
Characterization of the Nuclear Proteome of Chlamydomonas in Response to Salt Stress



Abstract
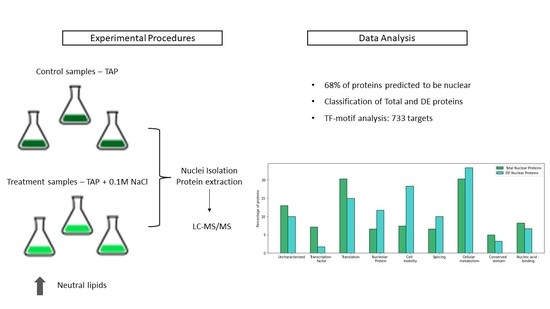
1. Introduction
2. Material and Methods
2.1. Cell Culture and Growth Conditions
2.2. Neutral Lipid Quantification
2.3. Nucleus Isolation and Extraction of Nuclear Proteins
2.4. Mass Spectrometry Analysis
2.5. Data Analysis
2.6. Microscopy
2.7. Prediction of Transcription Factor Target Genes
3. Results
3.1. The Impact of 0.1 M NaCl on Growth and Lipid Accumulation in C. reinhardtii
3.2. Proteomics Analysis
3.3. Protein Identification and Subcellular Localization
3.4. Enrichment of Nuclei Sample
3.5. Differentially Expressed and Exclusive Nuclear Proteins
3.6. Prediction of Nuclear Transcription Factor Target Genes in C. reinhardtii under Salt Stress
4. Discussion
4.1. Proteomic Analysis of C. reinhardtii Nuclear Proteins in Salt Stress
4.2. Transcription Factors
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hoegh-Guldberg, O.; Bruno, J.F. The Impact of Climate Change on the World’s Marine Ecosystems. Science 2010, 328, 1523–1528. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.C.; Yung, Y.L.; Lacis, A.A.; Mo, T.; Hansc, J.E. Greenhouse Effects Due to Man-Mad Perturbations of Trace Gases: Anthropogenic Gases May Alter Our Climate by Plugging an Atmospheric Window for Escaping Thermal Radiation. Science 1976, 194, 4266. [Google Scholar] [CrossRef] [PubMed]
- Merchant, S.S.; Kropat, J.; Liu, B.; Shaw, J.; Warakanont, J. TAG, You’re It! Chlamydomonas as a Reference Organism for Understanding Algal Triacylglycerol Accumulation. Curr. Opin. Biotechnol. 2012, 23, 352–363. [Google Scholar] [PubMed]
- Ryan Georgianna, D.; Mayfield, S.P. Exploiting Diversity and Synthetic Biology for the Production of Algal Biofuels. Nature 2012, 488, 329–335. [Google Scholar] [CrossRef] [PubMed]
- García, J.L.; de Vicente, M.; Galán, B. Microalgae, Old Sustainable Food and Fashion Nutraceuticals. Microb. Biotechnol. 2017, 10, 1017–1024. [Google Scholar] [CrossRef]
- Fan, J.; Zheng, L. Acclimation to NaCl and Light Stress of Heterotrophic Chlamydomonas reinhardtii for Lipid Accumulation. J. Biosci. Bioeng. 2017, 124, 302–308. [Google Scholar] [CrossRef]
- Lin, Q.; Zhuo, W.H.; Wang, X.W.; Chen, C.P.; Gao, Y.H.; Liang, J.R. Effects of Fundamental Nutrient Stresses on the Lipid Accumulation Profiles in Two Diatom Species Thalassiosira weissflogii and Chaetoceros muelleri. Bioprocess Biosyst. Eng. 2018, 41, 1213–1224. [Google Scholar] [CrossRef]
- He, Q.; Yang, H.; Wu, L.; Hu, C. Effect of Light Intensity on Physiological Changes, Carbon Allocation and Neutral Lipid Accumulation in Oleaginous Microalgae. Bioresour. Technol. 2015, 191, 219–228. [Google Scholar] [CrossRef]
- Fan, J.; Cui, Y.; Wan, M.; Wang, W.; Li, Y. Lipid Accumulation and Biosynthesis Genes Response of the Oleaginous Chlorella pyrenoidosa under Three Nutrition Stressors. Biotechnol. Biofuels 2014, 7, 17. [Google Scholar] [CrossRef]
- Hu, Q.; Sommerfeld, M.; Jarvis, E.; Ghirardi, M.; Posewitz, M.; Seibert, M.; Darzins, A. Microalgal Triacylglycerols as Feedstocks for Biofuel Production: Perspectives and Advances. Plant J. 2008, 54, 621–639. [Google Scholar] [CrossRef]
