
Loss of 4.1B Drives PRMT3-Mediated Regulation of GBM Brain Tumour Stem Cell Growth

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Drug Screening and Cell Viability Assays
2.3. EdU Cell Cycle Analysis
2.4. CRISPR-cas9 Knockout Procedures
2.5. Stable Lentiviral Knockdown and Overexpression Methods
2.6. RNA Sequencing and Bioinformatic Analysis
2.7. Real-Time Quantitative PCR
- PCR cycling conditions
| Steps | Temperature | Time | Cycles |
| Initial denaturation | 98 °C | 30 s | 1 |
| Denaturation | 98 °C | 10 s | 25–35 cycles |
| Annealing | 55 °C | 20 s | |
| Extension | 72 °C | 10 min | |
| Final extension | 72 °C | X min | 1 |
| Hold | 4 °C | Forever | |
2.8. Bisulfite Conversion and Promoter/Enhancer Methylation Assay
2.9. Western Blot
2.10. Nuclear Versus Cytoplasmic Extraction
2.11. Immunofluorescence Staining
2.12. Co-Immunoprecipitation Assays
2.13. Intracranial Xenografts
2.14. Statistical Analysis
3. Results
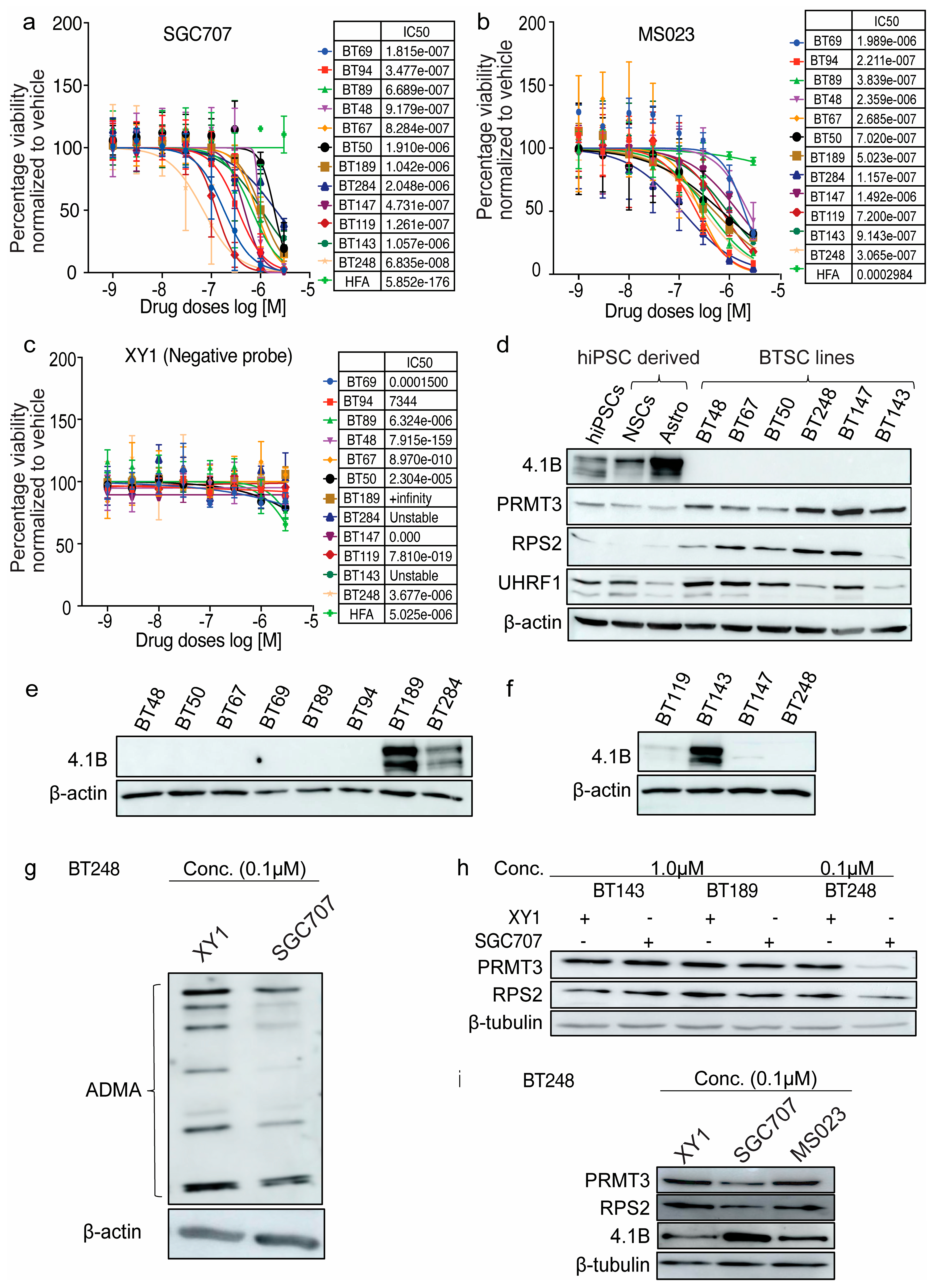
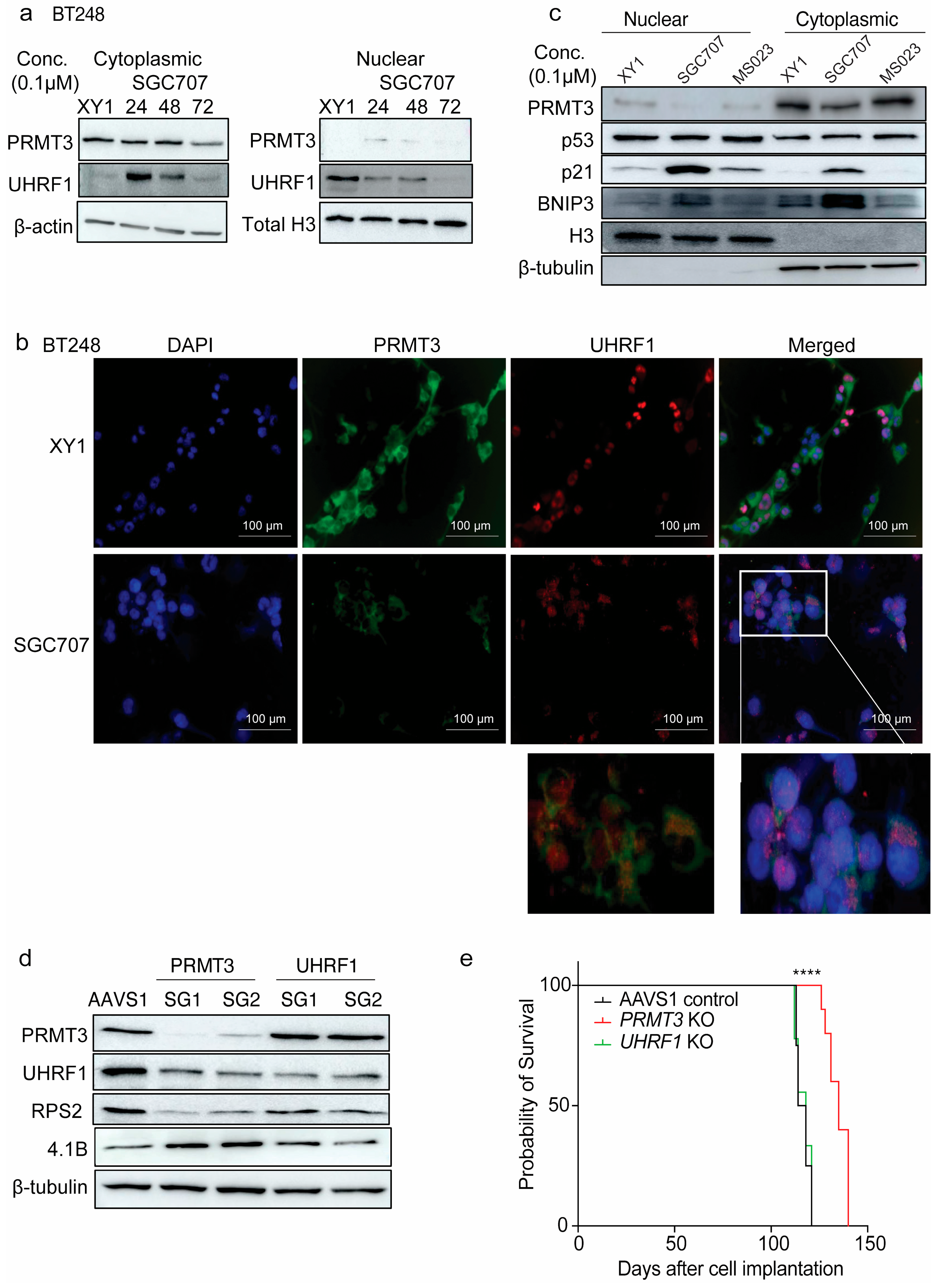
3.1. Expression Levels of Tumour-Suppressor Protein, 4.1B, Predict BTSCs’ Response to the PRMT3 Chemical Probe, SGC707
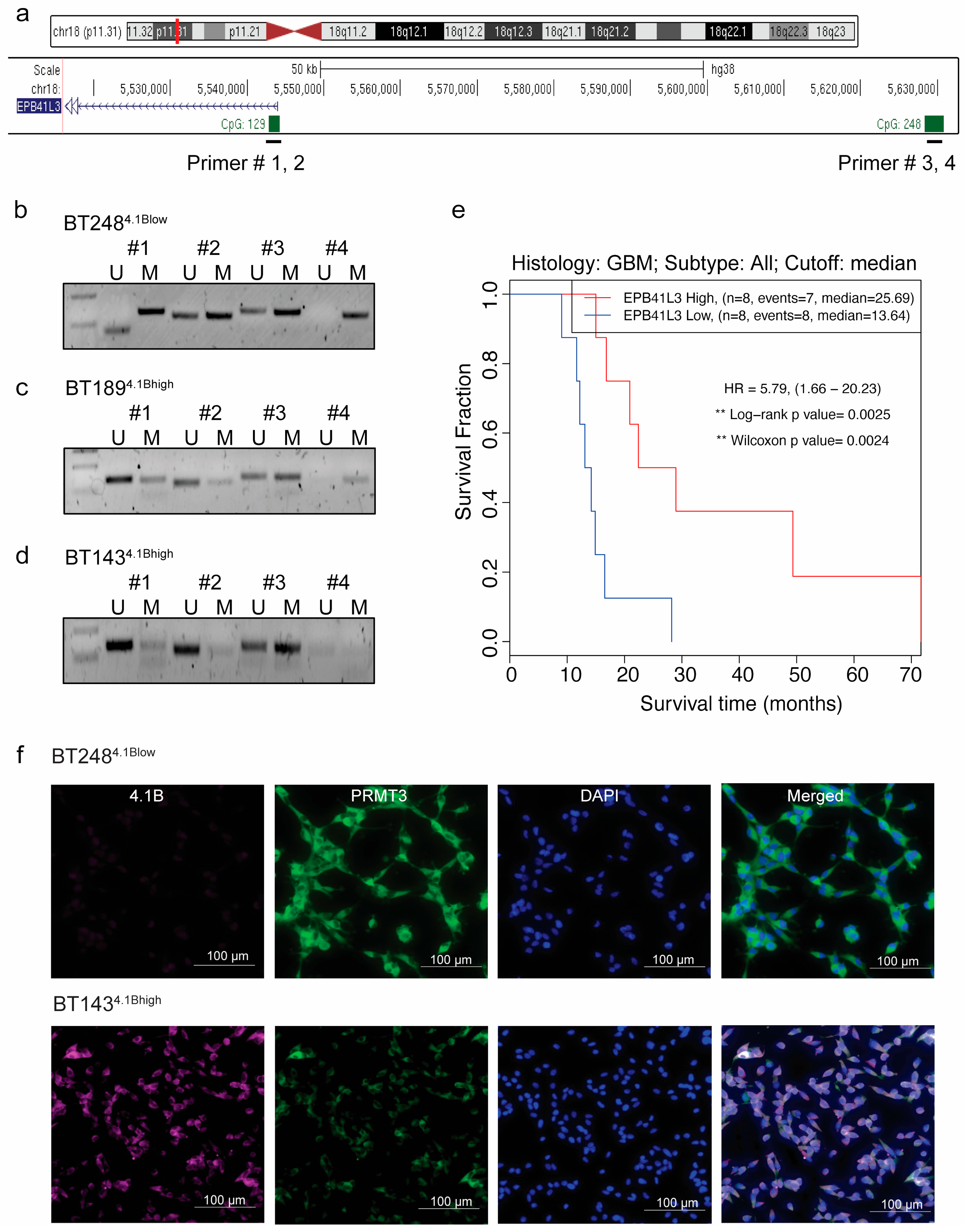
3.2. Hypermethylation of 5′ Regulatory Regions Results in a Loss of EPB41L3 Expression in BTSCs
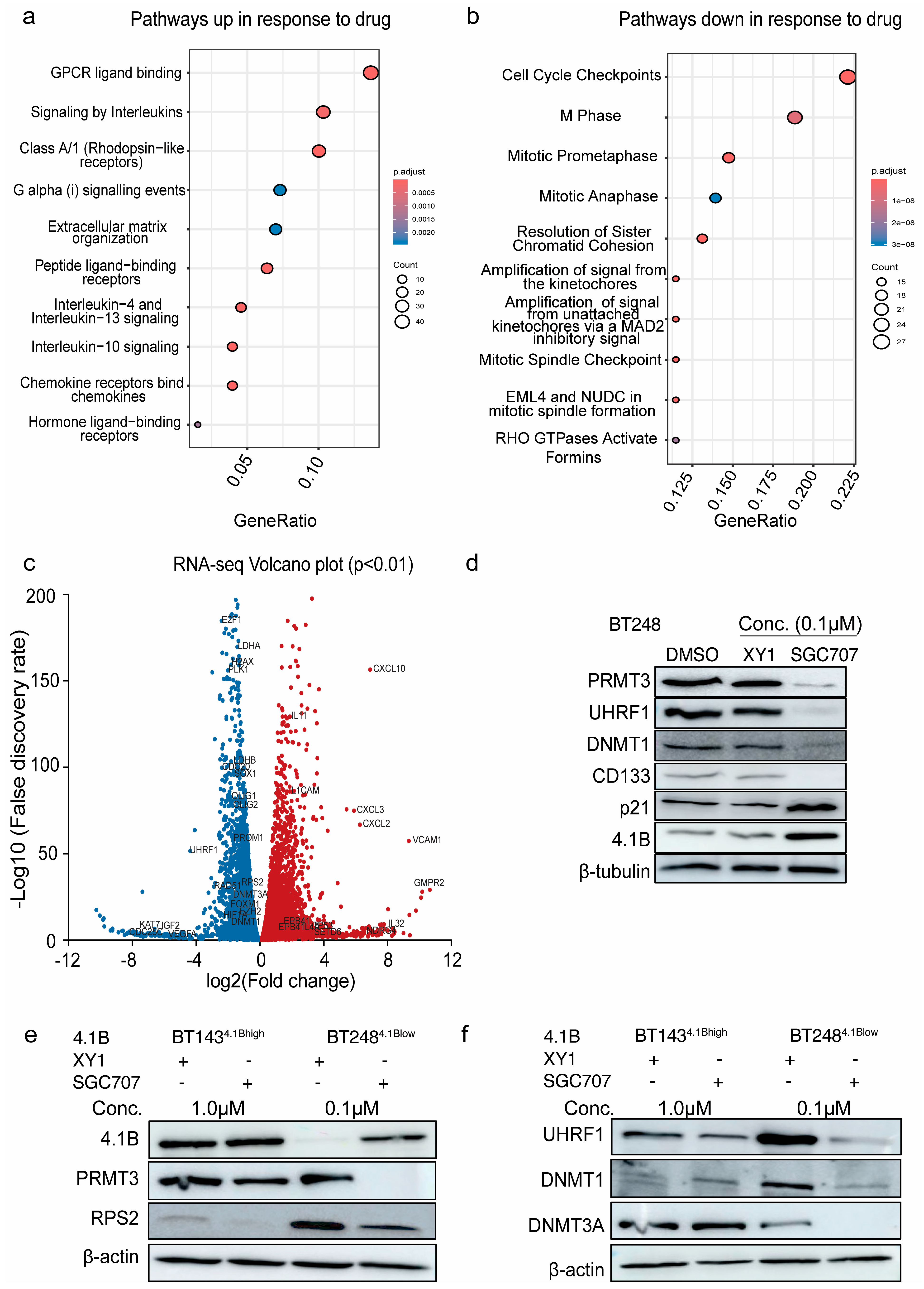
3.3. Inhibition of PRMT3 Activity with SGC707 Impacts the Expression of the Members of the DNA Methylation Complex, UHRF1/DNMT1
3.4. Induced Expression of 4.1B Impacts the Activity of PRMT3 and Its Downstream Effectors, UHRF1/DNMT1
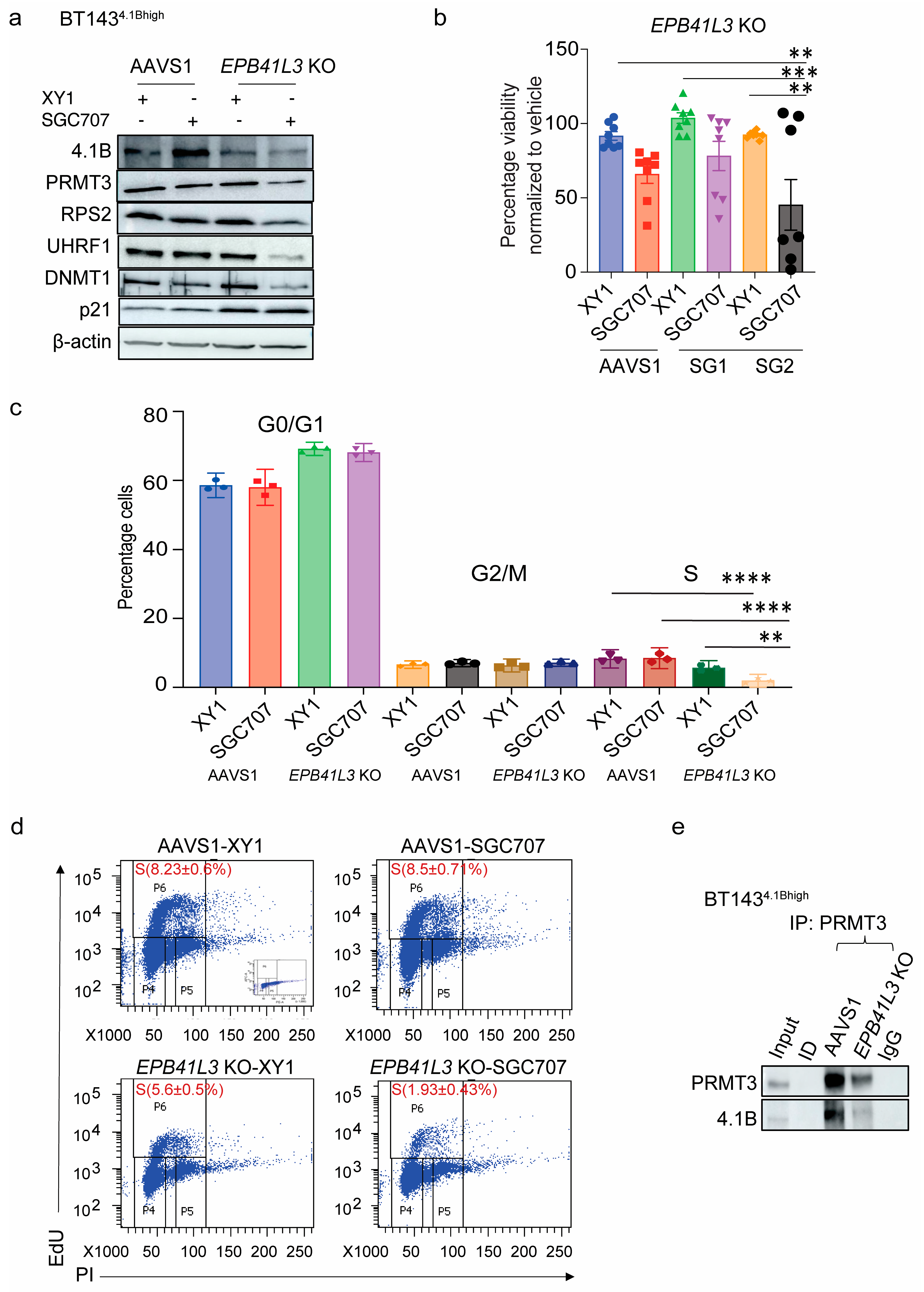
3.5. EPB41L3 Knockout Increases the Sensitivity of BTSCs to PRMT3 Inhibition
3.6. Inhibition of PRMT3 with SGC707 Changes the Subcellular Localization of Downstream Effector Molecules
