
Protein Engineering Paving the Way for Next-Generation Therapies in Cancer



Abstract
1. Introduction
2. Foundations of Protein Engineering
2.1. Directed Evolution: Mimicking Natural Selection in the Lab
2.2. Rational Design: Precision Engineering Based on Structural Insights
2.3. Hybrid Approaches: Combining the Best of Both Worlds
2.4. Tools and Technologies in Protein Engineering
2.4.1. CRISPR-Based Systems for Directed Modifications
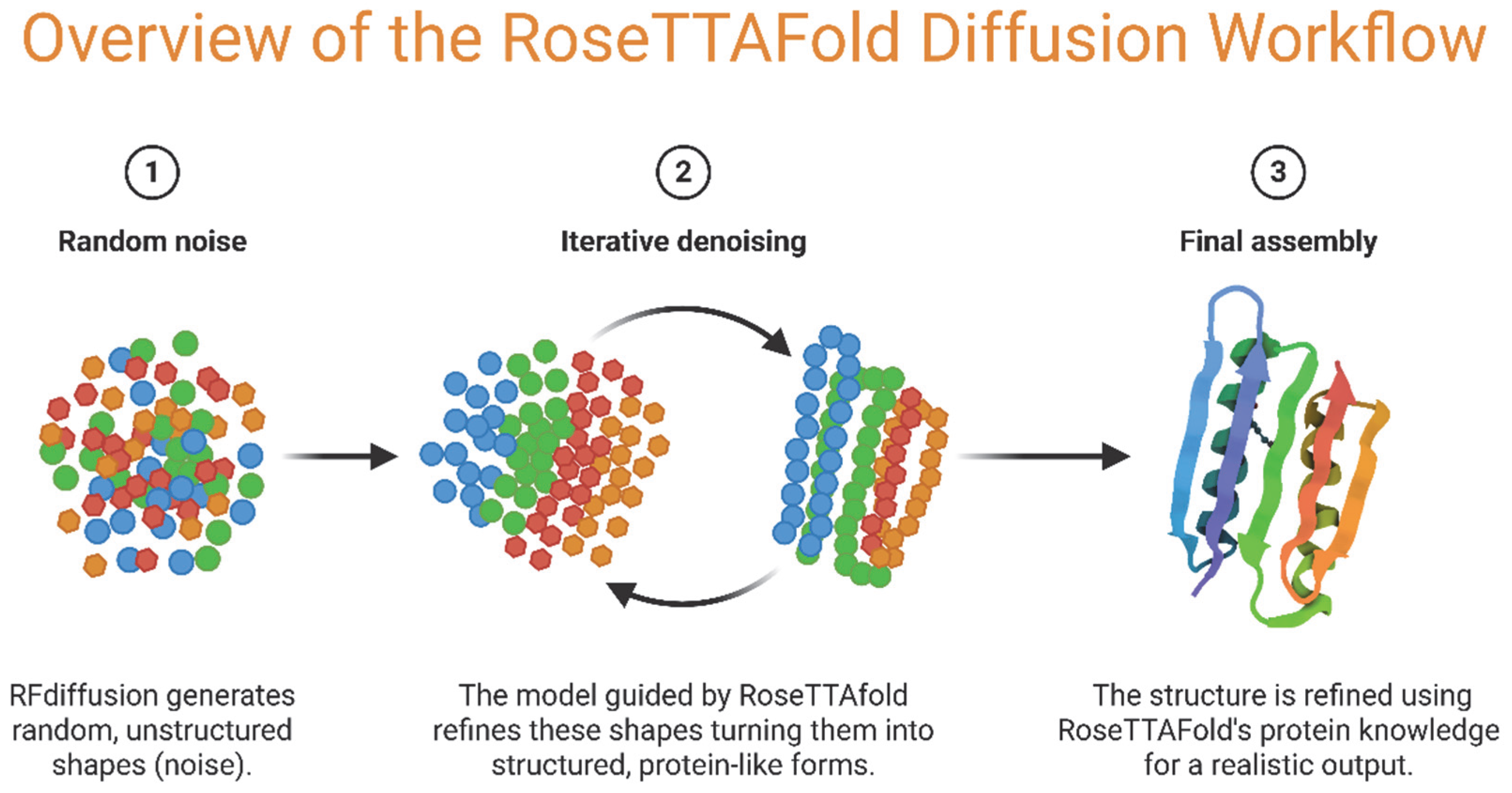
2.4.2. Advances in AI and Machine Learning for Protein Design
2.4.3. High-Throughput Screening and Automation
2.4.4. Synthetic Biology and De Novo Protein Design
3. Key Applications of Engineered Proteins in Cancer Therapeutics
3.1. Monoclonal Antibodies
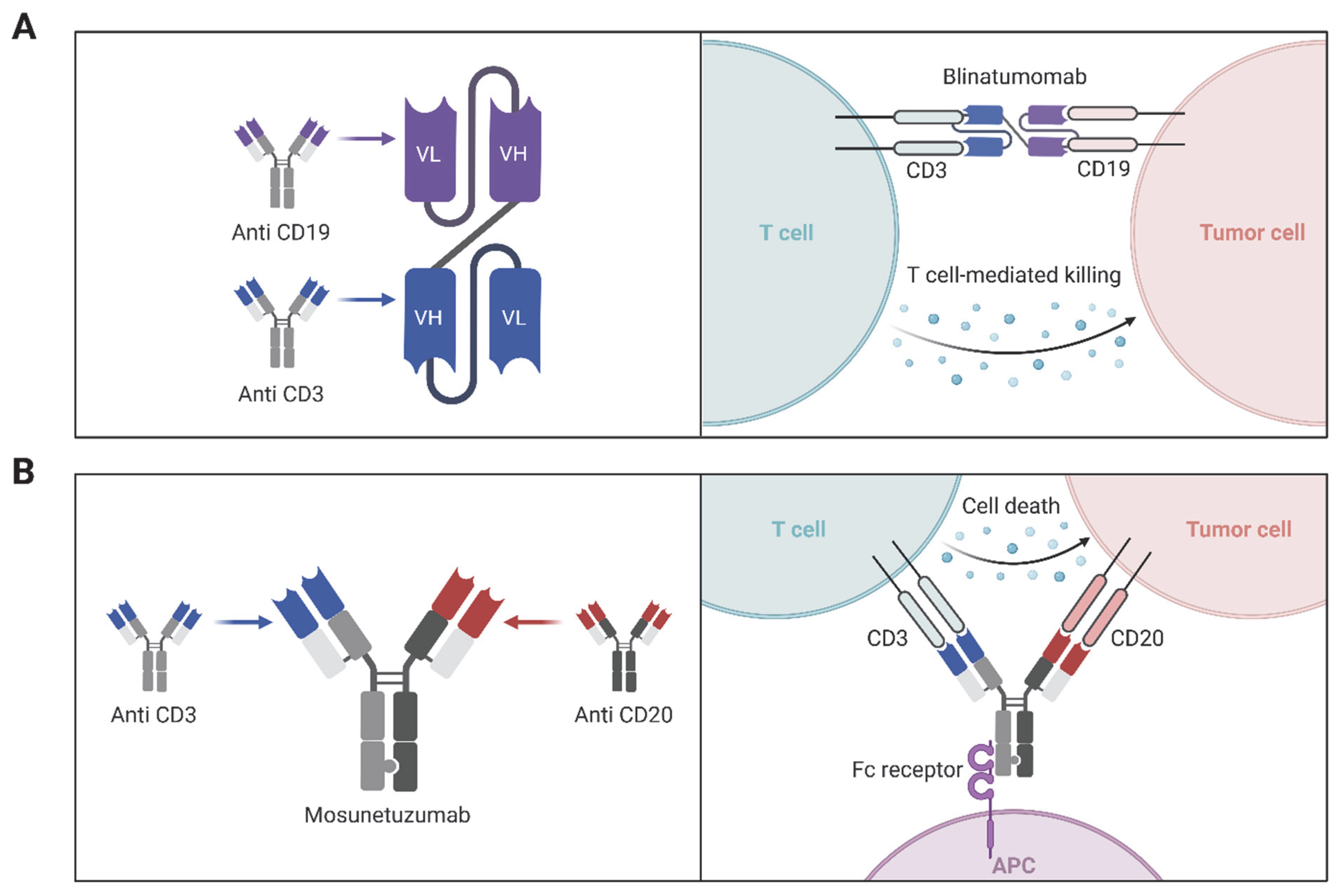
3.2. Bispecific and Multispecific Antibodies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody Name | Engineering Strategy | Target(s) | Mechanism/Engineering Purpose | Cancer Indication(s) | FDA Approval Year | Ref. |
|---|---|---|---|---|---|---|
| Trastuzumab (Herceptin) | Humanized IgG1 | HER2 | Humanization to reduce immunogenicity | HER2+ breast, gastric cancer | 1998 | [148] |
| Atezolizumab (Tecentriq) | Fc-engineered IgG1 | PD-L1 | Fc mutation reduces ADCC to preserve immune cells | NSCLC, urothelial carcinoma, | 2016 | [149] |
| Durvalumab (Imfinzi) | Fc-engineered IgG1 | PD-L1 | Reduced Fc effector function | NSCLC, SCLC, bladder cancer | 2017 | [150] |
| Blinatumomab (Blincyto) | Bispecific T cell engager | CD3/CD19 | BiTE format links T cells to B cells | B-cell ALL | 2014 | [151] |
| Mosunetuzumab | Bispecific antibody | CD20/CD3 | Redirects T cells to B cells | Follicular lymphoma | 2022 | [152] |
| Glofitamab | Bispecific antibody | CD20/CD3 | 2:1 binding format for enhanced avidity | DLBCL | 2023 | [153] |
| Brentuximab vedotin (Adcetris) | ADC | CD30 | MMAE cytotoxin via cleavable linker | Hodgkin lymphoma | 2011 | [154] |
| Trastuzumab emtansine (Kadcyla) | ADC | HER2 | DM1 payload conjugated to trastuzumab | HER2+ breast cancer | 2013 | [155] |
| Trastuzumab deruxtecan (Enhertu) | ADC | HER2 | Topoisomerase I inhibitor payload | HER2+ breast, gastric, lung cancers | 2019 | [156] |
| Sacituzumab govitecan (Trodelvy) | ADC | Trop-2 | SN-38 (irinotecan active form) conjugated | TNBC, urothelial carcinoma | 2020 | [157] |
| Margetuximab | Fc-engineered anti-HER2 | HER2 | Fc domain optimized for better FcγRIIIa binding (enhanced ADCC) | HER2+ breast cancer | 2020 | [158] |
| Elranatamab | Bispecific antibody | CD3/BCMA | Engages T cells with BCMA-expressing myeloma cells | Multiple myeloma | 2023 | [159] |
3.3. Engineered Cytokines and Fusion Proteins
3.4. Nanobodies and Single-Domain Antibodies
3.5. Protein-Based Drug Delivery Systems
4. Challenges and Limitations and Future
4.1. Protein Stability and Folding in Therapeutic Contexts
4.2. Immunogenicity and Strategies for Immune Evasion
4.3. Manufacturing and Scalability Issues
| Challenge | Underlying Issue | Example Case | Proposed Engineering Strategies |
|---|---|---|---|
| Protein Stability | Aggregation, misfolding, or degradation during production or storage | Bispecific antibodies prone to aggregation | Stabilizing mutations; computational folding models; optimized expression systems and formulations |
| Immunogenicity | Host immune system recognizes engineered protein as foreign | Anti-drug antibodies (ADAs) generated against cytokine fusion proteins | Deimmunization; humanization; epitope masking; computational T-cell epitope mapping |
| Manufacturing Scale-Up | Low yield, complex purification, batch variability | CAR T-cell therapies; bispecific antibody production | Automation; closed-system bioreactors; mammalian expression platforms; advanced chromatography |
| Regulatory Complexity | Need for extensive safety, efficacy, and stability data | Approval of bispecifics like blinatumomab required extended trials | Early engagement with regulators; adaptive trial design; real-world evidence generation |
| Cost and Accessibility | High development and production costs limit patient access | Limited access to ADCs and CAR T-cell therapies in low-resource settings | Biosimilar development; value-based pricing; process optimization for cost reduction |
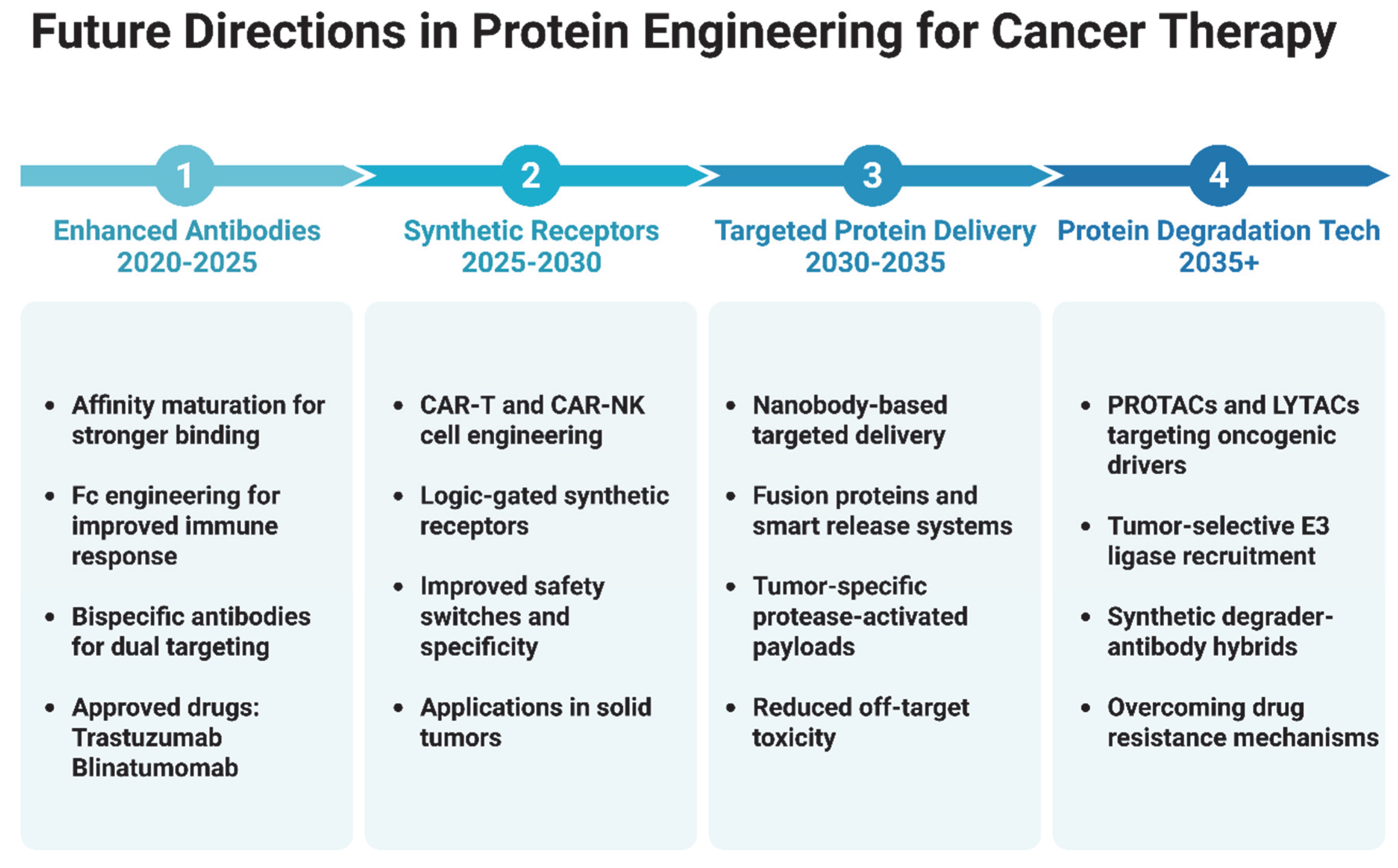
4.4. Future Perspectives and Research Directions
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Jacquemin, V.; Antoine, M.; Dom, G.; Detours, V.; Maenhaut, C.; Dumont, J.E. Dynamic Cancer Cell Heterogeneity: Diagnostic and Therapeutic Implications. Cancers 2022, 14, 280. [Google Scholar] [CrossRef]
- Liu, B.L.; Zhou, H.Y.; Tan, L.C.; Siu, K.T.H.; Guan, X.Y. Exploring treatment options in cancer: Tumor treatment strategies. Signal Transduct. Target. Ther. 2024, 9, 175. [Google Scholar] [CrossRef] [PubMed]
- El-Tanani, M.; Rabbani, S.A.; Babiker, R.; Rangraze, I.; Kapre, S.; Palakurthi, S.S.; Alnuqaydan, A.M.; Aljabali, A.A.; Rizzo, M.; El-Tanani, Y.; et al. Unraveling the tumor microenvironment: Insights into cancer metastasis and therapeutic strategies. Cancer Lett. 2024, 591, 216894. [Google Scholar] [CrossRef] [PubMed]
- Ghemrawi, R.; Abuamer, L.; Kremesh, S.; Hussien, G.; Ahmed, R.; Mousa, W.; Khoder, G.; Khair, M. Revolutionizing Cancer Treatment: Recent Advances in Immunotherapy. Biomedicines 2024, 12, 2158. [Google Scholar] [CrossRef]
- Papiez, M.A.; Krzysciak, W. Biological Therapies in the Treatment of Cancer-Update and New Directions. Int. J. Mol. Sci. 2021, 22, 11694. [Google Scholar] [CrossRef]
- Zahavi, D.; Weiner, L. Monoclonal Antibodies in Cancer Therapy. Antibodies 2020, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Eskafi, A.H.; Oghalaei, A.; Mahboudi, F.; Ghaderi, H.; Behdani, M.; Shoari, A.; Kazemi-Lomedasht, F. Investigation of the therapeutic potential of recombinant bispecific bivalent anti-PD-L1/VEGF nanobody in inhibition of angiogenesis. Immunopharm. Immunot. 2023, 45, 197–202. [Google Scholar] [CrossRef]
- Naderiyan, Z.; Sotoudeh, N.; Shoari, A.; Ghaderi, H.; Habibi-Anbouhi, M.; Moazzami, R.; Cohan, R.A.; Behdani, M. In Vitro and In Vivo Studies of a Heminecrolysin Toxin-VEGF Fusion Protein as a Novel Therapeutic for Solid Tumor Targeting. Mol. Biotechnol. 2023, 65, 766–773. [Google Scholar] [CrossRef]
- Mercogliano, M.F.; Bruni, S.; Mauro, F.L.; Schillaci, R. Emerging Targeted Therapies for HER2-Positive Breast Cancer. Cancers 2023, 15, 1987. [Google Scholar] [CrossRef]
- Salles, G.; Barrett, M.; Foa, R.; Maurer, J.; O’Brien, S.; Valente, N.; Wenger, M.; Maloney, D.G. Rituximab in B-Cell Hematologic Malignancies: A Review of 20 Years of Clinical Experience. Adv. Ther. 2017, 34, 2232–2273. [Google Scholar] [CrossRef]
