
Quality Control of Cell Lines Using DNA as Target


Abstract

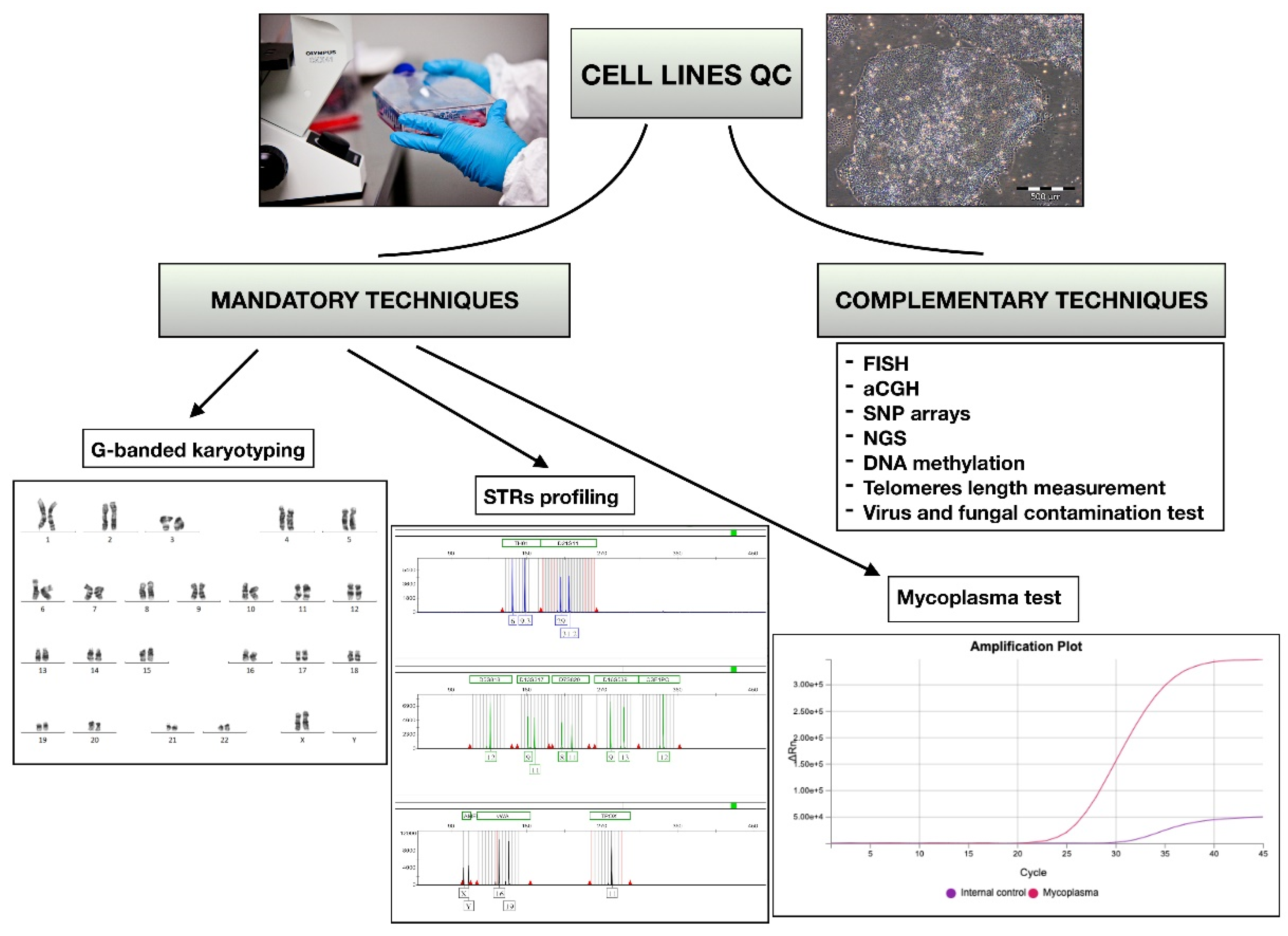
1. Introduction
2. Conventional Cytogenetics: Karyotyping
3. Fluorescence In Situ Hybridization (FISH)
4. Copy Number Variations (CNVs) Detection
5. DNA Methylation
6. Telomeres Length Measurement
7. DNA Fingerprinting and Short Tandem Repeats (STRs) Profiling
8. Microbiological Controls
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Geraghty, R.J.; Capes-Davis, A.; Davis, J.M.; Downward, J.; Freshney, R.I.; Knezevic, I.; Lovell-Badge, R.; Masters, J.R.W.; Meredith, J.; Stacey, G.N.; et al. Guidelines for the use of cell lines in biomedical research. Br. J. Cancer 2014, 111, 1021–1046. [Google Scholar] [CrossRef] [PubMed]
- Almeida, J.L.; Cole, K.D.; Plant, A.L. Standards for Cell Line Authentication and Beyond. PLoS Biol. 2016, 14, e1002476. [Google Scholar] [CrossRef]
- O’Shea, O.; Steeg, R.; Chapman, C.; Mackintosh, P.; Stacey, G.N. Development and implementation of large-scale quality control for the European bank for induced Pluripotent Stem Cells. Stem Cell Res. 2020, 45, 101773. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, S.; Stacey, G.N.; Akazawa, C.; Aoyama, N.; Baptista, R.; Bedford, P.; Griscelli, A.B.; Chandra, A.; Elwood, N.; Girard, M.; et al. Quality control guidelines for clinical-grade human induced pluripotent stem cell lines. Regen. Med. 2018, 13, 859–866. [Google Scholar] [CrossRef] [PubMed]
- ISBER, Best Practices: Recommendations for Repositories. 2018, pp. 1–104. Available online: https://www.isber.org/page/BPDownload4ed (accessed on 19 December 2021).
- Tigges, J.; Bielec, K.; Brockerhoff, G.; Hildebrandt, B.; Hübenthal, U.; Kapr, J.; Koch, K.; Teichweyde, N.; Wieczorek, D.; Rossi, A.; et al. Academic application of Good Cell Culture Practice for induced pluripotent stem cells. ALTEX 2021, 38, 595–614. [Google Scholar] [CrossRef] [PubMed]
- FDA. Good Clinical Practice: Consolidated Guideline; Office of the Federal Register, National Archives and Records Administration; FDA: White Oak, MY, USA, 1997.
- Coecke, S.; Balls, M.; Bowe, G.; Davis, J.; Gstraunthaler, G.; Hartung, T.; Hay, R.; Merten, O.-W.; Price, A.; Schechtman, L.; et al. Guidance on Good Cell Culture Practice. A report of the second ECVAM task force on good cell culture practice. Altern. Lab. Anim. 2005, 33, 261–287. [Google Scholar] [CrossRef] [PubMed]
- Pamies, D.; Bal-Price, A.; Chesne, C.; Coecke, S.; Dinnyes, A.; Eskes, C.; Grillari, R.; Gstraunthaler, G.; Hartung, T.; Jennings, P.; et al. Advanced Good Cell Culture Practice for human primary, stem cell-derived and organoid models as well as microphysiological systems. ALTEX 2018, 35, 353–378. [Google Scholar] [CrossRef]
- Capes-Davis, A.; Theodosopoulos, G.; Atkin, I.; Drexler, H.G.; Kohara, A.; MacLeod, R.A.; Masters, J.R.; Nakamura, Y.; Reid, Y.A.; Reddel, R.; et al. Check your cultures! A list of cross-contaminated or misidentified cell lines. Int. J. Cancer 2010, 127, 1–8. [Google Scholar] [CrossRef]
