
Nucleases and Co-Factors in DNA Replication Stress Responses

Abstract
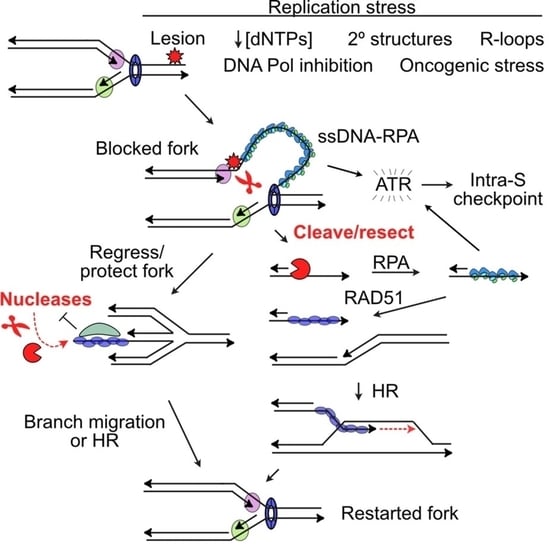
1. Introduction
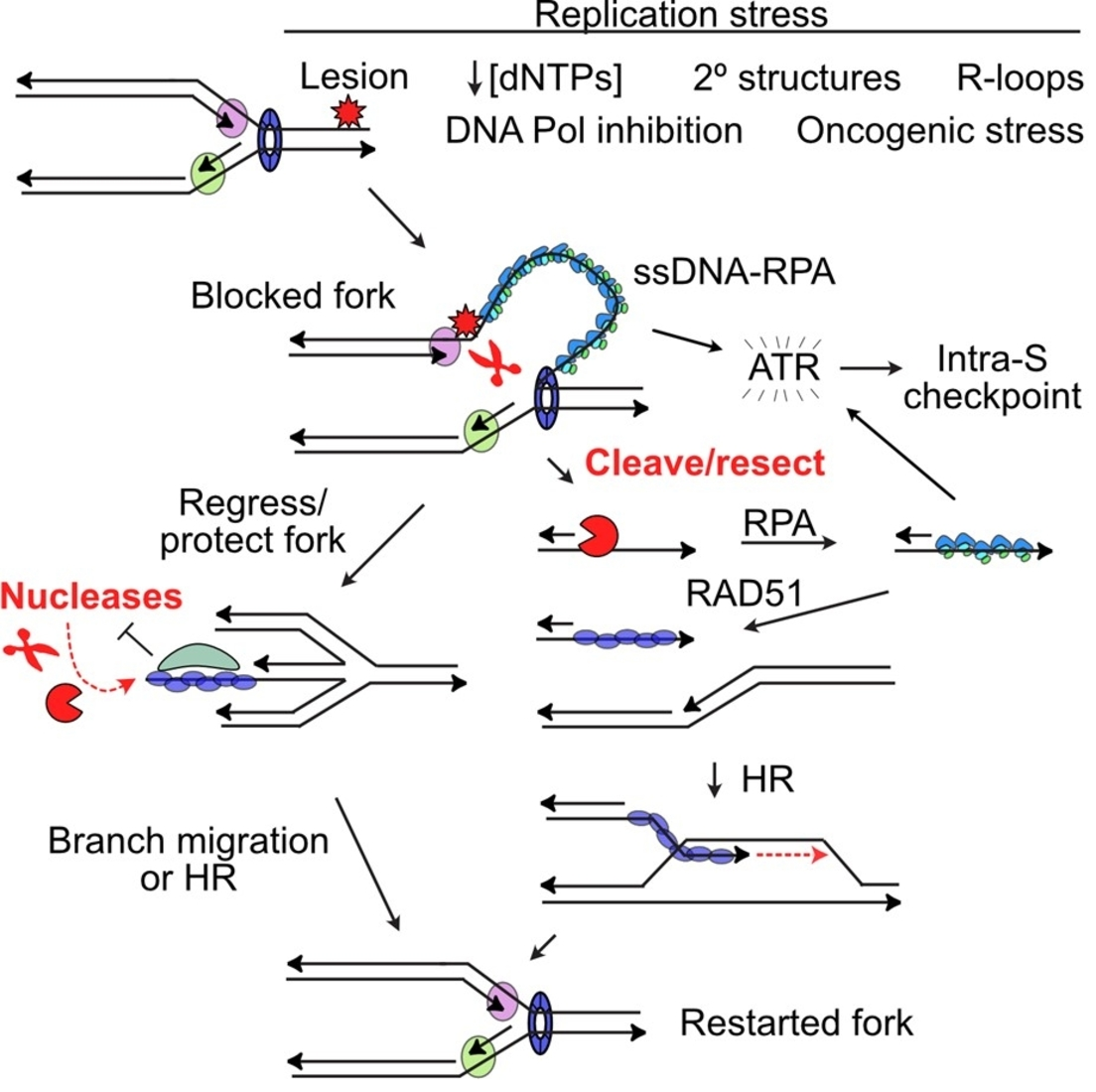
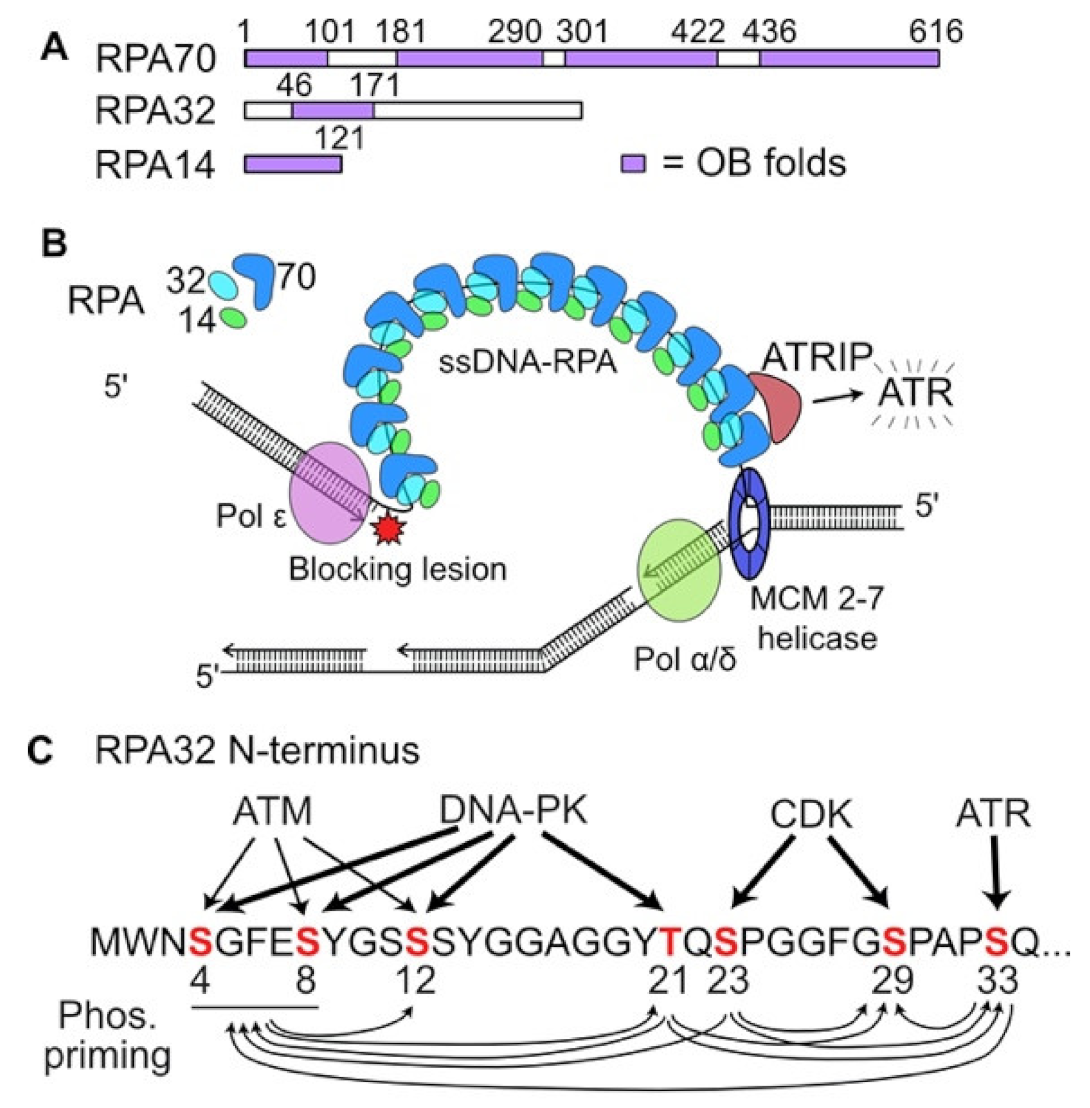
2. DDR Signaling in Response to Replication Stress
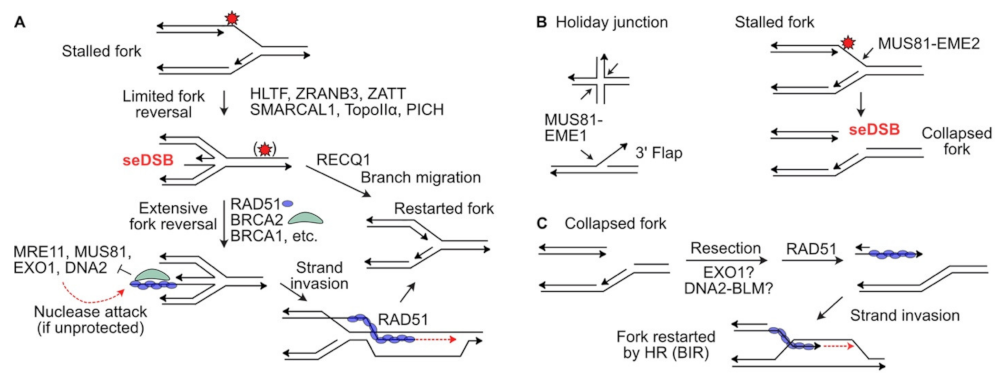
3. Protecting and Rescuing Blocked Replication Forks
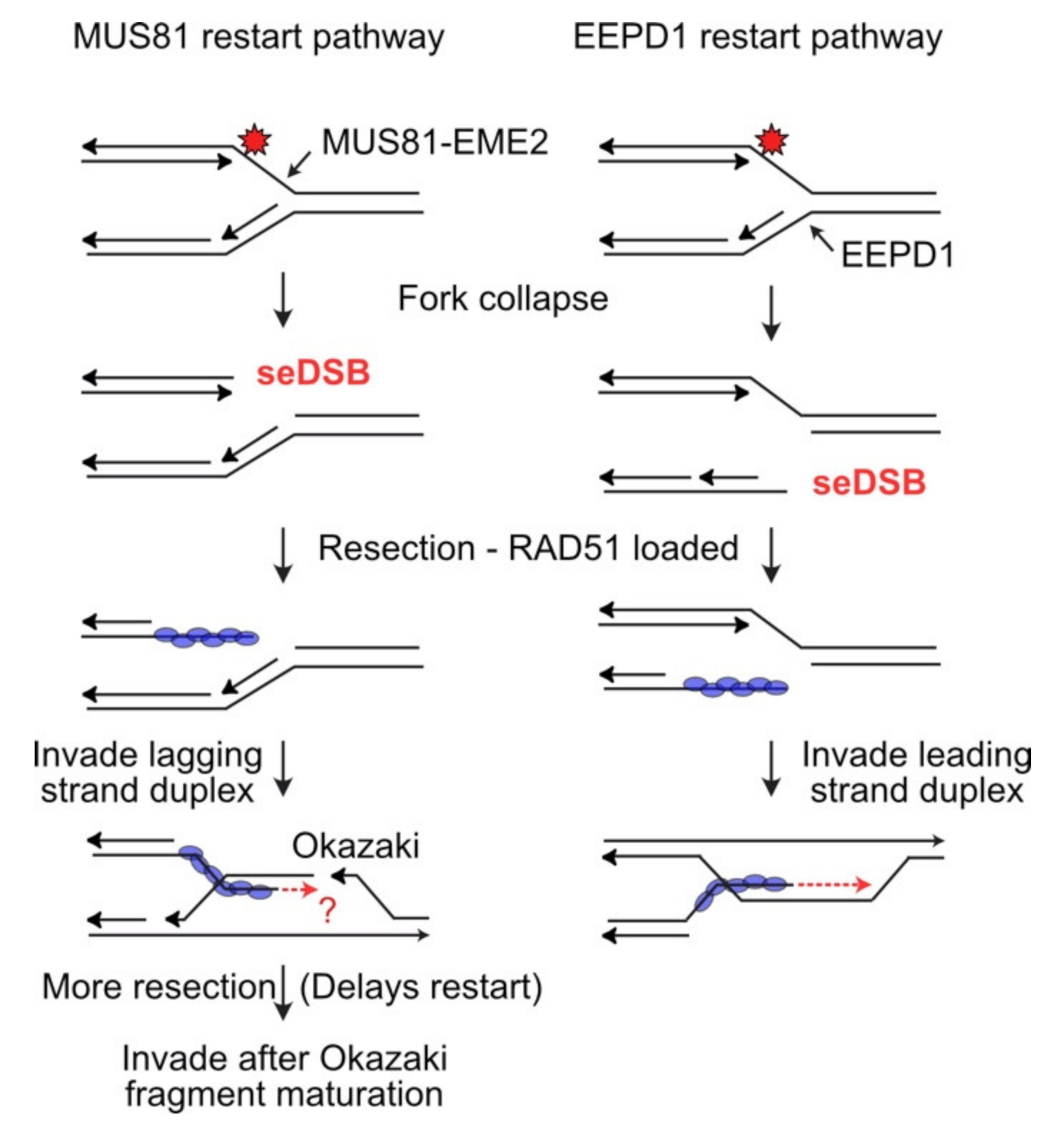
4. MUS81: An Ancient Structure-Specific Nuclease Involved in HR and Restart of Stressed Replication Forks
5. EEPD1: A 5′ Structure-Specific Endonuclease That Complements the 3′ MUS81 Nuclease
6. Metnase: A Recently Evolved Nuclease-Protein Methyl Transferase That Promotes Replication Fork Restart
7. Other Nucleases with Known or Potential Roles in Replication Stress Responses: CtIP, MRE11, EXO1, DNA2-BLM, SLX1-SLX4, XPF-ERCC1-SLX4, Artemis, XPG, and FEN1
8. Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Löb, D.; Lengert, N.; Chagin, V.O.; Reinhart, M.; Casas-Delucchi, C.S.; Cardoso, M.C.; Drossel, B. 3D replicon distributions arise from stochastic initiation and domino-like DNA replication progression. Nat. Commun. 2016, 7, 11207. [Google Scholar] [CrossRef] [PubMed]
- Chagin, V.O.; Casas-Delucchi, C.S.; Reinhart, M.; Schermelleh, L.; Markaki, Y.; Maiser, A.; Bolius, J.J.; Bensimon, A.; Fillies, M.; Domaing, P.; et al. 4D Visualization of replication foci in mammalian cells corresponding to individual replicons. Nat. Commun. 2016, 7, 11231. [Google Scholar] [CrossRef] [PubMed]
- IIyer, D.R.; Rhind, N. The Intra-S Checkpoint Responses to DNA Damage. Genes 2017, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento-Salinas, F.L.; Perez-Gonzalez, A.; Acosta-Casique, A.; Ix-Ballote, A.; Diaz, A.; Trevino, S.; Rosas-Murrieta, N.H.; Millán-Perez-Peña, L.; Maycotte., P. Reactive Oxygen Species: Role in Carcinogenesis, Cancer Cell Signaling and Tumor Progression. Life Sci. 2021, 284, 119942. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef]
- Nickoloff, J.A.; Sharma, N.; Taylor, L.; Allen, S.J.; Hromas, R. The Safe Path at the Fork: Ensuring Replication-Associated DNA Double-Strand Breaks are Repaired by Homologous Recombination. Front. Genet. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Conti, B.A.; Smogorzewska, A. Mechanisms of direct replication restart at stressed replisomes. DNA Repair 2020, 95, 102947. [Google Scholar] [CrossRef]
- Lyu, X.; Chastain, M.; Chai, W. Genome-Wide Mapping and Profiling of Gammah2ax Binding Hotspots in Response to Different Replication Stress Inducers. BMC Genom. 2019, 20, 579. [Google Scholar] [CrossRef]
- Poggi, L.; Richard, G.F. Alternative DNA Structures in Vivo: Molecular Evidence and Remaining Questions. Microbiol. Mol. Biol. Rev. 2020, 85, e00110–e00120. [Google Scholar] [CrossRef]
- Spiegel, J.; Adhikari, S.; Balasubramanian, S. The Structure and Function of DNA G-Quadruplexes. Trends Chem. 2019, 2, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Barlow, J.H.; Faryabi, R.B.; Callen, E.; Wong, N.; Malhowski, A.; Chen, H.T.; Gutierrez-Cruz, G.; Sun, H.W.; McKinnon, P.; Wright, G.; et al. Identification of Early Replicating Fragile Sites That Contribute to Genome Instability. Cell 2013, 152, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, S.; Freudenreich, C.H. The role of fork stalling and DNA structures in causing chromosome fragility. Genes Chromosom. Cancer 2018, 58, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Bochman, M.L.; Paeschke, K.; Zakian, V.A. DNA secondary structures: Stability and function of G-quadruplex structures. Nat. Rev. Genet. 2012, 13, 770–780. [Google Scholar] [CrossRef]
- Gadaleta, M.C.; Noguchi, E. Regulation of DNA Replication through Natural Impediments in the Eukaryotic Genome. Genes 2017, 8, 98. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2013, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Cicconi, A.; Chang, S. Shelterin and the replisome: At the intersection of telomere repair and replication. Curr. Opin. Genet. Dev. 2020, 60, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, H.; Garcia-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Santos-Pereira, J.M.; Aguilera, A. R loops: New modulators of genome dynamics and function. Nat. Rev. Genet. 2015, 16, 583–597. [Google Scholar] [CrossRef]
- Hamperl, S.; Bocek, M.J.; Saldivar, J.C.; Swigut, T.; Cimprich, K.A. Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell 2017, 170, 774–786. [Google Scholar] [CrossRef]
- Freudenreich, C.H. R-loops: Targets for nuclease cleavage and repeat instability. Curr. Genet. 2018, 64, 789–794. [Google Scholar] [CrossRef]
- Ivessa, A.; Lenzmeier, B.A.; Bessler, J.B.; Goudsouzian, L.K.; Schnakenberg, S.L.; Zakian, V.A. The Saccharomyces cerevisiae Helicase Rrm3p Facilitates Replication Past Nonhistone Protein-DNA Complexes. Mol. Cell 2003, 12, 1525–1536. [Google Scholar] [CrossRef]
- Billard, P.; A Poncet, D. Replication Stress at Telomeric and Mitochondrial DNA: Common Origins and Consequences on Ageing. Int. J. Mol. Sci. 2019, 20, 4959. [Google Scholar] [CrossRef]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.; Folkes, L.; Gromak, N.; Petermann, E. Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 2016, 7, 13087. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, R.; Lai, M.S.; Foiani, M. Preventing Replication Stress to Maintain Genome Stability: Resolving Conflicts between Replication and Transcription. Mol. Cell 2012, 45, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Gómez-González, B.; Aguilera, A. Transcription-mediated replication hindrance: A major driver of genome instability. Genes Dev. 2019, 33, 1008–1026. [Google Scholar] [CrossRef]