- Tobin, P.H.; Richards, D.H.; Callender, R.A.; Wilson, C.J. Protein engineering: A new frontier for biological therapeutics. Curr. Drug Metab. 2014, 15, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Ndochinwa, G.O.; Wang, Q.Y.; Okoro, N.O.; Amadi, O.C.; Nwagu, T.N.; Nnamchi, C.I.; Moneke, A.N.; Odiba, A.S. New advances in protein engineering for industrial applications: Key takeaways. Open Life Sci. 2024, 19, 20220856. [Google Scholar] [CrossRef] [PubMed]
- Yuen, C.M.; Liu, D.R. Dissecting protein structure and function using directed evolution. Nat. Methods 2007, 4, 995–997. [Google Scholar] [CrossRef]
- Son, A.; Park, J.; Kim, W.; Yoon, Y.; Lee, S.; Park, Y.; Kim, H. Revolutionizing Molecular Design for Innovative Therapeutic Applications through Artificial Intelligence. Molecules 2024, 29, 4626. [Google Scholar] [CrossRef]
- Ocana, A.; Pandiella, A.; Privat, C.; Bravo, I.; Luengo-Oroz, M.; Amir, E.; Gyorffy, B. Integrating artificial intelligence in drug discovery and early drug development: A transformative approach. Biomark. Res. 2025, 13, 45. [Google Scholar] [CrossRef]
- Kintzing, J.R.; Filsinger Interrante, M.V.; Cochran, J.R. Emerging Strategies for Developing Next-Generation Protein Therapeutics for Cancer Treatment. Trends Pharmacol. Sci. 2016, 37, 993–1008. [Google Scholar] [CrossRef]
- Zhou, S.; Liu, M.; Ren, F.; Meng, X.; Yu, J. The landscape of bispecific T cell engager in cancer treatment. Biomark. Res. 2021, 9, 38. [Google Scholar] [CrossRef]
- Akram, F.; Ali, A.M.; Akhtar, M.T.; Fatima, T.; Shabbir, I.; ul Haq, I. The journey of antibody-drug conjugates for revolutionizing cancer therapy: A review. Bioorgan Med. Chem. 2025, 117, 118010. [Google Scholar] [CrossRef] [PubMed]
- Roybal, K.T.; Lim, W.A. Synthetic Immunology: Hacking Immune Cells to Expand Their Therapeutic Capabilities. Annu. Rev. Immunol. 2017, 35, 229–253. [Google Scholar] [CrossRef]
- Liao, H.C.; Liu, S.J. Advances in nucleic acid-based cancer vaccines. J. Biomed. Sci. 2025, 32, 10. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Barwal, A.; Sharma, N.; Mir, D.S.; Kumar, P.; Kumar, V. Therapeutic proteins: Developments, progress, challenges, and future perspectives. 3 Biotech 2024, 14, 112. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Morales, J.; Vanella, R.; Appelt, E.A.; Whillock, S.; Paulk, A.M.; Shusta, E.V.; Hackel, B.J.; Liu, C.C.; Nash, M.A. Protein Engineering and High-Throughput Screening by Yeast Surface Display: Survey of Current Methods. Small Sci. 2023, 3, 2300095. [Google Scholar] [CrossRef]
- McLure, R.J.; Radford, S.E.; Brockwell, D.J. High-throughput directed evolution: A golden era for protein science. Trends Chem. 2022, 4, 378–391. [Google Scholar] [CrossRef]
- Korendovych, I.V. Rational and Semirational Protein Design. Methods Mol. Biol. 2018, 1685, 15–23. [Google Scholar] [CrossRef]
- Selles Vidal, L.; Isalan, M.; Heap, J.T.; Ledesma-Amaro, R. A primer to directed evolution: Current methodologies and future directions. RSC Chem. Biol. 2023, 4, 271–291. [Google Scholar] [CrossRef]
- Yao, J.W.; Wang, X.G. Artificial intelligence in de novo protein design. Med. Nov. Technol. Devices 2025, 26, 100366. [Google Scholar] [CrossRef]
- Cobb, R.E.; Chao, R.; Zhao, H. Directed Evolution: Past, Present and Future. AIChE J. 2013, 59, 1432–1440. [Google Scholar] [CrossRef]
- Romero, P.A.; Arnold, F.H. Exploring protein fitness landscapes by directed evolution. Nat. Rev. Mol. Cell Biol. 2009, 10, 866–876. [Google Scholar] [CrossRef]
- Xiao, H.; Bao, Z.; Zhao, H. High Throughput Screening and Selection Methods for Directed Enzyme Evolution. Ind. Eng. Chem. Res. 2015, 54, 4011–4020. [Google Scholar] [CrossRef]
- Nannemann, D.P.; Birmingham, W.R.; Scism, R.A.; Bachmann, B.O. Assessing directed evolution methods for the generation of biosynthetic enzymes with potential in drug biosynthesis. Future Med. Chem. 2011, 3, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Cobb, R.E.; Sun, N.; Zhao, H. Directed evolution as a powerful synthetic biology tool. Methods 2013, 60, 81–90. [Google Scholar] [CrossRef]
- Boder, E.T.; Midelfort, K.S.; Wittrup, K.D. Directed evolution of antibody fragments with monovalent femtomolar antigen-binding affinity. Proc. Natl. Acad. Sci. USA 2000, 97, 10701–10705. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Chandley, P.; Rohatgi, S. Recent Advances in the Development of Monoclonal Antibodies and Next-Generation Antibodies. Immunohorizons 2023, 7, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Hibbert, E.G.; Dalby, P.A. Directed evolution strategies for improved enzymatic performance. Microb. Cell Factories 2005, 4, 29. [Google Scholar] [CrossRef]
- Neylon, C. Chemical and biochemical strategies for the randomization of protein encoding DNA sequences: Library construction methods for directed evolution. Nucleic Acids Res. 2004, 32, 1448–1459. [Google Scholar] [CrossRef]
- Marshall, S.A.; Lazar, G.A.; Chirino, A.J.; Desjarlais, J.R. Rational design and engineering of therapeutic proteins. Drug Discov. Today 2003, 8, 212–221. [Google Scholar] [CrossRef]
- Tiwari, M.K.; Singh, R.; Singh, R.K.; Kim, I.W.; Lee, J.K. Computational approaches for rational design of proteins with novel functionalities. Comput. Struct. Biotechnol. J. 2012, 2, e201209002. [Google Scholar] [CrossRef]
- Maveyraud, L.; Mourey, L. Protein X-ray Crystallography and Drug Discovery. Molecules 2020, 25, 1030. [Google Scholar] [CrossRef]
- Ntountaniotis, D. Reactions in NMR Tubes as Key Weapon in Rational Drug Design. Methods Mol. Biol. 2018, 1824, 417–430. [Google Scholar] [CrossRef]
- Cebi, E.; Lee, J.; Subramani, V.K.; Bak, N.; Oh, C.; Kim, K.K. Cryo-electron microscopy-based drug design. Front. Mol. Biosci. 2024, 11, 1342179. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, Z.; Scheuring, S. A structural biology compatible file format for atomic force microscopy. Nat. Commun. 2025, 16, 1671. [Google Scholar] [CrossRef] [PubMed]
- Salo-Ahen, O.M.H.; Alanko, I.; Bhadane, R.; Bonvin, A.M.J.J.; Honorato, R.V.; Hossain, S.; Juffer, A.H.; Kabedev, A.; Lahtela-Kakkonen, M.; Larsen, A.S.; et al. Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development. Processes 2021, 9, 71. [Google Scholar] [CrossRef]
- Ferreira, P.; Fernandes, P.A.; Ramos, M.J. Modern computational methods for rational enzyme engineering. Chem Catal. 2022, 2, 2481–2498. [Google Scholar] [CrossRef]
- Harris, C.T.; Cohen, S. Reducing Immunogenicity by Design: Approaches to Minimize Immunogenicity of Monoclonal Antibodies. BioDrugs 2024, 38, 205–226. [Google Scholar] [CrossRef] [PubMed]
- Pongsupasa, V.; Anuwan, P.; Maenpuen, S.; Wongnate, T. Rational-Design Engineering to Improve Enzyme Thermostability. Methods Mol. Biol. 2022, 2397, 159–178. [Google Scholar] [CrossRef]
- Shi, H.; Fu, M.; Zhang, T.; Zhang, X.; Yao, L.; Xue, C.; Tang, C. Rational Design of Formate Dehydrogenase for Enhanced Thermal Stability and Catalytic Activity in Bioelectrocatalysis. J. Agric. Food Chem. 2024, 72, 23333–23344. [Google Scholar] [CrossRef]