- Pamies, D. Good Cell Culture Practice for stem cells and stem-cell-derived models. ALTEX 2017, 34, 95–132. [Google Scholar] [CrossRef]
- Cabrera, C.M.; Cobo, F.; Nieto, A.; Cortés, J.L.; Montes, R.M.; Catalina, P.; Concha, A. Identity tests: Determination of cell line cross-contamination. Cytotechnology 2006, 51, 45–50. [Google Scholar] [CrossRef]
- Ntai, A.; Baronchelli, S.; La Spada, A.; Moles, A.; Guffanti, A.; De Blasio, P.; Biunno, I. A Review of Research-Grade Human Induced Pluripotent Stem Cells Qualification and Biobanking Processes. Biopreserv. Biobank. 2017, 15, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Bolck, H.; Pauli, C.; Göbel, E.; Mühlbauer, K.; Dettwiler, S.; Moch, H.; Schraml, P. Cancer Sample Biobanking at the Next Level: Combining Tissue with Living Cell Repositories to Promote Precision Medicine. Front. Cell Dev. Biol. 2019, 7, 246. [Google Scholar] [CrossRef]
- Harper, P.S. The discovery of the human chromosome number in Lund, 1955–1956. Hum. Genet. 2006, 119, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Hook, E.B. Exclusion of chromosomal mosaicism: Tables of 90%, 95% and 99% confidence limits and comments on use. Am. J. Hum. Genet. 1977, 29, 94–97. [Google Scholar]
- Cooley, L.D.; Morton, C.C.; Sanger, W.G.; Saxe, D.F.; Mikhail, F.M. Section E6.5–6.8 of the ACMG technical standards and guidelines: Chromosome studies of lymph node and solid tumor–acquired chromosomal abnormalities. Genet. Med. 2016, 18, 643–648. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mikhail, F.M.; Heerema, N.A.; Rao, K.W.; Burnside, R.D.; Cherry, A.M.; Cooley, L.D. Section E6.1–6.4 of the ACMG technical standards and guidelines: Chromosome studies of neoplastic blood and bone marrow–acquired chromosomal abnormalities. Genet. Med. 2016, 18, 635–642. [Google Scholar] [CrossRef]
- Maciejewski, J.P.; Tiu, R.V.; O’Keefe, C. Application of array-based whole genome scanning technologies as a cytogenetic tool in haematological malignancies. Br. J. Haematol. 2009, 146, 479–488. [Google Scholar] [CrossRef]
- McGowan-Jordan, J.; Hastings, R.; Moore, S. Re: International System for Human Cytogenetic or Cytogenomic Nomenclature (ISCN): Some Thoughts, by T. Liehr. Cytogenet. Genome Res. 2021, 161, 225–226. [Google Scholar] [CrossRef]
- Tariq Ahmad Bhat, A.A.W. Fluorescence In Situ Hybridization (FISH) and Its Applications. Chromosome Struct. Aberrations 2017, 10, 343–367. [Google Scholar]
- Catalina, P.; Cobo, F.; Cortés, J.L.; Nieto, A.I.; Cabrera, C.; Montes, R.; Concha, Á.; Menendez, P. Conventional and molecular cytogenetic diagnostic methods in stem cell research: A concise review. Cell Biol. Int. 2007, 31, 861–869. [Google Scholar] [CrossRef]
