Abstract
Background/Objectives: DNA methylation profiles have emerged as robust biomarkers of ageing, leading to the development of “epigenetic clocks” that estimate biological age. Most established clocks (e.g., Horvath’s 353-CpG pan-tissue clock and Hannum’s 71-CpG blood clock) require dozens to hundreds of CpG sites. This study presents a novel saliva-specific epigenetic clock built on 10 sites identified from Illumina MethylationEPIC (850 k) array data. Methods: Saliva DNA methylation was analysed from 3408 individuals (age range 15–89 years, 68% male, 32% female, no diagnosed disease) from the Muhdo Health Ltd. dataset (2022–2024), and 10 CpG sites were selected where methylation levels showed the strongest positive correlations with chronological age (Pearson r = 0.48–0.66, p < 1 × 10−20). These CpGs map to genes involved in developmental and metabolic pathways (including ELOVL2, CHGA, OTUD7A, PRLHR, ZYG11A, and GPR158). A linear combination of the 10 methylation sites was used to calculate a “DNA methylation age”. Results: The 10-CpG clock’s predictions were highly correlated with chronological age (r = 0.80, R2 = 0.64), with a mean absolute error of ~5.5 years. Its performance, while slightly less precise than Horvath’s or Hannum’s multi-CpG clocks, is notable given the minimal marker set. It was observed that all 10 clock CpGs undergo age-related hypermethylation. The biological significance of these loci is discussed, along with the potential health and forensic applications of a saliva-based epigenetic age predictor. Conclusions: This study demonstrates that a saliva-specific epigenetic clock using only 10 CpG sites can capture a substantial portion of age-related DNA methylation changes, providing a cost-effective tool for age estimation.
1. Introduction
Epigenetic alterations are a hallmark of ageing, with DNA methylation changes at specific CpG sites accumulating over time [1,2]. Numerous studies have identified sets of CpGs whose methylation levels correlate strongly with chronological age (number of years lived as a function of time) [3]. This has enabled the creation of “epigenetic clocks”—mathematical models that translate methylation data into an estimated biological age (cell and tissue status) [2]. The first generation of epigenetic clocks, such as the multi-tissue clock by Horvath [1] and the blood-specific clock by Hannum [1], demonstrated that chronological age can be predicted with significant accuracy from DNA methylation signatures. Horvath’s pan-tissue clock utilizes 353 CpG sites and can predict age across multiple tissues (blood, saliva, brain, etc.) with a Pearson correlation of ~0.96 and average error of ~3–4 years. Similarly, Hannum’s blood-based clock uses 71 CpGs to achieve ~0.96 correlation and ~3.9-year error in whole blood [1]. These pioneering clocks established that DNA methylation patterns are tightly coupled with the ageing process.
However, most first-generation clocks rely on dozens or hundreds of CpGs, typically measured on arrays (450 k or 850 k) or sequencing platforms. This scale, while boosting accuracy, can be a barrier for practical applications where cost or DNA input is limited. In recent years, there has been growing interest in minimalist epigenetic clocks—models that use only a handful of CpG sites to predict age. The success of such small-panel clocks is exemplified by studies showing that even a single CpG (for example, in ELOVL2) can serve as a rough age indicator [4,5]. Indeed, ELOVL2 methylation alone has been reported to explain up to ~70% of age-related methylation variance in some tissues. Early efforts in forensic epigenetics demonstrated that 2–5 CpG models could estimate age [5] within about 5-year accuracy from blood or saliva. For instance, a study in 2011 first showed that saliva DNA methylation profiles could predict age with a mean error of ~5.2 years using just a few CpGs [3]. These findings motivate the development of simplified clocks tailored to specific sample types.
Saliva is an attractive tissue for epigenetic age assessment. It can be obtained non-invasively and is often available in biobanks or forensic casework (e.g., saliva on envelopes or cups). Saliva contains a mix of buccal epithelial cells and leukocytes, whose DNA methylation patterns reflect both systemic ageing and oral environmental exposures. Notably, Horvath’s multi-tissue clock included saliva in its training and achieved high accuracy in saliva samples (e.g., r = 0.83–0.89, error ~2.7–2.9 years) [6]. However, saliva-specific ageing markers may not have been captured effectively in earlier clocks, which predominantly used array panels limited to ~450 k CpGs. The newer Illumina MethylationEPIC array released in 2016 (~850 k sites) covers additional CpGs (including non-coding regions and enhancers) that could yield novel age-associated loci in saliva. Indeed, recent epigenome-wide association studies (EWAS) using the EPIC array have reported new age-linked CpGs in salivary DNA [7]. Leveraging such novel loci might improve age prediction specifically for saliva, or shed light on tissue-specific ageing mechanisms.
In this study, we describe the development of a novel saliva-based epigenetic clock using only 10 CpG sites. We used EPIC array data from thousands of saliva samples spanning the teen years through to old age, finding that 10 CpG loci exhibit strong age-related hypermethylation. We then constructed an age prediction model based on these 10 markers and evaluated its performance. This clock’s accuracy was compared with other clocks and the biological context of each CpG site was explored—particularly whether hypermethylation at these loci might be associated with age-related functional changes or health risks. To our knowledge, this is one of the smallest CpG-marker sets proposed for an epigenetic clock in saliva to date. By highlighting a minimal signature, the goal is to facilitate the development of cost-effective, tissue-specific age assays (for example, via targeted bisulfite sequencing or PCR assays) and to deepen our understanding of which gene loci are most tightly linked to the ageing process in oral tissues.
2. Methods
2.1. Sample Collection and DNA Methylation Profiling
This study used secondary data that had previously been collected through the normal day-to-day operations of Muhdo Health. Clients can opt in and provide informed consent for their anatomized data to be used for research purposes as an open source. This study follows the guidelines set by the institutional review board, in line with ethical considerations and the Declaration of Helsinki. For more information, refer to the “Institutional Review Board Statement” below. Saliva samples were obtained from 3408 individuals who are consumers of the Muhdo Health application, which allows for informed consent and the anonymisation of data for research purposes as an open source. In total, 2317 males and 1091 females aged 15.2 to 88.7 years, with no prior diagnosed disease, were included (see Supplementary Material Table S1). Genomic DNA was extracted from saliva using standard protocols. DNA methylation was assayed using the Illumina MethylationEPIC BeadChip array, which quantifies methylation (β-value, fraction methylated from 0 to 1) at >850,000 CpG sites across the genome (see Supplementary Material Table S2). All sample processing and hybridisation steps were performed according to manufacturer’s instructions via Eurofins Labs. Raw intensity data were normalised to correct for type I/II probe biases. Probes with known SNP interference or low reliability were removed. All samples passed quality control metrics (e.g., call rate > 98%). This study contains data obtained via the Muhdo Health Mobile Phone App with informed consent for use in DNA research, and all participants are anonymised.