- Huggins, D.J.; Sherman, W.; Tidor, B. Rational approaches to improving selectivity in drug design. J. Med. Chem. 2012, 55, 1424–1444. [Google Scholar] [CrossRef]
- Li, Y.C.; Zhang, R.F.; Yan, X.Y.; Fan, K.L. Machine learning facilitating the rational design of nanozymes. J. Mater. Chem. B 2023, 11, 6466–6477. [Google Scholar] [CrossRef]
- de Oliveira, T.M.; van Beek, L.; Shilliday, F.; Debreczeni, J.E.; Phillips, C. Cryo-EM: The Resolution Revolution and Drug Discovery. SLAS Discov. 2021, 26, 17–31. [Google Scholar] [CrossRef]
- Garibsingh, R.A.; Ndaru, E.; Garaeva, A.A.; Shi, Y.; Zielewicz, L.; Zakrepine, P.; Bonomi, M.; Slotboom, D.J.; Paulino, C.; Grewer, C.; et al. Rational design of ASCT2 inhibitors using an integrated experimental-computational approach. Proc. Natl. Acad. Sci. USA 2021, 118, e2104093118. [Google Scholar] [CrossRef] [PubMed]
- Nepali, K.; Sharma, S.; Sharma, M.; Bedi, P.M.; Dhar, K.L. Rational approaches, design strategies, structure activity relationship and mechanistic insights for anticancer hybrids. Eur. J. Med. Chem. 2014, 77, 422–487. [Google Scholar] [CrossRef]
- Phintha, A.; Chaiyen, P. Rational and mechanistic approaches for improving biocatalyst performance. Chem. Catal. 2022, 2, 2614–2643. [Google Scholar] [CrossRef]
- Molina-Espeja, P.; Vina-Gonzalez, J.; Gomez-Fernandez, B.J.; Martin-Diaz, J.; Garcia-Ruiz, E.; Alcalde, M. Beyond the outer limits of nature by directed evolution. Biotechnol. Adv. 2016, 34, 754–767. [Google Scholar] [CrossRef]
- Spirov, A.; Holloway, D. Using evolutionary computations to understand the design and evolution of gene and cell regulatory networks. Methods 2013, 62, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Huttanus, H.M.; Triola, E.H.; Velasquez-Guzman, J.C.; Shin, S.M.; Granja-Travez, R.S.; Singh, A.; Dale, T.; Jha, R.K. Targeted mutagenesis and high-throughput screening of diversified gene and promoter libraries for isolating gain-of-function mutations. Front. Bioeng. Biotechnol. 2023, 11, 1202388. [Google Scholar] [CrossRef]
- Shi, J.H.; Yuan, B.; Yang, H.Q.; Sun, Z.T. Recent advances on protein engineering for improved stability. Biodesign Res. 2025, 7, 100005. [Google Scholar] [CrossRef]
- Iwasaki, Y.W.; Tharakaraman, K.; Subramanian, V.; Khongmanee, A.; Hatas, A.; Fleischer, E.; Rurak, T.T.; Ngok-Ngam, P.; Tit-Oon, P.; Ruchirawat, M.; et al. Generation of bispecific antibodies by structure-guided redesign of IgG constant regions. Front. Immunol. 2022, 13, 1063002. [Google Scholar] [CrossRef]
- Chen, X.; Zaro, J.L.; Shen, W.C. Fusion protein linkers: Property, design and functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef]
- Alanazi, W.; Meng, D.; Pollastri, G. Advancements in one-dimensional protein structure prediction using machine learning and deep learning. Comput. Struct. Biotechnol. J. 2025, 27, 1416–1430. [Google Scholar] [CrossRef]
- Malhotra, Y.; John, J.; Yadav, D.; Sharma, D.; Rawal, K.; Mishra, V.; Chaturvedi, N. Advancements in protein structure prediction: A comparative overview of AlphaFold and its derivatives. Comput. Biol. Med. 2025, 188, 109842. [Google Scholar] [CrossRef]
- Ye, L.; Yang, C.; Yu, H. From molecular engineering to process engineering: Development of high-throughput screening methods in enzyme directed evolution. Appl. Microbiol. Biotechnol. 2018, 102, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Bhokisham, N.; Laudermilch, E.; Traeger, L.L.; Bonilla, T.D.; Ruiz-Estevez, M.; Becker, J.R. CRISPR-Cas System: The Current and Emerging Translational Landscape. Cells 2023, 12, 1103. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.A. CRISPR-Cas9: A fascinating journey from bacterial immune system to human gene editing. Prog. Mol. Biol. Transl. Sci. 2021, 178, 63–83. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.; Shirguppe, S.; Perez-Pinera, P. Protein Engineering Technologies for Development of Next-Generation Genome Editors. Curr. Opin. Biomed. Eng. 2023, 28, 100514. [Google Scholar] [CrossRef]
- Asmamaw, M.; Zawdie, B. Mechanism and Applications of CRISPR/Cas-9-Mediated Genome Editing. Biol. Targets Ther. 2021, 15, 353–361. [Google Scholar] [CrossRef]
- Liao, H.; Wu, J.; VanDusen, N.J.; Li, Y.; Zheng, Y. CRISPR-Cas9-mediated homology-directed repair for precise gene editing. Mol. Ther. Nucleic Acids 2024, 35, 102344. [Google Scholar] [CrossRef]
- Lei, T.; Wang, Y.; Zhang, Y.; Yang, Y.; Cao, J.; Huang, J.; Chen, J.; Chen, H.; Zhang, J.; Wang, L.; et al. Leveraging CRISPR gene editing technology to optimize the efficacy, safety and accessibility of CAR T-cell therapy. Leukemia 2024, 38, 2517–2543. [Google Scholar] [CrossRef]
- Dimitri, A.; Herbst, F.; Fraietta, J.A. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol. Cancer 2022, 21, 78. [Google Scholar] [CrossRef]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef]
- Pickar-Oliver, A.; Gersbach, C.A. The next generation of CRISPR-Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 2019, 20, 490–507. [Google Scholar] [CrossRef]
- Fu, Y.; He, X.; Gao, X.D.; Li, F.; Ge, S.; Yang, Z.; Fan, X. Prime editing: Current advances and therapeutic opportunities in human diseases. Sci. Bull. 2023, 68, 3278–3291. [Google Scholar] [CrossRef] [PubMed]
- Son, A.; Park, J.; Kim, W.; Lee, W.; Yoon, Y.; Ji, J.; Kim, H. Integrating Computational Design and Experimental Approaches for Next-Generation Biologics. Biomolecules 2024, 14, 1073. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, Q.; Nasif, K.F.A.; Xie, Y.; Deng, B.; Niu, S.; Pouriyeh, S.; Dai, Z.; Chen, J.; Xie, C.Y. AI-Driven Deep Learning Techniques in Protein Structure Prediction. Int. J. Mol. Sci. 2024, 25, 8426. [Google Scholar] [CrossRef] [PubMed]
- Pearce, R.; Zhang, Y. Deep learning techniques have significantly impacted protein structure prediction and protein design. Curr. Opin. Struct. Biol. 2021, 68, 194–207. [Google Scholar] [CrossRef]
- Yang, Z.; Zeng, X.; Zhao, Y.; Chen, R. AlphaFold2 and its applications in the fields of biology and medicine. Signal Transduct. Target. Ther. 2023, 8, 115. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Agarwal, V.; McShan, A.C. The power and pitfalls of AlphaFold2 for structure prediction beyond rigid globular proteins. Nat. Chem. Biol. 2024, 20, 950–959. [Google Scholar] [CrossRef]
- Bouatta, N.; Sorger, P.; AlQuraishi, M. Protein structure prediction by AlphaFold2: Are attention and symmetries all you need? Biol. Crystallogr. 2021, 77, 982–991. [Google Scholar] [CrossRef]
- Krishna, R.; Wang, J.; Ahern, W.; Sturmfels, P.; Venkatesh, P.; Kalvet, I.; Lee, G.R.; Morey-Burrows, F.S.; Anishchenko, I.; Humphreys, I.R.; et al. Generalized biomolecular modeling and design with RoseTTAFold All-Atom. Science 2024, 384, eadl2528. [Google Scholar] [CrossRef] [PubMed]
- You, Y.J.; Lai, X.; Pan, Y.; Zheng, H.R.; Vera, J.; Liu, S.; Deng, S.; Zhang, L. Artificial intelligence in cancer target identification and drug discovery. Signal Transduct. Target. Ther. 2022, 7, 156. [Google Scholar] [CrossRef]