- Winck, F.V.; Páez Melo, D.O.; González Barrios, A.F. Carbon Acquisition and Accumulation in Microalgae Chlamydomonas: Insights from “Omics” Approaches. J. Proteomics 2013, 94, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Mettler, T.; Mühlhaus, T.; Hemme, D.; Schöttler, M.-A.; Rupprecht, J.; Idoine, A.; Veyel, D.; Pal, S.K.; Yaneva-Roder, L.; Winck, F.V.; et al. Systems Analysis of the Response of Photosynthesis, Metabolism, and Growth to an Increase in Irradiance in the Photosynthetic Model Organism Chlamydomonas reinhardtii. Plant Cell 2014, 26, 2310–2350. [Google Scholar] [CrossRef] [PubMed]
- Merchant, S.S.; Prochnik, S.E.; Vallon, O.; Harris, E.H.; Karpowicz, S.J.; Witman, G.B.; Terry, A.; Salamov, A.; Fritz-Laylin, L.K.; Maréchal-Drouard, L.; et al. The Chlamydomonas Genome Reveals the Evolution of Key Animal and Plant Functions. Science 2007, 318, 245–250. [Google Scholar] [CrossRef]
- May, P.; Wienkoop, S.; Kempa, S.; Usadel, B.; Christian, N.; Rupprecht, J.; Weiss, J.; Recuenco-Munoz, L.; Ebenhöh, O.; Weckwerth, W.; et al. Metabolomics- and Proteomics-Assisted Genome Annotation and Analysis of the Draft Metabolic Network of Chlamydomonas reinhardtii. Genetics 2008, 179, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Mastrobuoni, G.; Irgang, S.; Pietzke, M.; Aßmus, H.E.; Wenzel, M.; Schulze, W.X.; Kempa, S. Proteome Dynamics and Early Salt Stress Response of the Photosynthetic Organism Chlamydomonas reinhardtii. BMC Genom. 2012, 13, 215. [Google Scholar] [CrossRef]
- Hounslow, E.; Vijay Kapoore, R.; Vaidyanathan, S.; Gilmour, D.J.; Wright, P.C. The Search for a Lipid Trigger: The Effect of Salt Stress on the Lipid Profile of the Model Microalgal Species Chlamydomonas reinhardtii for Biofuels Production. Curr. Biotechnol. 2016, 5, 305–313. [Google Scholar] [CrossRef]
- Wang, N.; Qian, Z.; Luo, M.; Fan, S.; Zhang, X.; Zhang, L. Identification of Salt Stress Responding Genes Using Transcriptome Analysis in Green Alga Chlamydomonas reinhardtii. Int. J. Mol. Sci. 2018, 19, 3359. [Google Scholar] [CrossRef]
- Arias, C.; Obudulu, O.; Zhao, X.; Ansolia, P.; Zhang, X.; Paul, S.; Bygdell, J.; Pirmoradian, M.; Zubarev, R.A.; Samuelsson, G.; et al. Nuclear Proteome Analysis of Chlamydomonas with Response to CO2 Limitation. Algal Res. 2020, 46, 101765. [Google Scholar] [CrossRef]
- Winck, F.V.; Riaño-Pachón, D.M.; Sommer, F.; Rupprecht, J.; Mueller-Roeber, B. The Nuclear Proteome of the Green Alga Chlamydomonas reinhardtii. PROTEOMICS 2012, 12, 95–100. [Google Scholar] [CrossRef]
- Gorman, D.S.; Levine, R.P. Cytochrome f and Plastocyanin: Their Sequence in the Photosynthetic Electron Transport Chain of Chlamydomonas reinhardi. Proc. Natl. Acad. Sci. USA 1965, 54, 1665–1669. [Google Scholar] [CrossRef]
- Kou, Z.; Bei, S.; Sun, J.; Pan, J. Fluorescent Measurement of Lipid Content in the Model Organism Chlamydomonas reinhardtii. J. Appl. Phycol. 2013, 25, 1633–1641. [Google Scholar] [CrossRef]
- Winck, F.V.; Kwasniewski, M.; Wienkoop, S.; Mueller-Roeber, B. AN OPTIMIZED METHOD FOR THE ISOLATION OF NUCLEI FROM CHLAMYDOMONAS REINHARDTII (CHLOROPHYCEAE)1. J. Phycol. 2011, 47, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate Proteome-Wide Label-Free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [PubMed]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A Comparative Platform for Green Plant Genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus Computational Platform for Comprehensive Analysis of (Prote)Omics Data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape Plugin to Assess Overrepresentation of Gene Ontology Categories in Biological Networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef]