2.2. Identification of Age-Associated CpGs
An epigenome-wide association analysis was conducted to find CpG sites whose methylation correlates with chronological age, which was input via date of birth (DOB) when registering a DNA methylation (DNAm) test for each probe on the array. The Pearson correlation coefficient (r) was computed between methylation β-value and age, in addition to p-values via linear regression (adjusting for sex and disease status, although for this clock development there was a focus on the raw age effect). There was particular attention given to CpGs showing increased methylation with age (hypermethylation), since age-related hypermethylation tends to occur at gene promoters and CpG islands, which might be more conserved across individuals. From this analysis, the top candidates based on effect size (correlation) and significance were selected. The list was narrowed down to 10 CpG sites that (1) showed among the highest positive correlations with age in saliva and (2) were present in at least 95% of samples (to avoid probes with missing data). Finally, (3) the 10 CpG sites chosen had a higher mean linear R2 when compared to the most significant hypomethylation CpG sites found; they also had a superior R2 when compared to less or more CpG sites, within a range of 1–50 sites (kept small for cost-effective small array technology). These 10 CpGs formed the basis of the clock method. Genomic annotations for these CpGs (gene association and CpG island context) were retrieved from the Illumina EPIC manifest and confirmed via the UCSC Genome Browser.
2.3. Clock Construction
An epigenetic age predictor was constructed using the 10 selected CpG sites. For simplicity and robustness, a linear model approach was chosen. Firstly, we explored whether a simple average of the 10 CpG β-values could serve as an age indicator. Additionally, to improve accuracy, a multivariate linear regression model on the training data was also fitted:
Predicted Age = β0 + Σi=110 βᵢ(CpGᵢ β-value)
The coefficients were determined by least squares optimisation to minimise the difference between predicted age and chronological age in the training set. The resulting model provides a DNA methylation age (DNAm age) for each saliva sample. Because the group cohort was large (n = 3408), the entire dataset was used to fit the final model to maximise its precision; however, a 5-fold cross-validation was employed to internally validate the model (each fold showed similar performance).
2.4. Performance Evaluation
The clock’s accuracy was assessed by several metrics. The primary measure was the Pearson correlation (r) between DNAm age and chronological age, as well as the coefficient of determination (R2). The mean absolute error (MAE) and root mean square error (RMSE) of the age predictions were also calculated. These metrics were compared and cross-referenced with published performances of other established clocks. All statistical analyses were performed in R (4.4.3) and Python (3.13.3), and figures were generated using Seaborn (0.13.2).
2.5. Comparative Analysis
To contextualise the 10-CpG clock, its performance was compared to that of Horvath’s and Hannum’s clocks using published correlations and errors on relevant tissues. Specifically, the performance of Horvath’s clock on saliva, reported in the original publication [2] and elsewhere, and Hannum’s performance on blood were noted. Overlaps in CpG sites were also explored: cross-referencing of the 10 CpGs with the lists of CpGs used in Horvath [1] and Hannum [1] was conducted to see if any were shared.
3. Results
3.1. Selection of a 10-CpG Saliva Clock
The epigenome-wide analysis of saliva DNA methylation identified numerous CpGs significantly correlated with age (overall, several thousand CpGs had p < 1 × 10−7). The characteristic pattern seen in other tissues was observed: some loci gain methylation (hypermethylation), while others lose methylation (hypomethylation) with age. To construct a stable saliva clock, the focus was on the top hypermethylated CpGs due to the reasoning that these might reside in crucial regulatory regions. Table 1 lists the 10 CpG sites ultimately selected for the clock, along with their nearest gene annotation and correlation with age in the dataset. Each of these 10 CpGs showed a strong positive correlation with chronological age (Pearson r ranging from ~0.48 to 0.66 in saliva). All correlations were highly significant (p-values in the order of 1 × 10−20 or smaller), given the large sample size.

Table 1.
Top 10 age-associated CpG sites in saliva selected from over 850 k CpG sites for the epigenetic clock. All sites show increasing methylation with age (positive correlation). Gene annotations are based on proximity to the closest gene (within ~<5 kb) according to Illumina EPIC manifest. “(none)” indicates no annotated gene in the vicinity (likely intergenic or in an undefined region).
All 10 clock CpGs are hypermethylated with age—that is, their β-values increase in older individuals compared to younger ones (see Figure 1). Notably, several of these CpGs are located in gene promoter regions within CpG islands. For example, cg21620282 lies in a CpG island in the 5′UTR/first exon of the CHGA gene (Chromogranin A) and cg05991454 resides in a CpG island on chromosome 4, with no nearby genes [8]. The CpG cg16867657 is located in the promoter of ELOVL2 (Elongation of Very Long Chain Fatty Acids Protein 2), a well-known age marker often highlighted in previous studies [2]. Other sites (cg06784991 in ZYG11A; cg11705975 in PRLHR) are within or near gene bodies, but may still affect regulatory regions. The presence of multiple clock CpGs in CpG islands is consistent with the tendency of age-related hypermethylation to occur at CpG-dense promoters, often of developmental genes [1]. It is worth noting that some of these CpGs were only interrogated in the newer EPIC array and were not present in the older 450 k array; for instance, cg21620282 (CHGA) and cg12841266 (LHFPL4) have recently been reported as novel age-associated CpGs in EPIC-based studies [8]. Thus, this clock captures both well-known and newly identified age markers.