- Rehman, A.U.; Li, M.; Wu, B.; Ali, Y.; Rasheed, S.; Shaheen, S.; Liu, X.; Luo, R.; Zhang, J. Role of artificial intelligence in revolutionizing drug discovery. Fundam. Res. 2024, 5, 1273–1287. [Google Scholar] [CrossRef]
- Grewal, S.; Hegde, N.; Yanow, S.K. Integrating machine learning to advance epitope mapping. Front. Immunol. 2024, 15, 1463931. [Google Scholar] [CrossRef]
- Fan, J.; Shi, S.; Xiang, H.; Fu, L.; Duan, Y.; Cao, D.; Lu, H. Predicting Elimination of Small-Molecule Drug Half-Life in Pharmacokinetics Using Ensemble and Consensus Machine Learning Methods. J. Chem. Inf. Model. 2024, 64, 3080–3092. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Mahdi, M.S.; Radi, U.K.; Jihad, A.; Abdulhussein, A.H.; Ahmad, I.; Mansuri, N.; Abdalrahman, M.A.; Alkhayyat, A.; Faisal, A. Machine learning based modeling for estimation of drug solubility in supercritical fluid by adjusting important parameters. Chemom. Intell. Lab. Syst. 2024, 254, 105241. [Google Scholar] [CrossRef]
- Eugster, R.; Orsi, M.; Buttitta, G.; Serafini, N.; Tiboni, M.; Casettari, L.; Reymond, J.L.; Aleandri, S.; Luciani, P. Leveraging machine learning to streamline the development of liposomal drug delivery systems. J. Control. Release 2024, 376, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Vanella, R.; Kovacevic, G.; Doffini, V.; Fernandez de Santaella, J.; Nash, M.A. High-throughput screening, next generation sequencing and machine learning: Advanced methods in enzyme engineering. Chem. Commun. 2022, 58, 2455–2467. [Google Scholar] [CrossRef]
- Khakzad, H.; Igashov, I.; Schneuing, A.; Goverde, C.; Bronstein, M.; Correia, B. A new age in protein design empowered by deep learning. Cell Syst. 2023, 14, 925–939. [Google Scholar] [CrossRef]
- Minot, M.; Reddy, S.T. Meta learning addresses noisy and under-labeled data in machine learning-guided antibody engineering. Cell Syst. 2024, 15, 4–18.e14. [Google Scholar] [CrossRef]
- Shoari, A.; Khalili-Tanha, G.; Coban, M.A.; Radisky, E.S. Structure and computation-guided yeast surface display for the evolution of TIMP-based matrix metalloproteinase inhibitors. Front. Mol. Biosci. 2023, 10, 1321956. [Google Scholar] [CrossRef] [PubMed]
- Lutz, S. Beyond directed evolution-semi-rational protein engineering and design. Curr. Opin. Biotechnol. 2010, 21, 734–743. [Google Scholar] [CrossRef]
- Vasina, M.; Velecky, J.; Planas-Iglesias, J.; Marques, S.M.; Skarupova, J.; Damborsky, J.; Bednar, D.; Mazurenko, S.; Prokop, Z. Tools for computational design and high-throughput screening of therapeutic enzymes. Adv. Drug Deliv. Rev. 2022, 183, 114143. [Google Scholar] [CrossRef]
- Zhu, Z.; Cuozzo, J. Review article: High-throughput affinity-based technologies for small-molecule drug discovery. SLAS Discov. 2009, 14, 1157–1164. [Google Scholar] [CrossRef] [PubMed]
- Bommarius, A.S.; Broering, J.M.; Chaparro-Riggers, J.F.; Polizzi, K.M. High-throughput screening for enhanced protein stability. Curr. Opin. Biotechnol. 2006, 17, 606–610. [Google Scholar] [CrossRef]
- Cheong, R.; Paliwal, S.; Levchenko, A. High-content screening in microfluidic devices. Expert Opin. Drug Discov. 2010, 5, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Murthy, T.; Lim, J. Life Sciences discovery and technology highlights. SLAS Technol. 2025, 30, 100235. [Google Scholar] [CrossRef]
- Battersby, B.J.; Trau, M. Novel miniaturized systems in high-throughput screening. Trends Biotechnol. 2002, 20, 167–173. [Google Scholar] [CrossRef]
- Dörr, M.; Fibinger, M.P.C.; Last, D.; Schmidt, S.; Santos-Aberturas, J.; Böttcher, D.; Hummel, A.; Vickers, C.; Voss, M.; Bornscheuer, U.T. Fully automatized high-throughput enzyme library screening using a robotic platform. Biotechnol. Bioeng. 2016, 113, 1421–1432. [Google Scholar] [CrossRef]
- Leet, J.E.; Belcastro, J.V.; Dowling, C.J.; Nemeth, G.A.; Weller, H.N. HPLC Biogram Analysis: A Powerful Tool Used for Hit Confirmation in Early Drug Discovery. J. Biomol. Screen. 2015, 20, 681–687. [Google Scholar] [CrossRef]
- Weng, L.; Spoonamore, J.E. Droplet Microfluidics-Enabled High-Throughput Screening for Protein Engineering. Micromachines 2019, 10, 734. [Google Scholar] [CrossRef] [PubMed]
- Kouba, P.; Kohout, P.; Haddadi, F.; Bushuiev, A.; Samusevich, R.; Sedlar, J.; Damborsky, J.; Pluskal, T.; Sivic, J.; Mazurenko, S. Machine Learning-Guided Protein Engineering. ACS Catal. 2023, 13, 13863–13895. [Google Scholar] [CrossRef]
- Zhang, Y.; Minagawa, Y.; Kizoe, H.; Miyazaki, K.; Iino, R.; Ueno, H.; Tabata, K.V.; Shimane, Y.; Noji, H. Accurate high-throughput screening based on digital protein synthesis in a massively parallel femtoliter droplet array. Sci. Adv. 2019, 5, eaav8185. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Luo, Y.; Zhao, H. Synthetic biology: Putting synthesis into biology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011, 3, 7–20. [Google Scholar] [CrossRef]
- Zhou, W.; Smidlehner, T.; Jerala, R. Synthetic biology principles for the design of protein with novel structures and functions. FEBS Lett. 2020, 594, 2199–2212. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Kortemme, T. Recent advances in de novo protein design: Principles, methods, and applications. J. Biol. Chem. 2021, 296, 100558. [Google Scholar] [CrossRef]
- Korendovych, I.V.; DeGrado, W.F. De novo protein design, a retrospective. Q. Rev. Biophys. 2020, 53, e3. [Google Scholar] [CrossRef]
- Miyamoto, K.; Tadokoro, T.; Matsumoto, A. Unique E2-binding specificity of artificial RING fingers in cancer cells. Sci. Rep. 2024, 14, 2545. [Google Scholar] [CrossRef]
- Lu, H.; Cheng, Z.; Hu, Y.; Tang, L.V. What Can De Novo Protein Design Bring to the Treatment of Hematological Disorders? Biology 2023, 12, 166. [Google Scholar] [CrossRef]
- Quijano-Rubio, A.; Ulge, U.Y.; Walkey, C.D.; Silva, D.A. The advent of de novo proteins for cancer immunotherapy. Curr. Opin. Chem. Biol. 2020, 56, 119–128. [Google Scholar] [CrossRef]
- Shirzadian, M.; Moori, S.; Rabbani, R.; Rahbarizadeh, F. SynNotch CAR-T cell, when synthetic biology and immunology meet again. Front. Immunol. 2025, 16, 1545270. [Google Scholar] [CrossRef] [PubMed]
- Allen, G.M.; Frankel, N.W.; Reddy, N.R.; Bhargava, H.K.; Yoshida, M.A.; Stark, S.R.; Purl, M.; Lee, J.; Yee, J.L.; Yu, W.; et al. Synthetic cytokine circuits that drive T cells into immune-excluded tumors. Science 2022, 378, eaba1624. [Google Scholar] [CrossRef] [PubMed]
- Crunkhorn, S. Synthetic cytokine circuit targets solid tumours. Nat. Rev. Drug Discov. 2023, 22, 99. [Google Scholar] [CrossRef]
- Plaper, T.; Rihtar, E.; Zeleznik Ramuta, T.; Forstneric, V.; Jazbec, V.; Ivanovski, F.; Bencina, M.; Jerala, R. The art of designed coiled-coils for the regulation of mammalian cells. Cell Chem. Biol. 2024, 31, 1460–1472. [Google Scholar] [CrossRef]
