1. Introduction
Every year for the past decade, thousands of undergraduates design new genetic circuits as part of the International Genetically Engineered Machine competition (iGEM) [
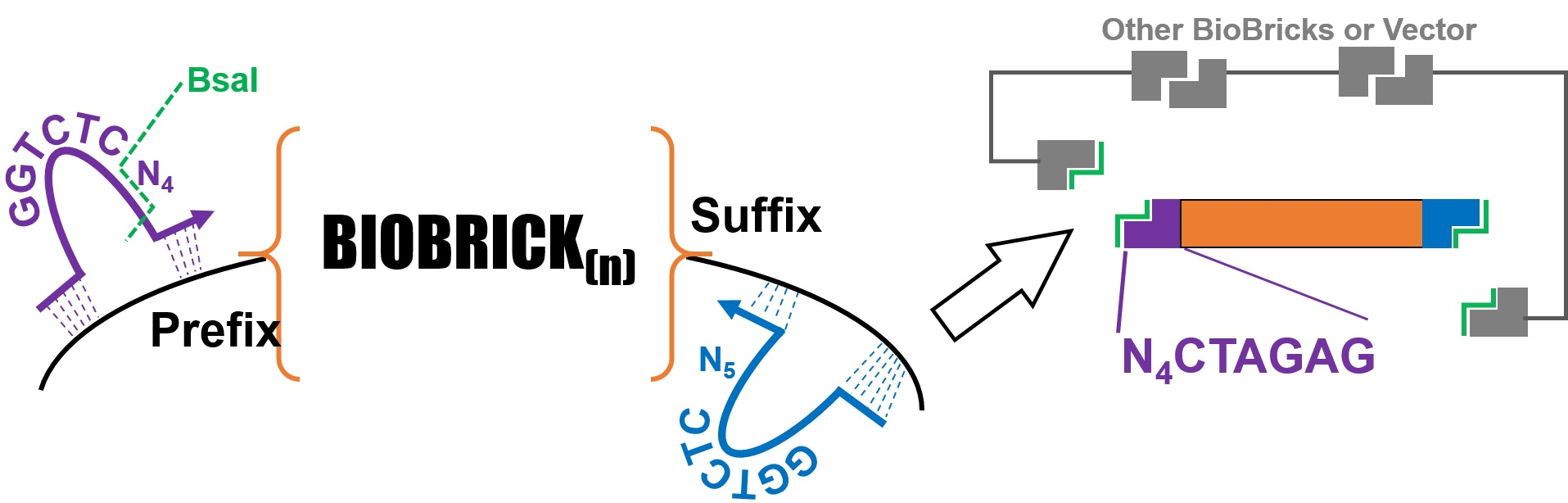
1]. These designs are assembled through the use of BioBricks: a collection of DNA parts in a standardized format. BioBricks are constructed and characterized in a way to make assembly of larger constructs possible and their behavior predictable, with relevant information for each part available at the iGEM registry of standard biological parts (parts.igem.org, last accessed 14 February 2023). Assembly of two or more parts together can be accomplished through a variety of means, including standard 3A assembly, Gibson assembly, or Golden Gate assembly. An advantage of Golden Gate assembly is that it can allow for flexible and modular assembly strategies, since the only requirement for matching two BioBricks is a short, user defined single-stranded overhang. To generate these overhangs, Type IIS restriction enzymes such as BsaI or BsmBI are used, which cut adjacent to their recognition sequence. A plethora of variations in Golden Gate cloning exist, including but not limited to MoClo, Mobius Assembly, Golden-Braid, Gold-Bricks, Jump Vectors, and Loop assembly [
2,
3,
4,
5,
6,
7,
8]. These are strategies which increase flexibility due to common sequences used in a set of plasmids, and sometimes are directed for use in specific organisms [
9].