- Kwasny, R.; Vedarethinam, I.; Shah, P.; Dimaki, M.; Silahtaroglu, A.; Tumer, Z.; Svendsen, W.E. Advanced microtechnologies for detection of chromosome abnormalities by fluorescent in situ hybridization. Biomed. Microdevices 2012, 14, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Onozato, M.L.; Yapp, C.; Richardson, D.; Sundaresan, T.; Chahal, V.; Lee, J.; Sullivan, J.P.; Madden, M.W.; Shim, H.S.; Liebers, M.; et al. Highly Multiplexed Fluorescence in Situ Hybridization for in Situ Genomics. J. Mol. Diagn. 2019, 21, 390–407. [Google Scholar] [CrossRef] [PubMed]
- SenGupta, S.; Dhanjal, S.; Harper, J. Quality control standards in PGD and PGS. Reprod. Biomed. Online 2016, 32, 263–270. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zarrei, M.; MacDonald, J.; Merico, D.; Scherer, S. A copy number variation map of the human genome. Nat. Rev. Genet. 2015, 16, 172–183. [Google Scholar] [CrossRef]
- Itsara, A.; Cooper, G.M.; Baker, C.; Girirajan, S.; Li, J.; Absher, D.; Krauss, R.M.; Myers, R.M.; Ridker, P.M.; Chasman, D.I.; et al. Population Analysis of Large Copy Number Variants and Hotspots of Human Genetic Disease. Am. J. Hum. Genet. 2009, 84, 148–161. [Google Scholar] [CrossRef]
- Macé, A.; Kutalik, Z.; Valsesia, A. Copy Number Variation. Methods Mol. Biol. 2018, 1793, 231–258. [Google Scholar] [CrossRef]
- Harel, T.; Lupski, J. Genomic disorders 20 years on-mechanisms for clinical manifestations. Clin. Genet. 2018, 93, 439–449. [Google Scholar] [CrossRef]
- Hanemaaijer, N.M.; Sikkema-Raddatz, B.; Van Der Vries, G.; Dijkhuizen, T.; Hordijk, R.; Van Essen, A.J.; Veenstra-Knol, H.E.; Kerstjens, M.; Herkert, J.C.; Gerkes, E.H.; et al. Practical guidelines for interpreting copy number gains detected by high-resolution array in routine diagnostics. Eur. J. Hum. Genet. 2012, 20, 161–165. [Google Scholar] [CrossRef]
- De Leeuw, N.; Hehir-Kwa, J.; Simons, A.; Van Kessel, A.G.; Smeets, D.; Faas, B.; Pfundt, R. SNP Array Analysis in Constitutional and Cancer Genome Diagnostics—Copy Number Variants, Genotyping and Quality Control. Cytogenet. Genome Res. 2011, 135, 212–221. [Google Scholar] [CrossRef]
- Cheung, S.W.; Bi, W. Novel applications of array comparative genomic hybridization in molecular diagnostics. Expert Rev. Mol. Diagn. 2018, 18, 531–542. [Google Scholar] [CrossRef]
- Mak, A.C.; Lai, Y.Y.; Lam, E.T.; Kwok, T.P.; Leung, A.K.; Poon, A.; Mostovoy, Y.; Hastie, A.R.; Stedman, W.; Anantharaman, T.; et al. Genome-Wide Structural Variation Detection by Genome Mapping on Nanochannel Arrays. Genetics 2016, 202, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Demichelis, F.; Greulich, H.; Macoska, J.A.; Beroukhim, R.; Sellers, W.R.; Garraway, L.; Rubin, M.A. SNP panel identification assay (SPIA): A genetic-based assay for the identification of cell lines. Nucleic Acids Res. 2008, 36, 2446–2456. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.; Li, Y.; Robertson, K.D. DNA Methylation: Superior or Subordinate in the Epigenetic Hierarchy? Genes Cancer 2011, 2, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Sant, K.E.; Nahar, M.S.; Dolinoy, D.C. DNA Methylation Screening and Analysis. Methods Mol. Biol. 2012, 889, 385–406. [Google Scholar] [CrossRef]