3.7. Dual-Targeting of UHRF1 and DNMT1 Impacts Cell Viability in BTSCs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gayatri, S.; Bedford, M.T. Readers of histone methylarginine marks. Biochim. Biophys. Acta 2014, 1839, 702–710. [Google Scholar] [CrossRef]
- Wu, Q.; Schapira, M.; Arrowsmith, C.H.; Barsyte-Lovejoy, D. Protein arginine methylation: From enigmatic functions to therapeutic targeting. Nat. Rev. Drug Discov. 2021, 20, 509–530. [Google Scholar] [CrossRef]
- Miyata, S.; Mori, Y.; Tohyama, M. PRMT3 is essential for dendritic spine maturation in rat hippocampal neurons. Brain Res. 2010, 1352, 11–20. [Google Scholar] [CrossRef]
- Swiercz, R.; Cheng, D.; Kim, D.; Bedford, M.T. Ribosomal protein RPS2 is hypomethylated in PRMT3-deficient mice. J. Biol. Chem. 2007, 282, 16917–16923. [Google Scholar] [CrossRef]
- Hsu, S.-H.; Hung, W.-C. Protein arginine methyltransferase 3: A crucial regulator in metabolic reprogramming and gene expression in cancers. Cancer Lett. 2023, 554, 216008. [Google Scholar] [CrossRef]
- Liao, Y.; Luo, Z.; Lin, Y.; Chen, H.; Chen, T.; Xu, L.; Orgurek, S.; Berry, K.; Dzieciatkowska, M.; Reisz, J.A. PRMT3 drives glioblastoma progression by enhancing HIF1A and glycolytic metabolism. Cell Death Dis. 2022, 13, 943. [Google Scholar] [CrossRef]
- Hu, Y.; Su, Y.; He, Y.; Liu, W.; Xiao, B. Arginine methyltransferase PRMT3 promote tumorigenesis through regulating c-MYC stabilization in colorectal cancer. Gene 2021, 791, 145718. [Google Scholar] [CrossRef]
- Shi, Y.; Niu, Y.; Yuan, Y.; Li, K.; Zhong, C.; Qiu, Z.; Li, K.; Lin, Z.; Yang, Z.; Zuo, D.; et al. PRMT3-mediated arginine methylation of IGFBP1 promotes oxaliplatin resistance in liver cancer. Nat. Commun. 2023, 14, 1932. [Google Scholar] [CrossRef]