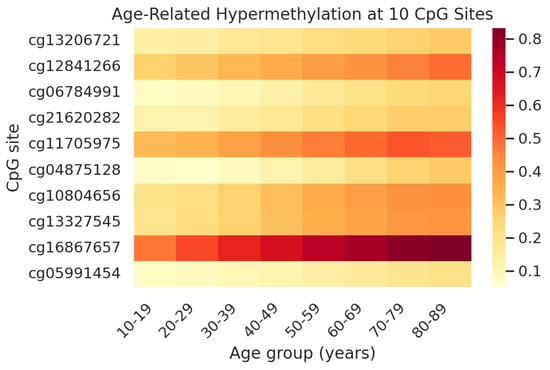
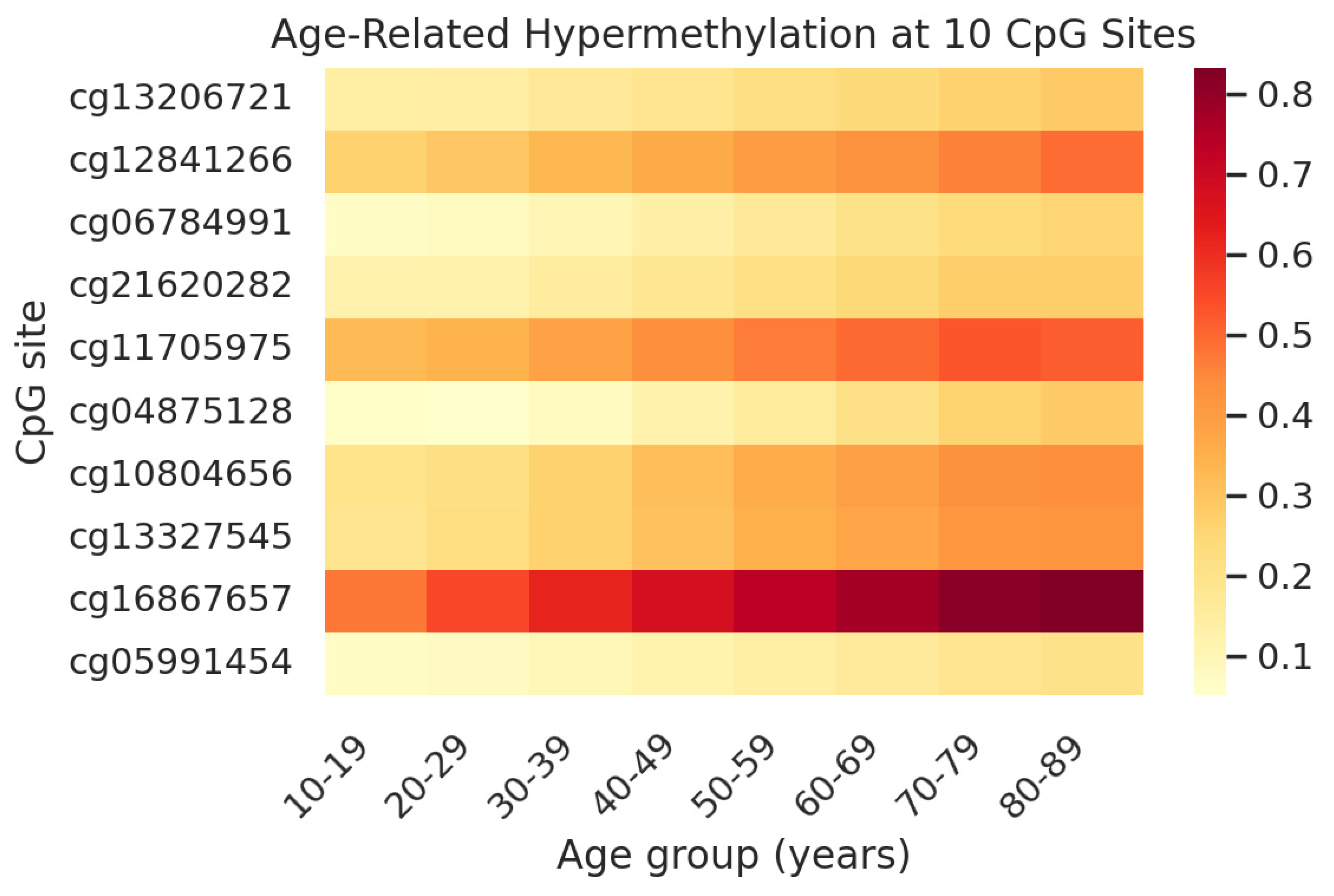
Figure 1.
Heatmap of average DNA methylation (β-value) at the 10 clock CpG sites across age groups in saliva. Rows correspond to the CpG sites (labelled by Illumina ID) and columns correspond to age groups (in years). Methylation levels are color-coded from low (yellow, ~0.1 or 10%) to high (dark red, ~0.8 or 80%). All 10 CpGs show an increasing methylation trend with age. For instance, the ELOVL2 site (cg16867657) is barely methylated in youth (light orange) but becomes heavily methylated by age 50+ (red). This age-related hypermethylation pattern is consistent across the panel, illustrating why the average of these CpGs serves as an effective age indicator. The concordant increase in methylation suggests that these loci might be controlled by ageing mechanisms common to most individuals.
3.2. Epigenetic Age Prediction Accuracy
Using the 10 selected CpGs, a linear model to compute DNA methylation age was built. The results uncovered that the 10-CpG clock predicts chronological age with reasonably high accuracy in this dataset. The Pearson correlation between DNAm age and chronological (actual) age was r = 0.80 (p < 1 × 10−50), indicating that about 64% of variance in age can be explained by methylation at these 10 sites (see Figure 2). The regression line between DNAm age and chronological age was equal to 0.85–0.90 in cross-validation, though a slight underestimation was observed at the oldest ages. The average error of the clock, measured as mean absolute deviation from chronological age, was ~5.5 years. The root mean square error (RMSE) was ~7.3 years. For context, the standard deviation of ages in the sample was ~18 years; an error of ~5–7 years represents a substantial reduction in uncertainty.
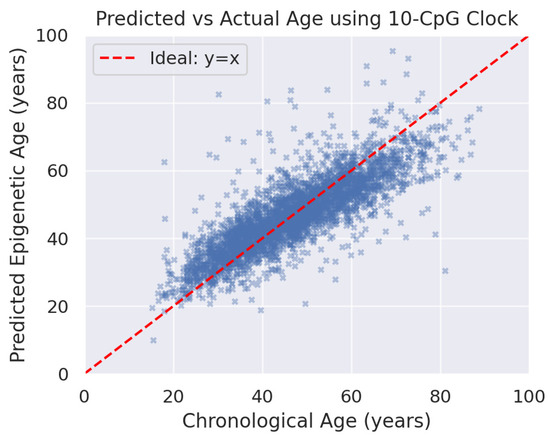
Figure 2.
