
Evolution of Acquired Drug Resistance in BRAF-Mutant Melanoma


Abstract

1. Introduction
2. The Melanoma Genome
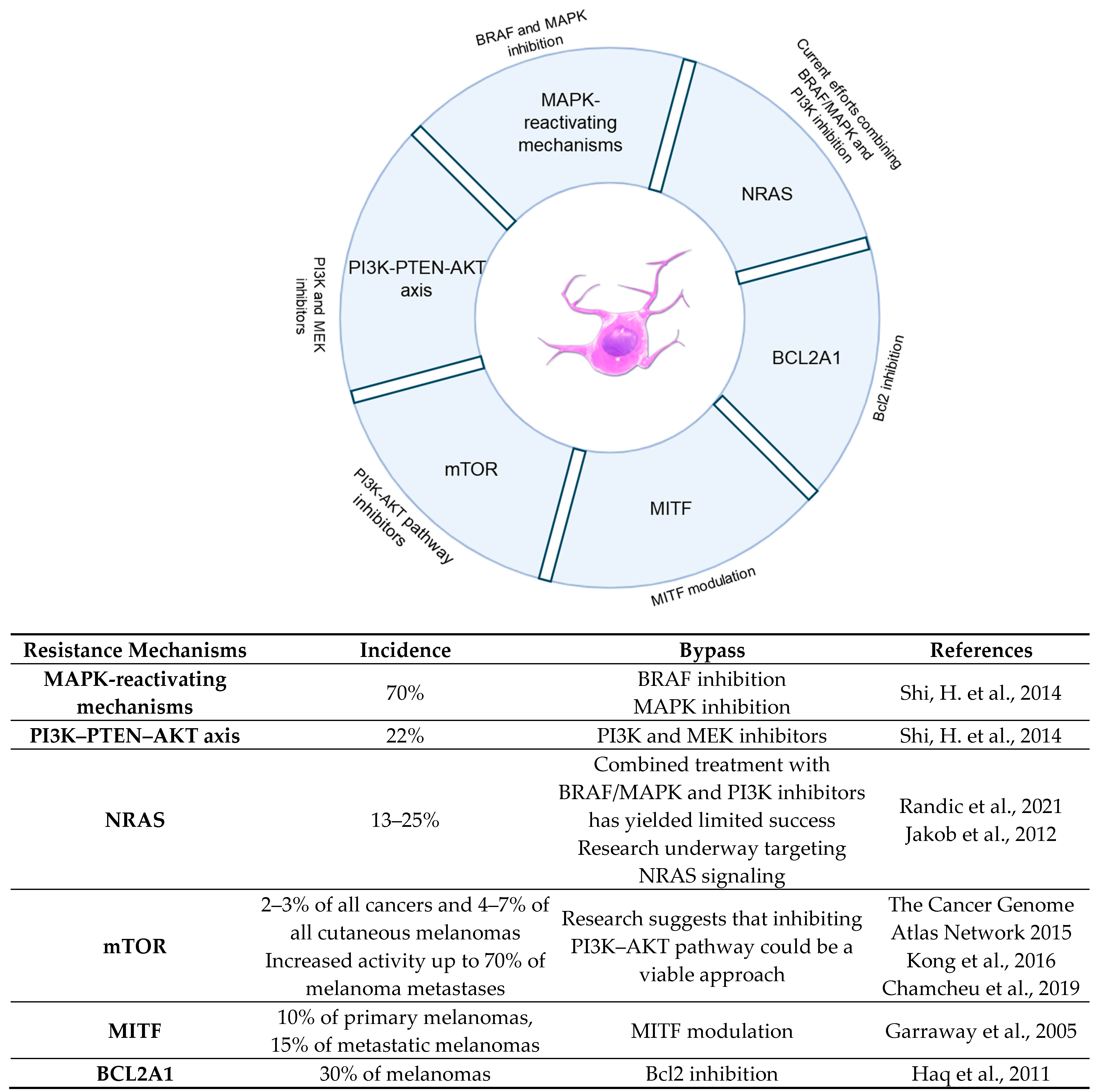
3. The BRAF–MEK–ERK Highway to Melanoma
3.1. BRAF
3.2. PI3K-PTEN-AKT
3.3. NRAS
3.4. mTOR
3.5. MITF: Directing the Melanoma Orchestra

{kind=link}
{kind=link}
{kind=link}
| MITF-Driven Resistance Mechanism | Ref. | Effect | Comments |
|---|---|---|---|
| PAX3-mediated upregulation of MITF | [96] | Drug resistance in response to BRAF/MEK inhibition | - |
| MITF focal amplification | [97,98,99,100] | Associated with tumor relapse and resistance to BRAF/MEK inhibition | Counteraction of BRAF-induced reduction of MITF |
| E318K mutation of MITF | [105,106,107] | Bypass of BRAF-induced senescence | E318K MITF variant increases its transcriptional activity |
| MITF-mediated upregulation of BCL2A1 | [110] | Reduced apoptosis of melanoma cells | Combined treatment with BRAF and BCL2A1 inhibitors increase apoptosis and reduce tumor volume |
| BRN2-mediated downregulation of MITF | [111] | Promotes migration and cell survival | Intratumor heterogeneity with proliferative or invasive cell subpopulations |
4. Final Remarks
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saginala, K.; Barsouk, A.; Aluru, J.S.; Rawla, P.; Barsouk, A. Epidemiology of Melanoma. Med. Sci. 2021, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, D.C.; Green, A.C.; Olsen, C.M. The Growing Burden of Invasive Melanoma: Projections of Incidence Rates and Numbers of New Cases in Six Susceptible Populations through 2031. J. Investig. Dermatol. 2016, 136, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Facts & Figures 2019; American Cancer Society: Atlanta, GA, USA, 2019. [Google Scholar]
- Leiter, U.; Eigentler, T.; Garbe, C. Epidemiology of skin cancer. Adv. Exp. Med. Biol. 2014, 810, 120–140. [Google Scholar] [PubMed]
- Thirlwell, C.; Nathan, P. Melanoma—Part 2: Management. BMJ 2008, 337, a2488. [Google Scholar] [CrossRef]
- Lapkina, E.Z.; Esimbekova, A.R.; Beleniuk, V.D.; Savchenko, A.A.; Ruksha, T.G. The Distribution of B16 Melanoma Cells in Cell-Cycle Phases under the Influence of Dacarbazine. Cell Tissue Biol. 2023, 17, 161–168. [Google Scholar] [CrossRef]
- Kaunitz, G.J.; Cottrell, T.R.; Lilo, M.; Muthappan, V.; Esandrio, J.; Berry, S.; Taube, J.M. Melanoma subtypes demonstrate distinct PD-L1 expression profiles. Lab. Investig. 2017, 97, 1063–1071. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Long, G.V.; Robert, C.; Brady, B.; Dutriaux, C.; Di Giacomo, A.M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; et al. Survival Outcomes in Patients with Previously Untreated BRAF Wild-Type Advanced Melanoma Treated with Nivolumab Therapy. JAMA Oncol. 2019, 5, 187. [Google Scholar] [CrossRef]
- Martincorena, I. Somatic mutation and clonal expansions in human tissues. Genome Med. 2019, 11, 35. [Google Scholar] [CrossRef]
- Pei, X.M.; Yeung, M.H.Y.; Wong, A.N.N.; Tsang, H.F.; Yu, A.C.S.; Yim, A.K.Y.; Wong, S.C.C. Targeted Sequencing Approach and Its Clinical Applications for the Molecular Diagnosis of Human Diseases. Cells 2023, 12, 493. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.-P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Colombino, M.; Paliogiannis, P.; Cossu, A.; De Re, V.; Miolo, G.; Botti, G.; Scognamiglio, G.; Ascierto, P.A.; Santeufemia, D.A.; Fraggetta, F.; et al. BRAF Mutations and Dysregulation of the MAP Kinase Pathway Associated to Sinonasal Mucosal Melanomas. J. Clin. Med. 2019, 8, 1577. [Google Scholar] [CrossRef] [PubMed]