- Singh, V.; Miranda, T.B.; Jiang, W.; Frankel, A.E.; Roemer, M.A.; Robb, V.; Gutmann, D.H.; Herschman, H.R.; Clarke, S.; Newsham, I.F. DAL-1/4.1B tumor suppressor interacts with protein arginine N-methyltransferase 3 (PRMT3) and inhibits its ability to methylate substrates in vitro and in vivo. Oncogene 2004, 23, 7761–7777. [Google Scholar] [CrossRef]
- Perez-Janices, N.; Blanco-Luquin, I.; Tuñón, M.T.; Barba-Ramos, E.; Ibáñez, B.; Zazpe-Cenoz, I.; Martinez-Aguillo, M.; Hernandez, B.; Martínez-Lopez, E.; Fernández, A.F.; et al. EPB41L3, TSP-1 and RASSF2 as new clinically relevant prognostic biomarkers in diffuse gliomas. Oncotarget 2015, 6, 368–380. [Google Scholar] [CrossRef]
- Zeng, R.; Liu, Y.; Jiang, Z.-J.; Huang, J.-P.; Wang, Y.; Li, X.-F.; Xiong, W.-B.; Wu, X.-C.; Zhang, J.-R.; Wang, Q.-E.; et al. EPB41L3 is a potential tumor suppressor gene and prognostic indicator in esophageal squamous cell carcinoma. Int. J. Oncol. 2018, 52, 1443–1454. [Google Scholar] [CrossRef]
- Cai, M.; Guo, H.; Wang, D.; Zhao, T.; Liang, X.; Li, J.; Cui, X.; Fu, S.; Yu, J. Expression, DNA methylation pattern and transcription factor EPB41L3 in gastric cancer: A study of 262 cases. Cell Commun. Signal. 2024, 22, 470. [Google Scholar] [CrossRef]
- Bernkopf, D.B.; Williams, E.D. Potential role of EPB41L3 (protein 4.1B/Dal-1) as a target for treatment of advanced prostate cancer. Expert Opin. Ther. Targets 2008, 12, 845–853. [Google Scholar] [CrossRef]
- Werren, E.A.; Rodriguez Bey, G.; Majethia, P.; Kaur, P.; Patil, S.J.; Kekatpure, M.V.; Afenjar, A.; Qebibo, L.; Burglen, L.; Tomoum, H.; et al. Biallelic EPB4.1L3 variants underlie a developmental disorder with seizures and myelination defects. Brain 2024, 147, 4033–4042. [Google Scholar] [CrossRef]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1081. [Google Scholar] [CrossRef]
