
A Comparison of Methods for the Production of Kilobase-Length Single-Stranded DNA

Abstract
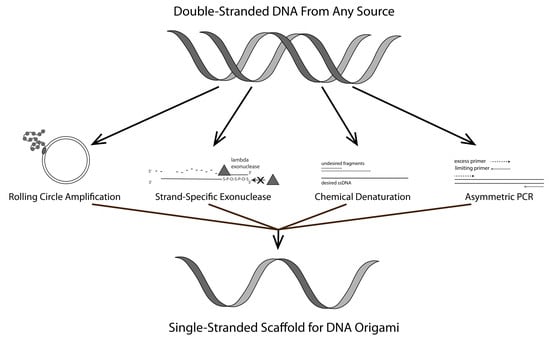
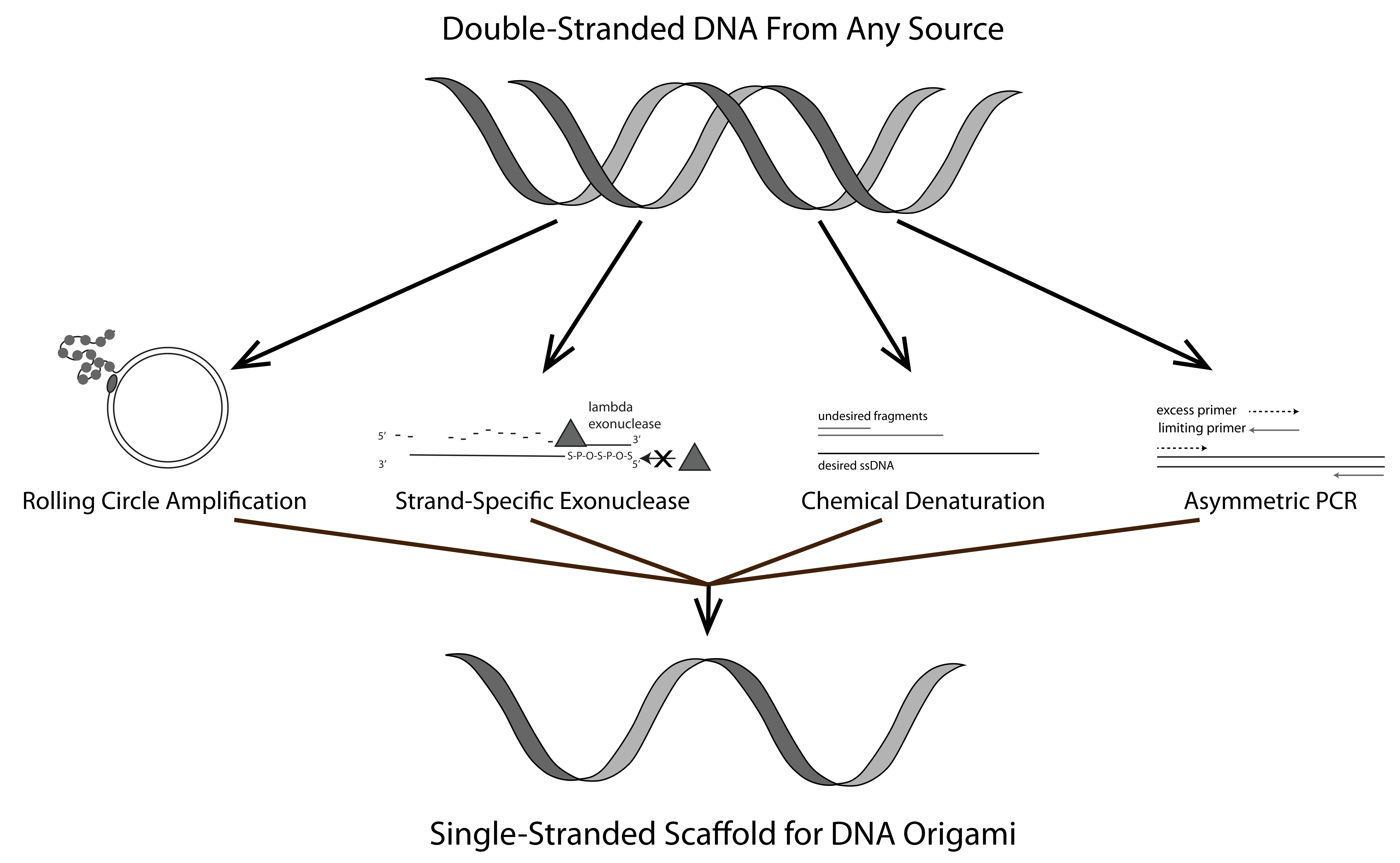
:
1. Introduction
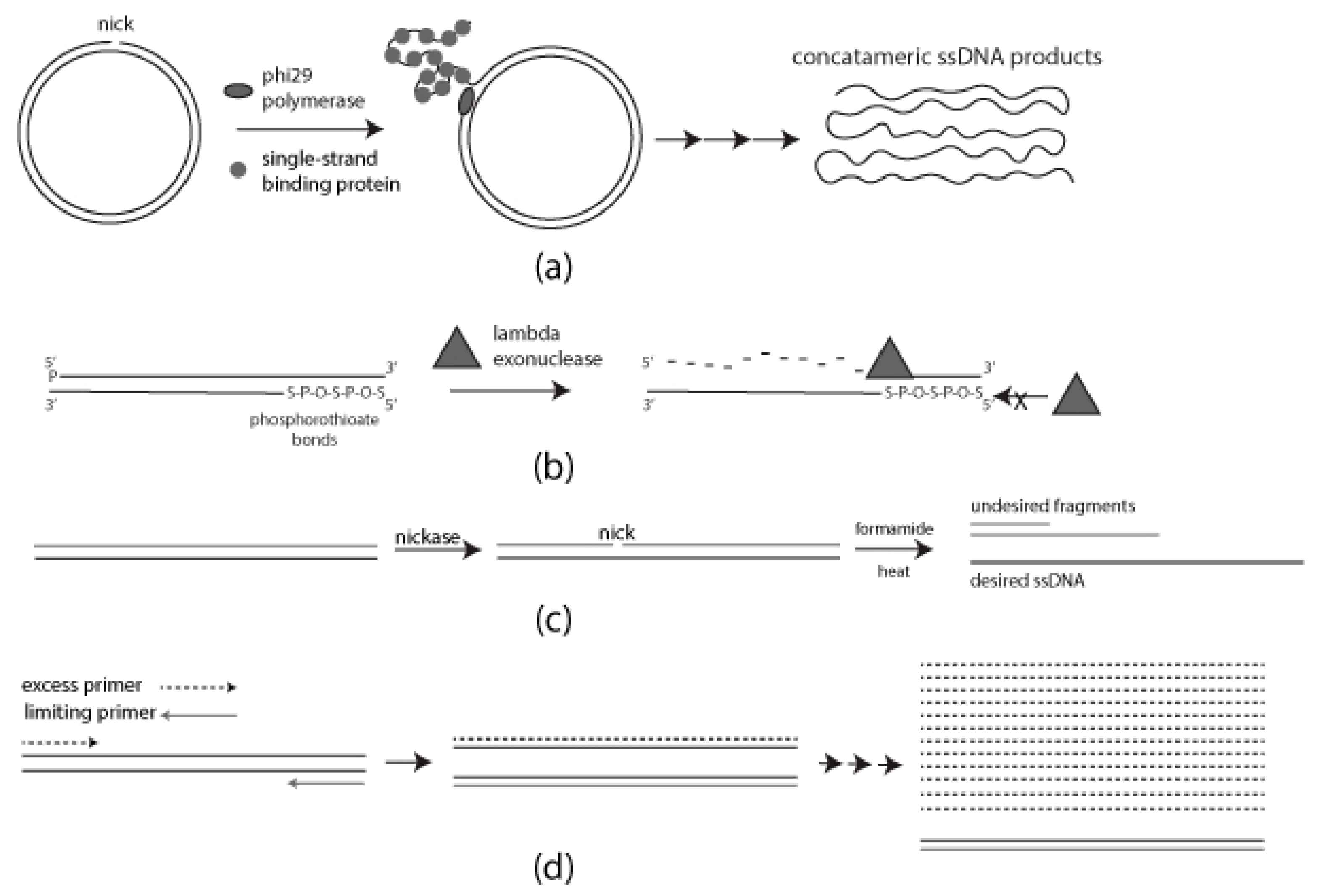
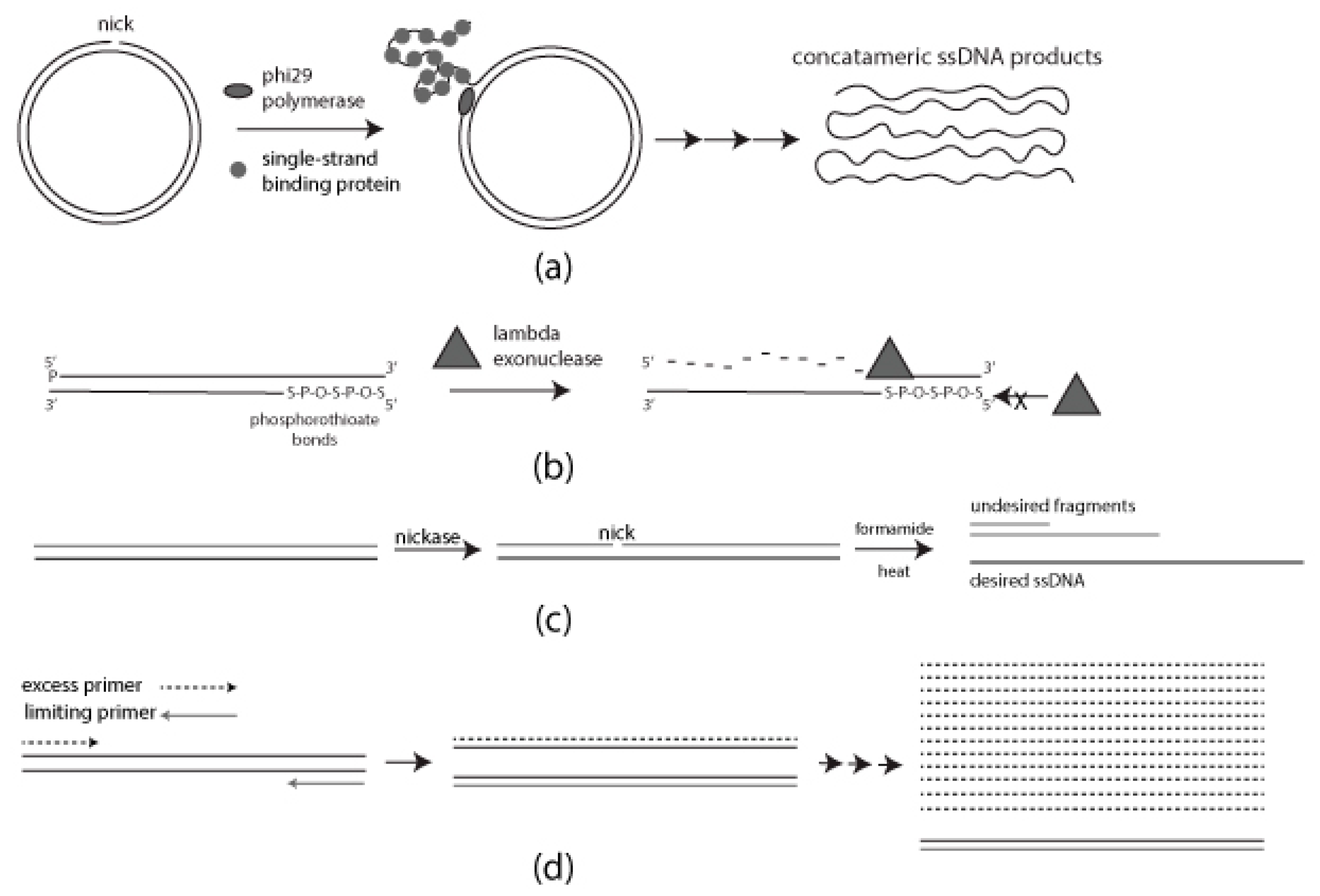
1.1. Rolling Circle Amplification (RCA)
1.2. Strand-Specific Lambda Exonuclease Digestion
1.3. Chemical Denaturation
1.4. Asymmetric PCR (aPCR)
2. Materials and Methods
2.1. Rolling Circle Amplification
2.1.1. Preparation of Nicked Template for RCA
2.1.2. RCA Mediated ssDNA Production
2.1.3. Digestion of RCA Products—XmnI Was Purchased from NEB
2.2. Strand-Specific Lambda Exonuclease Degradation
2.3. Formamide Separation
2.4. Asymmetric PCR (aPCR)
2.5. DNA Purification
2.5.1. dsDNA Purification
2.5.2. ssDNA Purification
3. Results
3.1. Rolling Circle Amplification (RCA)
3.2. Strand-Specific Lambda Exonuclease Degradation
3.3. Chemical Denaturation
3.4. Asymmetric PCR (aPCR)
4. Discussion
4.1. Rolling Circle Amplification (RCA)
4.2. Strand-Specific Lambda Exonuclease Degradation
4.3. Chemical Denaturation