- Burgos-Morales, O.; Gueye, M.; Lacombe, L.; Nowak, C.; Schmachtenberg, R.; Horner, M.; Jerez-Longres, C.; Mohsenin, H.; Wagner, H.J.; Weber, W. Synthetic biology as driver for the biologization of materials sciences. Mater. Today Bio 2021, 11, 100115. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Yin, H.; Zhang, D.; Zhang, M.; Chao, X.; Scimeca, L.; Wu, M.R. Synthetic biology approaches for improving the specificity and efficacy of cancer immunotherapy. Cell. Mol. Immunol. 2024, 21, 436–447. [Google Scholar] [CrossRef]
- Valabrega, G.; Montemurro, F.; Aglietta, M. Trastuzumab: Mechanism of action, resistance and future perspectives in HER2-overexpressing breast cancer. Ann. Oncol. 2007, 18, 977–984. [Google Scholar] [CrossRef]
- Hoffman, L.M.; Gore, L. Blinatumomab, a Bi-Specific Anti-CD19/CD3 BiTE((R)) Antibody for the Treatment of Acute Lymphoblastic Leukemia: Perspectives and Current Pediatric Applications. Front. Oncol. 2014, 4, 63. [Google Scholar] [CrossRef]
- Mebrahtu, A.; Lauren, I.; Veerman, R.; Akpinar, G.G.; Lord, M.; Kostakis, A.; Astorga-Wells, J.; Dahllund, L.; Olsson, A.; Andersson, O.; et al. A bispecific CD40 agonistic antibody allowing for antibody-peptide conjugate formation to enable cancer-specific peptide delivery, resulting in improved T proliferation and anti-tumor immunity in mice. Nat. Commun. 2024, 15, 9542. [Google Scholar] [CrossRef]
- Diab, A.; Tannir, N.M.; Bentebibel, S.E.; Hwu, P.; Papadimitrakopoulou, V.; Haymaker, C.; Kluger, H.M.; Gettinger, S.N.; Sznol, M.; Tykodi, S.S.; et al. Bempegaldesleukin (NKTR-214) plus Nivolumab in Patients with Advanced Solid Tumors: Phase I Dose-Escalation Study of Safety, Efficacy, and Immune Activation (PIVOT-02). Cancer Discov. 2020, 10, 1158–1173. [Google Scholar] [CrossRef]
- Ongaro, T.; Gouyou, B.; Stringhini, M.; Corbellari, R.; Neri, D.; Villa, A. A novel format for recombinant antibody-interleukin-2 fusion proteins exhibits superior tumor-targeting properties in vivo. Oncotarget 2020, 11, 3698–3711. [Google Scholar] [CrossRef]
- Minnar, C.M.; Lui, G.; Gulley, J.L.; Schlom, J.; Gameiro, S.R. Preclinical and clinical studies of a tumor targeting IL-12 immunocytokine. Front. Oncol. 2023, 13, 1321318. [Google Scholar] [CrossRef]
- Gondry, O.; Caveliers, V.; Xavier, C.; Raes, L.; Vanhoeij, M.; Verfaillie, G.; Fontaine, C.; Glorieus, K.; De Greve, J.; Joris, S.; et al. Phase II Trial Assessing the Repeatability and Tumor Uptake of [68Ga]Ga-HER2 Single-Domain Antibody PET/CT in Patients with Breast Carcinoma. J. Nucl. Med. 2024, 65, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Narbona, J.; Hernandez-Baraza, L.; Gordo, R.G.; Sanz, L.; Lacadena, J. Nanobody-Based EGFR-Targeting Immunotoxins for Colorectal Cancer Treatment. Biomolecules 2023, 13, 1042. [Google Scholar] [CrossRef]
- Palombarini, F.; Di Fabio, E.; Boffi, A.; Macone, A.; Bonamore, A. Ferritin Nanocages for Protein Delivery to Tumor Cells. Molecules 2020, 25, 825. [Google Scholar] [CrossRef]
- Delgado, M.; Garcia-Sanz, J.A. Therapeutic Monoclonal Antibodies against Cancer: Present and Future. Cells 2023, 12, 2837. [Google Scholar] [CrossRef] [PubMed]
- Harding, F.A.; Stickler, M.M.; Razo, J.; DuBridge, R.B. The immunogenicity of humanized and fully human antibodies: Residual immunogenicity resides in the CDR regions. MAbs 2010, 2, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Oldham, R.J.; Teal, E.; Beers, S.A.; Cragg, M.S. Fc-Engineering for Modulated Effector Functions-Improving Antibodies for Cancer Treatment. Antibodies 2020, 9, 64. [Google Scholar] [CrossRef]
- Almawash, S. Revolutionary Cancer Therapy for Personalization and Improved Efficacy: Strategies to Overcome Resistance to Immune Checkpoint Inhibitor Therapy. Cancers 2025, 17, 880. [Google Scholar] [CrossRef]
- Kwok, G.; Yau, T.C.; Chiu, J.W.; Tse, E.; Kwong, Y.L. Pembrolizumab (Keytruda). Hum. Vaccin. Immunother. 2016, 12, 2777–2789. [Google Scholar] [CrossRef]
- Lim, J.S.; Soo, R.A. Nivolumab in the treatment of metastatic squamous non-small cell lung cancer: A review of the evidence. Ther. Adv. Respir. Dis. 2016, 10, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.; Weber, J.S.; et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann. Oncol. 2019, 30, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Payandeh, Z.; Bahrami, A.A.; Hoseinpoor, R.; Mortazavi, Y.; Rajabibazl, M.; Rahimpour, A.; Taromchi, A.H.; Khalil, S. The applications of anti-CD20 antibodies to treat various B cells disorders. Biomed. Pharmacother. 2019, 109, 2415–2426. [Google Scholar] [CrossRef]
- Hauptrock, B.; Hess, G. Rituximab in the treatment of non-Hodgkin’s lymphoma. Biol. Targets Ther. 2008, 2, 619–633. [Google Scholar] [CrossRef]
- Crescioli, S.; Kaplon, H.; Chenoweth, A.; Wang, L.; Visweswaraiah, J.; Reichert, J.M. Antibodies to watch in 2024. MAbs 2024, 16, 2297450. [Google Scholar] [CrossRef]
- Herrera, M.; Pretelli, G.; Desai, J.; Garralda, E.; Siu, L.L.; Steiner, T.M.; Au, L. Bispecific antibodies: Advancing precision oncology. Trends Cancer 2024, 10, 893–919. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.M.; Lee, J.H.; Ko, S.; Hong, S.S.; Jin, H.E. Mechanism of Action and Pharmacokinetics of Approved Bispecific Antibodies. Biomol. Ther. 2024, 32, 708–722. [Google Scholar] [CrossRef]
- Huehls, A.M.; Coupet, T.A.; Sentman, C.L. Bispecific T-cell engagers for cancer immunotherapy. Immunol. Cell Biol. 2015, 93, 290–296. [Google Scholar] [CrossRef]
- Wu, J.; Fu, J.; Zhang, M.; Liu, D. Blinatumomab: A bispecific T cell engager (BiTE) antibody against CD19/CD3 for refractory acute lymphoid leukemia. J. Hematol. Oncol. 2015, 8, 104. [Google Scholar] [CrossRef]
- Kantarjian, H.; Stein, A.; Gokbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.M.; Wei, A.; Dombret, H.; Foa, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Marischen, L.; Fritsch, J.; Ilic, J.; Wahl, L.; Bertsch, T.; Knop, S.; Bold, A. Two Are Better than One: The Bi-Specific Antibody Mosunetuzumab Reveals an Improved Immune Response of Vgamma9Vdelta2 T Cells Targeting CD20 in Malignant B Cells in Comparison to the Mono-Specific Antibody Obinutuzumab. Int. J. Mol. Sci. 2025, 26, 1262. [Google Scholar] [CrossRef]
- Tapia-Galisteo, A.; Compte, M.; Alvarez-Vallina, L.; Sanz, L. When three is not a crowd: Trispecific antibodies for enhanced cancer immunotherapy. Theranostics 2023, 13, 1028–1041. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Seung, E.; Xu, L.; Rao, E.; Lord, D.M.; Wei, R.R.; Cortez-Retamozo, V.; Ospina, B.; Posternak, V.; Ulinski, G.; et al. Trispecific antibodies enhance the therapeutic efficacy of tumor-directed T cells through T cell receptor co-stimulation. Nat. Cancer 2020, 1, 86–98. [Google Scholar] [CrossRef]
- Grab, A.L.; Kim, P.S.; John, L.; Bisht, K.; Wang, H.; Baumann, A.; Van de Velde, H.; Sarkar, I.; Shome, D.; Reichert, P.; et al. Pre-Clinical Assessment of SAR442257, a CD38/CD3xCD28 Trispecific T Cell Engager in Treatment of Relapsed/Refractory Multiple Myeloma. Cells 2024, 13, 879. [Google Scholar] [CrossRef]