- Hamperl, S.; Cimprich, K.A. Conflict Resolution in the Genome: How Transcription and Replication Make It Work. Cell 2016, 167, 1455–1467. [Google Scholar] [CrossRef]
- Garcia-Muse, T.; Aguilera, A. Transcription–replication conflicts: How they occur and how they are resolved. Nat. Rev. Mol. Cell Biol. 2016, 17, 553–563. [Google Scholar] [CrossRef]
- Minchell, N.E.; Keszthelyi, A.; Baxter, J. Cohesin Causes Replicative DNA Damage by Trapping DNA Topological Stress. Mol. Cell 2020, 78, 739–751.e8. [Google Scholar] [CrossRef]
- Qiu, S.; Jiang, G.; Cao, L.; Huang, J. Replication Fork Reversal and Protection. Front. Cell Dev. Biol. 2021, 9, 670392. [Google Scholar] [CrossRef]
- Rickman, K.; Smogorzewska, A. Advances in understanding DNA processing and protection at stalled replication forks. J. Cell Biol. 2019, 218, 1096–1107. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Toledo, F. Mechanisms Generating Cancer Genome Complexity: Back to the Future. Cancers 2020, 12, 3783. [Google Scholar] [CrossRef]
- Nickoloff, J.A.; Jones, D.; Lee, S.-H.; Williamson, E.A.; Hromas, R. Drugging the Cancers Addicted to DNA Repair. JNCI J. Natl. Cancer Inst. 2017, 109, djx059. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA Damage, Repair, and Mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Jossen, R.; Bermejo, R. The DNA damage checkpoint response to replication stress: A Game of Forks. Front. Genet. 2013, 4, 26. [Google Scholar] [CrossRef]
- Hills, S.A.; Diffley, J.F. DNA Replication and Oncogene-Induced Replicative Stress. Curr. Biol. 2014, 24, R435–R444. [Google Scholar] [CrossRef] [PubMed]
- Primo, L.M.F.; Teixeira, L.K. DNA replication stress: Oncogenes in the spotlight. Genet. Mol. Biol. 2020, 43, e20190138. [Google Scholar] [CrossRef]
- Kotsantis, P.; Petermann, E.; Boulton, S.J. Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place. Cancer Discov. 2018, 8, 537–555. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D. Unwind and slow down: Checkpoint activation by helicase and polymerase uncoupling: Figure 1. Genes Dev. 2005, 19, 1007–1012. [Google Scholar] [CrossRef]
- Berti, M.; Cortez, D.; Lopes, M. The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat. Rev. Mol. Cell Biol. 2020, 21, 633–651. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Niu, H.; Miller, A.S.; Sung, P. Biochemical mechanism of DSB end resection and its regulation. DNA Repair 2015, 32, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S. Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 195–212. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Kim, W.; Kloeber, J.A.; Lou, Z. DNA end resection and its role in DNA replication and DSB repair choice in mammalian cells. Exp. Mol. Med. 2020, 52, 1705–1714. [Google Scholar] [CrossRef] [PubMed]
- Yazinski, S.A.; Zou, L. Functions, Regulation, and Therapeutic Implications of the ATR Checkpoint Pathway. Annu. Rev. Genet. 2016, 50, 155–173. [Google Scholar] [CrossRef]
- Shiotani, B.; Nguyen, H.D.; Håkansson, P.; Maréchal, A.; Tse, A.; Tahara, H.; Zou, L. Two Distinct Modes of ATR Activation Orchestrated by Rad17 and Nbs1. Cell Rep. 2013, 3, 1651–1662. [Google Scholar] [CrossRef]
- Haahr, P.; Hoffmann, S.; Tollenaere, M.; Ho, T.; Toledo, L.; Mann, M.; Bekker-Jensen, S.; Räschle, M.; Mailand, N. Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat. Cell Biol. 2016, 18, 1196–1207. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Jeggo, P.A. ATM’s Role in the Repair of DNA Double-Strand Breaks. Genes 2021, 12, 1370. [Google Scholar] [CrossRef]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The Essential Kinase ATR: Ensuring Faithful Duplication of a Challenging Genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef]
- Liu, S.; Opiyo, S.O.; Manthey, K.; Glanzer, J.G.; Ashley, A.K.; Amerin, C.; Troksa, K.; Shrivastav, M.; Nickoloff, J.A.; Oakley, G.G. Distinct roles for DNA-PK, ATM and ATR in RPA phosphorylation and checkpoint activation in response to replication stress. Nucleic Acids Res. 2012, 40, 10780–10794. [Google Scholar] [CrossRef] [PubMed]
- Anantha, R.W.; Vassin, V.M.; Borowiec, J.A. Sequential and Synergistic Modification of Human RPA Stimulates Chromosomal DNA Repair. J. Biol. Chem. 2007, 282, 35910–35923. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.; Nievera, C.J.; Klimovich, V.; Fanning, E.; Wu, X. RPA2 Is a Direct Downstream Target for ATR to Regulate the S-phase Checkpoint. J. Biol. Chem. 2006, 281, 39517–39533. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, A.; Zou, L. RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res. 2014, 25, 9–23. [Google Scholar] [CrossRef]
- Zuazua-Villar, P.; Ganesh, A.; Phear, G.; Gagou, M.E.; Meuth, M. Extensive RPA2 hyperphosphorylation promotes apoptosis in response to DNA replication stress in CHK1 inhibited cells. Nucleic Acids Res. 2015, 43, 9776–9787. [Google Scholar] [CrossRef]
