of Messina, 98166 S Agata-Messina, Italy.
Summary
Hydrogen sulphide (H2S) is a naturally occurring compound which has received more attention the recent years due to its central role as a gasotransmitter within many physiological systems, including teleosts. Endogenously generated H2S controls several important metabolic functions, while when encountered exogenously the effect of the gas can be detrimental. Exposure to the gas exogenously can occur because of the production of H2S by anaerobic bacterial decomposition of protein and other sulphur-containing organic matter is a water-soluble and colourless gas with the distinct odour of rotten eggs. In recent years, H2S-related mortality has become a major challenge in saline recirculating aquaculture systems (RAS) of Atlantic salmon. Significant advances have been made in elucidating its formation in these systems; however, the biological processes underlying the adaptive responses of salmon to this molecule remains elusive. Mucosal surfaces are considered the first line of defence and biological sensors of the changing environment. Because of these, they can potentially shed light on how salmon respond to H2S. This study investigates how mucosal organs of Atlantic salmon, such as the skin, gills, gut and olfactory organ, respond to H2S. Using in vitro and in vivo H2S exposure models, we found that molecules involved in sulphide detoxification, stress and immunity are modulated in a concentration- and tissue-specific manner. The olfactory organ, particularly the nasal leukocytes, responded to H2S and transcriptional profiling revealed that the VEGF pathway was a potential target. Pharmacological blocking of mucous production resulted in differential regulation of mucosal responses to H2S. Metabolomics further elucidated that low, sub-lethal concentrations of H2S dysregulated the circulating and mucosal metabolomes. A number of the H2S-responsive metabolites have not been implicated earlier in H2S detoxification, indicating that they may have novel functions in salmon. These results offer insights into how fish interact with H2S and are expected to contribute to resolving the issues of the biological functions of H2S in fish.
P33—Metabolomics Elucidates the Adaptive Responses of Atlantic Salmon to Stress and Infection
Lazado, C.C.1, Alipio, H.R.1,2, Timmerhaus, G.1, Breiland, M.W.3, Johansen, L.H.3, Burgerhout, E.3, Pedersen, L.F.4 and Strand, D.5
1 Nofima, Norwegian Institute of Food Fisheries and Aquaculture Research, 1433 Ås, Norway.
2 Aquaculture and Fisheries Group, Wageningen University and Research, 6708 PB Wageningen, Netherlands.
3 Nofima, Norwegian Institute of Food, Fisheries and Aquaculture Research, 9019 Tromsø, Norway.
4 Technical University of Denmark, DTU Aqua, Section for Aquaculture, The North Sea Research Centre, PO Box 101, 9850 Hirtshals, Denmark.
5 Norwegian Veterinary Institute, 1433 Ås, Norway.
Summary
Metabolomics can provide high-throughput insights into the dynamic small-molecule fluctuations occurring in response to infection and stress. However, its applications in fish as a tool in studying the physiological adaptive responses to different stressors and infectious agents have only gained prominence in recent years. In a collection of studies, we applied untargeted and targeted metabolomics by ultra-performance liquid chromatography coupled with a high-resolution quadrupole-orbitrap mass spectrometer to profile the metabolomic responses of Atlantic salmon (Salmo salar) to different relevant stressors during production, including 1) amoebic gill disease (AGD), a parasitic infection; 2) chemotherapeutic treatments; and 3) hydrogen sulphide (H2S), a toxic gas in a recirculating aquaculture system. Using different biological matrices, such as plasma, blood, and skin mucus, we have identified at least 500 metabolites responding to the different stressors. In AGD-affected fish, the severity of infection which is commonly assessed by the gross pathology of the gills was reflected in plasma metabolomic profiles. Metabolomic dysregulation was substantial in fish with a low gill score. Plasma metabolome analysis of salmon exposed to oxidative therapeutics identified several metabolites important for the regulation of oxidative stress response. The response of several metabolites (e.g., guanosine, inosine, tyrosine) to oxidative therapeutics has been validated in several trials, suggesting their potential role as biomarkers for oxidative stress concerning chemotherapeutic treatment. The physiological impacts of H2S, a toxic gas produced in RAS, have not been resolved in salmon. Using metabolomics, we have provided the first evidence that even at low, sub-lethal concentrations, H2S could alter the circulating and mucosal metabolomes of salmon. Many of the H2S-responsive metabolites have not been implicated in H2S detoxification in other organisms hence, suggesting their novel roles in fish. Metabolomics provided a high-throughput atlas of metabolites that were orchestrated by Atlantic salmon in response to stress and infection. The metabolites that were responsive to the different production stressors could be explored as potential biomarkers for the health status of fish.