For most assembly projects that utilize a Golden Gate approach, primers specific to each BioBrick or assembly intermediates must be designed and synthesized. Alternatively, specialized vectors must be procured that align with the aforementioned strategies and provide a repertoire of BsaI recognition sites. While the cost of DNA synthesis has decreased dramatically in recent years, purchasing new primers still represents an expense that is significant in certain contexts, such as laboratories in impoverished areas or synthetic biology programs in high schools and community colleges. Participation in iGEM provides these institutions with a distribution kit of BioBricks, often with several thousand of the most useful parts in the registry, and tasks students with assembling them into useful combinations. Many of these BioBricks are compatible with Golden Gate assembly (indicated by iGEM’s type IIS assembly standard, RFC1000), but only a select few can be readily used in MoClo or other existing Golden Gate assembly strategies. The need for designing and ordering primers to use these BioBricks also introduces opportunities for errors and creates at least a small delay in the flow of an assembly project.
This challenge has already been addressed in the context of one specific BioBrick, BBa_B0034 (a ribosome binding site, or RBS). To expedite addition of this common RBS to a coding region, an innovative PCR-based approach was developed that relied on a primer that had a short 3′ region matching the pSB1C3 backbone [
10]. This primer also contained the ribosome binding site and a 5′ anchor to ensure hybridization during PCR. We were inspired by this approach to create a strategy for appending any BioBrick with a Golden Gate cloning sequence. Instead of the addition of an RBS directly upstream of the BioBrick, our primers add a BsaI recognition site and a unique overhang. In this report, we describe the utility of these primers (hereafter referred to as ‘GEM-Gate’), which should facilitate quicker and cheaper assembly of DNA fragments by synthetic biologists.
2. Materials and Methods
2.1. PCR reactions
PCR reactions were generally carried out as follows: 10 μL of Q5 polymerase mix (New England Biolabs, Ipswitch, MA, USA) was mixed with 1 μL of each primer at 10 μM, 7.5 μL of distilled H
2O (dH
2O), and 0.5 μL of template typically at 0.1–1 ng/μL. Products were amplified by 28–32 cycles of 10″ at 94 °C, 20″ at 57–60 °C, and 30–60″ at 72 °C. Purified products were then sequenced and/or included in assembly reactions (see
supplemental methods). Reactions were scaled as needed.
2.2. Gel electrophoresis
PCR products were resolved on a 0.8% agarose gel in LAB buffer (10 mM lithium acetate and 10 mM boric acid) with ethidium bromide by running for 30–45 min at 12 V/cm. Gel images were obtained by scanning for 600 nm light for 30 s on an Odyssey-Fc (LICOR, Lincoln, NE, USA).
2.3. Golden Gate Assembly
Products were assembled using the Golden Gate cloning kit (New England Biolabs), generally as follows: DNA fragments, representing both BioBricks and the processed vector, were pooled together in appropriate ratios. To this mixture, dH2O, T4 DNA Ligase buffer, and NEB Golden Gate Enzyme mix was added and incubated according to the supplier instructions (or scaled), and then transformed and sequenced.
2.4. Transformation, Miniprep, and Sequencing
An amount of 2 μL of the assembled product was transformed into chemically competent Escherichia coli DH5-alpha using methods with either CCBM80 buffer (10 mM KOAc, 80 mM CaCl2, 20 mM MnCl2, 10 mM MgCl2, and 10% glycerol; pH 6.4) or TSS solution (10% PEG-8000, 5% DMSO, and 30 mM MgCl2 in LB). Commercially available alternatives were also used in some experiments (NEB 5-Alpha, New England Biolabs). After heat shock of 45″ and recovery, cells were incubated at 37 °C overnight on LB plates with 30 μg/mL chloramphenicol. Colonies were subcultured, then plasmid DNA was column purified (QuickLyse kit from QIAgen, Hilden, Germany) and assayed using appropriate primers in Sanger sequencing.