- Vassin, V.M.; Anantha, R.W.; Sokolova, E.; Kanner, S.; Borowiec, J.A. Human RPA phosphorylation by ATR stimulates DNA synthesis and prevents ssDNA accumulation during DNA-replication stress. J. Cell Sci. 2009, 122, 4070–4080. [Google Scholar] [CrossRef]
- Soniat, M.M.; Myler, L.R.; Kuo, H.-C.; Paull, T.T.; Finkelstein, I.J. RPA Phosphorylation Inhibits DNA Resection. Mol. Cell 2019, 75, 145–153.e5. [Google Scholar] [CrossRef]
- Patrick, S.M.; Oakley, G.G.; Dixon, K.; Turchi, J.J. DNA Damage Induced Hyperphosphorylation of Replication Protein A. 2. Characterization of DNA Binding Activity, Protein Interactions, and Activity in DNA Replication and Repair. Biochemistry 2005, 44, 8438–8448. [Google Scholar] [CrossRef]
- Oakley, G.G.; Patrick, S.M.; Yao, J.; Carty, M.P.; Turchi, J.J.; Dixon, K. RPA Phosphorylation in Mitosis Alters DNA Binding and Protein−Protein Interactions. Biochemistry 2003, 42, 3255–3264. [Google Scholar] [CrossRef]
- Marechal, A.; Li, J.M.; Ji, X.Y.; Wu, C.S.; Yazinski, S.A.; Nguyen, H.D.; Liu, S.; Jimenez, A.E.; Jin, J.; Zou, L. PRP19 Transforms into a Sensor of RPA-ssDNA after DNA Damage and Drives ATR Activation Via a Ubiquitin-Mediated Circuitry. Mol. Cell 2014, 53, 235–246. [Google Scholar] [CrossRef]
- Mishra, A.K.; Dormi, S.S.; Turchi, A.M.; Woods, D.S.; Turchi, J.J. Chemical inhibitor targeting the replication protein A–DNA interaction increases the efficacy of Pt-based chemotherapy in lung and ovarian cancer. Biochem. Pharmacol. 2014, 93, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Glanzer, J.G.; Liu, S.; Wang, L.; Mosel, A.; Peng, A.; Oakley, G.G. RPA Inhibition Increases Replication Stress and Suppresses Tumor Growth. Cancer Res. 2014, 74, 5165–5172. [Google Scholar] [CrossRef] [PubMed]
- Sadoughi, F.; Hallajzadeh, J.; Asemi, Z.; Mansournia, M.A.; Alemi, F.; Yousefi, B. Signaling pathways involved in cell cycle arrest during the DNA breaks. DNA Repair 2021, 98, 103047. [Google Scholar] [CrossRef] [PubMed]
- Brambati, A.; Zardoni, L.; Achar, Y.J.; Piccini, D.; Galanti, L.; Colosio, A.; Foiani, M.; Liberi, G. Dormant origins and fork protection mechanisms rescue sister forks arrested by transcription. Nucleic Acids Res. 2017, 46, 1227–1239. [Google Scholar] [CrossRef]
- Courtot, L.; Hoffmann, J.-S.; Bergoglio, V. The Protective Role of Dormant Origins in Response to Replicative Stress. Int. J. Mol. Sci. 2018, 19, 3569. [Google Scholar] [CrossRef]
- Williams, R.M.; Zhang, X. Roles of ATM and ATR in DNA double strand breaks and replication stress. Prog. Biophys. Mol. Biol. 2021, 163, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.W.; Botchan, M.R.; Berger, J.M. Mechanisms and regulation of DNA replication initiation in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 107–144. [Google Scholar] [CrossRef]
- Mukherjee, C.; Tripathi, V.; Manolika, E.M.; Heijink, A.M.; Ricci, G.; Merzouk, S.; De Boer, H.R.; Demmers, J.; Van Vugt, M.A.T.M.; Chaudhuri, A.R. RIF1 promotes replication fork protection and efficient restart to maintain genome stability. Nat. Commun. 2019, 10, 3287. [Google Scholar] [CrossRef]
- Bennett, L.G.; Wilkie, A.M.; Antonopoulou, E.; Ceppi, I.; Sanchez, A.; Vernon, E.G.; Gamble, A.; Myers, K.N.; Collis, S.J.; Cejka, P.; et al. MRNIP is a replication fork protection factor. Sci. Adv. 2020, 6, eaba5974. [Google Scholar] [CrossRef]
- Lim, K.S.; Li, H.; Roberts, E.; Gaudiano, E.F.; Clairmont, C.; Sambel, L.A.; Ponnienselvan, K.; Liu, J.C.; Yang, C.; Kozono, D.; et al. USP1 Is Required for Replication Fork Protection in BRCA1-Deficient Tumors. Mol. Cell 2018, 72, 925–941.e4. [Google Scholar] [CrossRef]
- Rickman, K.A.; Noonan, R.J.; Lach, F.; Sridhar, S.; Wang, A.; Abhyankar, A.; Huang, A.; Kelly, M.; Auerbach, A.D.; Smogorzewska, A. Distinct roles of BRCA2 in replication fork protection in response to hydroxyurea and DNA interstrand cross-links. Genes Dev. 2020, 34, 832–846. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Wu, H.; Jasin, M. A Distinct Replication Fork Protection Pathway Connects Fanconi Anemia Tumor Suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef]
- Bhat, K.; Krishnamoorthy, A.; Dungrawala, H.; Garcin, E.B.; Modesti, M.; Cortez, D. RADX Modulates RAD51 Activity to Control Replication Fork Protection. Cell Rep. 2018, 24, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Berti, M.; Teloni, F.; Mijic, S.; Ursich, S.; Fuchs, J.; Palumbieri, M.D.; Krietsch, J.; Schmid, J.; Garcin, E.B.; Gon, S.; et al. Sequential role of RAD51 paralog complexes in replication fork remodeling and restart. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tye, S.; Ronson, G.E.; Morris, J.R. A fork in the road: Where homologous recombination and stalled replication fork protection part ways. Semin. Cell Dev. Biol. 2020, 113, 14–26. [Google Scholar] [CrossRef]
- Tian, T.; Bu, M.; Chen, X.; Ding, L.; Yang, Y.; Han, J.; Feng, X.-H.; Xu, P.; Liu, T.; Ying, S.; et al. The ZATT-TOP2A-PICH Axis Drives Extensive Replication Fork Reversal to Promote Genome Stability. Mol. Cell 2020, 81, 198–211.e6. [Google Scholar] [CrossRef]
- Thangavel, S.; Berti, M.; Levikova, M.; Pinto, C.; Gomathinayagam, S.; Vujanovic, M.; Zellweger, R.; Moore, H.; Lee, E.H.; Hendrickson, E.A.; et al. DNA2 Drives Processing and Restart of Reversed Replication Forks in Human Cells. J. Cell Biol. 2015, 208, 545–562. [Google Scholar] [CrossRef]