- Dumaz, N.; Jouenne, F.; Delyon, J.; Mourah, S.; Bensussan, A.; Lebbé, C. Atypical BRAF and NRAS Mutations in Mucosal Melanoma. Cancers 2019, 11, 1133. [Google Scholar] [CrossRef] [PubMed]
- Magana-Garcia, M.; Ackerman, A.B. What are nevus cells? Am. J. Dermatopathol. 1990, 12, 93–102. [Google Scholar] [CrossRef]
- Shreberk-Hassidim, R.; Ostrowski, S.M.; Fisher, D.E. The Complex Interplay between Nevi and Melanoma: Risk Factors and Precursors. Int. J. Mol. Sci. 2023, 24, 3541. [Google Scholar] [CrossRef]
- Pollock, P.M.; Harper, U.L.; Hansen, K.S.; Yudt, L.M.; Stark, M.; Robbins, C.M.; Moses, T.Y.; Hostetter, G.; Wagner, U.; Kakareka, J.; et al. High frequency of BRAF mutations in nevi. Nat. Genet. 2003, 33, 19–20. [Google Scholar] [CrossRef]
- Yazdi, A.S.; Palmedo, G.; Flaig, M.J.; Puchta, U.; Reckwerth, A.; Rütten, A.; Mentzel, T.; Hügel, H.; Hantschke, M.; Schmid-Wendtner, M.-H.; et al. Mutations of the BRAF gene in benign and malignant melanocytic lesions. J. Investig. Dermatol. 2003, 121, 1160–1162. [Google Scholar] [CrossRef]
- Francis, J.H.; Grossniklaus, H.E.; Habib, L.A.; Marr, B.; Abramson, D.H.; Busam, K.J. BRAF, NRAS, and GNAQ Mutations in Conjunctival Melanocytic Nevi. Investig. Opthalmology Vis. Sci. 2018, 59, 117. [Google Scholar] [CrossRef]
- Bauer, J.; Curtin, J.A.; Pinkel, D.; Bastian, B.C. Congenital melanocytic nevi frequently harbor NRAS mutations but no BRAF mutations. J. Investig. Dermatol. 2007, 127, 179–182. [Google Scholar] [CrossRef]
- da Silva, V.M.; Martinez-Barrios, E.; Tell-Marti, G.; Dabad, M.; Carrera, C.; Aguilera, P.; Brualla, D.; Esteve-Codina, A.; Vicente, A.; Puig, S.; et al. Genetic Abnormalities in Large to Giant Congenital Nevi: Beyond NRAS Mutations. J. Investig. Dermatol. 2019, 139, 900–908. [Google Scholar] [CrossRef]
- Stark, M.S.; Tan, J.-M.; Tom, L.; Jagirdar, K.; Lambie, D.; Schaider, H.; Soyer, H.P.; Sturm, R.A. Whole-Exome Sequencing of Acquired Nevi Identifies Mechanisms for Development and Maintenance of Benign Neoplasms. J. Investig. Dermatol. 2018, 138, 1636–1644. [Google Scholar] [CrossRef] [PubMed]
- Mackiewicz-Wysocka, M.; Czerwińska, P.; Filas, V.; Bogajewska, E.; Kubicka, A.; Przybyła, A.; Dondajewska, E.; Kolenda, T.; Marszałek, A.; Mackiewicz, A. Oncogenic BRAF mutations and p16 expression in melanocytic nevi and melanoma in the Polish population. Adv. Dermatol. Allergol. 2017, 5, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Dhomen, N.; Reis-Filho, J.S.; Dias, S.d.R.; Hayward, R.; Savage, K.; Delmas, V.; Larue, L.; Pritchard, C.; Marais, R. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell 2009, 15, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, J.B.; Raimundo, L.; Calheiros, J.; Carvalho, C.; Barcherini, V.; Lima, N.R.; Gomes, C.; Almeida, M.I.; Alves, M.G.; Costa, J.L.; et al. Targeting p53 for Melanoma Treatment: Counteracting Tumour Proliferation, Dissemination and Therapeutic Resistance. Cancers 2021, 13, 1648. [Google Scholar] [CrossRef] [PubMed]
- Valverde, P.; Healy, E.; Jackson, I.; Rees, J.L.; Thody, A.J. Variants of the melanocyte-stimulating hormone receptor gene are associated with red hair and fair skin in humans. Nat. Genet. 1995, 11, 328–330. [Google Scholar] [CrossRef]
- Frändberg, P.-A.; Doufexis, M.; Kapas, S.; Chhajlani, V. Human pigmentation phenotype: A point mutation generates nonfunctional MSH receptor. Biochem. Biophys. Res. Commun. 1998, 245, 490–492. [Google Scholar] [CrossRef]