P34—Transcriptomic Changes in Pituitary and Testis of Maturing Atlantic Salmon (Salmo salar) Post-Smolts
Dessen, J-E., Skjold, V., Sveen, L., Bou, M., Johansson, G.S., Timmerhaus, G., Mota, V.C., Rørvik, K-A., Krasnov, A. and Burgerhout, E.
Nofima AS, PO Box 6122, 9292 Tromsø, Norway.
Summary
Male precocious maturation in the Atlantic salmon post-smolt production is an increasing problem as it influences fish health, welfare and flesh quality, and therefore has a large economic impact. As sexual maturation is influenced by photoperiod, it was hypothesised that by using different photoperiodic regimes the number of early mature males could be reduced.
In an overall study, three regimes were applied over 18 weeks, a continuous photoperiod of 24:0 (L:D) light (constant light - CL), an increasing photoperiod from 12:12 to 24:0 (increasing light - IL) and a decreasing photoperiod from 24:0 to 12:12 (decreasing light—DL). Fish were analysed for phenotypic changes (e.g., colouration and spotting), gonad development using histology and blood plasma sex steroids by LC-MS/MS. In this study, microarray analysis was used to further investigate the molecular pathways determining the onset of maturation by analysing the transcriptome profile of the pituitary and testis of males selected with a high or low gonadosomatic index (GSI).
Male salmon in the constant and increasing light groups showed clear signs of early maturation, indicated externally by increased amounts of spotting and a darker colouration. In addition, GSI and blood plasma steroid levels, such as androstenedione, testosterone and 11-ketotestosterone, were enhanced for the mentioned groups, coinciding with the early signs for maturation. The decreasing light group, on the other hand, showed much lower levels of early maturation based on these indicators, without affecting overall body growth or energy stores.
The pituitary and testis transcriptomes showed that 30 and 3941 differentially expressed genes (DEG), respectively, were specifically associated with maturity status. Gene ontology analysis showed that upregulated DEGs in the pituitary were mainly related to endocrine and neural processes. For testis of maturing fish, a large number of upregulated DEGs were involved in cell replication, tissue gametogenesis and metabolic pathways, indicative of the increase in testis size and the production of gametes. Downregulated DEGs were mainly involved in immune-related pathways. In agreement with previous studies, we also found large downregulation of anti-Mullerian hormone and dead-end protein, indicating that these genes play an important role in the onset of early maturation in Atlantic salmon post-smolts.
P36—A ddRAD Based Linkage Map of the Pacaman Lophiosilurus alexandri, a Promising Aquaculture Catfish Species
Coimbra, M.R.M.1, Farias, R.S.1, Silva, B.C.N.R.1, Bekaert, M.2 and Taggart, J.2
1 Federal Rural University of Pernambuco, Dept. of Fisheries and Aquaculture, Recife, Brazil.
2 University of Stirling, Institute of Aquaculture, Stirling, Scotland-United Kingdom.
Summary
The order Siluriformes is a group of ray-finned catfishes of relevance in the aquaculture industry. Around 50% of the catfish families are native to the Central and South America; however, Asia produces almost 90% of the world aquaculture production, while South America accounts for less than 0.5% of the total production. A Brazilian native catfish candidate species for aquaculture diversification is the Lophiosilurus alexandri (Pseudopimelodidae), which is also a threatened species subjected to supplementation aquaculture actions. They are known for successful artificial spawning, low dissolved oxygen requirements, high resistance to infectious disease and high feed conversion efficiency. Given the importance in terms of conservation and aquaculture, the use of genetic linkage maps might be extremely helpful toward time and cost-efficient breeding programs as well as for genetic traceability. Three full-sib families and 264 offspring of L. alexandri were used to construct ddRAD library that produced 234,182,556 high-quality paired-end reads. The reads were assembled de novo and genotypes for all samples were obtained using Stacks. The key parameter values employed in identifying marker loci were a maximum of three mismatches allowed in a locus (M) in an individual and up to two mismatches between loci when building the catalogue (n). Informative RAD markers were kept only when presenting at least two alleles with a minor allele frequency > 0.01 and were present in the three families and in at least 25% of the samples yielding 2,510,030 unique loci. Out of them, 5271 loci contained the candidate regions that fulfilled our SNP criteria. All of these markers were subsequently used to construct genetic linkage maps with Lep-Map3. SNPs deviating from the expected Mendelian segregation (p < 0.001) were excluded. Based on available karyotyping data, the number of linkage groups was set to 27 (LOD = 10). Within each linkage group, the sequence of SNPs was obtained by using the OrderMarkers module. The total length of the map in centimorgans was estimated using Kosambi mapping functions and three maps were generated, one including only the males, one female only and one sex averaged. A total of 2351 informative SNPs were mapped, being 1467 in family A, 1037 in family B and 765 in family C, respectively, to the 27 linkage groups (each comprising at least 10 SNPs). The proportion of polymorphic RADtags observed across all parental samples was 3–6 times lower than that observed for most other fish species studied by the same methodology. The maps spanned a total distance of 2201.3 cM, 2481.9 cM, 2824.9 cM for the averaged, female and male map, respectively. These resources constitute an important step toward the identification of economically relevant characteristics in the aquaculture of this potential species.