3. Results
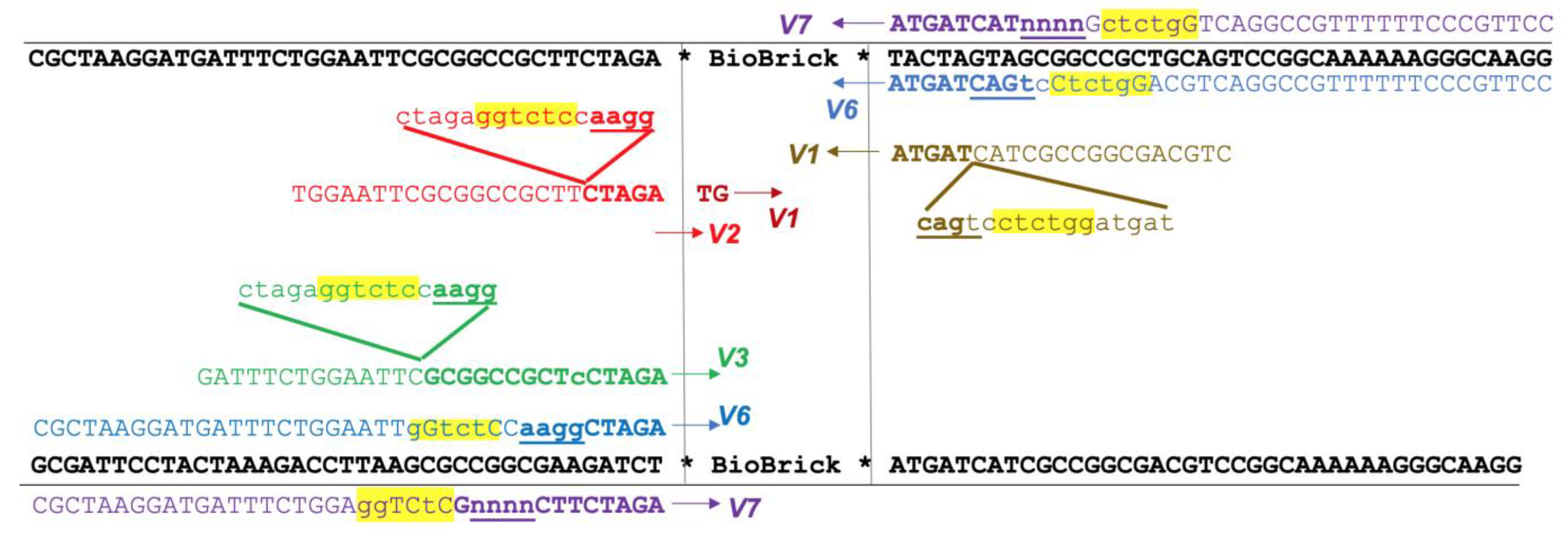
Our initial version (V1) of the GEM-Gate primers mimics the previously reported design used for the addition of ribosome binding sites (
Figure 1). Instead of adding a ribosome binding site, our primers add an 11 bp sequence that includes a BsaI recognition site and one of several different overhang sequences. These overhangs can be modified as necessary, but our initial selections were based upon the pSB1C3 prefix and suffix and 3A assembly sites, as well as the existing Golden Gate assembly-based methods. They were further validated using the ligase fidelity viewer (
Table S4) [
11,
12,
13].