Predicted epigenetic age versus chronological age for the 3408 saliva samples using the 10-CpG clock. Each blue cross (×) represents one individual. The red dashed line denotes the identity line (predicted age = actual age). The clock estimates are tightly clustered around the identity line (Pearson r ~0.80), demonstrating a strong linear relationship. At younger ages (<20 years), the clock tends to slightly over-predict (many points are above the line in the teens and 20s), whereas at the oldest ages (>80 years) there is a mild under-prediction (points below the line). This suggests that the model may compress the age range slightly at extremes, a common observation in age predictors. Overall, the high correlation and near-diagonal trend indicate that the 10-CpG clock captures the progression of methylation ageing.
The results highlight that even with only 10 methylation markers, it is possible to achieve a robust age predictor using saliva. However, the accuracy is, as expected, somewhat lower than that of multi-CpG clocks. For example, Horvath’s 353-CpG clock, if applied to saliva, has been reported to attain errors <3 years in some datasets, which is substantially better than this ~5.5-year mean absolute error (MAE). Horvath’s model achieves an error equal to ~2.7–2.9 years. Likewise, the Hannum 71-CpG clock has a ~3.9-year error in blood. Thus, there is a trade-off: the 10-CpG clock sacrifices some precision for the sake of parsimony, cost, and ability for mass utilisation. Nonetheless, the precision achieved is comparable to earlier simpler models [3]. For instance, a 3-CpG model for blood had a ~5-year error, similar to the 10 CpGs in saliva (albeit a different tissue) [9]. In this study, roughly 80% of samples had a DNAm age within ±7 years of the actual age, and ~50% were within ±5 years, which is adequate for many practical purposes (such as distinguishing age deciles or identifying outliers with accelerated ageing).
In summary, the 10-CpG saliva clock demonstrates the following: (a) high correlation with chronological age—indicating reliable tracking of ageing signals; (b) systematic hypermethylation with age at each included CpG—consistent with the known biology of epigenetic ageing; and (c) moderate prediction error in absolute terms—in the order of 5–6 years, which, while larger than flagship clocks, is impressive given the minimal markers. These results validate this study’s approach and set the stage for further exploration of the clock’s properties.
4. Discussion
This study aimed to use specific CpG sites via DNA methylation to estimate someone’s epigenetic age and compare this to someone’s chronological age. In undertaking this, a saliva-specific epigenetic clock that requires only 10 CpG sites yet captures a large portion of age-related methylation changes was created. This work demonstrates that saliva DNA methylation age can be estimated with a small, targeted panel of loci, making it feasible to implement in clinical or field settings where array-based methods may be impractical. In the context of epigenetic clock research, the 10-CpG model represents a middle ground between complex, highly accurate clocks and ultra-simplistic single-marker predictors.
4.1. Comparison with Established Clocks
The performance of our saliva clock is encouraging given its simplicity. With an R2 of ~0.64 and MAE ~5.5 years, it is not as precise as Horvath’s pan-tissue clock or Hannum’s blood clock, which both report R2 > 0.90 and errors of ~3–4 years. This gap is expected: these clocks use 71–353 CpGs and were trained on very large datasets, optimizing their fit. In contrast, the model intentionally restricts to 10 CpGs, which inherently limits the amount of age-related variance it can explain. Interestingly, the Horvath and Hannum clocks themselves share only a few common CpGs (6 out of 353 and 71), implying that multiple distinct subsets of CpGs can achieve high accuracy. This study’s approach essentially identifies one such subset tailored to saliva. The accuracy achieved (r~0.80) is on par with some earlier tissue-specific clocks that used a handful of markers. For example, as noted, studies that used 2–3 CpGs in saliva achieved ~5-year accuracy [3], and other forensic age prediction studies have used small panels with similar error margins. Thus, while this clock does not outperform the large multi-CpG models in sheer accuracy, it significantly reduces complexity with only a modest loss of precision. This makes it attractive for scenarios where measuring hundreds of CpGs is not feasible or necessary, or for mass-market projects, such as usage in forensics in police forces.
One advantage of using only 10 CpGs is the ability to develop low-cost, rapid assays. In a forensic context, for instance, one could design primers for these 10 loci and estimate the age of an unknown donor from a saliva trace. The small number of sites also facilitates interpretation and quality control: each CpG can be examined individually for consistency. However, a limitation to this specific 10-CpG clock, being trained specifically on saliva, is that it may not directly apply to other tissues, albeit it may demonstrate cross-over for blood [10]. Horvath’s clock is pan-tissue by design, meaning one model works for blood, saliva, brain, etc., possibly with an offset. The 10 CpGs might not even be variable with age in other tissues (for example, CHGA methylation changes might be saliva-specific). Thus, the current clock is intended for saliva (and potentially buccal swabs, which have similar cell types). The development of such tissue-specific clocks is still valuable, as it acknowledges that each tissue can have unique ageing methylation patterns.
4.2. Biological Significance of Clock CpGs
An interesting aspect of epigenetic clocks is that they often include CpGs in genes related to development, growth, or stress response. The 10-CpG clock is no exception. The genes associated with these CpGs hint at pathways that might be influenced by ageing.
ELOVL2 (cg16867657): ELOVL2 is perhaps the most famous epigenetic age marker. It encodes an enzyme involved in elongating polyunsaturated fatty acids. Hypermethylation of the ELOVL2 promoter with age has been observed in multiple tissues (blood, saliva, even non-human species) [11]. Recent work suggests ELOVL2 methylation is not just a passive clock: it may contribute to ageing phenotypes. For instance, it has been found that the DNA methylation of ELOVL2 could explain ~70% of age-related methylation variance, and it is posited as a universal ageing marker [12]. Moreover, ELOVL2 downregulation is linked to retinal aging and macular degeneration; experiments show that the loss of ELOV2 in mice leads to retinal dysfunction and accelerated ageing in the eye [13]. Thus, the hypermethylation of ELOVL2 with age (likely silencing its expression) could have functional consequences, potentially reducing the synthesis of long-chain fatty acids important for cell membranes and vision. In the saliva data, ELOVL2 methylation increases dramatically with age (from ~47% to 83%), reinforcing its role as a methylation clock cornerstone. While saliva itself may not reflect retinal changes, the systemic nature of ELOVL2 methylation makes it a reliable age indicator.