4.4. Asymmetric PCR (aPCR)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ramakrishnan, S.; Ijäs, H.; Linko, V.; Keller, A. Structural stability of DNA origami nanostructures under application-specific conditions. Comput. Struct. Biotechnol. J. 2018, 16, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Roodhuizen, J.A.L.; Hendrikx, P.J.T.M.; Hilbers, P.A.J.; de Greef, T.F.A.; Markvoort, A.J. Counterion-Dependent Mechanisms of DNA Origami Nanostructure Stabilization Revealed by Atomistic Molecular Simulation. ACS Nano 2019, 13, 10798–10809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, S.; Fan, C.; Gothelf, K.; Li, J.; Lin, C.; Liu, L.; Liu, N.; Nijenhuis, M.; Saccà, B.; Simmel, F.; et al. DNA origami. Nat. Rev. Methods Primers 2021, 1, 13. [Google Scholar] [CrossRef]
- Rothermund, P. Folding DNA to create nanoscale shapes and patterns. Nature 2006, 440, 297–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, Q.; Yang, Z.; Xu, K.; Wang, C.; Liang, H. DNA Nanostructure as an Efficient Drug Delivery Platform for Immunotherapy. Front. Pharmacol. 2020, 10, 1585. [Google Scholar] [CrossRef]
- Chandrasekaran, A.R. Nuclease resistance of DNA nanostructures. Nat. Rev. Chem. 2021, 5, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Liu, J.; Wu, X.; Ding, B. Multifunctional DNA Origami Nanoplatforms for Drug Delivery. Chem. Asian J. 2019, 14, 2193–2202. [Google Scholar] [CrossRef]