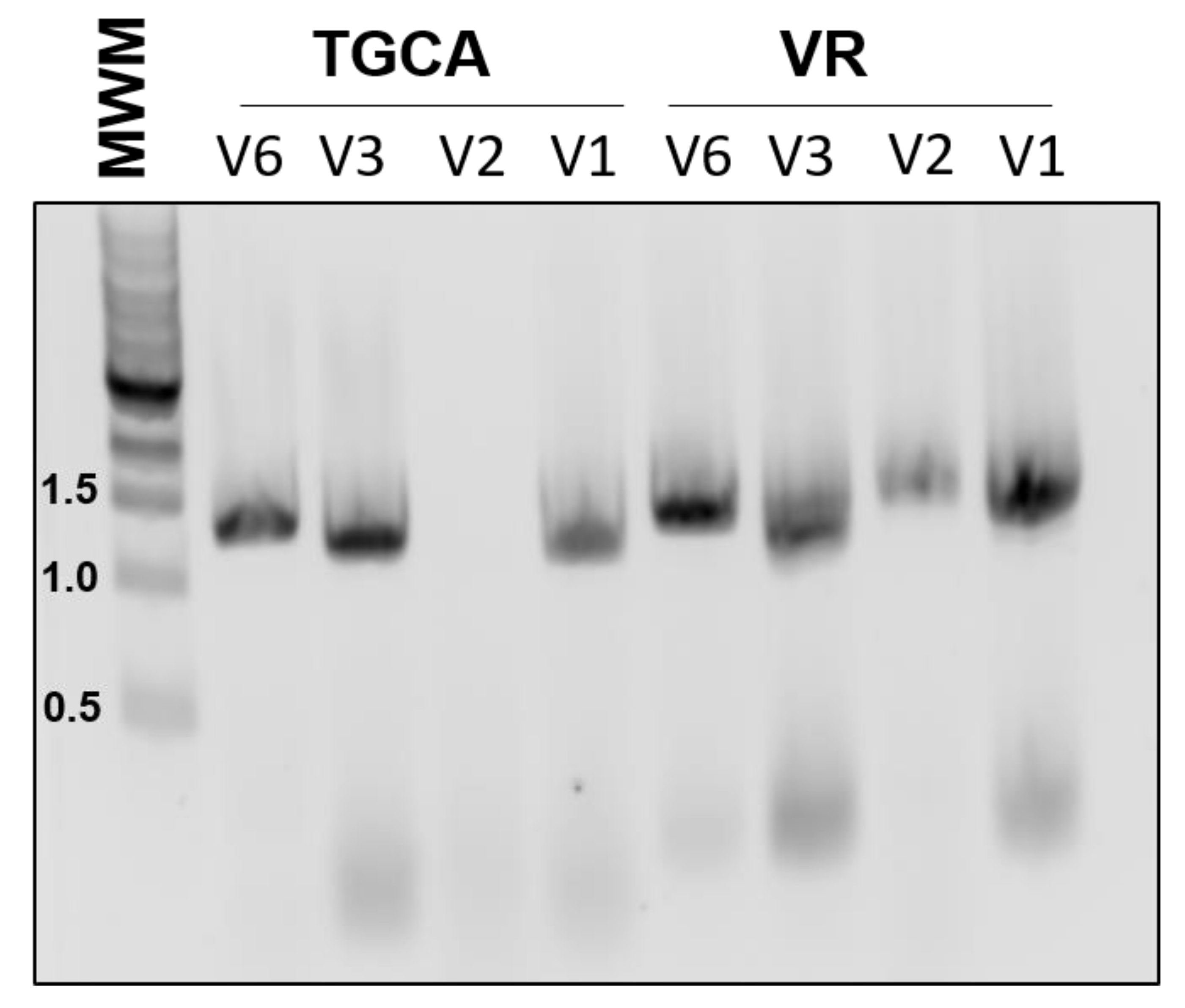
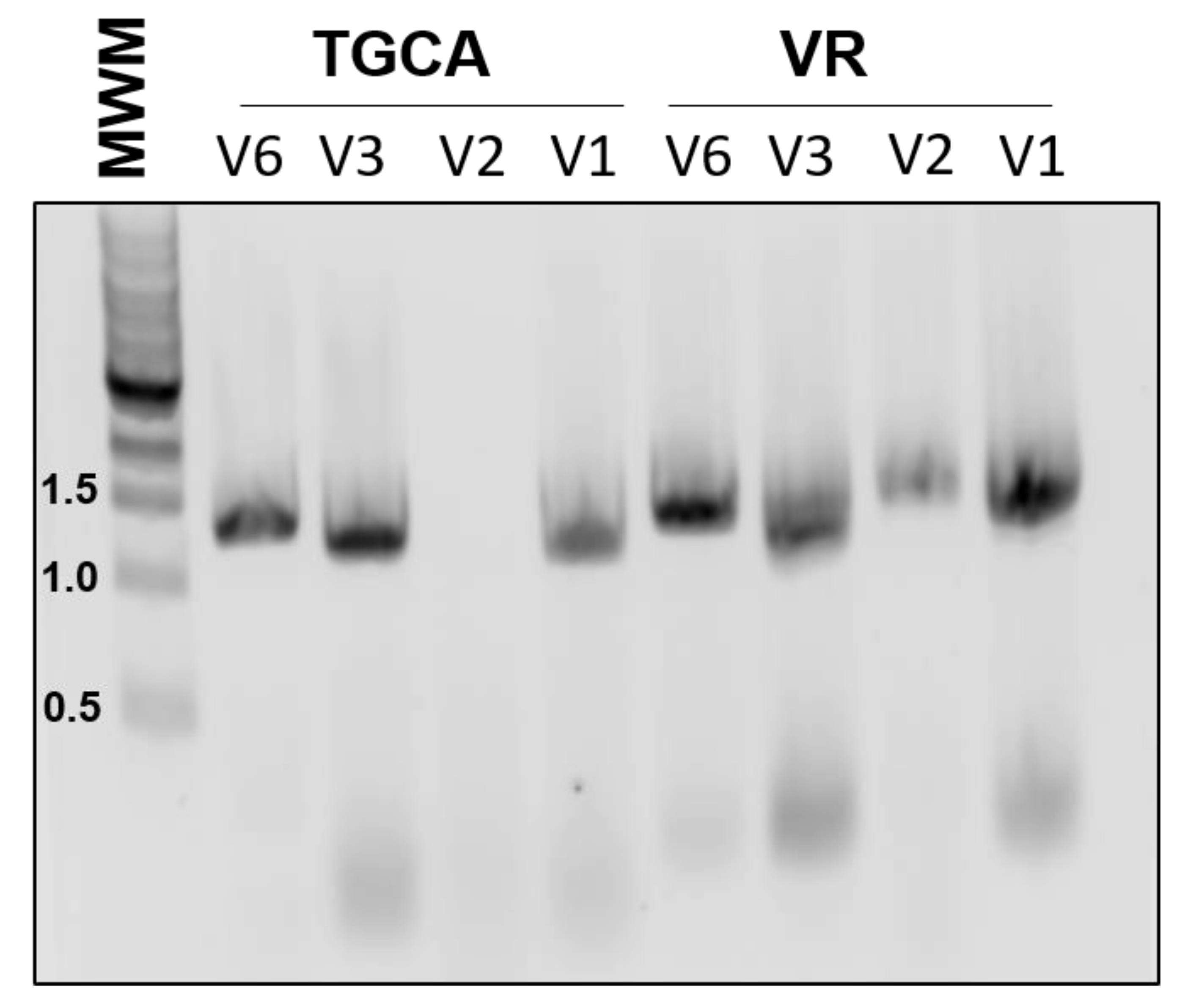
One drawback to our initial approach is that it limited the choice of BioBricks to coding regions, since the primer design relied on the presence of a start codon. Therefore, we designed another version of the primers, V2, which does not hybridize to the start codon, and instead has an even shorter 3′ region of homology to the template. Unfortunately, this version of the primers was not as robust and would sometimes fail to amplify the target (
Figure 2). This is not altogether surprising, since mismatches close to the 3′ end of a primer can drastically lower PCR efficiency and are the basis of allele-specific PCR diagnostic assays [
14,
15].
Both of these designs also have the potential drawback of amplifying composite BioBricks incorrectly. For example, amplification of the BioBrick BBa_K081014, which contains an RBS and the coding region for red fluorescent protein (RFP), should proceed from the beginning of the BioBrick. However, it is also possible for the 3′ end of the primer to attach to the scar sequence between the RBS and RFP, essentially acting to remove the RBS from the final product. To reduce this unwanted binding, we also tested primers that contained an extended 3′ region of homology (V3). These primers were robust and routinely gave efficient amplification. However, using these primers in assembly would introduce much larger scar sequences which would not conform to BioBrick standards.