- Kucera, M.; Isserlin, R.; Arkhangorodsky, A.; Bader, G.D. AutoAnnotate: A Cytoscape App for Summarizing Networks with Semantic Annotations. F1000Research 2016, 5, 1717. [Google Scholar] [CrossRef]
- Carnielli, C.M.; Winck, F.V.; Paes Leme, A.F. Functional Annotation and Biological Interpretation of Proteomics Data. Biochim. Biophys. Acta BBA-Proteins Proteom. 2015, 1854, 46–54. [Google Scholar] [CrossRef]
- Hooper, C.M.; Castleden, I.R.; Tanz, S.K.; Aryamanesh, N.; Millar, A.H. SUBA4: The Interactive Data Analysis Centre for Arabidopsis Subcellular Protein Locations. Nucleic Acids Res. 2017, 45, D1064–D1074. [Google Scholar] [CrossRef]
- Swarbreck, D.; Wilks, C.; Lamesch, P.; Berardini, T.Z.; Garcia-Hernandez, M.; Foerster, H.; Li, D.; Meyer, T.; Muller, R.; Ploetz, L.; et al. The Arabidopsis Information Resource (TAIR): Gene Structure and Function Annotation. Nucleic Acids Res. 2008, 36, D1009–D1014. [Google Scholar] [CrossRef]
- Brameier, M.; Krings, A.; MacCallum, R.M. NucPred—Predicting Nuclear Localization of Proteins. Bioinformatics 2007, 23, 1159–1160. [Google Scholar] [CrossRef] [PubMed]
- Tardif, M.; Atteia, A.; Specht, M.; Cogne, G.; Rolland, N.; Brugière, S.; Hippler, M.; Ferro, M.; Bruley, C.; Peltier, G.; et al. Predalgo: A New Subcellular Localization Prediction Tool Dedicated to Green Algae. Mol. Biol. Evol. 2012, 29, 3625–3639. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, P.; Riaño-Pachón, D.M.; Corrêa, L.G.G.; Rensing, S.A.; Kersten, B.; Mueller-Roeber, B. PlnTFDB: Updated Content and New Features of the Plant Transcription Factor Database. Nucleic Acids Res. 2010, 38, D822–D827. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.Y.; Krishnakumar, V.; Chan, A.P.; Thibaud-Nissen, F.; Schobel, S.; Town, C.D. Araport11: A Complete Reannotation of the Arabidopsis thaliana Reference Genome. Plant J. Cell Mol. Biol. 2017, 89, 789–804. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Tian, F.; Yang, D.C.; Meng, Y.Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a Central Hub for Transcription Factors and Regulatory Interactions in Plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Gribskov, M. Combining Evidence Using P-Values: Application to Sequence Homology Searches. Bioinforma. Oxf. Engl. 1998, 14, 48–54. [Google Scholar] [CrossRef]
- Hang, L.T.; Mori, K.; Tanaka, Y.; Morikawa, M.; Toyama, T. Enhanced Lipid Productivity of Chlamydomonas reinhardtii with Combination of NaCl and CaCl2 Stresses. Bioprocess Biosyst. Eng. 2020, 43, 971–980. [Google Scholar] [CrossRef]
- McClure-Begley, T.D.; Klymkowsky, M.W. Nuclear Roles for Cilia-Associated Proteins. Cilia 2017, 6, 8. [Google Scholar] [CrossRef]
- Hessen, D.O.; Van Donk, E.; Andersen, T. Growth Responses, P-Uptake and Loss of Flagellae in Chlamydomonas reinhardtii Exposed to UV-B. J. Plankton Res. 1995, 17, 17–27. [Google Scholar] [CrossRef]