- Eisenbarth, D.; Wang, Y.A. Glioblastoma heterogeneity at single cell resolution. Oncogene 2023, 42, 2155–2165. [Google Scholar] [CrossRef]
- Greenwald, A.C.; Darnell, N.G.; Hoefflin, R.; Simkin, D.; Mount, C.W.; Castro, L.N.G.; Harnik, Y.; Dumont, S.; Hirsch, D.; Nomura, M.; et al. Integrative spatial analysis reveals a multi-layered organization of glioblastoma. Cell 2024, 187, 2485–2502.e26. [Google Scholar] [CrossRef]
- Shen, Y.; Grisdale, C.J.; Islam, S.A.; Bose, P.; Lever, J.; Zhao, E.Y.; Grinshtein, N.; Ma, Y.; Mungall, A.J.; Moore, R.A.; et al. Comprehensive genomic profiling of glioblastoma tumors, BTICs, and xenografts reveals stability and adaptation to growth environments. Proc. Natl. Acad. Sci. USA 2019, 116, 19098–19108. [Google Scholar] [CrossRef]
- Bahia, R.K.; Hao, X.; Hassam, R.; Cseh, O.; Bozek, D.A.; Luchman, H.A. Epigenetic and molecular coordination between HDAC2 and SMAD3-SKI regulates essential brain tumour stem cell characteristics. Nat. Commun. 2023, 14, 5051. [Google Scholar] [CrossRef]
- Hao, X.; Bahia, R.K.; Cseh, O.; Bozek, D.A.; Blake, S.; Rinnenthal, J.; Weyer-Czernilofsky, U.; Rudolph, D.; Artee Luchman, H. BI-907828, a novel potent MDM2 inhibitor, inhibits glioblastoma brain tumor stem cells in vitro and prolongs survival in orthotopic xenograft mouse models. Neuro Oncol. 2023, 25, 913–926. [Google Scholar] [CrossRef]
- Luchman, H.A.; Stechishin, O.D.; Nguyen, S.A.; Lun, X.Q.; Cairncross, J.G.; Weiss, S. Dual mTORC1/2 blockade inhibits glioblastoma brain tumor initiating cells in vitro and in vivo and synergizes with temozolomide to increase orthotopic xenograft survival. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 5756–5767. [Google Scholar] [CrossRef]