- Schiöth, H.B.; Phillips, S.R.; Rudzishd, R.; Birch-Machin, M.A.; Wikberg, J.E.; Rees, J.L. Loss of function mutations of the human melanocortin 1 receptor are common and are associated with red hair. Biochem. Biophys. Res. Commun. 1999, 260, 488–491. [Google Scholar] [CrossRef]
- Palmer, J.S.; Duffy, D.L.; Box, N.F.; Aitken, J.F.; O’Gorman, L.E.; Green, A.C.; Hayward, N.K.; Martin, N.G.; Sturm, R.A. Melanocortin-1 receptor polymorphisms and risk of melanoma: Is the association explained solely by pigmentation phenotype? Am. J. Hum. Genet. 2000, 66, 176–186. [Google Scholar] [CrossRef]
- Rohrer, L.; Spohr, C.; Beha, C.; Griffin, R.; Braun, S.; Halbach, S.; Brummer, T. Analysis of RAS and drug induced homo- and heterodimerization of RAF and KSR1 proteins in living cells using split Nanoluc luciferase. Cell Commun. Signal. 2023, 21, 136. [Google Scholar] [CrossRef]
- Cronin, R.; Brooke, G.N.; Prischi, F. The role of the p90 ribosomal S6 kinase family in prostate cancer progression and therapy resistance. Oncogene 2021, 40, 3775–3785. [Google Scholar] [CrossRef]
- Yi, Y.W.; You, K.S.; Park, J.-S.; Lee, S.-G.; Seong, Y.-S. Ribosomal Protein S6: A Potential Therapeutic Target against Cancer? Int. J. Mol. Sci. 2021, 23, 48. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Blenis, J.; Yuan, J. Activation of PI3K/Akt and MAPK pathways regulates Myc-mediated transcription by phosphorylating and promoting the degradation of Mad1. Proc. Natl. Acad. Sci. USA 2008, 105, 6584–6589. [Google Scholar] [CrossRef] [PubMed]
- Cutler, R.E.; Stephens, R.M.; Saracino, M.R.; Morrison, D.K. Autoregulation of the Raf-1 serine/threonine kinase. Proc. Natl. Acad. Sci. USA 1998, 95, 9214–9219. Available online: https://www.ncbi.nlm.nih.gov/pubmed/9689060 (accessed on 25 July 2024). [CrossRef] [PubMed]
- Zhang, M.; Jang, H.; Li, Z.; Sacks, D.B.; Nussinov, R. B-Raf autoinhibition in the presence and absence of 14-3-3. Structure 2021, 29, 768–777.e2. [Google Scholar] [CrossRef]
- Maurer, G.; Tarkowski, B.; Baccarini, M. Raf kinases in cancer-roles and therapeutic opportunities. Oncogene 2011, 30, 3477–3488. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.H.; Frost, J.A. Phosphorylation of Raf-1 by p21-activated kinase 1 and Src regulates Raf-1 autoinhibition. J. Biol. Chem. 2003, 278, 11221–11226. [Google Scholar] [CrossRef]
- Tran, N.H.; Wu, X.; Frost, J.A. B-Raf and Raf-1 are regulated by distinct autoregulatory mechanisms. J. Biol. Chem. 2005, 280, 16244–16253. [Google Scholar] [CrossRef]
- Marais, R.; Light, Y.; Paterson, H.F.; Mason, C.S.; Marshall, C.J. Differential regulation of Raf-1, A-Raf, and B-Raf by oncogenic ras and tyrosine kinases. J. Biol. Chem. 1997, 272, 4378–4383. Available online: https://www.ncbi.nlm.nih.gov/pubmed/9020159 (accessed on 25 July 2024). [CrossRef]
- Wellbrock, C.; Karasarides, M.; Marais, R. The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol. 2004, 5, 875–885. [Google Scholar] [CrossRef]
- Takahashi, M.; Li, Y.; Dillon, T.J.; Kariya, Y.; Stork, P.J.S. Phosphorylation of the C-Raf N Region Promotes Raf Dimerization. Mol. Cell. Biol. 2017, 37, e00132-17. [Google Scholar] [CrossRef]
- TShabaneh, T.B.; Molodtsov, A.K.; Steinberg, S.M.; Zhang, P.; Torres, G.M.; Mohamed, G.A.; Boni, A.; Curiel, T.J.; Angeles, C.V.; Turk, M.J. Oncogenic BRAFV600E Governs Regulatory T-cell Recruitment during Melanoma Tumorigenesis. Cancer Res. 2018, 78, 5038–5049. [Google Scholar] [CrossRef] [PubMed]
- AKnight, A.; Karapetyan, L.; Kirkwood, J.M. Immunotherapy in Melanoma: Recent Advances and Future Directions. Cancers 2023, 15, 1106. [Google Scholar] [CrossRef]