- Lemaçon, D.; Jackson, J.; Quinet, A.; Brickner, J.R.; Li, S.; Yazinski, S.; You, Z.; Ira, G.; Zou, L.; Mosammaparast, N.; et al. MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Mijic, S.; Zellweger, R.; Chappidi, N.; Berti, M.; Jacobs, K.; Mutreja, K.; Ursich, S.; Chaudhuri, A.R.; Nussenzweig, A.; Janscak, P.; et al. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 2017, 8, 859. [Google Scholar] [CrossRef]
- Rondinelli, B.; Gogola, E.; Yücel, H.; Duarte, A.A.; Van De Ven, M.; Van Der Sluijs, R.; Konstantinopoulos, P.A.; Jonkers, J.; Ceccaldi, R.; Rottenberg, S.; et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat. Cell Biol. 2017, 19, 1371–1378. [Google Scholar] [CrossRef]
- Porebski, B.; Wild, S.; Kummer, S.; Scaglione, S.; Gaillard, P.-H.L.; Gari, K. WRNIP1 Protects Reversed DNA Replication Forks from SLX4-Dependent Nucleolytic Cleavage. iScience 2019, 21, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Quinet, A.; Tirman, S.; Cybulla, E.; Meroni, A.; Vindigni, A. To skip or not to skip: Choosing repriming to tolerate DNA damage. Mol. Cell 2021, 81, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.F.; Woodgate, R. Translesion DNA Polymerases. Cold Spring Harb. Perspect. Biol. 2013, 5, a010363. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Tang, T.; Guo, C. Regulation of translesion DNA synthesis in mammalian cells. Environ. Mol. Mutagen. 2020, 61, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Yekezare, M.; Gomez-Gonzalez, B.; Diffley, J.F. Controlling DNA Replication Origins in Response to DNA Damage—Inhibit Globally, Activate Locally. J. Cell Sci. 2013, 126, 1297–1306. [Google Scholar] [CrossRef]
- Lehmann, C.P.; Jiménez-Martín, A.; Branzei, D.; Tercero, J.A. Prevention of unwanted recombination at damaged replication forks. Curr. Genet. 2020, 66, 1045–1051. [Google Scholar] [CrossRef]
- Vaisman, A.; Woodgate, R. Translesion DNA Polymerases in Eukaryotes: What Makes Them Tick? Crit. Rev. Biochem. Mol. Biol. 2017, 52, 274–303. [Google Scholar] [CrossRef]
- Fabre, F.; Chan, A.; Heyer, W.D.; Gangloff, S. Alternate Pathways Involving Sgs1/Top3, Mus81/Mms4, and Srs2 Prevent Formation of Toxic Recombination Intermediates from Single-Stranded Gaps Created by DNA Replication. Proc. Natl. Acad. Sci. USA 2002, 99, 16887–16892. [Google Scholar] [CrossRef]
- Interthal, H.; Heyer, W.-D. MUS81 encodes a novel Helix-hairpin-Helix protein involved in the response to UV- and methylation-induced DNA damage in Saccharomyces cerevisiae. J. Cell Biol. 2000, 263, 812–827. [Google Scholar] [CrossRef]
- Boddy, M.N.; Gaillard, P.H.L.; McDonald, W.H.; Shanahan, P.; Yates, J.R., 3rd; Russell., P. Mus81-Eme1 Are Essential Components of a Holliday Junction Resolvase. Cell 2001, 107, 537–548. [Google Scholar] [CrossRef]
- Chen, X.-B.; Melchionna, R.; Aliouat-Denis, C.-M.; Gaillard, P.-H.; Blasina, A.; Van de Weyer, I.; Boddy, M.N.; Russell, P.; Vialard, J.; McGowan, C.H. Human Mus81-Associated Endonuclease Cleaves Holliday Junctions In Vitro. Mol. Cell 2001, 8, 1117–1127. [Google Scholar] [CrossRef]
- Dehé, P.M.; Coulon, S.; Scaglione, S.; Shanahan, P.; Takedachi, A.; Wohlschlegel, J.A.; Yates, J.R., 3rd; Llorente, B.; Russell, P.; Gaillard, P.H. Regulation of Mus81-Eme1 Holliday Junction Resolvase in Response to DNA Damage. Nat. Struct. Mol. Biol. 2013, 20, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Naim, V.; Wilhelm, T.; Debatisse, M.; Rosselli, F. Ercc1 and Mus81-Eme1 Promote Sister Chromatid Separation by Processing Late Replication Intermediates at Common Fragile Sites During Mitosis. Nat. Cell Biol. 2013, 15, 1008–1015. [Google Scholar] [CrossRef]
- Wyatt, H.D.; Sarbajna, S.; Matos, J.; West, S.C. Coordinated Actions of SLX1-SLX4 and MUS81-EME1 for Holliday Junction Resolution in Human Cells. Mol. Cell 2013, 52, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Sarbajna, S.; Davies, D.; West, S.C. Roles of SLX1–SLX4, MUS81–EME1, and GEN1 in avoiding genome instability and mitotic catastrophe. Genes Dev. 2014, 28, 1124–1136. [Google Scholar] [CrossRef]
- Amangyeld, T.; Shin, Y.-K.; Lee, M.; Kwon, B.; Seo, Y.-S. Human MUS81-EME2 can cleave a variety of DNA structures including intact Holliday junction and nicked duplex. Nucleic Acids Res. 2014, 42, 5846–5862. [Google Scholar] [CrossRef]
- Pepe, A.; West, S.C. MUS81-EME2 Promotes Replication Fork Restart. Cell Rep. 2014, 7, 1048–1055. [Google Scholar] [CrossRef]
- Pepe, A.; West, S.C. Substrate Specificity of the Mus81-Eme2 Structure Selective Endonuclease. Nucleic Acids Res. 2014, 42, 3833–3845. [Google Scholar] [CrossRef]
- Gao, H.; Chen, X.-B.; McGowan, C.H. Mus81 Endonuclease Localizes to Nucleoli and to Regions of DNA Damage in Human S-phase Cells. Mol. Biol. Cell 2003, 14, 4826–4834. [Google Scholar] [CrossRef]
- Kramara, J.; Osia, B.; Malkova, A. Break-Induced Replication: The Where, the Why, and the How. Trends Genet. 2018, 34, 518–531. [Google Scholar] [CrossRef]
- Pardo, B.; Moriel-Carretero, M.; Vicat, T.; Aguilera, A.; Pasero, P. Homologous recombination and Mus81 promote replication completion in response to replication fork blockage. EMBO Rep. 2020, 21, e49367. [Google Scholar] [CrossRef] [PubMed]