- Stechishin, O.D.; Luchman, H.A.; Ruan, Y.; Blough, M.D.; Nguyen, S.A.; Kelly, J.J.; Cairncross, J.G.; Weiss, S. On-target JAK2/STAT3 inhibition slows disease progression in orthotopic xenografts of human glioblastoma brain tumor stem cells. Neuro-Oncology 2013, 15, 198–207. [Google Scholar] [CrossRef]
- Chesnelong, C.; Restall, I.; Weiss, S. Isolation and culture of glioblastoma brain tumor stem cells. Methods Mol. Biol. 2019, 1869, 11–21. [Google Scholar]
- Cusulin, C.; Chesnelong, C.; Bose, P.; Bilenky, M.; Kopciuk, K.; Chan, J.A.; Cairncross, J.G.; Jones, S.J.; Marra, M.A.; Luchman, H.A.; et al. Precursor states of brain tumor initiating cell lines are predictive of survival in xenografts and associated with glioblastoma subtypes. Stem Cell Rep. 2015, 15, 1–9. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 2017, 32, 42–46. Available online: http://recur.bioinfo.cnio.es/ (accessed on 1 July 2025). [CrossRef]
- Kaur, R.; Aiken, C.; Morrison, L.C.; Rao, R.; Del Bigio, M.R.; Rampalli, S. OTX2 exhibit cell-context-dependent effects on cellular and molecular properties of human embryonic neural precursors and medulloblastoma cells. Dis. Model Mech. 2015, 8, 1295–1309. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Yu, G.; He, Q. ReactomePA: An RE/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 2016, 12, 477–479. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- UCSC Genome Browser. Available online: http://genome.ucsc.edu/ (accessed on 22 March 2024).
- Blanchart, A.; Navis, A.C.; Assaife-Lopes, N.; Usoskin, D.; Aranda, S.; Sontheimer, J. UHRF1 licensed self-renewal of active adult neural stem cells. Stem Cells 2018, 36, 1736–1751. [Google Scholar] [CrossRef]
- Kim, J.K.; Estève, P.O.; Jacobsen, S.E.; Pradhan, S. UHRF1 binds G9a and participates in p21 transcriptional regulation in mammalian cells. Nucleic Acids Res. 2009, 37, 493–505. [Google Scholar] [CrossRef]