- Rosenbaum, J.L.; Moulder, J.E.; Ringo, D.L. FLAGELLAR ELONGATION AND SHORTENING IN CHLAMYDOMONAS: The Use of Cycloheximide and Colchicine to Study the Synthesis and Assembly of Flagellar Proteins. J. Cell Biol. 1969, 41, 600. [Google Scholar] [CrossRef] [PubMed]
- Siaut, M.; Cuiné, S.; Cagnon, C.; Fessler, B.; Nguyen, M.; Carrier, P.; Beyly, A.; Beisson, F.; Triantaphylidès, C.; Li-Beisson, Y.; et al. Oil Accumulation in the Model Green Alga Chlamydomonas reinhardtii: Characterization, Variability between Common Laboratory Strains and Relationship with Starch Reserves. BMC Biotechnol. 2011, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Pascual, J.; Alegre, S.; Nagler, M.; Escandón, M.; Annacondia, M.L.; Weckwerth, W.; Valledor, L.; Cañal, M.J. The Variations in the Nuclear Proteome Reveal New Transcription Factors and Mechanisms Involved in UV Stress Response in Pinus radiata. J. Proteom. 2016, 143, 390–400. [Google Scholar] [CrossRef]
- Petibon, C.; Malik Ghulam, M.; Catala, M.; Abou Elela, S. Regulation of Ribosomal Protein Genes: An Ordered Anarchy. Wiley Interdiscip. Rev. RNA 2021, 12, e1632. [Google Scholar] [CrossRef]
- Tiruneh, B.S.; Kim, B.H.; Gallie, D.R.; Roy, B.; Von Arnim, A.G. The Global Translation Profile in a Ribosomal Protein Mutant Resembles That of an EIF3 Mutant. BMC Biol. 2013, 11, 123. [Google Scholar] [CrossRef]
- Carroll, A.J. The Arabidopsis Cytosolic Ribosomal Proteome: From Form to Function. Front. Plant Sci. 2013, 4, 32. [Google Scholar] [CrossRef]
- Wang, J.; Lan, P.; Gao, H.; Zheng, L.; Li, W.; Schmidt, W. Expression Changes of Ribosomal Proteins in Phosphate- and Iron-Deficient Arabidopsis Roots Predict Stress-Specific Alterations in Ribosome Composition. BMC Genom. 2013, 14, 783. [Google Scholar] [CrossRef]
- Rosado, A.; Li, R.; Van De Ven, W.; Hsu, E.; Raikhel, N.V. Arabidopsis Ribosomal Proteins Control Developmental Programs through Translational Regulation of Auxin Response Factors. Proc. Natl. Acad. Sci. USA 2012, 109, 19537–19544. [Google Scholar] [CrossRef]
- Schippers, J.H.M.; Mueller-Roeber, B. Ribosomal Composition and Control of Leaf Development. Plant Sci. 2010, 179, 307–315. [Google Scholar] [CrossRef]
- Takeuchi, T.; Benning, C. Nitrogen-Dependent Coordination of Cell Cycle, Quiescence and TAG Accumulation in Chlamydomonas. Biotechnol. Biofuels 2019, 12, 292. [Google Scholar] [CrossRef] [PubMed]
- Couso, I.; Pérez-Pérez, M.E.; Martínez-Force, E.; Kim, H.S.; He, Y.; Umen, J.G.; Crespo, J.L. Autophagic Flux Is Required for the Synthesis of Triacylglycerols and Ribosomal Protein Turnover in Chlamydomonas. J. Exp. Bot. 2018, 69, 1355–1367. [Google Scholar] [CrossRef] [PubMed]
- Park, J.J.; Wang, H.; Gargouri, M.; Deshpande, R.R.; Skepper, J.N.; Holguin, F.O.; Juergens, M.T.; Shachar-Hill, Y.; Hicks, L.M.; Gang, D.R. The Response of Chlamydomonas reinhardtii to Nitrogen Deprivation: A Systems Biology Analysis. Plant J. 2015, 81, 611–624. [Google Scholar] [CrossRef]
- Blaby, I.K.; Blaby-Haas, C.E.; Tourasse, N.; Hom, E.F.Y.; Lopez, D.; Aksoy, M.; Grossman, A.; Umen, J.; Dutcher, S.; Porter, M.; et al. The Chlamydomonas Genome Project: A Decade On. Trends Plant Sci. 2014, 19, 672–680. [Google Scholar] [CrossRef]