- Dendouga, N.; Gao, H.; Moechars, D.; Janicot, M.; Vialard, J.; McGowan, C.H. Disruption of Murine Mus81 Increases Genomic Instability and DNA Damage Sensitivity but Does Not Promote Tumorigenesis. Mol. Cell. Biol. 2005, 25, 7569–7579. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Zheng, H.; Wen, X.; Sun, J.; Wang, Y.; Gao, X.; Guo, L.; Lu, R. MUS81 is associated with cell proliferation and cisplatin sensitivity in serous ovarian cancer. Biochem. Biophys. Res. Commun. 2016, 476, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Zhong, A.; Zhang, H.; Xie, S.; Deng, M.; Zheng, H.; Wang, Y.; Chen, M.; Lu, R.; Guo, L. Inhibition of MUS81 improves the chemical sensitivity of olaparib by regulating MCM2 in epithelial ovarian cancer. Oncol. Rep. 2018, 39, 1747–1756. [Google Scholar] [CrossRef]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a Trap to Kill Cancer Cells: Parp Inhibitors and Their Mechanisms of Action. Sci. Transl. Med. 2016, 8, 362ps17. [Google Scholar] [CrossRef]
- Ho, S.S.W.; Zhang, W.Y.L.; Tan, N.Y.J.; Khatoo, M.; Suter, M.A.; Tripathi, S.; Cheung, F.S.G.; Lim, W.K.; Tan, P.H.; Ngeow, J.; et al. The DNA Structure-Specific Endonuclease MUS81 Mediates DNA Sensor STING-Dependent Host Rejection of Prostate Cancer Cells. Immunity 2016, 44, 1177–1189. [Google Scholar] [CrossRef]
- Lai, X.; Broderick, R.; Bergoglio, V.; Zimmer, J.; Badie, S.; Niedzwiedz, W.; Hoffmann, J.S.; Tarsounas., M. Mus81 Nuclease Activity Is Essential for Replication Stress Tolerance and Chromosome Segregation in Brca2-Deficient Cells. Nat. Commun. 2017, 8, 15983. [Google Scholar] [CrossRef]
- van Wietmarschen, N.; Sridharan, S.; Nathan, W.J.; Tubbs, A.; Chan, E.M.; Callen, E.; Wu, W.; Belinky, F.; Tripathi, V.; Wong, N.; et al. Repeat expansions confer WRN dependence in microsatellite-unstable cancers. Nature 2020, 586, 292–298. [Google Scholar] [CrossRef]
- Calzetta, N.L.; Besteiro, M.A.G.; Gottifredi, V. Mus81-Eme1–dependent aberrant processing of DNA replication intermediates in mitosis impairs genome integrity. Sci. Adv. 2020, 6, eabc8257. [Google Scholar] [CrossRef]
- Epum, E.A.; Haber, J.E. DNA replication: The recombination connection. Trends Cell Biol. 2021, 32, 45–57. [Google Scholar] [CrossRef]
- Özer, Ö.; Hickson, I.D. Pathways for maintenance of telomeres and common fragile sites during DNA replication stress. Open Biol. 2018, 8, 180018. [Google Scholar] [CrossRef] [PubMed]
- Brower, V. Tracking Chemotherapy’s Effects on Secondary Cancers. JNCI J. Natl. Cancer Inst. 2013, 105, 1421–1422. [Google Scholar] [CrossRef] [PubMed]
- Kamran, S.; De Gonzalez, A.B.; Ng, A.; Haas-Kogan, D.; Viswanathan, A.N. Therapeutic radiation and the potential risk of second malignancies. Cancer 2016, 122, 1809–1821. [Google Scholar] [CrossRef]
- Wu, Y.; Lee, S.H.; Williamson, E.A.; Reinert, B.L.; Cho, J.H.; Xia, F.; Jaiswal, A.S.; Srinivasan, G.; Patel, B.; Brantley, A.; et al. Eepd1 Rescues Stressed Replication Forks and Maintains Genome Stability by Promoting End Resection and Homologous Recombination Repair. PLoS Genet. 2015, 11, e1005675. [Google Scholar] [CrossRef]
- Sharma, N.; Speed, M.C.; Allen, C.P.; Maranon, D.G.; Williamson, E.; Singh, S.; Hromas, R.; Nickoloff, J.A. Distinct roles of structure-specific endonucleases EEPD1 and Metnase in replication stress responses. NAR Cancer 2020, 2, zcaa008. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D. Proteomic Analyses of the Eukaryotic Replication Machinery. Methods Enzymol. 2017, 591, 33–53. [Google Scholar] [CrossRef]
- Sirbu, B.M.; Couch, F.B.; Feigerle, J.T.; Bhaskara, S.; Hiebert, S.W.; Cortez., D. Analysis of Protein Dynamics at Active, Stalled, and Collapsed Replication Forks. Genes. Dev. 2011, 25, 1320–1327. [Google Scholar] [CrossRef]
- Kim, H.-S.; Nickoloff, J.A.; Wu, Y.; Williamson, E.A.; Sidhu, G.S.; Reinert, B.L.; Jaiswal, A.S.; Srinivasan, G.; Patel, B.; Kong, K.; et al. Endonuclease EEPD1 Is a Gatekeeper for Repair of Stressed Replication Forks. J. Biol. Chem. 2017, 292, 2795–2804. [Google Scholar] [CrossRef]
- Chun, C.; Wu, Y.; Lee, S.-H.; Williamson, E.A.; Reinert, B.L.; Jaiswal, A.S.; Nickoloff, J.A.; Hromas, R.A. The homologous recombination component EEPD1 is required for genome stability in response to developmental stress of vertebrate embryogenesis. Cell Cycle 2016, 15, 957–962. [Google Scholar] [CrossRef]
- Panopoulou, G.; Hennig, S.; Groth, D.; Krause, A.; Poustka, A.J.; Herwig, R.; Vingron, M.; Lehrach, H. New Evidence for Genome-Wide Duplications at the Origin of Vertebrates Using an Amphioxus Gene Set and Completed Animal Genomes. Genome Res. 2003, 13, 1056–1066. [Google Scholar] [CrossRef]
- Kim, H.-S.; Kim, S.-K.; Hromas, R.; Lee, S.-H. The Set Domain Is Essential for Metnase Functions in Replication Restart and the 5′ End of Ss-Overhang Cleavage. PLoS ONE 2015, 10, e0139418. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-S.; Chen, Q.; Kim, S.-K.; Nickoloff, J.A.; Hromas, R.; Georgiadis, M.M.; Lee, S.-K. The Ddn Catalytic Motif Is Required for Metnase Functions in Nhej Repair and Replication Restart. J. Biol. Chem. 2014, 289, 10930–10938. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-J.; Yoon, B.-H.; Kim, S.-Y. GENT2: An updated gene expression database for normal and tumor tissues. BMC Med Genom. 2019, 12, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Puigvert, J.C.; Sanjiv, K.; Helleday, T. Targeting DNA repair, DNA metabolism and replication stress as anti-cancer strategies. FEBS J. 2015, 283, 232–245. [Google Scholar] [CrossRef]