- Suelves, M.; Carrio, E.; Núñez-Álvarez, Y.; Peinado, M.A. DNA methylation dynamics in cellular commitment and differentiation. Brief. Funct. Genom. 2016, 15, 443–453. [Google Scholar] [CrossRef]
- Lenz, M.; Goetzke, R.; Schenk, A.; Schubert, C.; Veeck, J.; Hemeda, H.; Koschmieder, S.; Zenke, M.; Schuppert, A.; Wagner, W. Epigenetic Biomarker to Support Classification into Pluripotent and Non-Pluripotent Cells. Sci. Rep. 2015, 5, 8973. [Google Scholar] [CrossRef]
- De Almeida, D.C.; Ferreira, M.R.; Franzen, J.; Weidner, C.I.; Frobel, J.; Zenke, M.; Costa, I.G.; Wagner, W. Epigenetic Classification of Human Mesenchymal Stromal Cells. Stem Cell Rep. 2016, 6, 168–175. [Google Scholar] [CrossRef]
- Unnikrishnan, A.; Freeman, W.M.; Jackson, J.; Wren, J.D.; Porter, H.; Richardson, A. The role of DNA methylation in epigenetics of aging. Pharmacol. Ther. 2019, 195, 172–185. [Google Scholar] [CrossRef]
- Schellenberg, A.; Mauen, S.; Koch, C.M.; Jans, R.; de Waele, P.; Wagner, W. Proof of principle: Quality control of therapeutic cell preparations using senescence-associated DNA-methylation changes. BMC Res. Notes 2014, 7, 254. [Google Scholar] [CrossRef]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA methylation landscape of cancer. Trends Genet. 2021, 37, 1012–1027. [Google Scholar] [CrossRef]
- Van Ly, D.; Low, R.R.J.; Frölich, S.; Bartolec, T.K.; Kafer, G.R.; Pickett, H.A.; Gaus, K.; Cesare, A.J. Telomere Loop Dynamics in Chromosome End Protection. Mol. Cell 2018, 71, 510–525.e6. [Google Scholar] [CrossRef] [PubMed]
- Aubert, G.; Hills, M.; Lansdorp, P.M. Telomere length measurement—Caveats and a critical assessment of the available technologies and tools. Mutat. Res. Mol. Mech. Mutagen. 2012, 730, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Maciejowski, J.; De Lange, T. Telomeres in cancer: Tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 2017, 18, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Pickett, H.A.; Henson, J.D.; Au, A.Y.; Neumann, A.A.; Reddel, R.R. Normal mammalian cells negatively regulate telomere length by telomere trimming. Hum. Mol. Genet. 2011, 20, 4684–4692. [Google Scholar] [CrossRef] [PubMed]
- Kahl, V.F.S.; Allen, J.A.M.; Nelson, C.B.; Sobinoff, A.; Lee, M.; Kilo, T.; Vasireddy, R.S.; Pickett, H.A. Telomere Length Measurement by Molecular Combing. Front. Cell Dev. Biol. 2020, 8, 493. [Google Scholar] [CrossRef]
- Hiraishi, N.; Terai, M.; Fujiwara, M.; Aida, J.; Izumiyama-Shimomura, N.; Ishikawa, N.; Tomita, K.-I.; Matsuda, Y.; Arai, T.; Takubo, K.; et al. Quantitative fluorescence in situ hybridization for investigation of telomere length dynamics in the pituitary gland using samples from 128 autopsied patients. Tissue Cell 2018, 53, 1–7. [Google Scholar] [CrossRef]