P37—Occurrence of the Denitrification Genes nirS and nosZ in the Microbiome of the Aquaculture Extractive Species Holothuria tubulosa
Martínez- Moreno, S.1, León-Palmero, E.1, Cabello, A.M.2, Pula, H.J.3, Ferrera, I.2 and Reche, I.1
1 Dpto. Ecología-Universidad de Granada, España.
2 Instituto Español de Oceanografía, España.
3 Aula del Mar-Universidad de Granada, España.
Summary
Sea cucumbers (holothurians) are benthic invertebrates inhabiting shallow and deep waters in the oceans and key species processing sediment organic matter. Previous studies have shown that holothurians are able to remove nitrates from seawater in aquaculture tanks, consequently being an excellent extractive species for integrated multitrophic aquaculture. However, the ultimate players of this role of holothurians might be related to their bacterial symbionts. Here, we explored the microbiomes associated with the subcuticle and the mucus of Holothuria tubulosa. We performed cuticle biopsies in various specimens and took mucus using cotton swabs. In these samples, we extracted DNA, amplified the V4 hypervariable region of 16S RNA gene, and tested for the presence of the genes that code the nitrite reductase (i.e., nirS) and the nitrous oxide reductase (i.e., nosZ) enzymes in the denitrification. We describe the taxonomic composition of holothurian bacterial symbionts and note the presence of Alphaproteobacteria (Rhodobacterales) and Gammaproteobacteria (Pseudomonadales) in abundance. Furthermore, we confirmed the presence of the abovementioned functional genes in the mucus samples. These results suggest that the microbiome associated with sea cucumbers might have a relevant role in nitrogen removal in integrated multitrophic aquaculture tanks. This knowledge can also be useful for understanding the potential ecosystem services of sea cucumbers in eutrophicated coastal areas.
Acknowledgments: This research was founded by Junta de Andalucía—Consejería de conocimiento, investigación y universidad. Secretaría General de Universidades, Investigación y Tecnología. Ref: P20_00705.
P39—Intergenerational Transfer of Persistent Communities in Female Nile tilapia
Abdelhafiz, Y.1, Fernandes, J.M.O.1, Donati, C.2, Pindo M.2 and Kiron, V.1
1 Faculty of Biosciences and Aquaculture, Nord University, Bodø, Norway.
2 Unit of Computational Biology, Research and Innovation Centre, Fondazione Edmund Mach, San Michele all’Adige, Italy.
Summary
Resident microbial communities that can support various host functions play a key role in host development and health. For instance, certain microorganisms harbored in the host gut can produce vitamins and valuable metabolites, such as short chain fatty acids. In addition, host-associated microbes train and modulate the immune system to establish tolerance to commensal bacteria. In aquatic animals, microbial symbionts can be vertically transferred from the parent fish to their progeny. Such transfer of microbes in the mouthbrooder fish species has not yet been reported. Here, we employed Nile tilapia (Oreochromis niloticus), which is an important aquaculture species and by far the most farmed tropical fish, to investigate the vertical transmission of microbes across generations, using a 16S rRNA amplicon sequencing approach. To this aim, the Nile tilapia used in this study were produced from fertilized eggs that were being incubated by wild mouthbrooders caught from Nile River, Egypt. We examined the microbiota by collecting samples from the buccal cavity and posterior intestine of wild and captive Nile tilapia reared at the Bukta research station (Bodø, Norway) for three generations. Employing a 16S rRNA gene sequencing technique, we found that the core microbiome in the buccal cavity and the intestine contains beneficial microbes such as Nocardioides, Propionibacterium and Sphingomonas. These microbes have a broad spectrum of antimicrobial properties. In addition, Propionibacterium can produce propionic acids, which are a type of short chain fatty acid. Interestingly, our analysis revealed that these microbes could be transferred across generations. Here, we report for the first time the buccal cavity microbial profile in wild female Nile tilapia. The present study provides novel information regarding the buccal cavity microbiome and its potential role in egg incubation in the mouthbrooder fish species.