- Feng, Z.; Scott, S.P.; Bussen, W.; Sharma, G.G.; Guo, G.; Pandita, T.K.; Powell, S.N. Rad52 Inactivation Is Synthetically Lethal with Brca2 Deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Lok, B.; Carley, A.C.; Tchang, B.; Powell, S.N. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene 2012, 32, 3552–3558. [Google Scholar] [CrossRef] [PubMed]
- Hromas, R.; Kim, H.-S.; Sidhu, G.; Williamson, E.; Jaiswal, A.; Totterdale, T.A.; Nole, J.; Lee, S.-H.; Nickoloff, J.A.; Kong, K.Y. The endonuclease EEPD1 mediates synthetic lethality in RAD52-depleted BRCA1 mutant breast cancer cells. Breast Cancer Res. 2017, 19, 122. [Google Scholar] [CrossRef] [PubMed]
- Cordaux, R.; Udit, S.; Batzer, M.A.; Feschotte, C. Birth of a Chimeric Primate Gene by Capture of the Transposase Gene from a Mobile Element. Proc. Natl. Acad. Sci. USA 2006, 103, 8101–8106. [Google Scholar] [CrossRef]
- De Haro, L.P.; Wray, J.; Williamson, E.A.; Durant, S.T.; Corwin, L.; Gentry, A.C.; Osheroff, N.; Lee, S.-H.; Hromas, R.; Nickoloff, J.A. Metnase promotes restart and repair of stalled and collapsed replication forks. Nucleic Acids Res. 2010, 38, 5681–5691. [Google Scholar] [CrossRef]
- Lee, S.-H.; Oshige, M.; Durant, S.T.; Rasila, K.K.; Williamson, E.A.; Ramsey, H.; Kwan, L.; Nickoloff, J.A.; Hromas, R. The SET domain protein Metnase mediates foreign DNA integration and links integration to nonhomologous end-joining repair. Proc. Natl. Acad. Sci. USA 2005, 102, 18075–18080. [Google Scholar] [CrossRef] [PubMed]
- Nickoloff, J.A.; Sharma, N.; Taylor, L.; Allen, S.J.; Lee, S.-H.; Hromas, R. Metnase and EEPD1: DNA Repair Functions and Potential Targets in Cancer Therapy. Front. Oncol. 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-S.; Williamson, E.A.; Nickoloff, J.A.; Hromas, R.A.; Lee, S.-H. Metnase Mediates Loading of Exonuclease 1 onto Single Strand Overhang DNA for End Resection at Stalled Replication Forks. J. Biol. Chem. 2017, 292, 1414–1425. [Google Scholar] [CrossRef] [PubMed]
- Fnu, S.; Williamson, E.A.; De Haro, L.P.; Brenneman, M.; Wray, J.; Shaheen, M.; Radhakrishnan, K.; Lee, S.-H.; Nickoloff, J.A.; Hromas, R. Methylation of histone H3 lysine 36 enhances DNA repair by nonhomologous end-joining. Proc. Natl. Acad. Sci. USA 2010, 108, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Williamson, E.A.; Rasila, K.K.; Corwin, L.K.; Wray, J.; Beck, B.D.; Severns, V.; Mobarak, C.; Lee, S.-H.; Nickoloff, J.A.; Hromas, R. The SET and transposase domain protein Metnase enhances chromosome decatenation: Regulation by automethylation. Nucleic Acids Res. 2008, 36, 5822–5831. [Google Scholar] [CrossRef]
- Hromas, R.; Williamson, E.A.; Fnu, S.; Lee, Y.-J.; Park, S.-J.; Beck, B.D.; You, J.-S.; Laitao, A.; Nickoloff, J.A.; Lee, S.-H. Chk1 Phosphorylation of Metnase Enhances DNA Repair but Inhibits Replication Fork Restart. Oncogene 2012, 31, 4245–4254. [Google Scholar] [CrossRef]
- Williamson, E.A.; Wu, Y.; Singh, S.; Byrne, M.; Wray, J.; Lee, S.-H.; A Nickoloff, J.; Hromas, R. The DNA repair component Metnase regulates Chk1 stability. Cell Div. 2014, 9, 1. [Google Scholar] [CrossRef]
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. Blm-Dna2-Rpa-Mrn and Exo1-Blm-Rpa-Mrn Constitute Two DNA End Resection Machineries for Human DNA Break Repair. Genes Dev. 2011, 25, 350–362. [Google Scholar] [CrossRef]
- Lyu, X.; Lei, K.; Sang, P.B.; Shiva, O.; Chastain, M.; Chi, P.; Chai, W. Human CST complex protects stalled replication forks by directly blocking MRE11 degradation of nascent-strand DNA. EMBO J. 2020, 40, e103654. [Google Scholar] [CrossRef]
- Przetocka, S.; Porro, A.; Bolck, H.A.; Walker, C.; Lezaja, A.; Trenner, A.; von Aesch, C.; Himmels, S.F.; D’Andrea, A.D.; Ceccaldi, R.; et al. Ctip-Mediated Fork Protection Synergizes with Brca1 to Suppress Genomic Instability Upon DNA Replication Stress. Mol. Cell 2018, 72, 568–582.e6. [Google Scholar] [CrossRef]
- Locke, A.J.; Hossain, L.; McCrostie, G.; A Ronato, D.; Fitieh, A.; Rafique, T.A.; Mashayekhi, F.; Motamedi, M.; Masson, J.-Y.; Ismail, I.H. SUMOylation mediates CtIP’s functions in DNA end resection and replication fork protection. Nucleic Acids Res. 2021, 49, 928–953. [Google Scholar] [CrossRef] [PubMed]
- Young, S.J.; West, S.C. Coordinated roles of SLX4 and MutSβ in DNA repair and the maintenance of genome stability. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 157–177. [Google Scholar] [CrossRef] [PubMed]
- Payliss, B.J.; Patel, A.; Sheppard, A.C.; Wyatt, H.D.M. Exploring the Structures and Functions of Macromolecular SLX4-Nuclease Complexes in Genome Stability. Front. Genet. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Guervilly, J.-H.; Gaillard, P.-H.L. SLX4: Multitasking to maintain genome stability. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 475–514. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Lu, H.; Wang, Z.; Hu, Q.; Wang, H.; Xiang, R.; Chiba, T.; Wu, X. Ercc1/Xpf Is Important for Repair of DNA Double-Strand Breaks Containing Secondary Structures. iScience 2019, 16, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Bétous, R.; De Rugy, T.G.; Pelegrini, A.L.; Queille, S.; De Villartay, J.-P.; Hoffmann, J.-S. DNA replication stress triggers rapid DNA replication fork breakage by Artemis and XPF. PLoS Genet. 2018, 14, e1007541. [Google Scholar] [CrossRef]