CHGA (cg21620282): This CpG is located in the promoter of the CHGA gene, which encodes Chromogranin A—a protein that regulates catecholamine release in neuroendocrine cells. CHGA has roles in cardiovascular physiology; for example, it is a precursor to peptides that modulate blood pressure. The hypermethylation of CHGA with age could potentially downregulate its expression. Intriguingly, CHGA knockout mice develop a hypertensive, hyperadrenergic phenotype (high blood pressure and elevated adrenaline) [14,15], indicating the importance of CHGA in cardiovascular homeostasis. Thus, reduced CHGA expression in later life (possibly via promoter methylation) might contribute to the increased prevalence of hypertension or sympathetic nervous system overactivity in the elderly. Our data show that CHGA methylation roughly doubles from youth (~12%) to old age (~27%). While this needs functional validation, it aligns with the idea that age-related promoter hypermethylation can lead to endocrine changes. CHGA’s inclusion in the clock also underscores that some age markers discovered by EPIC array (and not in earlier 450 k studies) are biologically relevant.
OTUD7A (cg04875128): This CpG is in the gene body of OTUD7A (OTU Deubiquitinase 7A) on chromosome 15. OTUD7A is a deubiquitinating enzyme; interestingly, it lies in a region (15q13.3) where deletions cause developmental disorders (including intellectual disability and epilepsy), highlighting its importance in neural development. The function of OTUD7A in adults is less clear, but it may influence protein homeostasis through the ubiquitin–proteasome system. Hypermethylation in OTUD7A might reduce its expression with age. One could speculate that lower OTUD7A activity might impair protein quality control in cells, potentially contributing to age-related cellular stress. Although direct evidence of OTUD7A in ageing is lacking, the fact that it emerged as a top age-correlated site in multiple studies (including an EWAS that found OTUD7A loci among the most significant age-associated CpGs [16,17]) suggests that it is a consistent marker of ageing. Further research is needed to see if OTUD7A methylation correlates with cognitive ageing or neurodegeneration, given its link to neurodevelopment.
PRLHR (cg11705975): This site maps to PRLHR (prolactin-releasing hormone receptor), also known as the receptor for prolactin-releasing peptide (PrRP). PrRP and its receptor PRLHR are involved in neuroendocrine functions, including the regulation of stress responses, feeding behaviour, and blood pressure. The age-related hypermethylation of PRLHR could downregulate this receptor. If PrRP/PRLHR signalling is dampened in older individuals, there could be effects on hormonal balance or stress resilience. Although not widely studied in ageing, one could hypothesise that changes in this pathway might affect metabolism or hypothalamic functions with age. This study found that PRLHR CpG methylation steadily increases (from ~32% to ~52%), suggesting gradual silencing. It would be interesting to measure if PrRP levels or activity change with age in parallel. At a minimum, PRLHR being an age marker reinforces the theme that neuropeptide and hormonal signalling pathways undergo epigenetic changes over the lifespan.
ZYG11A (cg06784991): The ZYG11A gene (zyg-11 family member A) encodes a protein that acts as a substrate recruiter for an E3 ubiquitin ligase complex, targeting specific proteins for degradation [18]. ZYG11A has been implicated in cell-cycle regulation and has a known role in pancreatic beta-cell proliferation. Remarkably, a human ZYG11A mutation was recently linked to young-onset diabetes due to impaired beta-cell growth. This implies that ZYG11A is necessary for maintaining insulin-producing cells. With age, if ZYG11A expression decreases (possibly via promoter methylation), this could limit the regenerative capacity of beta-cells, contributing to glucose dysregulation or type 2 diabetes risk in older adults. Our clock’s ZYG11A CpG shows one of the largest relative methylation changes (~4-fold increase from teen to elderly). It has been identified that ZYG11A is one of the top age-associated genes in blood DNA as well [9]. This consistency underscores ZYG11A as having a core function in ageing epigenetic change. Functionally, age-related ZYG11A hypermethylation might reduce the turnover of certain cell-cycle proteins, possibly affecting cell proliferation or senescence.
GPR158 (cg13206721) and GPR158-AS1: Cg13206721 is linked to GPR158, a gene encoding an orphan G-protein-coupled receptor, and the antisense transcript GPR158-AS1. GPR158 is highly expressed in the brain and has garnered attention for its role in mood regulation and cognitive function [19]. Notably, GPR158 was shown to mediate the effects of osteocalcin, a bone-derived hormone, on cognitive ageing; in mice, osteocalcin improves memory in older animals via GPR158 receptors in the brain [19]. Moreover, GPR158 links chronic stress to depression; it is upregulated by stress, and its deletion in mice produces resilience to depression-like states [20]. This study found that GPR158 locus methylation increases with age, raising intriguing questions: If GPR158 expression is reduced in older individuals due to promoter methylation, could that impact age-related cognitive decline or stress responses? It might, for example, diminish the brain’s responsiveness to osteocalcin, potentially contributing to memory loss. Additionally, a recent study found that deleting Gpr158 in mice altered synaptic physiology and dendritic architecture in hippocampal neurons [21], suggesting a role in neural plasticity. Thus, age-related GPR158 methylation might reflect or even drive changes in brain function with age. GPR158 (cg13206721) methylation rose from ~14% to ~28%. While saliva cells are not neurons, the epigenetic drift at this locus could be systemic. GPR158 is also expressed in peripheral tissues and involved in endocrine processes. Therefore, GPR158 hypermethylation is another clue that neurological and endocrine regulatory genes undergo epigenetic ageing.
LHFPL4 (cg12841266): LHFPL4 (also known as FAM126A) belongs to a family of lipoma HMGIC fusion partner-like genes, some of which have roles in the nervous system. LHFPL4 specifically has been less studied, but interestingly, the nearby gene LHFPL3 is involved in synaptic transmission in auditory pathways. The CpG cg12841266 (in the LHFPL4 gene body) was highlighted as a novel age-associated CpG in an EPIC-array study [22], indicating that it consistently gains methylation with age. While the functional impact of LHFPL4 methylation is unknown, the gene’s potential involvement in cell membrane or synaptic functions warrants exploration. The results showed that LHFPL4 methylation changed from ~26% to ~49% over the age span. Given its robust correlation with age, it serves as a valuable clock component, even if its biological role remains to be elucidated.