- Sinha, R.; Allemand, E.; Zhang, Z.; Karni, R.; Myers, M.P.; Krainer, A.R. Arginine methylation controls the subcellular localization and functions of the oncoprotein splicing factor SF2/ASF. Mol. Cell Biol. 2010, 30, 2762–2774. [Google Scholar] [CrossRef]
- Kim, K.H.; Oprescu, S.N.; Snyder, M.M.; Kim, A.; Jia, Z.; Yue, F.; Kuang, S. PRMT5 mediates FoxO1 methylation and subcellular localization to regulate lipophagy in myogenic progenitors. Cell Rep. 2023, 42, 113329. [Google Scholar] [CrossRef]
- Hahm, J.Y.; Kim, J.-Y.; Park, J.W.; Kang, J.-Y.; Kim, K.-B.; Kim, S.-R.; Cho, H.; Seo, S.-B. Methylation of UHRF1 by SET7 is essential for DNA double-strand break repair. Nucleic Acids Res. 2019, 47, 184–196. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, Q.; Tan, S.; You, J.; Lyu, C.; Zhang, Y.; Han, M.; Chen, Z.; Li, J.; Wang, H.; et al. SET8 prevents excessive DNA methylation by methylation-mediated degradation of UHRF1 and DNMT. Nucleic Acids Res. 2019, 47, 9053–9068. [Google Scholar]
- Gu, L.; Fu, Y.; Li, X. Roles of post-translational modifications of UHRF1 in cancer. Epigenetics Chromatin 2024, 17, 15. [Google Scholar] [CrossRef]
- Wang, L.; Yang, X.; Zhao, K.; Huang, S.; Qin, Y.; Chen, Z.; Hu, X.; Jin, G.; Zhou, Z. MOF-mediated acetylation of UHRF1 enhances UHRF1 E3 ligase activity to facilitate DNA methylation maintenance. Cell Rep. 2024, 43, 113908. [Google Scholar] [CrossRef]
- Manu, K.S.; Chai, T.F.; The, J.T.; Zhu, W.L.; Casey, P.J.; Wang, M. Inhibition of Isoprenylcysteine Carboxymethyltransferase induces cell-cycle arrest and apoptosis through p21 and p21-regulated BNIP3 induction in pancreatic cancer. Mol. Cancer Ther. 2017, 16, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Seno, S.; Kimura, M.; Yashiro, Y.; Kimura, R.; Adachi, K.; Terabayashi, A.; Takahashi, M.; Oyama, T.; Abe, H.; Abe, T.; et