All of the previous versions were based on the design of the RBS addition primer previously reported, in which the sequence to be added was looped out. We also tested a different approach, in which the added Golden Gate sequences represented mismatches to the template (V6). In our experience, these primers were almost as dependable as the V3 set, yet did not introduce an undesirable scar sequence (
Figure 2). All of our subsequent amplification and assembly utilized this iteration of primer sequence, unless otherwise noted.
Several different BioBricks were amplified with these primers; most gave the desired product, although some templates and primer combinations occasionally failed to amplify the target. The products that were obtained were sequenced using internal primers and in most cases the Golden Gate sequences were present as expected (
Figure S1). In order to expand the utility of our method, we sought to overcome the difficulty in amplifying recalcitrant BioBricks (for a partial list, see Text S1). These problematic templates could be successfully amplified with the control VF2 and VR primers, but not when a forward and reverse GEM-Gate primer was used, presumably due to the unusual type of target binding these primers employ. However, we were eventually successful in amplifying all BioBricks tested if VF2 or VR was paired with only one GEM-Gate primer (i.e., AATT_F6 paired with VR). The product would be used as the template in a subsequent PCR in which the forward primer is replaced by one that binds to the 5′ end of the V6 primers, and VR is replaced by the desired reverse GEM-Gate primer (
Figure S2). In short, this two-stage approach builds upon the addition of the desired Golden Gate sequence on one end before adding that sequence to the other. This approach also introduces some added flexibly, since one can choose a range of reverse primers to use in the 2nd PCR reaction, depending on the assembly strategy being used.
To demonstrate the utility of this approach, we successfully amplified and assembled two BioBricks: BBa_R0010, the lac operon promoter, and the aforementioned BBa_K081014 (RFP). The junction between these BioBricks was confirmed by sequencing, and gave the expected ‘scar’ sequence, consisting of the overhang present in the selected primers. Moreover, this scar sequence did not impact or abolish the expected function, as the resulting colonies exhibited a characteristic red color (
Figure S3). As a control, we tested an improper selection of primers or BioBricks. Primers which lead to incompatible overhang sequences could amplify their target as expected but failed to produce colonies during assembly. Likewise, selection of the rare BioBricks that contain internal BsaI sites (such as K081012, which encodes GFP) failed to assemble properly.
We also screened the iGEM DNA distribution kit BioBricks for sequences that might match both the hybridizing scar sequence and the BsaI overhang. In most cases, there were no such sites, and off-target annealing was predicted to be minimal (
Tables S2 and S3). Several representative BioBricks, including the few predicted to be problematic, were tested by PCR to determine if undesired products would accumulate. In preliminary results, amplified BioBricks mostly resulted in a single product where expected, sometimes with a minor, low molecular weight product (presumably primer dimers, see
Figure S6).
Occasionally, we observed template sequences at the end of the PCR products instead of the expected BsaI recognition site and overhangs (
Figure S7). One explanation was that the proof-reading polymerase employed in our reaction repaired the primer-template mismatch in favor of the template, which has been noted to occur within 6–8 nucleotides of the 3′ end of a primer [
16]. Therefore, we designed another version of the primers (V7), in which the overhangs and potential mismatches were recessed a further three nucleotides. These new primers add a slightly larger scar, but are robust and can be used successfully in assembly of multiple fragments (
Figures S8–S11). Just as with the previous primer set, assembly attempts with incompatible overhangs failed to produce significant numbers of colonies.
4. Discussion
We were able to successfully use this approach to amplify several different BioBricks and assemble together a promoter and different reporter gene coding sequences. While the pilot experiments presented here assembled at most four DNA fragments (three BioBricks and a plasmid backbone), use of additional genes and primer overhangs should allow for larger, more complex assemblies, as is typical for most Golden Gate-based approaches. GEM-Gate provides an alternative to the use of traditional Golden Gate or Gibson assembly, which should allow researchers to go from a design to colonies within 24 h. Traditional assembly, if it requires a primer order from a commercial supplier, will often take 1–3 days more (
Figure S5). In research settings with deadlines (such as an iGEM team), this delay is a loss of valuable time.