Intergenic CpGs (cg13327545, cg10804656, cg05991454): Three of the clock’s CpGs are not annotated to a specific gene. Often, such CpGs reside in regulatory regions like enhancers or non-coding RNAs. For instance, cg13327545 and cg10804656 are both located on chromosome 10 in regions that might be distal enhancers (the former is near ~22.62 Mb, which could be an enhancer region for a developmental gene). Intergenic age-associated CpGs can still have functional relevance; they might regulate neighbouring genes in trans. Alternatively, they could be markers of heterochromatin changes with age. Cg05991454, which showed steady hypermethylation (from ~7% to ~20%), is in a CpG island on chr4 that is gene-desert. While we emphasize gene-linked sites in our interpretation, these “anonymous” CpGs are important precisely because they were among the top genes correlated with age—possibly indicating previously unrecognized regulatory elements. Future genome-wide analyses might reveal if these intergenic loci contact gene promoters via chromatin looping, thereby influencing gene expression.
In summary, the genes associated with the 10 CpGs are enriched in functions related to metabolism (ELOVL2, CHGA), hormone/neuropeptide signalling (CHGA, PRLHR, GPR158), and protein homeostasis/cell cycle (ZYG11A, OTUD7A). This mirrors the notion that ageing involves interconnected decline in metabolic, endocrine, and proteostatic processes. Hypermethylation at these loci likely represents the gradual epigenetic silencing of certain genes as part of the ageing program. It is tempting to speculate that such silencing might contribute to age-related phenotypes—for example, reduced ELOVL2 leading to lipid abnormalities, or reduced ZYG11A contributing to impaired cell regeneration. Our data cannot prove causation, but these CpGs warrant experimental follow-up. At a minimum, they serve as convenient sentinels of the ageing process in saliva DNA.
Saliva-Specific Considerations: An epigenetic clock derived from saliva inherently captures the ageing of the cell types present (oral epithelial cells and leukocytes). One might ask, do the 10 CpGs identified reflect ageing in epithelial cells, immune cells, or both? Some markers like ELOVL2 and ZYG11A are very robust across many tissues (likely present in both cell types). Others might be more cell-type-specific. For instance, CHGA is primarily expressed in neuroendocrine cells; interestingly, small salivary glands in the mouth express chromogranin, and circulating leukocytes do not, so CHGA methylation in saliva could be coming from minor salivary gland epithelial DNA, or perhaps neutrophils (some immune cells express chromogranin A as well). GPR158 is expressed in many tissues, including salivary glands possibly. Without cell-sorted data, we cannot pinpoint the cell of origin for each CpG’s signal. However, the clock’s accuracy suggests that whatever mixture of cells saliva has, the composite methylation of these 10 sites reliably shifts with the donor’s age. This bodes well for practical use: one does not need to correct for cell-type proportion. The selected CpGs likely change with age in a cell-autonomous way in both buccal and immune cells.
It is also noteworthy that the clock’s error was slightly larger in younger individuals (a common observation is that methylation age has more variability in children/teens). Some of the 10 CpGs may start changing only after puberty or in early adulthood, leading to less precision at younger ages. This could be addressed by adding a few CpGs that specifically capture paediatric developmental methylation changes, or by using an alternate model (e.g., with a non-linear fit) for the lower age range. In this case, since the youngest individual was 15 and most individuals were of post-puberty age, the linear model sufficed. Nonetheless, an avenue for improvement is a piecewise model that is age-conditional.
Applications and Future Directions: The 10-CpG saliva clock has several applications. In forensic science, being able to estimate the age of a person from saliva traces (left on, say, a bite mark or a discarded cup) can be invaluable. Traditional forensic age assays have used blood markers, but saliva is often more readily found at crime scenes. A small CpG panel can be developed into a forensic kit. Importantly, all 10 sites are CpG methylation markers, which are generally stable in stored DNA and can be analysed even in somewhat degraded DNA. Prior forensic age prediction models have included CpGs like ELOVL2, FHL2, KLF14, TRIM59, etc., many of which overlap in function with the chosen loci in this study.
In biomedical research, a saliva epigenetic age predictor could be useful for non-invasive health monitoring. For example, longitudinal changes in someone’s saliva DNAm age compared to their chronological age (often termed “epigenetic age acceleration”) could potentially indicate accelerated ageing or health deterioration. In blood, such age acceleration metrics have been linked to a higher risk of mortality, cancer, and neurodegenerative disease. It would be interesting to see if saliva DNAm age acceleration correlates with lifestyle factors (smoking, diet, stress) or oral health (periodontal disease might influence methylation in saliva). Since this clock is trained on chronological age, any discrepancy between DNAm age and actual age might reflect biological age acceleration.
Another future direction is refining the clock by adding or swapping CpGs to improve accuracy while maintaining parsimony. Perhaps a 5-CpG model could be nearly as good, or a 15-CpG model could breach the 5-year error barrier. Our selection was based on linear correlation; using a machine learning approach on the EPIC data might find combinations that capture non-linear effects or interactions. However, any added complexity must be weighed against ease of use. Ten CpGs already strikes a balance in our view: enough to capture a robust signal (dampening the noise of any single locus’s outliers), but few enough to be easily measured.
4.3. Limitations
We acknowledge several limitations. First, our clock was trained and tested on the same cohort (albeit with internal cross-validation). An independent validation on a separate set of saliva samples would strengthen confidence in its generalisability. Second, the cohort demographics were not deeply considered in this analysis, although the results were adjusted for sex, which should mitigate sex bias. Nonetheless, our clock might have a small systematic bias if applied to a population very different from this study set. Third, while we discussed biological significance, mechanistic conclusions cannot be drawn without experimental validation. The associations between the hypermethylation of these genes and functional decline are correlative. It is possible that many clock CpGs are “passengers” of ageing—they change because of epigenetic drift or environment, not because they themselves drive ageing. Indeed, a debate in the field is whether methylation changes cause ageing or are markers of other underlying processes. This clock, like others, is primarily a predictive tool, but it offers candidate loci for further investigation into causality.