- Porcelli, L.; Porcelli, L.; Di Fonte, R.; Di Fonte, R.; Pierri, C.L.; Pierri, C.L.; Fucci, L.; Fucci, L.; Saponaro, C.; Saponaro, C.; et al. BRAFV600E;K601Q metastatic melanoma patient-derived organoids and docking analysis to predict the response to targeted therapy. Pharmacol. Res. 2022, 182, 106323. [Google Scholar] [CrossRef]
- Gogas, H.J.; Kirkwood, J.M.; Sondak, V.K. Chemotherapy for metastatic melanoma: Time for a change? Cancer 2007, 109, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of Mutated, Activated BRAF in Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef]
- Proietti, I.; Skroza, N.; Bernardini, N.; Tolino, E.; Balduzzi, V.; Marchesiello, A.; Michelini, S.; Volpe, S.; Mambrin, A.; Mangino, G.; et al. Mechanisms of Acquired BRAF Inhibitor Resistance in Melanoma: A Systematic Review. Cancers 2020, 12, 2801. [Google Scholar] [CrossRef]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600-Mutant Advanced Melanoma Treated with Vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef]
- Kansler, E.R.; Verma, A.; Langdon, E.M.; Simon-Vermot, T.; Yin, A.; Lee, W.; Attiyeh, M.; Elemento, O.; White, R.M. Melanoma genome evolution across species. BMC Genom. 2017, 18, 136. [Google Scholar] [CrossRef]
- Johannessen, C.M.; Johnson, L.A.; Piccioni, F.; Townes, A.; Frederick, D.T.; Donahue, M.K.; Narayan, R.; Flaherty, K.T.; Wargo, J.A.; Root, D.E.; et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature 2013, 504, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Alabi, S.; Jaime-Figueroa, S.; Yao, Z.; Gao, Y.; Hines, J.; Samarasinghe, K.T.G.; Vogt, L.; Rosen, N.; Crews, C.M. Mutant-selective degradation by BRAF-targeting PROTACs. Nat. Commun. 2021, 12, 920. [Google Scholar] [CrossRef] [PubMed]
- Turajlic, S.; Furney, S.J.; Stamp, G.; Rana, S.; Ricken, G.; Oduko, Y.; Saturno, G.; Springer, C.; Hayes, A.; Gore, M.; et al. Whole-genome sequencing reveals complex mechanisms of intrinsic resistance to BRAF inhibition. Ann. Oncol. 2014, 25, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Andrade, J.R.; Gallagher, A.D.; Maharaj, J.; McClelland, S.E. Disentangling the roles of aneuploidy, chromosomal instability and tumour heterogeneity in developing resistance to cancer therapies. Chromosome Res. 2023, 31, 28. [Google Scholar] [CrossRef]
- Villanueva, J.; Vultur, A.; Lee, J.T.; Somasundaram, R.; Fukunaga-Kalabis, M.; Cipolla, A.K.; Wubbenhorst, B.; Xu, X.; Gimotty, P.A.; Kee, D.; et al. Acquired Resistance to BRAF Inhibitors Mediated by a RAF Kinase Switch in Melanoma Can Be Overcome by Cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010, 18, 683–695. [Google Scholar] [CrossRef]
- Muñoz-Maldonado, C.; Zimmer, Y.; Medová, M. A Comparative Analysis of Individual RAS Mutations in Cancer Biology. Front. Oncol. 2019, 9, 1088. [Google Scholar] [CrossRef]
- Randic, T.; Kozar, I.; Margue, C.; Utikal, J.; Kreis, S. NRAS mutant melanoma: Towards better therapies. Cancer Treat. Rev. 2021, 99, 102238. [Google Scholar] [CrossRef]
- Jakob, J.A.; Bassett, R.L.; Ng, C.S.; Curry, J.L.; Joseph, R.W.; Alvarado, G.C.; Rohlfs, M.L.; Richard, J.; Gershenwald, J.E.; Kim, K.B.; et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer 2012, 118, 4014–4023. [Google Scholar] [CrossRef]
- Takács, T.; Kudlik, G.; Kurilla, A.; Szeder, B.; Buday, L.; Vas, V. The effects of mutant Ras proteins on the cell signalome. Cancer Metastasis Rev. 2020, 39, 1051–1065. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Vakana, E.; Pratt, S.; Blosser, W.; Dowless, M.; Simpson, N.; Yuan, X.-J.; Jaken, S.; Manro, J.; Stephens, J.; Zhang, Y.; et al. LY3009120, a panRAF inhibitor, has significant anti-tumor activity in BRAF and KRAS mutant preclinical models of colorectal cancer. Oncotarget 2017, 8, 9251–9266. [Google Scholar] [CrossRef] [PubMed]