- Zhao, L.; Li, S.; Wei, X.; Qi, X.; Liu, D.; Liu, L.; Wen, F.; Zhang, J.S.; Wang, F.; Liu, Z.L.; et al. A novel CD19/CD22/CD3 trispecific antibody enhances therapeutic efficacy and overcomes immune escape against B-ALL. Blood 2022, 140, 1790–1802. [Google Scholar] [CrossRef]
- Sun, Y.; Yu, X.; Wang, X.; Yuan, K.; Wang, G.; Hu, L.; Zhang, G.; Pei, W.; Wang, L.; Sun, C.; et al. Bispecific antibodies in cancer therapy: Target selection and regulatory requirements. Acta Pharm. Sin. B 2023, 13, 3583–3597. [Google Scholar] [CrossRef] [PubMed]
- Firestone, R.; Lesokhin, A.M.; Usmani, S.Z. An Embarrassment of Riches: Three FDA-Approved Bispecific Antibodies for Relapsed Refractory Multiple Myeloma. Blood Cancer Discov. 2023, 4, 433–436. [Google Scholar] [CrossRef]
- Greenblatt, K.; Khaddour, K. Trastuzumab. In StatPearls; StatPearls: Treasure Island, FL, USA, 2025. [Google Scholar]
- Markham, A. Atezolizumab: First Global Approval. Drugs 2016, 76, 1227–1232. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Erratum to: Durvalumab: First Global Approval. Drugs 2017, 77, 1817. [Google Scholar] [CrossRef]
- Przepiorka, D.; Ko, C.W.; Deisseroth, A.; Yancey, C.L.; Candau-Chacon, R.; Chiu, H.J.; Gehrke, B.J.; Gomez-Broughton, C.; Kane, R.C.; Kirshner, S.; et al. FDA Approval: Blinatumomab. Clin. Cancer Res. 2015, 21, 4035–4039. [Google Scholar] [CrossRef]
- Kang, C. Mosunetuzumab: First Approval. Drugs 2022, 82, 1229–1234. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Glofitamab: First Approval. Drugs 2023, 83, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Richardson, N.C.; Kasamon, Y.L.; Chen, H.; de Claro, R.A.; Ye, J.; Blumenthal, G.M.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Brentuximab Vedotin in First-Line Treatment of Peripheral T-Cell Lymphoma. Oncologist 2019, 24, e180–e187. [Google Scholar] [CrossRef] [PubMed]
- Ballantyne, A.; Dhillon, S. Trastuzumab emtansine: First global approval. Drugs 2013, 73, 755–765. [Google Scholar] [CrossRef]
- Keam, S.J. Trastuzumab Deruxtecan: First Approval. Drugs 2020, 80, 501–508. [Google Scholar] [CrossRef]
- Syed, Y.Y. Sacituzumab Govitecan: First Approval. Drugs 2020, 80, 1019–1025. [Google Scholar] [CrossRef]
- Markham, A. Margetuximab: First Approval. Drugs 2021, 81, 599–604. [Google Scholar] [CrossRef]
- Dhillon, S. Elranatamab: First Approval. Drugs 2023, 83, 1621–1627. [Google Scholar] [CrossRef]
- Boersma, B.; Poinot, H.; Pommier, A. Stimulating the Antitumor Immune Response Using Immunocytokines: A Preclinical and Clinical Overview. Pharmaceutics 2024, 16, 974. [Google Scholar] [CrossRef]
- VanDyke, D.; Iglesias, M.; Tomala, J.; Young, A.; Smith, J.; Perry, J.A.; Gebara, E.; Cross, A.R.; Cheung, L.S.; Dykema, A.G.; et al. Engineered human cytokine/antibody fusion proteins expand regulatory T cells and confer autoimmune disease protection. Cell Rep. 2022, 41, 111478. [Google Scholar] [CrossRef]
- Shi, W.; Liu, N.; Lu, H. Advancements and challenges in immunocytokines: A new arsenal against cancer. Acta Pharm. Sin. B 2024, 14, 4649–4664. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Sun, J.; Yuan, Y.; Ji, D.; Sun, Y.; Liu, Y.; Li, S.; Zhu, X.; Wu, X.; Hu, J.; et al. Proximity-enabled covalent binding of IL-2 to IL-2Ralpha selectively activates regulatory T cells and suppresses autoimmunity. Signal Transduct. Target. Ther. 2023, 8, 28. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.M.; Rolig, A.S.; Charych, D.H.; Hoch, U.; Kasiewicz, M.J.; Rose, D.C.; McNamara, M.J.; Hilgart-Martiszus, I.F.; Redmond, W.L. NKTR-214 immunotherapy synergizes with radiotherapy to stimulate systemic CD8(+) T cell responses capable of curing multi-focal cancer. J. Immunother. Cancer 2020, 8, e000464. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Eto, M.; Motzer, R.; De Giorgi, U.; Buchler, T.; Basappa, N.S.; Mendez-Vidal, M.J.; Tjulandin, S.; Hoon Park, S.; Melichar, B.; et al. Lenvatinib plus pembrolizumab versus sunitinib as first-line treatment of patients with advanced renal cell carcinoma (CLEAR): Extended follow-up from the phase 3, randomised, open-label study. Lancet Oncol. 2023, 24, 228–238. [Google Scholar] [CrossRef]
- Yi, M.; Li, T.Y.; Niu, M.K.; Zhang, H.X.; Wu, Y.Z.; Wu, K.M.; Dai, Z.J. Targeting cytokine and chemokine signaling pathways for cancer therapy. Signal Transduct. Target. Ther. 2024, 9, 176. [Google Scholar] [CrossRef]
- Ma, W.W.; Saccardo, A.; Roccatano, D.; Aboagye-Mensah, D.; Alkaseem, M.; Jewkes, M.; Di Nezza, F.; Baron, M.; Soloviev, M.; Ferrari, E. Modular assembly of proteins on nanoparticles. Nat. Commun. 2018, 9, 1489. [Google Scholar] [CrossRef]
- Driscoll, C.L.; Keeble, A.H.; Howarth, M.R. SpyMask enables combinatorial assembly of bispecific binders. Nat. Commun. 2024, 15, 2403. [Google Scholar] [CrossRef]
- Baldo, B.A. Side effects of cytokines approved for therapy. Drug Saf. 2014, 37, 921–943. [Google Scholar] [CrossRef]
- Sznol, M.; Rizvi, N. Teaching an old dog new tricks: Re-engineering IL-2 for immuno-oncology applications. J. Immunother. Cancer 2023, 11, e006346. [Google Scholar] [CrossRef]
- Shoari, A.; Tahmasebi, M.; Khodabakhsh, F.; Cohan, R.A.; Oghalaie, A.; Behdani, M. Angiogenic biomolecules specific nanobodies application in cancer imaging and therapy; review and updates. Int. Immunopharmacol. 2022, 105, 108585. [Google Scholar] [CrossRef]
- Zhu, H.; Ding, Y. Nanobodies: From Discovery to AI-Driven Design. Biology 2025, 14, 547. [Google Scholar] [CrossRef] [PubMed]
- Jovcevska, I.; Muyldermans, S. The Therapeutic Potential of Nanobodies. BioDrugs 2020, 34, 11–26. [Google Scholar] [CrossRef]
- Keyaerts, M.; Xavier, C.; Heemskerk, J.; Devoogdt, N.; Everaert, H.; Ackaert, C.; Vanhoeij, M.; Duhoux, F.P.; Gevaert, T.; Simon, P.; et al. Phase I Study of 68Ga-HER2-Nanobody for PET/CT Assessment of HER2 Expression in Breast Carcinoma. J. Nucl. Med. 2016, 57, 27–33. [Google Scholar] [CrossRef]
- Fan, J.; Zhuang, X.; Yang, X.; Xu, Y.; Zhou, Z.; Pan, L.; Chen, S. A multivalent biparatopic EGFR-targeting nanobody drug conjugate displays potent anticancer activity in solid tumor models. Signal Transduct. Target. Ther. 2021, 6, 320. [Google Scholar] [CrossRef]
- Dieras, V.; Miles, D.; Verma, S.; Pegram, M.; Welslau, M.; Baselga, J.; Krop, I.E.; Blackwell, K.; Hoersch, S.; Xu, J.; et al. Trastuzumab emtansine versus capecitabine plus lapatinib in patients with previously treated HER2-positive advanced breast cancer (EMILIA): A descriptive analysis of final overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Luo, J.; Gui, X.; Zheng, Y.; Schaar, E.; Liu, G.; Shi, J. Bioengineered nanotechnology for nucleic acid delivery. J. Control. Release 2023, 364, 124–141. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.F.; Tang, L.X.; Xie, Y.Z.Y.; Xianyu, Y.L.; Zhang, L.M.; Wang, P.; Hamada, Y.; Jiang, K.; Zheng, W.F.; Jiang, X.Y. Gold nanoclusters-assisted delivery of siRNA for effective treatment of pancreatic cancer. Nat. Commun. 2017, 8, 15130. [Google Scholar] [CrossRef]