- Conomos, D.; Stutz, M.; Hills, M.; Neumann, A.A.; Bryan, T.M.; Reddel, R.R.; Pickett, H.A. Variant repeats are interspersed throughout the telomeres and recruit nuclear receptors in ALT cells. J. Cell Biol. 2012, 199, 893–906. [Google Scholar] [CrossRef]
- American Type Culture Collection Standards Development Organization Workgroup ASN-0002 Cell line misidentification: The beginning of the end. Nat. Cancer 2010, 10, 441–448. [CrossRef]
- Silvers, W.K.; Moorhead, P.; Defendi, V.; Billingham, R.E. Immunological and Karyological Criteria for Identification of Cell Lines. JNCI J. Natl. Cancer Inst. 1960, 25, 359–385. [Google Scholar] [CrossRef]
- Brand, K.G.; Syverton, J.T. Results of species-specific hemagglutination tests on ‘’transformed’’, nontransformed, and primary cell cultures. JNCI J. Natl. Cancer Inst. 1962, 28, 147–157. [Google Scholar]
- MacLeod, R.A.F.; Kaufmann, M.; Drexler, H.G. Cytogenetic harvesting of commonly used tumor cell lines. Nat. Protoc. 2007, 2, 372–382. [Google Scholar] [CrossRef][Green Version]
- Vořechovský, I.; Královičová, J.; Laycock, M.D.; Webster, A.D.B.; Marsh, S.G.; Madrigal, A.; Hammarström, L. Short tandem repeat (STR) haplotypes in HLA: An integrated 50-kb STR/linkage disequilibrium/gene map between the RING3 and HLA-B genes and identification of STR haplotype diversification in the class III region. Eur. J. Hum. Genet. 2001, 9, 590–598. [Google Scholar] [CrossRef][Green Version]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; Dewaard, J.R. Biological identifications through DNA barcodes. Proc. R. Soc. B Boil. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef]
- Jeffreys, A.J.; Wilson, V.; Thein, S.L. Hypervariable ‘minisatellite’ regions in human DNA. Nature 1985, 314, 67–73. [Google Scholar] [CrossRef]
- Koreth, J.; O’Leary, J.J.; James, O.D.M. Microsatellites and PCR genomic analysis. J. Pathol. 1996, 178, 239–248. [Google Scholar] [CrossRef]
- National Institute of Standards and Technology, Variant Allele Reports. Available online: https://strbase.nist.gov/var_tab.htm (accessed on 19 December 2021).
- Korch, C.T.; Hall, E.M.; Dirks, W.G.; Sykes, G.R.; Capes-Davis, A.; Barrett, T.; Butler, J.M.; Neve, R.M.; Nims, R.W.; Storts, D.R.; et al. Human Cell Line Authentication. Standardization of Short Tandem Repeat (STR) Profiling, ASN-0002 Revised 2021, April 2021 ed.; American National Standards Institute, American Type Culture Collection Standards Development Organization: New York, NY, USA, 2021. [Google Scholar]
- Korch, C.T.; Capes-Davis, A. The Extensive and Expensive Impacts of HEp-2 [HeLa], Intestine 407 [HeLa], and Other False Cell Lines in Journal Publications. SLAS Discov. Adv. Sci. Drug Discov. 2021, 26, 1268–1279. [Google Scholar] [CrossRef]
- ICLAC. Register of Misidentified Cell Lines. 2021. Available online: https://iclac.org/databases/cross-contaminations/ (accessed on 19 December 2021).