Acknowledgments: This study has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant 683210), from the Research Council of Norway under the Toppforsk programme (grant 250548/F20) and the Faculty of Biosciences and Aquaculture (Nord University, Norway) (grant no. 224000-124).
Validation of Oxford Nanopore Minion Technology for a Fast and Low-Cost Profiling of Mucosal Sea Bream (Sparus aurata) Microbiota
Toxqui, M.S.1,2, Naya-Catalá, F.2, Sitjà-Bobadilla, A.1, Piazzon, M.C.1 and Pérez-Sánchez, J2.
1 Fish Pathology Group, Institute of Aquaculture Torre de la Sal (CSIC), Castellón, Spain.
2 Nutrigenomics and Fish Growth Endocrinology Group, Institute of Aquaculture Torre de la Sal (CSIC), Castellón, Spain.
Summary
With the growth of the aquaculture industry, there has been a growing interest in manipulating fish gut microbiota to improve welfare and nutrition since it is critical for many host functions such as digestion, nutrient metabolism, disease resistance, and immune function. Accurate and rapid identification of microbial communities through the sequencing of the 16S ribosomal RNA (16S rRNA) gene is an approach widely used since the introduction of high-throughput sequencing technologies (HTS). Nowadays, several affordable methods involving different sequencing technologies exist, 16S amplicon Illumina sequencing being the most widely used. However, the choice of sequencing platform and the downstream analysis are known to yield somewhat different results. Of particular interest, Oxford Nanopore MinION Technology offers a low price, portability, and fast sequencing throughput compared to the Illumina MiSeq and PacBio sequencers. Nevertheless, the error rate of the generated amplicon data is higher, and differences in the experimental conditions for 16S rRNA-based PCR could bias microbiota assessments in the samples. To find a suitable solution that could offer reliable results considering the time, cost, and sequencing throughput, this study aimed to (1) evaluate the feasibility of MinION sequencing by comparing16S amplicon data of gilthead sea bream intestinal microbiota using the Illumina MiSeq, PacBio (performed with standard procedures by sequencing companies), and MinION (on-site sequencing) platforms, and (2) standardize the experimental 16S protocol for sample preparation and PCR conditions when sequencing microbiota samples with the Oxford Nanopore MinION Technology. Different 16S gene regions were amplified depending on the sequencing platform (V3–V4 for Illumina MiSeq and full 16S rRNA gene for MinION and PacBio), and bioinformatics pipelines were conducted according to each sequencer amplicon data. Concerning the MinION protocol, five PCR conditions were tested using the Nanopore 16S barcoding kit, combining different temperatures, cycling conditions, and nesting. For all samples, the taxonomic assignment was made using the SILVA (
http://www.arb-silva.de) database v. 138. The coming results will provide precise insights into the advantages and disadvantages of this specific sequencing platform, and their suitability depending on the experimental approach, expected output, and cost and time constraints of a given experiment.
Acknowledgments: EATFISH (H2020 #956697), AQUAEXCEL3.0 (H2020 #871108), RYC2018-024049-I & ESF.
SAMBA: A Bayesian Network Application to Predict Changes in the Composition and Function of Mucosal Microbiome in Farmed Fish
Soriano, B.1,2,3, Hafez, A.1,4, Naya-Català, F.2, Toxqui, M.S.2, Piazzon, M.C.2, Arnau, V.3, Sitjà-Bobadilla, A.2, Llorens, C.1 and Pérez-Sánchez, J2.