- Labadorf, A.; Link, A.; Rogers, M.F.; Thomas, J.; Reddy, A.S.N.; Ben-Hur, A. Genome-Wide Analysis of Alternative Splicing in Chlamydomonas reinhardtii. BMC Genom. 2010, 11, 114. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, Y.; Chen, M.; Xiao, P.; Hu, C.; Zeng, Z.; Wang, C.; Wang, J.; Hu, Z. Genome-Wide Long Non-Coding RNA Screening, Identification and Characterization in a Model Microorganism Chlamydomonas reinhardtii. Sci. Rep. 2016 61 2016, 6, 34109. [Google Scholar] [CrossRef]
- Lou, S.; Sun, T.; Li, H.; Hu, Z. Mechanisms of MicroRNA-Mediated Gene Regulation in Unicellular Model Alga Chlamydomonas reinhardtii. Biotechnol. Biofuels 2018, 11, 244. [Google Scholar] [CrossRef]
- Goldschmidt-Clermont, M.; Rahire, M.; Rochaix, J.D. Redundant Cis-Acting Determinants of 3′ Processing and RNA Stability in the Chloroplast RbcL MRNA of Chlamydomonas. Plant J. 2008, 53, 566–577. [Google Scholar] [CrossRef]
- Kück, U.; Schmitt, O. The Chloroplast Trans-Splicing RNA–Protein Supercomplex from the Green Alga Chlamydomonas reinhardtii. Cells 2021, 10, 290. [Google Scholar] [CrossRef]
- Riaño-Pachón, D.M.; Corrêa, L.G.G.; Trejos-Espinosa, R.; Mueller-Roeber, B. Green Transcription Factors: A Chlamydomonas Overview. Genetics 2008, 179, 31–39. [Google Scholar] [CrossRef]
- Anderson, M.S.; Muff, T.J.; Georgianna, D.R.; Mayfield, S.P. Towards a Synthetic Nuclear Transcription System in Green Algae: Characterization of Chlamydomonas reinhardtii Nuclear Transcription Factors and Identification of Targeted Promoters. Algal Res. 2017, 22, 47–55. [Google Scholar] [CrossRef]
- Ambawat, S.; Sharma, P.; Yadav, N.R.; Yadav, R.C. MYB Transcription Factor Genes as Regulators for Plant Responses: An Overview. Physiol. Mol. Biol. Plants Int. J. Funct. Plant Biol. 2013, 19, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB Transcription Factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Roy, S. Function of MYB Domain Transcription Factors in Abiotic Stress and Epigenetic Control of Stress Response in Plant Genome. Plant Signal. Behav. 2016, 11, e1117723. [Google Scholar] [CrossRef]
- Yang, C.; Hager, P.W.; Stiller, J.W. The Identification of Putative RNA Polymerase II C-Terminal Domain Associated Proteins in Red and Green Algae. Transcription 2014, 5, e970944. [Google Scholar] [CrossRef][Green Version]
- Torres-Romero, I.; Kong, F.; Légeret, B.; Beisson, F.; Peltier, G.; Li-Beisson, Y. Chlamydomonas Cell Cycle Mutant Crcdc5 Over-Accumulates Starch and Oil. Biochimie 2020, 169, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yang, S.; Yuan, L.; Cui, Y.; Wu, K. Comparative Analysis of SWIRM Domain-Containing Proteins in Plants. Comp. Funct. Genom. 2012, 2012, 310402. [Google Scholar] [CrossRef]
- March, E.; Farrona, S. Plant Deubiquitinases and Their Role in the Control of Gene Expression through Modification of Histones. Front. Plant Sci. 2018, 8, 2274. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, T.; Shinozaki, K. A Cdc5+ Homolog of a Higher Plant, Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 1996, 93, 13371–13376. [Google Scholar] [CrossRef]
- Antosch, M.; Mortensen, S.A.; Grasser, K.D. Plant Proteins Containing High Mobility Group Box DNA-Binding Domains Modulate Different Nuclear Processes. Plant Physiol. 2012, 159, 875–883. [Google Scholar] [CrossRef]
- Launholt, D.; Grasser, K.D. The HMG-Box: A Versatile Protein Domain Occurring in a Wide Variety of DNA-Binding Proteins. Cell. Mol. Life Sci. 2007, 64, 2590–2606. [Google Scholar] [CrossRef]