- Vaught, J.; Korch, C.T. The Continuing Saga of Cell Line Misidentification. Biopreserv. Biobank. 2021, 19, 357–358. [Google Scholar] [CrossRef]
- Cobo, F.; Stacey, G.N.; Hunt, C.; Cabrera, C.; Nieto, A.; Montes, R.; Cortés, J.L.; Catalina, P.; Barnie, A.; Concha, Á. Microbiological control in stem cell banks: Approaches to standardisation. Appl. Microbiol. Biotechnol. 2005, 68, 456–466. [Google Scholar] [CrossRef]
- Berthomé, M.; Gallot, G.; Vivien, R.; Clémenceau, B.; Nguyen, J.-M.; Coste-Burel, M.; Vié, H. Viral DNA contamination is responsible for Epstein–Barr virus detection in cytotoxic T lymphocytes stimulated in vitro with Epstein–Barr virus B-lymphoblastoid cell line. Cancer Immunol. Immunother. 2010, 59, 1867–1875. [Google Scholar] [CrossRef]
- Rusmevichientong, A.; Das Gupta, J.; Elias, P.S.; Silverman, R.H.; Chow, S.A. Analysis of Single-Nucleotide Polymorphisms in Patient-Derived Retrovirus Integration Sites Reveals Contamination from Cell Lines Acutely Infected by Xenotropic Murine Leukemia Virus-Related Virus. J. Virol. 2011, 85, 12830–12834. [Google Scholar] [CrossRef][Green Version]
- Stacey, G.; Possee, R. Safety aspects of insect cell cultures. In Current Applications of Cell Culture Engineering V2, Insect Cell Cultures-Fundamental and Applied Aspects; Vlak, J.M., Tramper, J., Mitenburger, H.G., Eds.; Kluwer Acad Publishers: Dordrecht, The Netherlands, 1996; pp. 299–303. [Google Scholar]
- Mahy, B.W.; Dykewicz, C.; Fisher-Hoch, S.; Ostroff, S.; Tipple, M.; Sanchez, A. Virus zoonoses and their potential for contamination of cell cultures. Dev. Biol. Stand. 1991, 75, 183–189. [Google Scholar]
- Uryvaev, L.V.; Dedova, A.V.; Ionova, K.S.; Parasjuk, N.A.; Selivanova, T.K.; Bunkova, N.I.; Gushina, E.A.; Grebennikova, V.; Podchernjaeva, R.J. Contamination of Cell Cultures with Bovine Viral Diarrhea Virus (BVDV). Bull. Exp. Biol. Med. 2012, 153, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Romero, N.; Velazquez-Salinas, L.; Ridpath, J.F.; Verdugo-Rodríguez, A.; Basurto-Alcántara, F.J. Detection and genotyping of bovine viral diarrhea virus found contaminating commercial veterinary vaccines, cell lines, and fetal bovine serum lots originating in Mexico. Arch. Virol. 2021, 166, 1999–2003. [Google Scholar] [CrossRef]
- Stacey, G.N. Cell Culture Contamination. Methods Mol. Biol. 2011, 731, 79–91. [Google Scholar] [CrossRef]
- Mirjalili, A.; Parmoor, E.; Bidhendi, S.M.; Sarkari, B. Microbial contamination of cell cultures: A 2 years study. Biologicals 2005, 33, 81–85. [Google Scholar] [CrossRef]
- Frommer, W.; Archer, L.; Boon, B.; Brunius, G.; Collins, C.H.; Crooy, P.; Doblhoff-Dier, O.; Donikian, R.; Economidis, J.; Frontali, C.; et al. Safe biotechnology (5). Recommendations for safe work with animal and human cell cultures concerning potential human pathogens. Appl. Microbiol. Biotechnol. 1993, 39, 141–147. [Google Scholar] [CrossRef]
- Nübling, C.M.; Baylis, S.A.; Hanschmann, K.-M.; Montag-Lessing, T.; Chudy, M.; Kreß, J.; Ulrych, U.; Czurda, S.; Rosengarten, R.; The Mycoplasma Collaborative Study Group. World Health Organization International Standard to Harmonize Assays for Detection of mycoplasma DNA. Appl. Environ. Microbiol. 2015, 81, 5694–5702. [Google Scholar] [CrossRef]
- Armstrong, S.E.; Mariano, J.A.; Lundin, D.J. The scope of mycoplasma contamination within the biopharmaceutical industry. Biologicals 2010, 38, 211–213. [Google Scholar] [CrossRef]