al. β-Thujaplicin enhances TRAIL-induced apoptosis via the dual effects of XIAP inhibition and degradation in NCI-H460 human lung cancer cells. Medicines 2021, 8, 26. [Google Scholar] [CrossRef]
- Lee, Y.S.; Choi, K.M.; Kim, W.; Jeon, Y.S.; Lee, Y.M.; Hong, J.T.; Yun, Y.P.; Yoo, H.S. Hinokitiol inhibits cell growth through induction of S-phase arrest and apoptosis in human colon cancer cells and suppresses tumor growth in a mouse xenograft experiment. J. Nat. Prod. 2013, 76, 2195–2202. [Google Scholar] [CrossRef]
- Zhang, G.; He, J.; Ye, X.; Zhu, J.; Hu, X.; Shen, M.; Ma, Y.; Mao, Z.; Song, H.; Chen, F. β-Thujaplicin induces autophagic cell death, apoptosis, and cell cycle arrest through ROS-mediated Akt and p38/ERK MAPK signaling in human hepatocellular carcinoma. Cell Death Dis. 2019, 10, 255. [Google Scholar] [CrossRef]
- Myrianthopoulos, V.; Cartron, P.F.; Liutkevičiūtė, Z.; Klimašauskas, S.; Matulis, D.; Bronner, C.; Martinet, N.; Mikros, E. Tandem virtual screening targeting the SRA domain of UHRF1 identifies a novel chemical toll modulating DNA methylation. Eur. J. Med. Chem. 2016, 114, 390–396. [Google Scholar] [CrossRef]
- Zhao, Q.; Rank, G.; Tan, Y.T.; Li, H.; Moritz, R.L.; Simpson, R.J.; Cerruti, L.; Curtis, D.J.; Patel, D.J.; Allis, C.D.; et al. PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nat. Struct. Mol. Biol. 2009, 16, 304–311. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BTSC ID | Age | Sex | Diagnosis | MGMT Status | EGFR Status | p53 Status | PTEN Status | IDH1/2 Status |
|---|---|---|---|---|---|---|---|---|
| BT48 | 68 | male | GBM | M | Mt | wt | het | wt/wt |
| BT50 | 61 | male | GBM | um | Wt | wt | het | wt/wt |