- Pedersen, D.S.; Coppens, F.; Ma, L.; Antosch, M.; Marktl, B.; Merkle, T.; Beemster, G.T.S.; Houben, A.; Grasser, K.D. The Plant-Specific Family of DNA-Binding Proteins Containing Three HMG-Box Domains Interacts with Mitotic and Meiotic Chromosomes. New Phytol. 2011, 192, 577–589. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phytozome ID | Phytozome Description | SUBA | PredAlgo | NucPred | Fold Change |
|---|---|---|---|---|---|
| Uncharacterized | |||||
| Cre08.g382150.t1.1 # | Uncharacterized | Nuclear | Not nuclear | −15.579728 | |
| Cre02.g118750.t1.1 # | Uncharacterized | Nuclear | Nuclear | −15.490067 | |
| Cre14.g610900.t1.1 # | Progesterone induced blocking factor 1 | Nuclear | Not nuclear | −14.833532 | |
| Cre14.g633950.t1.2 | Uncharacterized | Secretory pathway | Nuclear | −0.9458238 | |
| Cre02.g095146.t1.1 | Uncharacterized | Nuclear | Not nuclear | −0.9081868 | |
| Cre16.g687182.t1.1 | Uncharacterized | Mitochondrion | Nuclear | −0.8512069 | |
| Translation and ribosome biogenesis | |||||
| Cre02.g091100.t1.2 # | Cytosolic 80S ribosomal protein L15 | Cytosol | Nuclear | Not nuclear | −16.058077 |
| Cre08.g360900.t1.2 # | Cytosolic 80S ribosomal protein S15 | Cytosol | Nuclear | Nuclear | −15.928843 |
| Cre06.g264300.t1.2 # | Chloroplast ribosomal protein S15 | Mitochondrion | Chloroplast | Nuclear | −15.800218 |
| Cre12.g512600.t1.2 # | Cytosolic 80S ribosomal protein L18 | Cytosol | Chloroplast | Nuclear | −15.686391 |
| Cre10.g417700.t1.2 | Cytosolic 80S ribosomal protein L3 | Cytosol | Chloroplast | Nuclear | −0.9360618 |
| Cre09.g397697.t1.1 | Ribosomal protein L4 | Cytosol | Nuclear | Nuclear | −0.9273469 |
| Cre10.g420750.t1.2 | Cytosolic 80S ribosomal protein L30 | Cytosol | Nuclear | Nuclear | −0.9272525 |
| Cre12.g489153.t1.1 | Cytosolic 80S ribosomal protein L31 | Cytosol | Nuclear | Nuclear | −0.9179496 |
| Cre01.g011000.t1.2 | Cytosolic 80S ribosomal protein L6 | Cytosol | Nuclear | Nuclear | −0.8997668 |
| Nucleolar protein–nucleosome biogenesis | |||||
| Cre06.g260850.t1.2 # | Nucleolar protein | Nuclear | Chloroplast | Not nuclear | −14.923977 |
| Cre10.g442000.t1.2 | U3 small nucleolar RNA-associated protein 6 (UTP6) | Nuclear | Nuclear | Nuclear | −0.9217898 |
| Cre12.g548000.t1.2 | U3 small nucleolar RNA-associated protein 10 (UTP10) | Plasma Membrane | Mitochondrion | Not nuclear | −0.9089622 |
| Cre12.g525200.t1.2 | Nucleolar protein | Nuclear | Nuclear | Nuclear | −0.9059913 |
| Cre16.g656250.t1.1 | U1 small nuclear ribonucleoprotein | Nuclear | Nuclear | Not nuclear | −0.9035496 |
| Cre17.g737250.t1.2 | U5 small nuclear ribonucleoprotein protein | Cytosol | Nuclear | Nuclear | −0.8846926 |
| Cre01.g055400.t1.2 | Nucleolar and coiled body phosphoprotein 1 | Nuclear | Nuclear | Not nuclear | −0.8817377 |
| Cell mobility-related | |||||
| Cre02.g091700.t1.2 # | Protofilament ribbon protein of flagellar microtubules | Mitochondrion | Nuclear | Nuclear | −16.528622 |
| Cre16.g655750.t1.2 # | Tektin | Nuclear | Nuclear | −16.511042 | |
| Cre02.g141350.t1.2 | Nexin dynein regulatory complex 10 | Nuclear | Not nuclear | −0.8690188 | |
| Cre12.g536100.t1.2 # | Flagellar-associated protein 126 | Nuclear | Nuclear | −15.967271 | |