- Proietto, M.; Crippa, M.; Damiani, C.; Pasquale, V.; Sacco, E.; Vanoni, M.; Gilardi, M. Tumor heterogeneity: Preclinical models, emerging technologies, and future applications. Front. Oncol. 2023, 13, 1164535. [Google Scholar] [CrossRef]
- Rahban, M.; Ahmad, F.; Piatyszek, M.A.; Haertle, T.; Saso, L.; Saboury, A.A. Stabilization challenges and aggregation in protein-based therapeutics in the pharmaceutical industry. RSC Adv. 2023, 13, 35947–35963. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, Y.; Park, J.; Liu, X.; Hu, Y.; Wang, T.; McFarland, K.; Betenbaugh, M.J. Design and Production of Bispecific Antibodies. Antibodies 2019, 8, 43. [Google Scholar] [CrossRef]
- Ingavat, N.; Dzulkiflie, N.; Liew, J.M.; Wang, X.; Leong, E.; Loh, H.P.; Ng, S.K.; Yang, Y.; Zhang, W. Investigation on environmental factors contributing to bispecific antibody stability and the reversal of self-associated aggregates. Bioresour. Bioprocess. 2024, 11, 82. [Google Scholar] [CrossRef] [PubMed]
- Sumida, K.H.; Nunez-Franco, R.; Kalvet, I.; Pellock, S.J.; Wicky, B.I.M.; Milles, L.F.; Dauparas, J.; Wang, J.; Kipnis, Y.; Jameson, N.; et al. Improving Protein Expression, Stability, and Function with ProteinMPNN. J. Am. Chem. Soc. 2024, 146, 2054–2061. [Google Scholar] [CrossRef] [PubMed]
- Seeliger, D.; Schulz, P.; Litzenburger, T.; Spitz, J.; Hoerer, S.; Blech, M.; Enenkel, B.; Studts, J.M.; Garidel, P.; Karow, A.R. Boosting antibody developability through rational sequence optimization. MAbs 2015, 7, 505–515. [Google Scholar] [CrossRef]
- Warszawski, S.; Borenstein Katz, A.; Lipsh, R.; Khmelnitsky, L.; Ben Nissan, G.; Javitt, G.; Dym, O.; Unger, T.; Knop, O.; Albeck, S.; et al. Optimizing antibody affinity and stability by the automated design of the variable light-heavy chain interfaces. PLoS Comput. Biol. 2019, 15, e1007207. [Google Scholar] [CrossRef]
- Nugrahadi, P.P.; Hinrichs, W.L.J.; Frijlink, H.W.; Schoneich, C.; Avanti, C. Designing Formulation Strategies for Enhanced Stability of Therapeutic Peptides in Aqueous Solutions: A Review. Pharmaceutics 2023, 15, 935. [Google Scholar] [CrossRef] [PubMed]
- Svilenov, H.L.; Winter, G. Formulations That Suppress Aggregation During Long-Term Storage of a Bispecific Antibody are Characterized by High Refoldability and Colloidal Stability. J. Pharm. Sci. 2020, 109, 2048–2058. [Google Scholar] [CrossRef]
- Svilenov, H.; Winter, G. The ReFOLD assay for protein formulation studies and prediction of protein aggregation during long-term storage. Eur. J. Pharm. Biopharm. 2019, 137, 131–139. [Google Scholar] [CrossRef]
- Baker, M.P.; Reynolds, H.M.; Lumicisi, B.; Bryson, C.J. Immunogenicity of protein therapeutics: The key causes, consequences and challenges. Self/Nonself 2010, 1, 314–322. [Google Scholar] [CrossRef]
- Kuriakose, A.; Chirmule, N.; Nair, P. Immunogenicity of Biotherapeutics: Causes and Association with Posttranslational Modifications. J. Immunol. Res. 2016, 2016, 1298473. [Google Scholar] [CrossRef]
- Howard, E.L.; Goens, M.M.; Susta, L.; Patel, A.; Wootton, S.K. Anti-Drug Antibody Response to Therapeutic Antibodies and Potential Mitigation Strategies. Biomedicines 2025, 13, 299. [Google Scholar] [CrossRef]
- Rudman, S.M.; Jameson, M.B.; McKeage, M.J.; Savage, P.; Jodrell, D.I.; Harries, M.; Acton, G.; Erlandsson, F.; Spicer, J.F. A phase 1 study of AS1409, a novel antibody-cytokine fusion protein, in patients with malignant melanoma or renal cell carcinoma. Clin. Cancer Res. 2011, 17, 1998–2005. [Google Scholar] [CrossRef] [PubMed]
- Zinsli, L.V.; Stierlin, N.; Loessner, M.J.; Schmelcher, M. Deimmunization of protein therapeutics—Recent advances in experimental and computational epitope prediction and deletion. Comput. Struct. Biotechnol. J. 2021, 19, 315–329. [Google Scholar] [CrossRef]
- Parker, A.S.; Choi, Y.; Griswold, K.E.; Bailey-Kellogg, C. Structure-guided deimmunization of therapeutic proteins. J. Comput. Biol. 2013, 20, 152–165. [Google Scholar] [CrossRef]
- Lou, H.; Feng, M.; Hageman, M.J. Advanced Formulations/Drug Delivery Systems for Subcutaneous Delivery of Protein-Based Biotherapeutics. J. Pharm. Sci. 2022, 111, 2968–2982. [Google Scholar] [CrossRef]
- Bittner, B.; Richter, W.; Schmidt, J. Subcutaneous Administration of Biotherapeutics: An Overview of Current Challenges and Opportunities. BioDrugs 2018, 32, 425–440. [Google Scholar] [CrossRef] [PubMed]
- Beinfeld, M.; Atlas, S.J.; Touchette, D.; McKenna, A.; Rind, D.; Pearson, S.D. The effectiveness and value of nadofaragene firadenovec, oportuzumab monatox, and pembrolizumab for BCG-unresponsive non-muscle-invasive bladder cancer. J. Manag. Care Spec. Pharm. 2021, 27, 797–804. [Google Scholar] [CrossRef]
- Fesnak, A.D. The Challenge of Variability in Chimeric Antigen Receptor T cell Manufacturing. Regen. Eng. Transl. Med. 2020, 6, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Sifniotis, V.; Cruz, E.; Eroglu, B.; Kayser, V. Current Advancements in Addressing Key Challenges of Therapeutic Antibody Design, Manufacture, and Formulation. Antibodies 2019, 8, 36. [Google Scholar] [CrossRef]
- Gray, D. Overview of protein expression by mammalian cells. Curr. Protoc. Protein Sci. 1997, 10, 5.9.1–5.9.18. [Google Scholar] [CrossRef]
- Ito, T.; Lutz, H.; Tan, L.; Wang, B.; Tan, J.; Patel, M.; Chen, L.; Tsunakawa, Y.; Park, B.; Banerjee, S. Host cell proteins in monoclonal antibody processing: Control, detection, and removal. Biotechnol. Prog. 2024, 40, e3448. [Google Scholar] [CrossRef]
- Liang, X.; He, Q.; Qin, G.; Li, G.; Li, Q.; Tan, H.; Wang, Z.; Fan, M.; Xu, D. Effectively removing the homodimer in bispecific antibodies by weak partitioning mode of anion exchange chromatography. J. Chromatogr. B 2023, 1225, 123767. [Google Scholar] [CrossRef] [PubMed]
- Abou-El-Enein, M.; Elsallab, M.; Feldman, S.A.; Fesnak, A.D.; Heslop, H.E.; Marks, P.; Till, B.G.; Bauer, G.; Savoldo, B. Scalable Manufacturing of CAR T cells for Cancer Immunotherapy. Blood Cancer Discov. 2021, 2, 408–422. [Google Scholar] [CrossRef]
- Melocchi, A.; Schmittlein, B.; Sadhu, S.; Nayak, S.; Lares, A.; Uboldi, M.; Zema, L.; di Robilant, B.N.; Feldman, S.A.; Esensten, J.H. Automated manufacturing of cell therapies. J. Control. Release 2025, 381, 113561. [Google Scholar] [CrossRef]
- Lee, J.S.Z.; Prabhu, A.V.; Wu, Y.Y.; Bin Abdul Rahim, A.A.; Chen, S.; Naing, M.W.; Liu, D. Transition from manual to automated processes for autologous T cell therapy manufacturing using bioreactor with expandable culture area. Sci. Rep. 2025, 15, 15819. [Google Scholar] [CrossRef] [PubMed]
- Mirfakhraie, R.; Dehaghi, B.K.; Ghorbi, M.D.; Ghaffari-Nazari, H.; Mohammadian, M.; Salimi, M.; Ardakani, M.T.; Parkhideh, S. All about blinatumomab: The bispecific T cell engager immunotherapy for B cell acute lymphoblastic leukemia. Hematol. Transfus. Cell Ther. 2024, 46, 192–200. [Google Scholar] [CrossRef]