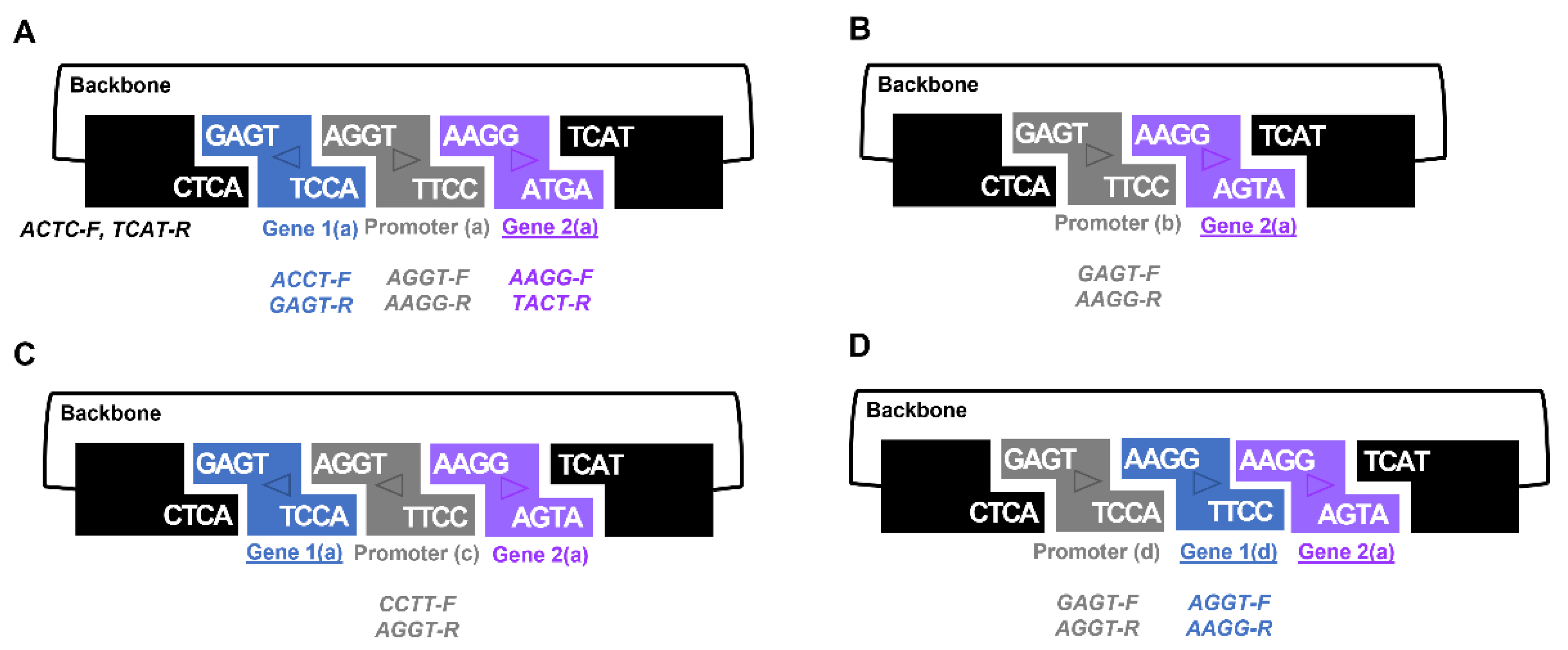
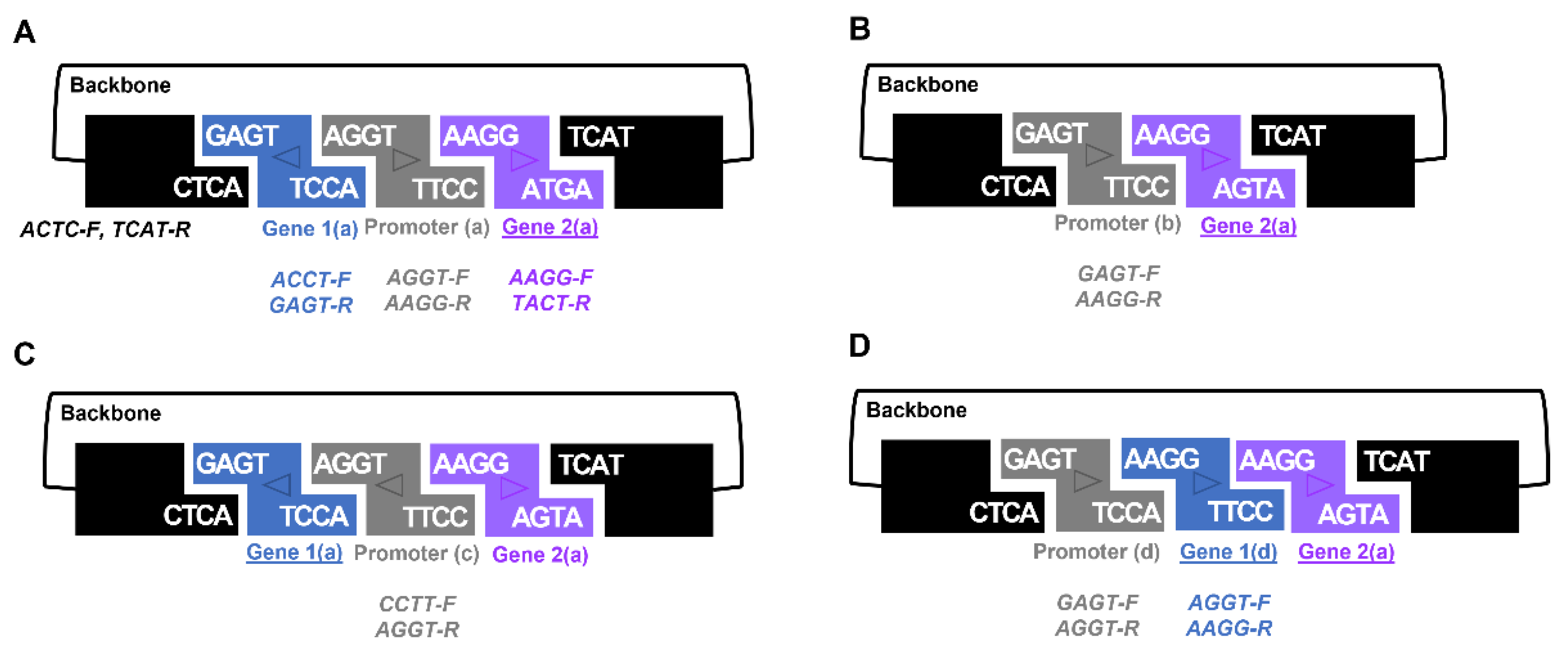
The GEM-gate approach also provides scientists with added flexibility. Using the same set of primers and shuffling which BioBrick received each overhang allows the production of different fusions (
Figure 3). Indeed, careful selection of these primers permits assembly of the same gene in either a forward or reverse orientation (
Figure S6). One notable caveat to the selection of overhang sequences in the primers used are those that seek to take advantage of 3A assembly sites, such as ‘AATT’, which are problematic palindromes and lead to inefficient assembly. As mentioned above, we successfully deployed the GEM-gate strategy to combine three fragments together (
Figures S9–S11).
Thus, we report here a small set of primers that can endow a lab with the ability to make endless BioBrick combinations. We demonstrated how the unusual binding of these primers and amplification can be achieved, and how this can be a prelude to Golden Gate assembly. This ‘GEM-Gate’ approach obviates the need for large primer orders, which requires both time and money. Recent work by others has made it easier to obtain the required polymerases and restriction enzymes for Golden Gate assembly, further expanding access to this technique [
17]. While alternative strategies and plasmids exist that utilize Golden Gate assembly, these approaches still require obtaining specific primers or plasmids. We hope that the GEM-gate approach will help expedite the work of synthetic biologists in resource-deprived settings, especially undergraduates or high school students in laboratories with a constrained budget and looming deadlines.



{kind=link}
{kind=link}
{kind=link}
{kind=link}