- Udomprasert, A.; Kangsamaksin, T. DNA origami applications in cancer therapy. Cancer Sci. 2017, 108, 1535–1543. [Google Scholar] [CrossRef]
- Choi, Y.; Schmidt, C.; Tinnefeld, P.; Bald, I.; Rödiger, S. A new reporter design based on DNA origami nanostructures for quantification of short oligonucleotides using microbeads. Sci. Rep. 2019, 9, 4769. [Google Scholar] [CrossRef]
- Chandrasekaran, A.R.; Punnoose, J.A.; Zhou, L.; Dey, P.; Dey, B.K.; Halvorsen, K. DNA nanotechnology approaches for microRNA detection and diagnosis. Nucleic Acids Res. 2019, 47, 10489–10505. [Google Scholar] [CrossRef]
- Bush, J.; Singh, S.; Vargas, M.; Oktay, E.; Hu, C.-H.; Veneziano, R. Synthesis of DNA Origami Scaffolds: Current and Emerging Strategies. Molecules 2020, 25, 3386. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, T.R.; Du, R.R.; Huang, H.; Wamhoff, E.-C.; Bathe, M. Bioproduction of pure, kilobase-scale single-stranded DNA. Sci. Rep. 2019, 9, 6121. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, F.A.S.; Praetorius, F.; Wachauf, C.H.; Brüggenthies, G.; Kohler, F.; Kick, B.; Kadletz, K.L.; Pham, P.N.; Behler, K.L.; Gerling, T.; et al. Custom-Size, Functional, and Durable DNA Origami with Design-Specific Scaffolds. ACS Nano 2019, 13, 5015–5027. [Google Scholar] [CrossRef]