Convergence with Other Ageing Hallmarks
It is intriguing to note that several genes highlighted by our saliva clock intersect with other ageing hallmarks beyond epigenetics. For instance, ZYG11A relates to proteostasis (protein degradation), ELOVL2 to metabolic dysfunction (lipid changes), and GPR158 to altered intercellular communication (hormonal signalling). This suggests that epigenetic changes at these loci might be part of a broader network of ageing changes. Such convergence reinforces the concept that epigenetic clocks are tapping into fundamental ageing processes.
In light of the above, our 10-CpG clock not only serves as a practical age estimator, but also a compact model of the ageing epigenome in saliva. Each CpG in the model could be viewed as a proxy for an “ageing module”—be it metabolic ageing (ELOVL2), neuroendocrine ageing (CHGA, GPR158), or replicative ageing (ZYG11A, OTUD7A). This is somewhat speculative, but future multi-omics studies (integrating methylation with transcriptomics and proteomics in saliva) could test these links. Saliva, interestingly, contains secreted factors from salivary glands and gingival crevices, so correlating saliva epigenetic age with saliva proteomic profiles (inflammation markers, etc.) would be an exciting angle.
5. Conclusions
This study identified that a novel epigenetic clock using just 10 CpG sites can effectively predict age in saliva DNA. The clock’s specificity to saliva underscores that tissue-tailored clocks can be developed to leverage the most informative markers for that tissue. While less complex than multi-tissue clocks, the 10-CpG model achieves a high correlation with age and offers practical advantages. Its constituent CpGs highlight key genes and pathways that may be entwined with the ageing process, such as lipid metabolism, neuroendocrine regulation, and cell-cycle control. The hypermethylation of these loci with age could serve as both a biomarker of ageing and a mechanistic clue to age-related dysfunction (e.g., hypertension, cognitive decline, or diabetes).
These findings encourage further research in several directions. First, external validation of the saliva clock in independent cohorts (possibly including younger children or very elderly individuals >90, as well as diverse ethnic groups) will be important to confirm its universality. Second, exploring modifications of the clock—for example, incorporating a few hypomethylated sites or adapting it to a semi-supervised model that can estimate “biological age”—could enhance its utility for predicting health outcomes. Third, investigating interventions (such as lifestyle changes) to see if they slow the tick of this epigenetic clock could provide insights into modifiable factors of ageing. If an intervention leads to a lower DNAm age than expected, this suggests a deceleration in epigenetic ageing. Given the non-invasive nature of saliva collection, this clock is well suited for longitudinal monitoring in large populations or clinical trials.
As a final note, the 10-CpG saliva epigenetic clock represents a step toward more accessible epigenetic age assessment. Its development illustrates that even as few as ten methylation sites, when carefully chosen, carry substantial information about the ageing state of an individual. This paves the way for cost-effective epigenetic clocks that could be deployed in routine healthcare or forensic investigations. Moreover, the specific CpGs in the clock offer a compact window into the biological changes accompanying ageing, pointing to candidate genes that merit further study in the quest to understand and perhaps ameliorate human ageing.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/dna5020028/s1, Table S1: Participants’ disease conditions; Table S2: Methylation Level of CpG Sites.
Author Contributions
Conceptualization: C.C.; Methodology: C.C. and J.B.; Software, C.C. and J.B.; Validation, H.C.C. and J.B.; Formal Analysis, H.C.C.; Investigation, C.C.; Resources, C.C. and H.C.C.; Data Curation, C.C. and J.B.; Writing—Original Draft Preparation, C.C.; Writing—Review and Editing, C.C. and H.C.C.; Visualization, C.C.; Supervision, H.C.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
This study requires no ethical approval. This study was completed in accordance with the Institutional statements outlined in the “Guidelines for ethical approval of research involving human participants”, page 7, of the University of Essex, which states that ethical approval is not required for studies based on “previously collected data for research”, which this work falls under. The data used within this study are defined as secondary due to the data being previously collected via Muhdo’s normal day-to-day operations; these data were not specifically collected for this study. This work was conducted according to the guidelines of the Declaration of Helsinki, and all participants gave written informed consent via Muhdo Health Ltd. for data to be anonymized and utilized for research and publication (clients chose to opt in). Muhdo Health company is GDPR-compliant and registered in the U.K. The dataset specified in this work is shared in the Supplementary Materials and is completely available and not withheld.
Informed Consent Statement
This work was conducted in accordance with the Declaration of Helsinki, and all participants gave written informed consent via Muhdo Health Ltd. for data to be anonymized and utilized for research and publication purposes.
Data Availability Statement
All data are available in the Supplementary Information.
Acknowledgments
We would like to thank Eurofins Denmark for conducting the laboratory analysis on the saliva samples and Tag.Bio for aiding in statistical analysis.
Conflicts of Interest
Christopher Collins (Data Analyst) and James Brown (Nutrition Director) are affiliated with Muhdo Health Ltd. and declare no conflicts of interest in relation to this study. This study uses data collected by Muhdo Health that were anonymized upon collection and consent was given for the use of these data for research and publication purposes. This study aims to allow open access to an epigenetic clock and the company does gain any commercial benefit as a result of any information disclosed. Henry C. Chung is external to the company and declares no conflicts of interest. No parties were directly paid, funded, or in any way compensated for their role in the data collection, analysis, methods, reporting, writing, or scrutiny of this paper. Data were made available via the Muhdo Health Data repository, and all authors freely used uncompensated personal time to complete the paper. All data are accessible via Muhdo Health Ltd. All data are available via the Supplemental Information supplied.