| Cre10.g427300.t1.2 # | Radial spoke protein 2 | Nuclear | Nuclear | −15.789686 | |
| Cre12.g528000.t1.2 # | Flagellar-associated protein 303 | Golgi | Mitochondrion | Nuclear | −15.748193 |
| Cre01.g025450.t1.2 # | Nexin dynein regulatory complex 9 | Nuclear | Nuclear | −15.415973 | |
| Cre10.g465250.t1.2 # | Radial spoke protein 11 | Nuclear | Nuclear | −15.266677 | |
| Cre16.g654300.t1.2 # | Radial spoke protein 23 | Extracellular | Nuclear | Nuclear | −15.187778 |
| Cre12.g556250.t1.2 # | Septin-like protein | Nuclear | Nuclear | −15.125212 | |
| Cre17.g703600.t2.1 | Basal body protein | Nuclear | Not nuclear | −0.9299278 | |
| Splicing | |||||
| Cre12.g513500.t1.2 # | Transcription coupled DNA repair protein | Nuclear | Nuclear | Nuclear | −16.807381 |
| Cre04.g226450.t1.1 | Nuclear pre-mRNA splicing factor | Nuclear | Nuclear | Nuclear | −0.9098076 |
| Cre08.g372800.t1.1 | Nuclear pre-mRNA splicing factor involved in polyadenylation | Nuclear | Nuclear | Nuclear | −0.907742 |
| Cre14.g621000.t1.2 | Pre-mRNA splicing factor | Nuclear | Nuclear | Not nuclear | −0.8972515 |
| Cre06.g259800.t1.2 | Cwc22 pre-mRNA splicing factor | Nuclear | Chloroplast | Not nuclear | −0.8688461 |
| Cre02.g075650.t1.2 | Pre-mRNA splicing factor RBM22/SLT11 (RBM22) | Nuclear | Nuclear | Nuclear | −0.8473909 |
| Cellular nitrogen compound metabolic process | |||||
| Cre07.g322176.t1.1 | DNA directed RNA polymerase I subunit RPA2 (RPA2 | Nuclear | Nuclear | Nuclear | −0.9289727 |
| Cre03.g172950.t1.2 | TruB family RNA pseudouridine synthase | Nuclear | Nuclear | Nuclear | −0.915942 |
| Cre01.g051100.t1.2 | SNW domain-containing protein 1 (SNW1 | Nuclear | Nuclear | Not nuclear | −0.8950347 |
| Cre06.g300750.t1.1 | Intron-binding protein aquarius (AQR) | Cytosol | Nuclear | Nuclear | −0.8681914 |
| Process metabolic cellular | |||||
| Cre06.g282800.t1.2 # | Isocitrate lyase | Peroxisome | Nuclear | Nuclear | −15.573973 |
| Cre06.g257601.t1.2 # | 2-Cys peroxiredoxin | Chloroplast | Chloroplast | Nuclear | −15.168163 |
| Cre01.g010900.t1.2 | Glyceraldehyde phosphate dehydrogenase | Chloroplast | Chloroplast | Nuclear | −0.945159 |
| Cre06.g250200.t1.2 | S-Adenosylmethionine synthase | Cytosol | Nuclear | Nuclear | −0.9164602 |
| Cre06.g278254.t1.1 | Serine/threonine protein kinase SRPK3 [EC:2.7.11.1] (SRPK3 | Nuclear | Chloroplast | Nuclear | −0.9144799 |
| Cre03.g149100.t1.2 | Citrate synthase | Peroxisome | Mitochondrion | Nuclear | −0.8881439 |
| Cre01.g044800.t1.2 | Pyruvate formate lyase | Chloroplast | Nuclear | −0.9242933 | |
| Cre06.g288750.t1.2 | Nuclear RNA cap-binding protein | Nuclear | Nuclear | Nuclear | −0.8752861 |
| Conserved Domain | |||||
| Cre01.g035000.t1.2 # | Periodic tryptophan protein 1 (PWP1) | Nuclear | Nuclear | Nuclear | −15.201243 |
| Ribosome biogenesis | |||||
| Cre10.g450879.t1.2 # | Ribosome biogenesis protein SSF1/2 (SSF1_2) | Nuclear | Nuclear | Not nuclear | −16.726006 |
| Cre07.g314100.t1.2 | Ribosome biogenesis protein UTP30 (UTP30 | Nuclear,cytosol | Nuclear | Nuclear | −0.8590539 |
| RNA binding/RNA metabolism | |||||
| Cre12.g528950.t1.1 # | Puf protein | Nuclear,cytosol | Nuclear | Nuclear | −16.174809 |
| Cre04.g213905.t2.1 # | RNA-binding protein 39 (RBM39) | Nuclear | Nuclear | Not nuclear | −15.477695 |