- AAl Mahi, A.; Ablain, J. RAS pathway regulation in melanoma. Dis. Model. Mech. 2022, 15, dmm049229. [Google Scholar] [CrossRef] [PubMed]
- Florent, L.; Saby, C.; Slimano, F.; Morjani, H. BRAF V600-Mutated Metastatic Melanoma and Targeted Therapy Resistance: An Update of the Current Knowledge. Cancers 2023, 15, 2607. [Google Scholar] [CrossRef]
- Dummer, R.; Schadendorf, D.; Ascierto, P.A.; Arance, A.; Dutriaux, C.; Di Giacomo, A.M.; Rutkowski, P.; Del Vecchio, M.; Gutzmer, R.; Mandala, M.; et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 435–445. [Google Scholar] [CrossRef]
- Dorard, C.; Estrada, C.; Barbotin, C.; Larcher, M.; Garancher, A.; Leloup, J.; Beermann, F.; Baccarini, M.; Pouponnot, C.; Larue, L.; et al. RAF proteins exert both specific and compensatory functions during tumour progression of NRAS-driven melanoma. Nat. Commun. 2017, 8, 15262. [Google Scholar] [CrossRef]
- Monaco, K.-A.; Delach, S.; Yuan, J.; Mishina, Y.; Fordjour, P.; Labrot, E.; McKay, D.; Guo, R.; Higgins, S.; Wang, H.Q.; et al. LXH254, a Potent and Selective ARAF-Sparing Inhibitor of BRAF and CRAF for the Treatment of MAPK-Driven Tumors. Clin. Cancer Res. 2021, 27, 2061–2073. [Google Scholar] [CrossRef]
- Teh, J.L.; Cheng, P.F.; Purwin, T.J.; Nikbakht, N.; Patel, P.; Chervoneva, I.; Ertel, A.; Fortina, P.M.; Kleiber, I.; HooKim, K.; et al. In Vivo E2F Reporting Reveals Efficacious Schedules of MEK1/2–CDK4/6 Targeting and mTOR–S6 Resistance Mechanisms. Cancer Discov. 2018, 8, 568–581. [Google Scholar] [CrossRef]
- Devitt, B.; Liu, W.; Salemi, R.; Wolfe, R.; Kelly, J.; Tzen, C.; Dobrovic, A.; McArthur, G. Clinical outcome and pathological features associated with NRAS mutation in cutaneous melanoma. Pigment. Cell Melanoma Res. 2011, 24, 666–672. [Google Scholar] [CrossRef]
- Ballesteros-Álvarez, J.; Andersen, J.K. mTORC2: The other mTOR in autophagy regulation. Aging Cell 2021, 20, e13431. [Google Scholar] [CrossRef]
- DGuertin, D.A.; Sabatini, D.M. Defining the Role of mTOR in Cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Romeo, Y.; Moreau, J.; Zindy, P.-J.; Saba-El-Leil, M.; Lavoie, G.; Dandachi, F.; Baptissart, M.; Borden, K.L.B.; Meloche, S.; Roux, P.P. RSK regulates activated BRAF signalling to mTORC1 and promotes melanoma growth. Oncogene 2013, 32, 2917–2926. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, W.; Zhang, G.; Kwong, L.; Lu, H.; Tan, J.; Sadek, N.; Xiao, M.; Zhang, J.; Labrie, M.; et al. Targeting mTOR signaling overcomes acquired resistance to combined BRAF and MEK inhibition in BRAF-mutant melanoma. Oncogene 2021, 40, 5590–5599. [Google Scholar] [CrossRef] [PubMed]
- Damsky, W.; Micevic, G.; Meeth, K.; Muthusamy, V.; Curley, D.P.; Santhanakrishnan, M.; Erdelyi, I.; Platt, J.T.; Huang, L.; Theodosakis, N.; et al. mTORC1 activation blocks BrafV600E-induced growth arrest but is insufficient for melanoma formation. Cancer Cell 2015, 27, 41–56. [Google Scholar] [CrossRef]
- Kong, Y.; Si, L.; Li, Y.; Wu, X.; Xu, X.; Dai, J.; Tang, H.; Ma, M.; Chi, Z.; Sheng, X.; et al. Analysis of mTOR Gene Aberrations in Melanoma Patients and Evaluation of Their Sensitivity to PI3K–AKT–mTOR Pathway Inhibitors. Clin. Cancer Res. 2016, 22, 1018–1027. [Google Scholar] [CrossRef]