- Zhao, W.; Ali, M.M.; Brook, M.A.; Li, Y. Rolling Circle Amplification: Applications in Nanotechnology and Biodetection with Functional Nucleic Acids. Angew. Chem. Int. Ed. 2008, 47, 6330–6337. [Google Scholar] [CrossRef]
- Gu, L.; Yan, W.; Liu, L.; Wang, S.; Zhang, X.; Lyu, M. Research Progress on Rolling Circle Amplification (RCA)-Based Biomedical Sensing. Pharmaceuticals 2018, 11, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducani, C.; Kaul, C.; Moche, M.; Shih, W.M.; Högberg, B. Enzymatic production of ‘monoclonal stoichiometric’ single-stranded DNA oligonucleotides. Nat. Methods 2013, 10, 647–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducani, C.; Bernardinelli, G.; Högberg, B. Rolling circle replication requires single-stranded DNA binding protein to avoid termination and production of double-stranded DNA. Nucleic Acids Res. 2014, 42, 10596–10604. [Google Scholar] [CrossRef] [Green Version]
- Van Emmerik, C.L.; Gachulincova, I.; Lobbia, V.R.; Daniëls, M.A.; Heus, H.A.; Soufi, A.; Nelissen, F.H.; van Ingen, H. Ramified rolling circle amplification for synthesis of nucleosomal DNA sequences. Anal. Biochem. 2019, 588, 113469. [Google Scholar] [CrossRef]