| Cre07.g349100.t1.1 # | ATP-dependent RNA helicase | Nuclear | Secretory pathway | Not nuclear | −15.236351 |
| Cre10.g439700.t1.2 | Conserved expressed putative RNA-binding protein | Nuclear | Nuclear | Not nuclear | −0.9235499 |
| Transcription factor | |||||
| Cre09.g393728.t1.1 # | SET domain-containing protein 6 (SE TD6) | Chloroplast | Nuclear | Nuclear | −14.438726 |
| Histone | |||||
| Cre13.g567450.t1.2 | Histone H1 | Nuclear | Nuclear | Nuclear | −0.9387458 |
| Cre06.g275900.t1.2 | Histone H1 | Nuclear | Nuclear | Nuclear | −0.895312 |
| C. reinhardtii Phytozome Gene ID | TF Family 1 | A. thaliana Homolog TAIR ID 2 | Motif ID 3 | Number of Targets 4 |
|---|---|---|---|---|
| Cre01.g062172.t1.1 | CCAAT | N/A | ||
| Cre02.g073650.t2.1 | Top | N/A | ||
| Cre02.g115250.t1.1 | Top | N/A | ||
| Cre03.g152150.t1.2 | C2H2 | AT5G04390.1 | TFmatrixID_0216, TFmatrixID_0953 | 3 |
| Cre03.g176651.t1.1 | MYB-related | AT3G09600.2 | TFmatrixID_1236, TFmatrixID_1247, TFmatrixID_1521 | 11 |
| Cre03.g197350.t1.2 | MYB | AT1G09770.1 | TFmatrixID_0099, TFmatrixID_0524, TFmatrixID_0547 | 115 |
| Cre06.g250950.t1.2 | C3H | AT5G42820.2 | TFmatrixID_0992 | 0 |
| Cre06.g261450.t1.2 | HMG | AT4G11080.1 | TFmatrixID_1071, TFmatrixID_1072 | 585 |
| Cre06.g264750.t1.2 | CCAAT | AT1G54830.3 | TF_motif_seq_0257 | 0 |
| Cre06.g268600.t1.2 | CSD | AT2G21060.1 | TFmatrixID_0222 | 0 |
| Cre07.g344050.t1.1 | Top | AT3G54320.3 | TFmatrixID_0516, TFmatrixID_0539 | 0 |
| Cre09.g393728.t1.1 | SET | AT1G12890.1 | TFmatrixID_0053, TFmatrixID_0075, TFmatrixID_0101 | 0 |
| Cre10.g446900.t1.2 | Top | AT2G31370.5 | TFmatrixID_0188 | 0 |
| Cre10.g455600.t1.1 | PHD | AT3G19510.1 | TFmatrixID_0285 | 0 |
| Cre10.g466250.t1.2 | Top | N/A | ||
| Cre11.g482700.t1.2 | HMG | AT1G04880.1 | TFmatrixID_0788, TFmatrixID_0793 | 17 |
| Cre12.g508150.t1.2 | SNF2 | AT5G08520.1 | TFmatrixID_0356, TFmatrixID_1252, TFmatrixID_1257 | 3 |
| Cre12.g520650.t1.2 | TUB | N/A | ||
| Cre13.g570050.t1.2 | CCAAT | N/A | ||
| Cre14.g620850.t1.2 | bHLH | AT1G69010.1 | TFmatrixID_0162, TFmatrixID_0801, TFmatrixID_0802 | 0 |
| Cre16.g672300.t1.2 | HMG | AT4G11080.1 | TFmatrixID_1071, TFmatrixID_1072 | 585 |
| Cre17.g702650.t1.1 | HMG | AT4G11080.1 | TFmatrixID_1071, TFmatrixID_1072 | 585 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Oliveira Magalhães, L.; Nunes de Mello, F.; Vischi Winck, F. Characterization of the Nuclear Proteome of Chlamydomonas in Response to Salt Stress. Phycology 2022, 2, 280-296. https://doi.org/10.3390/phycology2030015
de Oliveira Magalhães L, Nunes de Mello F, Vischi Winck F. Characterization of the Nuclear Proteome of Chlamydomonas in Response to Salt Stress. Phycology. 2022; 2(3):280-296. https://doi.org/10.3390/phycology2030015
Chicago/Turabian Stylede Oliveira Magalhães, Larissa, Fabio Nunes de Mello, and Flavia Vischi Winck. 2022. "Characterization of the Nuclear Proteome of Chlamydomonas in Response to Salt Stress" Phycology 2, no. 3: 280-296. https://doi.org/10.3390/phycology2030015
APA Stylede Oliveira Magalhães, L., Nunes de Mello, F., & Vischi Winck, F. (2022). Characterization of the Nuclear Proteome of Chlamydomonas in Response to Salt Stress. Phycology, 2(3), 280-296. https://doi.org/10.3390/phycology2030015