1 Biotechvana, Parc Científic Universitat de Valéncia, Spain.
2 Institute of Aquaculture Torre de la Sal (IATS-CSIC), Spain.
3 Institute for Integrative Systems Biology, UVEG-CSIC, Spain.
4 Faculty of Computers and Information, Minia University, Egypt.
Summary
SAMBA (Scanning Aquaculture Microbiome via Bayesian Approach) is a Guide User Interface application developed in R and powered by a Bayesian network model that indexes the frequencies of the bacterial taxa in a given biological system, correlating microbial architectures with a number of biotic and abiotic factors (e.g., sex, age, season, diet composition, genetic background, etc.). The application identifies conditional dependencies and interrelations among the distinct variables indexed in the network, allowing us to predict how the taxa abundance of the pan-microbiome will vary depending on experimental variables. The application permits us to predict and assign a metagenome to each inferred microbiome in order to determine its functional profile. SAMBA also permits us to interrogate the Bayesian network model to identify which experimental conditions are optimal for obtaining a given pan-microbiome and associated metagenome. At this stage, a locally run application of SAMBA was used to interrogate the pan-microbiome of farmed gilthead sea bream (a highly valued farmed fish in the Mediterranean region) with different nutritional and genetic backgrounds across the production cycle. The application can also be used to model the host, metagenome and environment interrelations in a wide range of organisms, including humans. Interfaces of SAMBA were wrapped by Shiny, a framework to build interactive web applications by R. The application uses distinct R dependencies such as (1)
bnlearn, for learning the structure of Bayesian networks and estimating their parameters based on their structure using data from 16s amplicon experiments, (2)
visNetwork and
bnviewer for network visualization and (3) PICRUSt2 to infer metagenomic functions from the microbiome population determined by 16s data-specific environmental conditions. SAMBA will be implemented to be accessible as a web server at
www.nutrigroup-iats.org.
Acknowledgments: This work was supported by a pre-doctoral research fellowship from Industrial Doctorate of MINECO to BS (Grant 659 DI-17-09134). Additional funding was obtained from National (BreamAquaINTECH, RTI2018-09412-B-I00; RYC2018-024049-I & ESF) and European (AquaIMPACT, H2020 #818367; EATFISH (H2020 #956697) Projects.



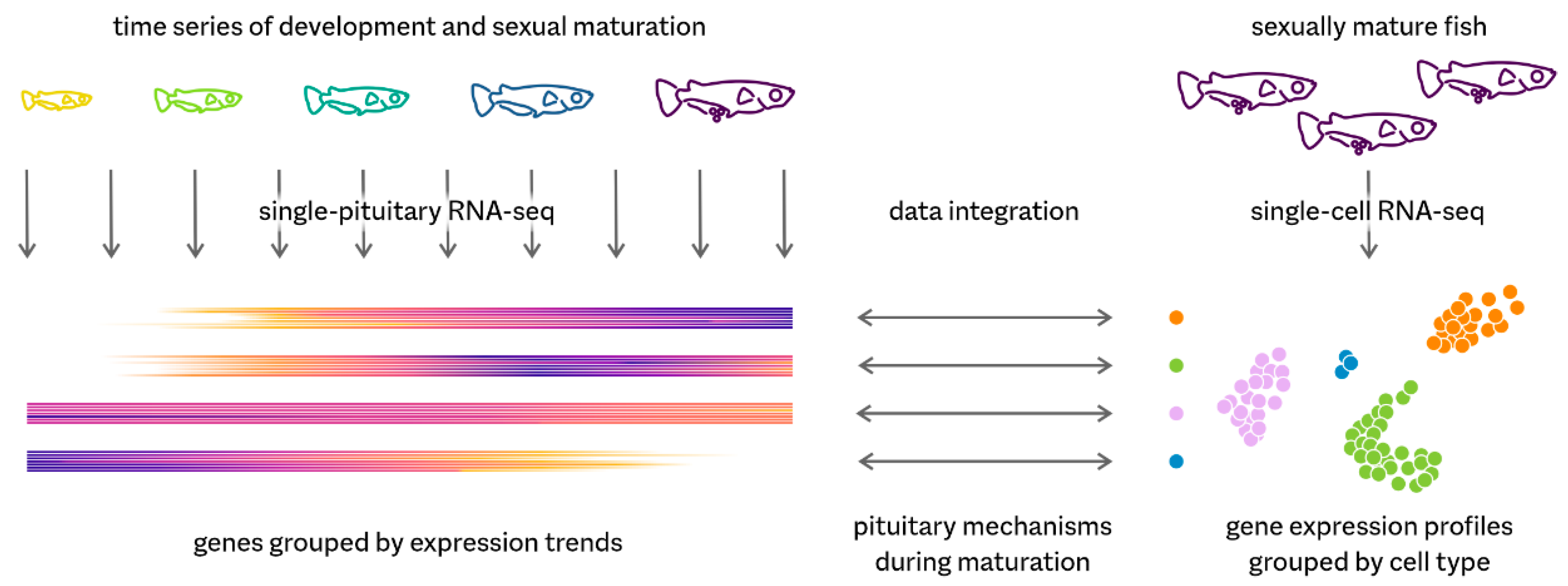{kind=link}
{kind=link}
{kind=link}