| BT67 | 44 | male | GBM | M | Wt | wt | het | wt/wt |
| BT69 | 50 | male | GBM/GS | um | Het | wt | mt | wt/wt |
| BT89 | 59 | female | GBM | M | Wt | wt | wt | wt/wt |
| BT94 | 60 | female | GBM | M | Wt | wt | mt | wt/wt |
| BT147 | 56 | male | GBM-r | U | vIII | mt | mt | Wt/wt |
| BT189 | 55 | female | GBM | U | Wt | mt | wt | wt/wt |
| BT248Z | 56 | female | GBM | M | Wt | mt | wt | wt/wt |
| BT284 | - | - | GBM | - | Wt | mt | - | wt/- |
| BT119 | - | female | GBM-r | U | Mt | mt | Het/Het | wt/wt |
| BT143 | 46 | female | GBM-r | M | Wt | mt | het | wt/wt |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bahia, R.K.; Heemskerk, K.; Assaf, S.; Cseh, O.; Hao, X.; Hassam, R.; Prinos, P.; Luchman, H.A.; Weiss, S. Loss of 4.1B Drives PRMT3-Mediated Regulation of GBM Brain Tumour Stem Cell Growth. Int. J. Transl. Med. 2025, 5, 29. https://doi.org/10.3390/ijtm5030029
Bahia RK, Heemskerk K, Assaf S, Cseh O, Hao X, Hassam R, Prinos P, Luchman HA, Weiss S. Loss of 4.1B Drives PRMT3-Mediated Regulation of GBM Brain Tumour Stem Cell Growth. International Journal of Translational Medicine. 2025; 5(3):29. https://doi.org/10.3390/ijtm5030029
Chicago/Turabian StyleBahia, Ravinder K., Kyle Heemskerk, Samir Assaf, Orsolya Cseh, Xiaoguang Hao, Rozina Hassam, Panagiotis Prinos, H. Artee Luchman, and Samuel Weiss. 2025. "Loss of 4.1B Drives PRMT3-Mediated Regulation of GBM Brain Tumour Stem Cell Growth" International Journal of Translational Medicine 5, no. 3: 29. https://doi.org/10.3390/ijtm5030029
APA StyleBahia, R. K., Heemskerk, K., Assaf, S., Cseh, O., Hao, X., Hassam, R., Prinos, P., Luchman, H. A., & Weiss, S. (2025). Loss of 4.1B Drives PRMT3-Mediated Regulation of GBM Brain Tumour Stem Cell Growth. International Journal of Translational Medicine, 5(3), 29. https://doi.org/10.3390/ijtm5030029