- Joffroy, B.; Uca, Y.; Prešern, D.; Doye, J.; Schmidt, T.L. Rolling circle amplification shows a sinusoidal template length-dependent amplification bias. Nucleic Acids Res. 2017, 46, 538–545. [Google Scholar] [CrossRef] [Green Version]
- Lim, B.; Choong, Y.; Ismail, A.; Glökler, J.; Konthur, Z.; Lim, T. Directed evolution of nucleotide-based libraries using lambda exonuclease. BioTechniques 2012, 53, 357–364. [Google Scholar] [CrossRef]
- Murgha, Y.; Rouillard, J.-M.; Gulari, E. Methods for the Preparation of Large Quantities of Complex Single-Stranded Oligonucleotide Libraries. PLoS ONE 2014, 9, e94752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Citartan, M.; Tang, T.-H.; Tan, S.-C.; Gopinath, S.C.B. Conditions optimized for the preparation of single-stranded DNA (ssDNA) employing lambda exonuclease digestion in generating DNA aptamer. World J. Microbiol. Biotechnol. 2010, 27, 1167–1173. [Google Scholar] [CrossRef]
- Lohman, G. The Effect of Nucleic Acid Modifications on Digestion by DNA Exonucleases. New England Biolabs. Available online: https://international.neb.com/tools-and-resources/feature-articles/the-effect-of-nucleic-acid-modifications-on-digestion-by-dna-exonucleases (accessed on 20 May 2021).