- Chamcheu, J.C.; Roy, T.; Uddin, M.B.; Banang-Mbeumi, S.; Chamcheu, R.-C.N.; Walker, A.L.; Liu, Y.-Y.; Huang, S. Role and Therapeutic Targeting of the PI3K/Akt/mTOR Signaling Pathway in Skin Cancer: A Review of Current Status and Future Trends on Natural and Synthetic Agents Therapy. Cells 2019, 8, 803. [Google Scholar] [CrossRef]
- Deng, W.; Gopal, Y.N.V.; Scott, A.; Chen, G.; Woodman, S.E.; Davies, M.A. Role and therapeutic potential of PI3K-mTOR signaling in de novo resistance to BRAF inhibition. Pigment. Cell Melanoma Res. 2012, 25, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Hertwig, P. Neue Mutationen und Kopplungsgruppen bei der Hausmaus. Z. Für Indukt. Abstamm.-Und Vererbungslehre 1942, 80, 220–246. [Google Scholar]
- Steingrímsson, E.; Copeland, N.G.; Jenkins, N.A. Melanocytes and the Microphthalmia transcription factor network. Annu. Rev. Genet. 2004, 38, 365–411. [Google Scholar] [CrossRef]
- Oppezzo, A.; Rosselli, F. The underestimated role of the microphthalmia-associated transcription factor (MiTF) in normal and pathological haematopoiesis. Cell Biosci. 2021, 11, 18. [Google Scholar] [CrossRef] [PubMed]
- Hodgkinson, C.A.; Moore, K.J.; Nakayama, A.; Steingrímsson, E.; Copeland, N.G.; Jenkins, N.A.; Arnheiter, H. Mutations at the mouse microphthalmia locus are associated with defects in a gene encoding a novel basic-helix-loop-helix-zipper protein. Cell 1993, 74, 395–404. Available online: https://www.ncbi.nlm.nih.gov/pubmed/8343963 (accessed on 25 July 2024). [CrossRef] [PubMed]
- Flesher, J.L.; Paterson-Coleman, E.K.; Vasudeva, P.; Ruiz-Vega, R.; Marshall, M.; Pearlman, E.; MacGregor, G.R.; Neumann, J.; Ganesan, A.K. Delineating the role of MITF isoforms in pigmentation and tissue homeostasis. Pigment. Cell Melanoma Res. 2020, 33, 279–292. [Google Scholar] [CrossRef]
- Kawakami, A.; Fisher, D.E. The master role of microphthalmia-associated transcription factor in melanocyte and melanoma biology. Lab. Investig. 2017, 97, 649–656. [Google Scholar] [CrossRef]
- Hershey, C.L.; Fisher, D.E. Genomic analysis of the Microphthalmia locus and identification of the MITF-J/Mitf-J isoform. Gene 2005, 347, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Yasumoto, K.-I.; Kawaguchi, N.; Udono, T.; Watanabe, K.-I.; Saito, H.; Takahashi, K.; Noda, M.; Shibahara, S. Mitf-D, a newly identified isoform, expressed in the retinal pigment epithelium and monocyte-lineage cells affected by Mitf mutations. Biochim. Biophys. Acta 2002, 1574, 15–23. [Google Scholar] [CrossRef]
- Maruotti, J.; Thein, T.; Zack, D.J.; Esumi, N. MITF-M, a ‘melanocyte-specific’ isoform, is expressed in the adult retinal pigment epithelium. Pigment. Cell Melanoma Res. 2012, 25, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Shibahara, S.; Takeda, K.; Yasumoto, K.-I.; Udono, T.; Watanabe, K.-I.; Saito, H.; Takahashi, K. Microphthalmia-associated transcription factor (MITF): Multiplicity in structure, function, and regulation. J. Investig. Dermatol. Symp. Proc. 2001, 6, 99–104. [Google Scholar] [CrossRef]
- Takemoto, C.M.; Yoon, Y.-J.; Fisher, D.E. The identification and functional characterization of a novel mast cell isoform of the microphthalmia-associated transcription factor. J. Biol. Chem. 2002, 277, 30244–30252. [Google Scholar] [CrossRef]
- Tshori, S.; Sonnenblick, A.; Yannay-Cohen, N.; Kay, G.; Nechushtan, H.; Razin, E. Microphthalmia transcription factor isoforms in mast cells and the heart. Mol. Cell. Biol. 2007, 27, 3911–3919. [Google Scholar] [CrossRef]
- Bismuth, K.; Maric, D.; Arnheiter, H. MITF and cell proliferation: The role of alternative splice forms. Pigment. Cell Res. 2005, 18, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Goding, C.R.; Arnheiter, H. MITF—The first 25 years. Genes Dev. 2019, 33, 983–1007. [Google Scholar] [CrossRef] [PubMed]