- Laborde, S.; Degrave, A.; Lehmann, D.; Jouette, S.; Rofel, C.; Muller, T.; Hertzog, N.; Rook, M.; Ribault, S. Detection of Mollicutes in bioreactor samples by real-time transcription-mediated amplification. Lett. Appl. Microbiol. 2010, 50, 633–638. [Google Scholar] [CrossRef]
- Volokhov, D.V.; Norris, T.; Rios, C.; Davidson, M.K.; Messick, J.B.; Gulland, F.M.; Chizhikov, V.E. Novel hemotrophic mycoplasma identified in naturally infected California sea lions (Zalophus californianus). Veter. Microbiol. 2011, 149, 262–268. [Google Scholar] [CrossRef]
- Falagan-Lotsch, P.; Lopes, T.S.; Ferreira, N.; Balthazar, N.; Monteiro, A.M.; Borojevic, R.; Granjeiro, J.M. Performance of PCR-based and Bioluminescent assays for mycoplasma detection. J. Microbiol. Methods 2015, 118, 31–36. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO expert committee on biological standardization. WHO Tech. Rep. Ser. 2014, 987, 42. [Google Scholar]


{kind=link}
{kind=link}
| Quality Control (QC) | DNA Target | Purpose or Attribute | Recommendation by the Application Field | References |
|---|---|---|---|---|
| G-banded karyotyping | Chromosomal metaphases analysis (alterations ≥5–10 Mb in size) | Stability, authentication, and species cross-contamination | Gold standard Mandatory for research and therapeutic use | [17,18,20,22] |
| FISH | Unique DNA sequences along the chromosomes | Stability | Complementary use if needed | [21,22,23,24] |
| aCGH, SNP arrays or NGS | CNVs | Stability and authentication | Highly recommended for clinical applications | [26,27,28,30,31,32,33,34] |
| DNA methylation | Methyl group to the C-5 position of the cytosine ring of DNA usually in a CpG dinucleotide | Identity and differentiation, senescence and tumorigenicity | Optional for research (if others QC for the same attributes are performed) Recommended for clinical applications | [38,39,41,42] |
| Telomeres length measurement | Nucleoprotein structures composed by repetitive hexanucleotide (TTAGGGn) combined with the protein complex shelterin | Tumorigenicity and senescence | Optional | [45,46,47,48,49] |
| STRs profiling | DNA hypervariable regions consisting of core sequences of 1–6 bp, repeated in a different number | Authentication | Gold standard Mandatory for research and therapeutic use | [1,10,59] |
| Microbiological tests | Microbial DNA detection | Mycoplasma contamination Virus and fungal contamination | Mandatory periodically for research and therapeutic use Mandatory in clinical applications | [1,63,70,71,73] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrillo-Ávila, J.A.; Catalina, P.; Aguilar-Quesada, R. Quality Control of Cell Lines Using DNA as Target. DNA 2022, 2, 44-55. https://doi.org/10.3390/dna2010004
Carrillo-Ávila JA, Catalina P, Aguilar-Quesada R. Quality Control of Cell Lines Using DNA as Target. DNA. 2022; 2(1):44-55. https://doi.org/10.3390/dna2010004
Chicago/Turabian StyleCarrillo-Ávila, José Antonio, Purificación Catalina, and Rocío Aguilar-Quesada. 2022. "Quality Control of Cell Lines Using DNA as Target" DNA 2, no. 1: 44-55. https://doi.org/10.3390/dna2010004
APA StyleCarrillo-Ávila, J. A., Catalina, P., & Aguilar-Quesada, R. (2022). Quality Control of Cell Lines Using DNA as Target. DNA, 2(1), 44-55. https://doi.org/10.3390/dna2010004