References
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, J.; Wang, L.; Gao, Y.; Feng, G.; Li, G.; Zou, J.; Yu, M.; Li, Y.F.; Liu, C.; et al. Lipid metabolism dysfunction induced by age-dependent DNA methylation accelerates aging. Signal Transduct. Target. Ther. 2022, 7, 162. [Google Scholar] [CrossRef] [PubMed]
- Bocklandt, S.; Lin, W.; Sehl, M.E.; Sánchez, F.J.; Sinsheimer, J.S.; Horvath, S.; Vilain, E. Epigenetic predictor of age. PLoS ONE 2011, 6, e14821. [Google Scholar] [CrossRef] [PubMed]
- El-Shishtawy, N.M.; El Marzouky, F.M.; El-Hagrasy, H.A. DNA methylation of ELOVL2 gene as an epigenetic marker of age among Egyptian population. Egypt. J. Med. Hum. Genet. 2024, 25, 14. [Google Scholar] [CrossRef]
- Garagnani, P.; Bacalini, M.G.; Pirazzini, C.; Gori, D.; Giuliani, C.; Mari, D.; Passarino, G.; Di Blasio, A.M.; Capri, M.; Salvioli, S. Methylation of ELOVL2 gene as a new epigenetic marker of age. Aging Cell 2012, 11, 1132–1134. [Google Scholar] [CrossRef]
- Feil, R.; Fraga, M.F. Epigenetics and the environment: Emerging patterns and implications. Nat. Rev. Genet. 2012, 13, 97–109. [Google Scholar] [CrossRef]
- Braun, P.R.; Han, S.; Hing, B.; Nagahama, Y.; Gaul, L.N.; Heinzman, J.T.; Grossbach, A.J.; Close, L.; Dlouhy, B.J.; Howard, M.A., III; et al. Genome-wide DNA methylation comparison between live human brain and peripheral tissues within individuals. Transl. Psychiatry 2019, 9, 47. [Google Scholar] [CrossRef]
- Tajuddin, S.M.; Hernandez, D.G.; Chen, B.H.; Noren Hooten, N.; Mode, N.A.; Nalls, M.A.; Singleton, A.B.; Ejiogu, N.; Chitrala, K.N.; Zonderman, A.B.; et al. Novel age-associated DNA methylation changes and epigenetic age acceleration in middle-aged African Americans and whites. Clin. Epigenetics 2019, 11, 119. [Google Scholar] [CrossRef]
- Weidner, C.I.; Lin, Q.; Koch, C.M.; Eisele, L.; Beier, F.; Ziegler, P.; Bauerschlag, D.O.; Jöckel, K.-H.; Erbel, R.; Mühleisen, T.W.; et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014, 15, R24. [Google Scholar] [CrossRef]
- Laboute, T.; Zucca, S.; Holcomb, M.; Patil, D.N.; Garza, C.; Wheatley, B.A.; Roy, R.N.; Forli, S.; Martemyanov, K.A. Orphan receptor GPR158 serves as a metabotropic glycine receptor: mGlyR. Science 2023, 379, 1352–1358. [Google Scholar] [CrossRef]
- Reynolds, L.M.; Taylor, J.R.; Ding, J.; Lohman, K.; Johnson, C.; Siscovick, D.; Burke, G.; Post, W.; Shea, S.; Jacobs, D.R., Jr.; et al. Age-related variations in the methylome associated with gene expression in human monocytes and T cells. Nat. Commun. 2014, 5, 5366. [Google Scholar] [CrossRef] [PubMed]
- Pevsner, J.; Sabunciyan, S.; Yolken, R.H.; Webster, M.J.; Dinkins, T.; Callinan, P.A.; Fan, J.B.; Potash, J.B.; Feinberg, A.P. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 2012, 13, R43. [Google Scholar]
- Chen, D.; Chao, D.L.; Rocha, L.; Kolar, M.; Nguyen Huu, V.A.; Krawczyk, M.; Dasyani, M.; Wang, T.; Jafari, M.; Jabari, M.; et al. The lipid elongation enzyme ELOVL2 is a molecular regulator of aging in the retina. Aging Cell 2020, 19, e13100. [Google Scholar] [CrossRef] [PubMed]
- Dev, N.B.; Mir, S.A.; Gayen, J.R.; Siddiqui, J.A.; Mustapic, M.; Vaingankar, S.M. Cardiac electrical activity in a genomically “humanized” chromogranin a monogenic mouse model with hyperadrenergic hypertension. J. Cardiovasc. Transl. Res. 2014, 7, 483–493. [Google Scholar] [CrossRef]
- Rakyan, V.K.; Down, T.A.; Maslau, S.; Andrew, T.; Yang, T.P.; Beyan, H.; Whittaker, P.; McCann, O.T.; Finer, S.; Valdes, A.M.; et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010, 20, 434–439. [Google Scholar] [CrossRef]
- Li, A.; Mueller, A.; English, B.; Arena, A.; Vera, D.; Kane, A.E.; Sinclair, D.A. Novel feature selection methods for construction of accurate epigenetic clocks. PLoS Comput. Biol. 2022, 18, e1009938. [Google Scholar] [CrossRef]
- Florath, I.; Butterbach, K.; Müller, H.; Bewerunge-Hudler, M.; Brenner, H. Cross-sectional and longitudinal changes in DNA methylation with age: An epigenome-wide analysis revealing over 60 novel age-associated CpG sites. Hum. Mol. Genet. 2014, 23, 1186–1201. [Google Scholar] [CrossRef]
- Tan, Q.; Heijmans, B.T.; Hjelmborg, J.V.B.; Soerensen, M.; Christensen, K.; Christiansen, L. Epigenetic drift in the aging genome: A ten-year follow-up in an elderly twin cohort. Int. J. Epidemiol. 2016, 45, 1146–1158. [Google Scholar] [CrossRef]
- Chang, J.; Song, Z.; Wei, S.; Zhou, Y.; Ju, J.; Yao, P.; Jiang, Y.; Jin, H.; Chi, X.; Li, N. Expression mapping and functional analysis of orphan G-protein-coupled receptor GPR158 in the adult mouse brain using a GPR158 transgenic mouse. Biomolecules 2023, 13, 479. [Google Scholar] [CrossRef]
- Sutton, L.P.; Orlandi, C.; Song, C.; Oh, W.C.; Muntean, B.S.; Xie, K.; Filippini, A.; Xie, X.; Satterfield, R.; Yaeger, J.D.W.; et al. Orphan receptor GPR158 controls stress-induced depression. eLife 2018, 7, e33273. [Google Scholar] [CrossRef]
- Çetereisi, D.; Kramvis, I.; Gebuis, T.; van der Loo, R.J.; Gouwenberg, Y.; Mansvelder, H.D.; Li, K.W.; Smit, A.B.; Spijker, S. Gpr158 Deficiency Impacts Hippocampal CA1 Neuronal Excitability, Dendritic Architecture, and Affects Spatial Learning. Front. Cell. Neurosci. 2019, 13, 465. [Google Scholar] [CrossRef] [PubMed]
- Alsaleh, H.; Haddrill, P.R. Identifying blood-specific age-related DNA methylation markers on the Illumina MethylationEPIC® BeadChip. Forensic Sci. Int. 2019, 303, 109944. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).