- Hallsson, J.H.; Favor, J.; Hodgkinson, C.; Glaser, T.; Lamoreux, M.L.; Magnúsdóttir, R.; Gunnarsson, G.J.; Sweet, H.O.; Copeland, N.G.; Jenkins, N.A.; et al. Genomic, transcriptional and mutational analysis of the mouse microphthalmia locus. Genetics 2000, 155, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Primot, A.; Mogha, A.; Corre, S.; Roberts, K.; Debbache, J.; Adamski, H.; Dreno, B.; Khammari, A.; Lesimple, T.; Mereau, A.; et al. ERK-regulated differential expression of the Mitf 6a/b splicing isoforms in melanoma. Pigment. Cell Melanoma Res. 2010, 23, 93–102. [Google Scholar] [CrossRef]
- Smith, M.P.; Wellbrock, C. Molecular Pathways: Maintaining MAPK Inhibitor Sensitivity by Targeting Nonmutational Tolerance. Clin. Cancer Res. 2016, 22, 5966–5970. [Google Scholar] [CrossRef]
- Garraway, L.A.; Widlund, H.R.; Rubin, M.A.; Getz, G.; Berger, A.J.; Ramaswamy, S.; Beroukhim, R.; Milner, D.A.; Granter, S.R.; Du, J.; et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 2005, 436, 117–122. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef]
- Wellbrock, C.; Marais, R. Elevated expression of MITF counteracts B-RAF-stimulated melanocyte and melanoma cell proliferation. J. Cell Biol. 2005, 170, 703–708. [Google Scholar] [CrossRef]
- Arozarena, I.; Wellbrock, C. Overcoming resistance to BRAF inhibitors. Ann. Transl. Med. 2017, 5, 387. [Google Scholar] [CrossRef]
- Seberg, H.E.; Van Otterloo, E.; Cornell, R.A. Beyond MITF: Multiple transcription factors directly regulate the cellular phenotype in melanocytes and melanoma. Pigment. Cell Melanoma Res. 2017, 30, 454–466. [Google Scholar] [CrossRef]
- Giuliano, S.; Cheli, Y.; Ohanna, M.; Bonet, C.; Beuret, L.; Bille, K.; Loubat, A.; Hofman, V.; Hofman, P.; Ponzio, G.; et al. Microphthalmia-Associated Transcription Factor Controls the DNA Damage Response and a Lineage-Specific Senescence Program in Melanomas. Cancer Res. 2010, 70, 3813–3822. [Google Scholar] [CrossRef] [PubMed]
- Ballotti, R.; Cheli, Y.; Bertolotto, C. The complex relationship between MITF and the immune system: A Melanoma ImmunoTherapy (response) Factor? Mol. Cancer 2020, 19, 170. [Google Scholar] [CrossRef] [PubMed]
- Vlčková, K.; Vachtenheim, J.; Réda, J.; Horák, P.; Ondrušová, L. Inducibly decreased MITF levels do not affect proliferation and phenotype switching but reduce differentiation of melanoma cells. J. Cell. Mol. Med. 2018, 22, 2240–2251. [Google Scholar] [CrossRef] [PubMed]
- Ballotti, R.; Bertolotto, C. Deregulated MITF sumoylation: A route to melanoma. Mol. Cell. Oncol. 2017, 4, e1331154. [Google Scholar] [CrossRef]
- Italian Melanoma Intergroup Italian Melanoma Intergroup (I.M.I.); Ciccarese, G.; Dalmasso, B.; Bruno, W.; Queirolo, P.; Pastorino, L.; Andreotti, V.; Spagnolo, F.; Tanda, E.; Ponti, G.; et al. Clinical, pathological and dermoscopic phenotype of MITF p.E318K carrier cutaneous melanoma patients. J. Transl. Med. 2020, 18, 78. [Google Scholar] [CrossRef]
- Guhan, S.M.; Artomov, M.; McCormick, S.; Njauw, C.N.; Stratigos, A.J.; Shannon, K.; Tsao, H. Cancer risks associated with the germline MITF(E318K) variant. Sci. Rep. 2020, 10, 17051. [Google Scholar] [CrossRef]
- Bronisz, A.; Carey, H.A.; Godlewski, J.; Sif, S.; Ostrowski, M.C.; Sharma, S.M. The multifunctional protein fused in sarcoma (FUS) is a coactivator of microphthalmia-associated transcription factor (MITF). J. Biol. Chem. 2014, 289, 326–334. [Google Scholar] [CrossRef]