- Trego, K.S.; Groesser, T.; Davalos, A.R.; Parplys, A.C.; Zhao, W.; Nelson, M.; Hlaing, A.; Shih, B.; Rydberg, B.; Pluth, J.M.; et al. Non-catalytic Roles for XPG with BRCA1 and BRCA2 in Homologous Recombination and Genome Stability. Mol. Cell 2016, 61, 535–546. [Google Scholar] [CrossRef]
- Zheng, L.; Zhou, M.; Chai, Q.; Parrish, J.; Xue, D.; Patrick, S.M.; Turchi, J.J.; Yannone, S.M.; Chen, D.; Shen, B. Novel function of the flap endonuclease 1 complex in processing stalled DNA replication forks. EMBO Rep. 2005, 6, 83–89. [Google Scholar] [CrossRef]
- Zheng, L.; Jia, J.; Finger, L.D.; Guo, Z.; Zer, C.; Shen, B. Functional regulation of FEN1 nuclease and its link to cancer. Nucleic Acids Res. 2010, 39, 781–794. [Google Scholar] [CrossRef]
- Nickoloff, J.A.; Taylor, L.; Sharma, N.; Kato, T.A. Exploiting DNA Repair Pathways for Tumor Sensitization, Mitigation of Resistance, and Normal Tissue Protection in Radiotherapy. Cancer Drug Resist. 2014, 4, 244–263. [Google Scholar] [CrossRef]
- Carrassa, L.; Damia, G. DNA damage response inhibitors: Mechanisms and potential applications in cancer therapy. Cancer Treat. Rev. 2017, 60, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Helleday, T. DNA REPAIR. Drugging DNA Repair. Science 2016, 352, 1178–1179. [Google Scholar] [CrossRef] [PubMed]
- Gavande, N.S.; VanderVere-Carozza, P.S.; Pawelczak, K.S.; Vernon, T.L.; Jordan, M.R.; Turchi, J.J. Structure-Guided Optimization of Replication Protein a (Rpa)-DNA Interaction Inhibitors. ACS Med. Chem. Lett. 2020, 11, 1118–1124. [Google Scholar] [CrossRef] [PubMed]
- Patrone, J.D.; Waterson, A.G.; Fesik, S.W. Recent advancements in the discovery of protein–protein interaction inhibitors of replication protein A. MedChemComm 2016, 8, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Neher, T.M.; Bodenmiller, D.; Fitch, R.; Jalal, S.I.; Turchi, J.J. Novel Irreversible Small Molecule Inhibitors of Replication Protein A Display Single-Agent Activity and Synergize with Cisplatin. Mol. Cancer Ther. 2011, 10, 1796–1806. [Google Scholar] [CrossRef]
- Dupre, A.; Boyer-Chatenet, L.; Sattler, R.M.; Modi, A.P.; Lee, J.H.; Nicolette, M.L.; Kopelovich, L.; Jasin, M.; Baer, R.; Paull, T.T.; et al. A Forward Chemical Genetic Screen Reveals an Inhibitor of the Mre11-Rad50-Nbs1 Complex. Nat. Chem. Biol. 2008, 4, 119–125. [Google Scholar] [CrossRef]
- Chow, T.Y.-K.; A Alaoui-Jamali, M.; Yeh, C.; Yuen, L.; Griller, D. The DNA Double-Stranded Break Repair Protein Endo-Exonuclease as a Therapeutic Target for Cancer. Mol. Cancer Ther. 2004, 3, 911–919. [Google Scholar]
- Wang, Y.-Y.; Hung, A.C.; Lo, S.; Hsieh, Y.-C.; Yuan, S.-S.F. MRE11 as a molecular signature and therapeutic target for cancer treatment with radiotherapy. Cancer Lett. 2021, 514, 1–11. [Google Scholar] [CrossRef]
- Lin, Z.P.; Ratner, E.S.; Whicker, M.E.; Lee, Y.; Sartorelli, A.C. Triapine Disrupts Ctip-Mediated Homologous Recombination Repair and Sensitizes Ovarian Cancer Cells to Parp and Topoisomerase Inhibitors. Mol. Cancer Res. 2014, 12, 381–393. [Google Scholar] [CrossRef]
- Kuster, A.; Mozaffari, N.L.; Wilkinson, O.J.; Wojtaszek, J.L.; Zurfluh, C.; Przetocka, S.; Zyla, D.; von Aesch, C.; Dillingham, M.S.; Williams, R.S.; et al. A stapled peptide mimetic of the CtIP tetramerization motif interferes with double-strand break repair and replication fork protection. Sci. Adv. 2021, 7, eabc6381. [Google Scholar] [CrossRef]
- Kuo, C.-H.; Leu, Y.-L.; Wang, T.-H.; Tseng, W.-C.; Feng, C.-H.; Wang, S.-H.; Chen, C.-C. A novel DNA repair inhibitor, diallyl disulfide (DADS), impairs DNA resection during DNA double-strand break repair by reducing Sae2 and Exo1 levels. DNA Repair 2019, 82, 102690. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhou, M.; Li, Z.; Li, H.; Polaczek, P.; Dai, H.; Wu, Q.; Liu, C.; Karanja, K.K.; Popuri, V.; et al. A Selective Small, Olecule Dna2 Inhibitor for Sensitization of Human Cancer Cells to Chemotherapy. EBioMedicine 2016, 6, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Peng, X.; Daley, J.; Yang, L.; Shen, J.; Nguyen, N.; Bae, G.; Niu, H.; Peng, Y.; Hsieh, H.-J.; et al. Inhibition of DNA2 nuclease as a therapeutic strategy targeting replication stress in cancer cells. Oncogenesis 2017, 6, e319. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ali, I.Y.; EL Fisher, C.; Arribas-Bosacoma, R.; Rajasekaran, M.B.; Williams, G.; Walker, S.; Booth, J.R.; Hudson, J.J.; Roe, S.M.; et al. Uncovering an allosteric mode of action for a selective inhibitor of human Bloom syndrome protein. eLife 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.H.; Dexheimer, T.S.; Rosenthal, A.S.; Chu, W.K.; Singh, D.K.; Mosedale, G.; Bachrati, C.Z.; Schultz, L.; Sakurai, M.; Savitsky, P.; et al. A Small Molecule Inhibitor of the BLM Helicase Modulates Chromosome Stability in Human Cells. Chem. Biol. 2013, 20, 55–62. [Google Scholar] [CrossRef]
- Yin, Q.-K.; Wang, C.-X.; Wang, Y.-Q.; Guo, Q.-L.; Zhang, Z.-L.; Ou, T.-M.; Huang, S.-L.; Li, D.; Wang, H.-G.; Tan, J.-H.; et al. Huang. Discovery of Isaindigotone Derivatives as Novel Bloom’s Syndrome Protein (BLM) Helicase Inhibitors That Disrupt the BLM/DNA Interactions and Regulate the Homologous Recombination Repair. J. Med. Chem. 2019, 62, 3147–3162. [Google Scholar] [CrossRef]