- Han, D.; Qi, X.; Myhrvold, C.; Wang, B.; Dai, M.; Jiang, S.; Bates, M.; Liu, Y.; An, B.; Zhang, F.; et al. Single-stranded DNA and RNA origami. Science 2017, 358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Högberg, B.; Liedl, T.; Shih, W.M. Folding DNA Origami from a Double-Stranded Source of Scaffold. J. Am. Chem. Soc. 2009, 131, 9154–9155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avci-Adali, M.; Paul, A.; Wilhelm, N.; Ziemer, G.; Wendel, H. Upgrading SELEX Technology by Using Lambda Exonuclease Di-gestion for Single-Stranded DNA Generation. Molecules 2010, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pagratis, N. Rapid preparation of single stranded DNA from PCR products by streptavidin induced electrophoretic mobility shift. Nucleic Acids Res. 1996, 24, 3645–3646. [Google Scholar] [CrossRef] [Green Version]
- Hegedüs, É.; Kókai, E.; Kotlyar, A.; Dombrádi, V.; Szabó, G. Separation of 1-23-kb complementary DNA strands by urea-agarose gel electrophoresis. Nucleic Acids Res. 2009, 37, e112. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Lim, H.J.; Son, A. Characterization of denaturation and renaturation of DNA for DNA hybridization. Environ. Health Toxicol. 2014, 29, e2014007. [Google Scholar] [CrossRef]
- Kilili, G.K.; Tilton, L.; Karbiwnyk, C.M. NaOH concentration and streptavidin bead type are key factors for optimal DNA aptamer strand separation and isolation. BioTechniques 2016, 61, 114–116. [Google Scholar] [CrossRef] [Green Version]
- Kuo, T.-C. Streamlined method for purifying single-stranded DNA from PCR products for frequent or high-throughput needs. BioTechniques 2005, 38, 700–702. [Google Scholar] [CrossRef]
- Veneziano, R.; Shepherd, T.R.; Ratanalert, S.; Bellou, L.; Tao, C.; Bathe, M. In vitro synthesis of gene-length single-stranded DNA. Sci. Rep. 2018, 8, 6548. [Google Scholar] [CrossRef] [Green Version]
- Tolnai, Z.; Harkai, Á.; Szeitner, Z.; Scholz, É.N.; Percze, K.; Gyurkovics, A.; Mészáros, T. A simple modification increases specificity and efficiency of asymmetric PCR. Anal. Chim. Acta 2018, 1047, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Fock, J.; Minero, G.A.S.; Garbarino, F.; Hansen, M.F. Ultrasensitive Real-Time Rolling Circle Amplification Detection Enhanced by Nicking-Induced Tandem-Acting Polymerases. Anal. Chem. 2019, 91, 10102–10109. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Gao, F.; Fock, J.; Dufva, M.; Hansen, M. Homogeneous circle-to-circle amplification for real-time optomagnetic detection of SARS-CoV-2 RdRp coding sequence. Biosens. Bioelectron. 2020, 165, 112356. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-H.; Zhang, X.-L.; Wu, J.; Lin, N.; Sun, W.-M.; Chen, M.; Ou, Q.-S.; Lin, Z.-Y. Hyperbranched rolling circle amplification (HRCA)-based fluorescence biosensor for ultrasensitive and specific detection of single-nucleotide polymorphism genotyping associated with the therapy of chronic hepatitis B virus infection. Talanta 2018, 191, 277–282. [Google Scholar] [CrossRef]