- Bas, T.G.; Duarte, V. Biosimilars in the Era of Artificial Intelligence-International Regulations and the Use in Oncological Treatments. Pharmaceuticals 2024, 17, 925. [Google Scholar] [CrossRef]
- Love, J.C.; Love, K.R.; Barone, P.W. Enabling global access to high-quality biopharmaceuticals. Curr. Opin. Chem. Eng. 2013, 2, 383–390. [Google Scholar] [CrossRef]
- Yang, J.; Carioto, J.; Pyenson, B.; Smith, R.; Jacobson, N.; Pittinger, S.; Shelbaya, A. Greater uptake, an alternative reimbursement methodology needed to realize cost-saving potential of oncology biosimilars in the United States. J. Manag. Care Spec. Pharm. 2021, 27, 1642–1651. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Garcia, Z.; Chen, D.; Liu, H.; Trelstad, P. Cost and supply considerations for antibody therapeutics. MAbs 2025, 17, 2451789. [Google Scholar] [CrossRef]
- Niazi, S.K.; Magoola, M. Advances in Escherichia coli-based therapeutic protein expression: Mammalian conversion, continuous manufacturing, and cell-free production. Biologics 2023, 3, 380–401. [Google Scholar] [CrossRef]
- Schutz, A.; Bernhard, F.; Berrow, N.; Buyel, J.F.; Ferreira-da-Silva, F.; Haustraete, J.; van den Heuvel, J.; Hoffmann, J.E.; de Marco, A.; Peleg, Y.; et al. A concise guide to choosing suitable gene expression systems for recombinant protein production. STAR Protoc. 2023, 4, 102572. [Google Scholar] [CrossRef] [PubMed]
- Geethakumari, P.R.; Ramasamy, D.P.; Dholaria, B.; Berdeja, J.; Kansagra, A. Balancing Quality, Cost, and Access During Delivery of Newer Cellular and Immunotherapy Treatments. Curr. Hematol. Malig. Rep. 2021, 16, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, A.W.; Hlavka, J.P.; Case, S.R. Biosimilar Cost Savings in the United States: Initial Experience and Future Potential. Rand Health Q. 2018, 7, 3. [Google Scholar] [PubMed]
- Sugiki, T.; Fujiwara, T.; Kojima, C. Latest approaches for efficient protein production in drug discovery. Expert Opin. Drug Dis. 2014, 9, 1189–1204. [Google Scholar] [CrossRef]
- Khatami, M.H.; Mendes, U.C.; Wiebe, N.; Kim, P.M. Gate-based quantum computing for protein design. PLoS Comput. Biol. 2023, 19, e1011033. [Google Scholar] [CrossRef]
- Doga, H.; Raubenolt, B.; Cumbo, F.; Joshi, J.; DiFilippo, F.P.; Qin, J.; Blankenberg, D.; Shehab, O. A Perspective on Protein Structure Prediction Using Quantum Computers. J. Chem. Theory Comput. 2024, 20, 3359–3378. [Google Scholar] [CrossRef]
- Ebrahimi, S.B.; Samanta, D. Engineering protein-based therapeutics through structural and chemical design. Nat. Commun. 2023, 14, 2411. [Google Scholar] [CrossRef]
- Peng, K.; Fu, Y.X.; Liang, Y. Engineering cytokines for tumor-targeting and selective T cell activation. Trends Mol. Med. 2025, 31, 373–387. [Google Scholar] [CrossRef]
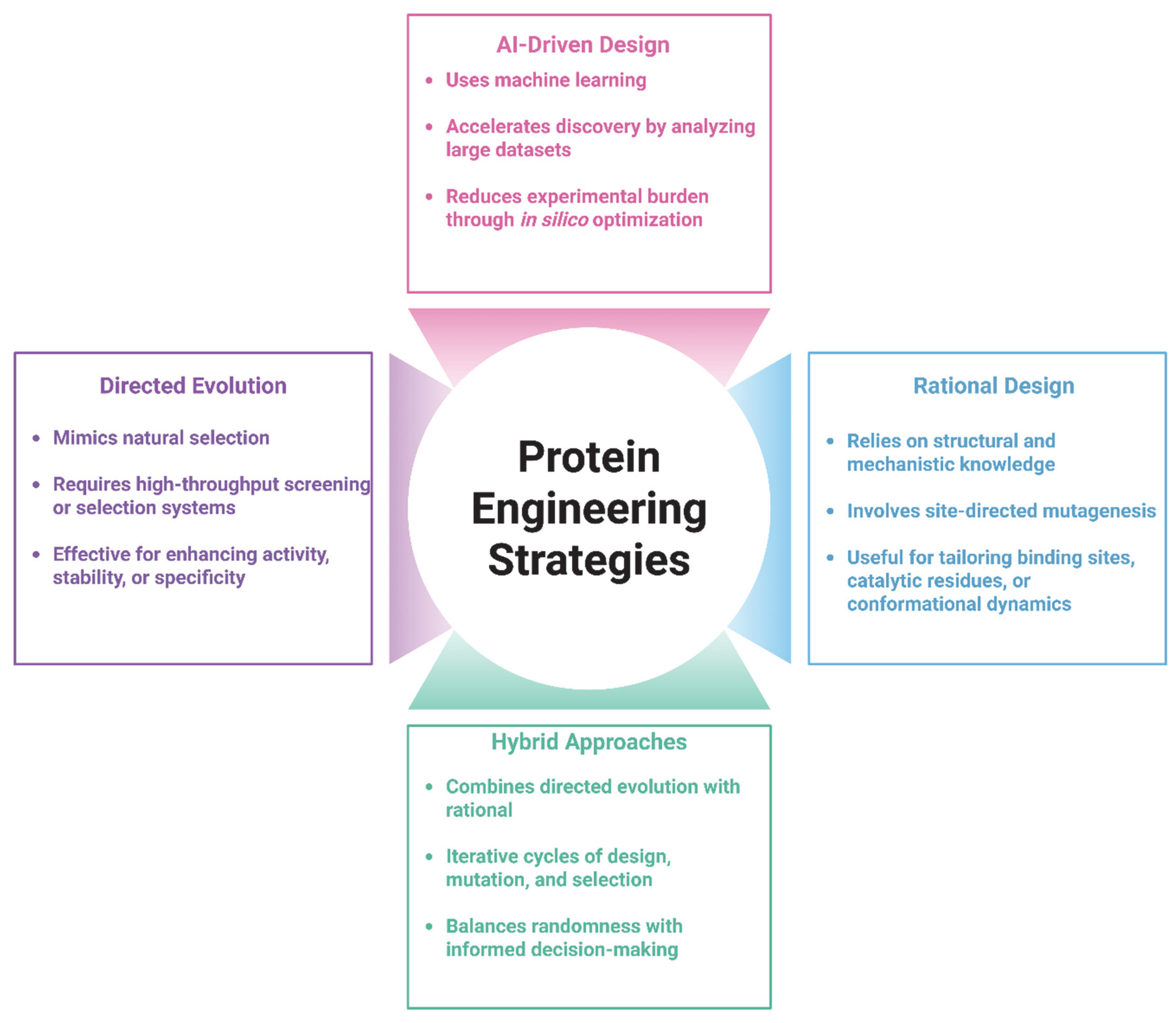
| Strategy | Key Features | Advantages | Limitations | Applications for Cancer Therapeutics |
|---|---|---|---|---|
| Directed Evolution | Iterative rounds of random mutagenesis and selection | Does not require structural knowledge; explores large sequence space | Time- and resource-intensive screening; less predictable outcomes | Optimizing antibody affinity; improving enzyme stability |
| Rational Design | Structure-guided site-directed mutagenesis | High precision; targeted functional improvements | Requires high-resolution structural data; limited by model accuracy | Reducing immunogenicity; improving thermal stability of therapeutic proteins |
| Hybrid Approaches | Combines rational design to guide focused libraries for directed evolution | Balances precision and diversity; more efficient than fully random libraries | Dependent on computational tools and effective screening strategies | Bispecific antibody design; fusion protein stability optimization |
| AI-Driven Design | Uses machine learning and deep learning to predict structure-function relations | Rapid in silico screening; enables de novo design; expands design possibilities | Requires large, high-quality datasets; experimental validation still needed | De novo protein design; optimizing stability, solubility, and immune evasion properties of therapeutics |
| Therapeutic Modality | Mechanism of Action | Representative Example(s) | Clinical/Application Highlights | Ref. |
|---|---|---|---|---|
| Monoclonal Antibodies | Bind specific tumor-associated antigens to mediate immune responses (ADCC, CDC) | Trastuzumab, Rituximab | HER2+ breast cancer, B-cell malignancies; immune checkpoint blockade (e.g., anti-PD-1/PD-L1) | [11,117] |
| Bispecific Antibodies | Engage two different targets (CD3 on T cells and CD19 on tumor cell) | Blinatumomab, BiA9*2_HF | Redirects T cells to tumor anddual signaling modulation in B-cell acute lymphoblastic leukemia and multiple myeloma | [118,119] |
| Engineered Cytokines | Modulate immune response; enhance effector cell activity | Bempegaldesleukin (NKTR-214), L19-IL-2 | Biased receptor targeting to reduce toxicity, synergy with checkpoint inhibitors and tumor-specific immune activation in renal cell carcinoma and melanoma | [120,121] |
| Fusion Proteins | Combine targeting and effector domains into one protein | VEGF-HNc, NHS-IL-12 | Tumor-specific delivery of toxins or cytokines, improved specificity and reduced systemic effects in solid malignancies | [9,122] |
| Nanobodies (Single-domain Abs) | Small antibody fragments target specific epitopes | 68Ga-labeled anti-HER2, EGFR nanobody-drug conjugates | Enhanced tumor penetration and clearance; suitable for imaging and ADCs in HER2+ breast cancer, EGFR-expressing solid tumors and glioblastoma | [123,124] |
| Protein-Based Delivery Systems | Targeted delivery of drugs or nucleic acids using protein carriers | Trastuzumab emtansine (T-DM1), Ferritin-based nanocages | Improved stability and bioavailability; siRNA and chemotherapy delivery with tumor selectivity in HER2+ breast cancer and | [10,125] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naderiyan, Z.; Shoari, A. Protein Engineering Paving the Way for Next-Generation Therapies in Cancer. Int. J. Transl. Med. 2025, 5, 28. https://doi.org/10.3390/ijtm5030028
Naderiyan Z, Shoari A. Protein Engineering Paving the Way for Next-Generation Therapies in Cancer. International Journal of Translational Medicine. 2025; 5(3):28. https://doi.org/10.3390/ijtm5030028
Chicago/Turabian StyleNaderiyan, Zahra, and Alireza Shoari. 2025. "Protein Engineering Paving the Way for Next-Generation Therapies in Cancer" International Journal of Translational Medicine 5, no. 3: 28. https://doi.org/10.3390/ijtm5030028
APA StyleNaderiyan, Z., & Shoari, A. (2025). Protein Engineering Paving the Way for Next-Generation Therapies in Cancer. International Journal of Translational Medicine, 5(3), 28. https://doi.org/10.3390/ijtm5030028