- Datta, A.; Brosh, R.M., Jr. New Insights Into DNA Helicases as Druggable Targets for Cancer Therapy. Front. Mol. Biosci. 2018, 5, 59. [Google Scholar] [CrossRef]
- Budke, B.; Lv, W.; Kozikowski, A.P.; Connell, P.P. Recent Developments Using Small Molecules to Target RAD51: How to Best Modulate RAD51 for Anticancer Therapy? ChemMedChem 2016, 11, 2468–2473. [Google Scholar] [CrossRef]
- King, H.O.; Brend, T.; Payne, H.L.; Wright, A.; Ward, T.; Patel, K.; Egnuni, T.; Stead, L.F.; Patel, A.; Wurdak, H.; et al. RAD51 Is a Selective DNA Repair Target to Radiosensitize Glioma Stem Cells. Stem Cell Rep. 2017, 8, 125–139. [Google Scholar] [CrossRef]
- Balbous, A.; Cortes, U.; Guilloteau, K.; Rivet, P.; Pinel, B.; Duchesne, M.; Godet, J.; Boissonnade, O.; Wager, M.; Bensadoun, R.J.; et al. A radiosensitizing effect of RAD51 inhibition in glioblastoma stem-like cells. BMC Cancer 2016, 16, 1–13. [Google Scholar] [CrossRef]
- Pastushok, L.; Fu, Y.; Lin, L.; Luo, Y.; DeCoteau, J.F.; Lee, K.; Geyer, C.R. A Novel Cell-Penetrating Antibody Fragment Inhibits the DNA Repair Protein RAD51. Sci. Rep. 2019, 9, 11227. [Google Scholar] [CrossRef] [PubMed]
- Turchick, A.; Hegan, D.C.; Jensen, R.B.; Glazer, P.M. A cell-penetrating antibody inhibits human RAD51 via direct binding. Nucleic Acids Res. 2017, 45, 11782–11799. [Google Scholar] [CrossRef] [PubMed]
- Turchick, A.; Liu, Y.; Zhao, W.; Cohen, I.; Glazer, P.M. Synthetic lethality of a cell-penetrating anti-RAD51 antibody in PTEN-deficient melanoma and glioma cells. Oncotarget 2019, 10, 1272–1283. [Google Scholar] [CrossRef] [PubMed]
- Ngo, S.T.; Van Vu, V.; Phung, H.T.T. Computational investigation of possible inhibitors of the winged-helix domain of MUS81. J. Mol. Graph. Model. 2020, 103, 107771. [Google Scholar] [CrossRef] [PubMed]
- Williamson, E.A.; Damiani, L.; Leitao, A.; Hu, C.; Hathaway, H.; Oprea, T.; Sklar, L.; Shaheen, M.; Bauman, J.; Wang, W.; et al. Targeting the Transposase Domain of the DNA Repair Component Metnase to Enhance Chemotherapy. Cancer Res. 2012, 72, 6200–6208. [Google Scholar] [CrossRef]
- Arora, S.; Heyza, J.; Zhang, H.; Kalman-Maltese, V.; Tillison, K.; Floyd, A.M.; Chalfin, E.M.; Bepler, G.; Patrick, S.M. Identification of small molecule inhibitors of ERCC1-XPF that inhibit DNA repair and potentiate cisplatin efficacy in cancer cells. Oncotarget 2016, 7, 75104–75117. [Google Scholar] [CrossRef]
- McNeil, E.M.; Astell, K.R.; Ritchie, A.-M.; Shave, S.; Houston, D.R.; Bakrania, P.; Jones, H.M.; Khurana, P.; Wallace, C.; Chapman, T.; et al. Inhibition of the ERCC1–XPF structure-specific endonuclease to overcome cancer chemoresistance. DNA Repair 2015, 31, 19–28. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Biochemical Activities | Biological Functions | Inhibitor References |
|---|---|---|---|
| RPA | Binds ssDNA, ATRIP, and itself | DNA replication and repair; activates ATR through ATRIP binding to RPA-bound ssDNA; replaced by RAD51 on ssDNA during HR | [61,62,153,154,155] |
| MRE11 | DSB end binding, 3′-5′ exonuclease, endonuclease | Early DSB sensor, ATM activation, promotes cNHEJ, initiates resection for HR | [156,157,158] |
| CtIP | Endonuclease | Promotes limited resection by MRE11 | [159,160] |
| EXO1 | 5′-3′ exonuclease | Extensive end resection | [161] * |
| DNA2 | 5′-3′ exonuclease | Extensive end resection | [162,163] |
| BLM | 3′-5′ helicase | Unwinds DNA structures during HR, promotes resection by DNA2 | [164,165,166,167] |
| RAD51 | Strand invasion (recombinase) | Binds dsDNA, ssDNA and itself, catalyzes HR | [168,169,170,171,172,173] |
| MUS81-EME2 | 3′ structure specific endonuclease | Cleaves stalled forks, promotes fork restart | [174] |
| EEPD1 | 5′ structure specific endonuclease | Cleaves stalled forks, promotes fork restart and fork resection by EXO1 | None † |
| Metnase | 5′ structure specific endonuclease, protein methylase | Cleaves stalled forks, promotes fork restart and fork resection by EXO1 | [175] |
| SLX1-SLX4 | 5′ structure specific endonuclease | Cleaves branched structures, promotes HR, crosslink repair, and telomere maintenance | None † |
| XPF-ERCC1 | 5′ structure specific endonuclease | Nucleotide excision repair, inter-strand crosslink repair, HR (replication stress?) | [176,177] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nickoloff, J.A.; Sharma, N.; Taylor, L.; Allen, S.J.; Hromas, R. Nucleases and Co-Factors in DNA Replication Stress Responses. DNA 2022, 2, 68-85. https://doi.org/10.3390/dna2010006
Nickoloff JA, Sharma N, Taylor L, Allen SJ, Hromas R. Nucleases and Co-Factors in DNA Replication Stress Responses. DNA. 2022; 2(1):68-85. https://doi.org/10.3390/dna2010006
Chicago/Turabian StyleNickoloff, Jac A., Neelam Sharma, Lynn Taylor, Sage J. Allen, and Robert Hromas. 2022. "Nucleases and Co-Factors in DNA Replication Stress Responses" DNA 2, no. 1: 68-85. https://doi.org/10.3390/dna2010006
APA StyleNickoloff, J. A., Sharma, N., Taylor, L., Allen, S. J., & Hromas, R. (2022). Nucleases and Co-Factors in DNA Replication Stress Responses. DNA, 2(1), 68-85. https://doi.org/10.3390/dna2010006