- Gao, Z.; Wu, C.; Lv, S.; Wang, C.; Zhang, N.; Xiao, S.; Han, Y.; Xu, H.; Zhang, Y.; Li, F.; et al. Nicking-enhanced rolling circle amplification for sensitive fluorescent detection of cancer-related microRNAs. Anal. Bioanal. Chem. 2018, 410, 6819–6826. [Google Scholar] [CrossRef]
- Chen, J.; Baker, Y.R.; Brown, A.; El-Sagheer, A.H.; Brown, T. Enzyme-free synthesis of cyclic single-stranded DNA constructs containing a single triazole, amide or phosphoramidate backbone linkage and their use as templates for rolling circle amplification and nanoflower formation. Chem. Sci. 2018, 9, 8110–8120. [Google Scholar] [CrossRef] [Green Version]
- Gu, H.; Breaker, R.R. Production of single-stranded DNAs by self-cleavage of rolling-circle amplification products. BioTechniques 2013, 54, 337–343. [Google Scholar] [CrossRef]
- Tian, B.; Fock, J.; Minero, G.; Hansen, M. Nicking-associated on-loop and off-loop enzymatic cascade amplification for opto-magnetic detection of a highly conserved dengue virus sequence. Biosens. Bioelectron. 2020, 160, 112219. [Google Scholar] [CrossRef]
- Zhu, X.; Feng, C.; Zhang, B.; Tong, H.; Gao, T.; Li, G. A netlike rolling circle nucleic acid amplification technique. Analyst 2014, 140, 74–78. [Google Scholar] [CrossRef]
- Minev, D.; Guerra, R.; Kishi, J.Y.; Smith, C.; Krieg, E.; Said, K.; Hornick, A.; Sasaki, H.M.; Filsinger, G.; Beliveau, B.J.; et al. Rapid in vitro production of single-stranded DNA. Nucleic Acids Res. 2019, 47, 11956–11962. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.; Pierce, K.; Rice, J.; Wangh, L. Linear-After-The-Exponential (LATE)-PCR: An advanced method of asymmetric PCR and its uses in quantitative real-time analysis. Proc. Natl. Acad. Sci. USA 2004, 101, 1933–1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, K.; Shi, X.; Zhu, X.; Cui, L.; Shao, Q.; Ma, D. Construction and optimization of an efficient amplification method of a random ssDNA library by asymmetric emulsion PCR. Biotechnol. Appl. Biochem. 2015, 64, 239–243. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequences |
|---|---|
| GFP exo-sense (undesired strand) | 5′-/5Phos/ATT AGT TCA TAG CCC ATA TAT GGA GTT CCG-3′ |
| GFP exo-anti (desired strand) | 5′-T *A * T * A * T *A TAC GCC TTA AGA TAC ATT GAT GAG TTT GGA C-3′ |
| Methods | Benefits | Limitations | Improvements |
|---|---|---|---|
| RCA |
|
|
|
| Strand-Specific Exonuclease |
|
|
|
| Chemical Denaturation |
|
|
|
| aPCR |
|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, C.-Y.; Henderson, E.R. A Comparison of Methods for the Production of Kilobase-Length Single-Stranded DNA. DNA 2022, 2, 56-67. https://doi.org/10.3390/dna2010005
Oh C-Y, Henderson ER. A Comparison of Methods for the Production of Kilobase-Length Single-Stranded DNA. DNA. 2022; 2(1):56-67. https://doi.org/10.3390/dna2010005
Chicago/Turabian StyleOh, Chang-Yong, and Eric R. Henderson. 2022. "A Comparison of Methods for the Production of Kilobase-Length Single-Stranded DNA" DNA 2, no. 1: 56-67. https://doi.org/10.3390/dna2010005
APA StyleOh, C.-Y., & Henderson, E. R. (2022). A Comparison of Methods for the Production of Kilobase-Length Single-Stranded DNA. DNA, 2(1), 56-67. https://doi.org/10.3390/dna2010005