- Mansky, K.C.; Sankar, U.; Han, J.; Ostrowski, M.C. Microphthalmia transcription factor is a target of the p38 MAPK pathway in response to receptor activator of NF-kappa B ligand signaling. J. Biol. Chem. 2002, 277, 11077–11083. [Google Scholar] [CrossRef]
- Haq, R.; Fisher, D.E. Biology and Clinical Relevance of the Micropthalmia Family of Transcription Factors in Human Cancer. J. Clin. Oncol. 2011, 29, 3474–3482. [Google Scholar] [CrossRef]
- Fane, M.E.; Chhabra, Y.; Smith, A.G.; Sturm, R.A. BRN 2, a POU erful driver of melanoma phenotype switching and metastasis. Pigment. Cell Melanoma Res. 2019, 32, 9–24. [Google Scholar] [CrossRef]
- Herbert, K.; Binet, R.; Lambert, J.-P.; Louphrasitthiphol, P.; Kalkavan, H.; Sesma-Sanz, L.; Robles-Espinoza, C.D.; Sarkar, S.; Suer, E.; Andrews, S.; et al. BRN2 suppresses apoptosis, reprograms DNA damage repair, and is associated with a high somatic mutation burden in melanoma. Genes Dev. 2019, 33, 310–332. [Google Scholar] [CrossRef] [PubMed]
- Gopal, P.; Sarihan, E.I.; Chie, E.K.; Kuzmishin, G.; Doken, S.; Pennell, N.A.; Raymond, D.P.; Murthy, S.C.; Ahmad, U.; Raja, S.; et al. Clonal selection confers distinct evolutionary trajectories in BRAF-driven cancers. Nat. Commun. 2019, 10, 5143. [Google Scholar] [CrossRef] [PubMed]
- Bokharaie, H.; Kolch, W.; Krstic, A. Analysis of Alternative mRNA Splicing in Vemurafenib-Resistant Melanoma Cells. Biomolecules 2022, 12, 993. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros-Álvarez, J. A Network of bHLHZip Transcription Factors in Melanoma: Interactions of MITF, TFEB and TFE3. Ph.D. Thesis, University of Iceland, School of Health Sciences, Faculty of Medicine, Reykjavik, Iceland, 2019. [Google Scholar]

| MAPK-Reactivating Event | Ref. | Sample | Incidence (Count) | Incidence (%) | Comments |
|---|---|---|---|---|---|
| NRAS mutation | [52] | Progressive tumors after vemurafenib treatment | 13/71 | 18% | G12D/R, G13R, Q61K/R/L |
| NRAS mutation | [53] | Biopsies from BRAFi-resistant patients | 4/19 | 21% | Mutually exclusive with BRAF splicing variants |
| KRAS mutation | [52] | Progressive tumors after vemurafenib treatment | 5/71 | 7% | G12C, G12R, Q61H |
| BRAF amplification | [52] | Progressive tumors after vemurafenib treatment | 11/57 | 19% | 2–15 fold |
| BRAF splice mutants | [52] | Progressive tumors after vemurafenib treatment | 6/48 | 13% | Deletion exons 2–8, 2–10, 4–8 |
| BRAF splice mutants | [53] | Biopsies from BRAFi-resistant patients | 6/19 | 32% | Deletion of exons 4–10 (1), exons 4–8, (1), exons 2–8 (1), or exons 2–10 (3) |
| MEK1 | [52] | Progressive tumors after vemurafenib treatment | 2/71 | 3% | K57N, C121S |
| CDKN2A loss | [52] | Progressive tumors after vemurafenib treatment | 3/44 | 7% | --- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ballesteros-Álvarez, J.; Blázquez-Medela, A.M. Evolution of Acquired Drug Resistance in BRAF-Mutant Melanoma. DNA 2024, 4, 355-369. https://doi.org/10.3390/dna4040025
Ballesteros-Álvarez J, Blázquez-Medela AM. Evolution of Acquired Drug Resistance in BRAF-Mutant Melanoma. DNA. 2024; 4(4):355-369. https://doi.org/10.3390/dna4040025
Chicago/Turabian StyleBallesteros-Álvarez, Josué, and Ana M. Blázquez-Medela. 2024. "Evolution of Acquired Drug Resistance in BRAF-Mutant Melanoma" DNA 4, no. 4: 355-369. https://doi.org/10.3390/dna4040025
APA StyleBallesteros-Álvarez, J., & Blázquez-Medela, A. M. (2024). Evolution of Acquired Drug Resistance in BRAF-Mutant Melanoma. DNA, 4(4), 355-369. https://doi.org/10.3390/dna4040025