

In Silico Evaluation of Different Flavonoids from Medicinal Plants for Their Potency against SARS-CoV-2

 ,
,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
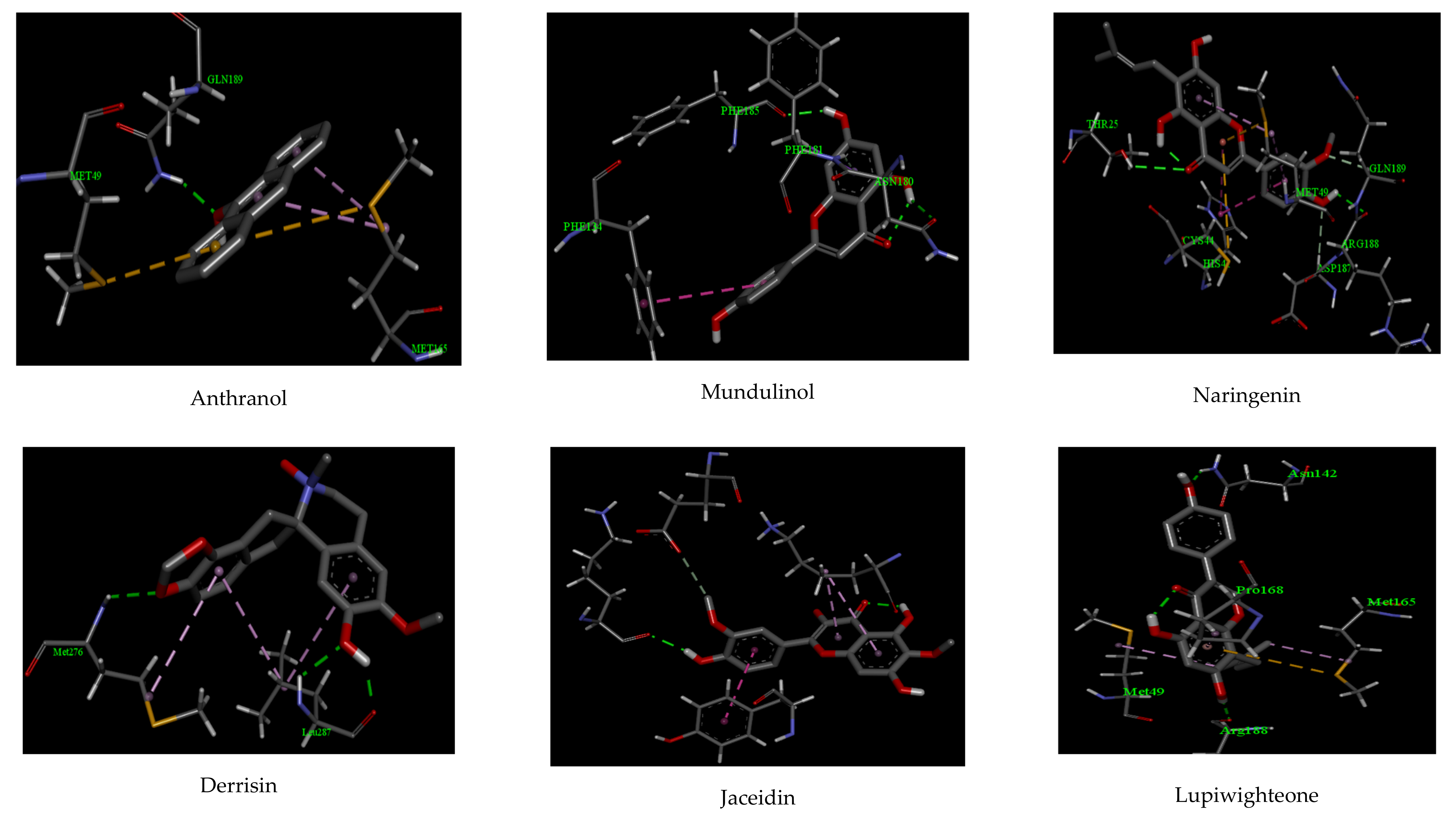
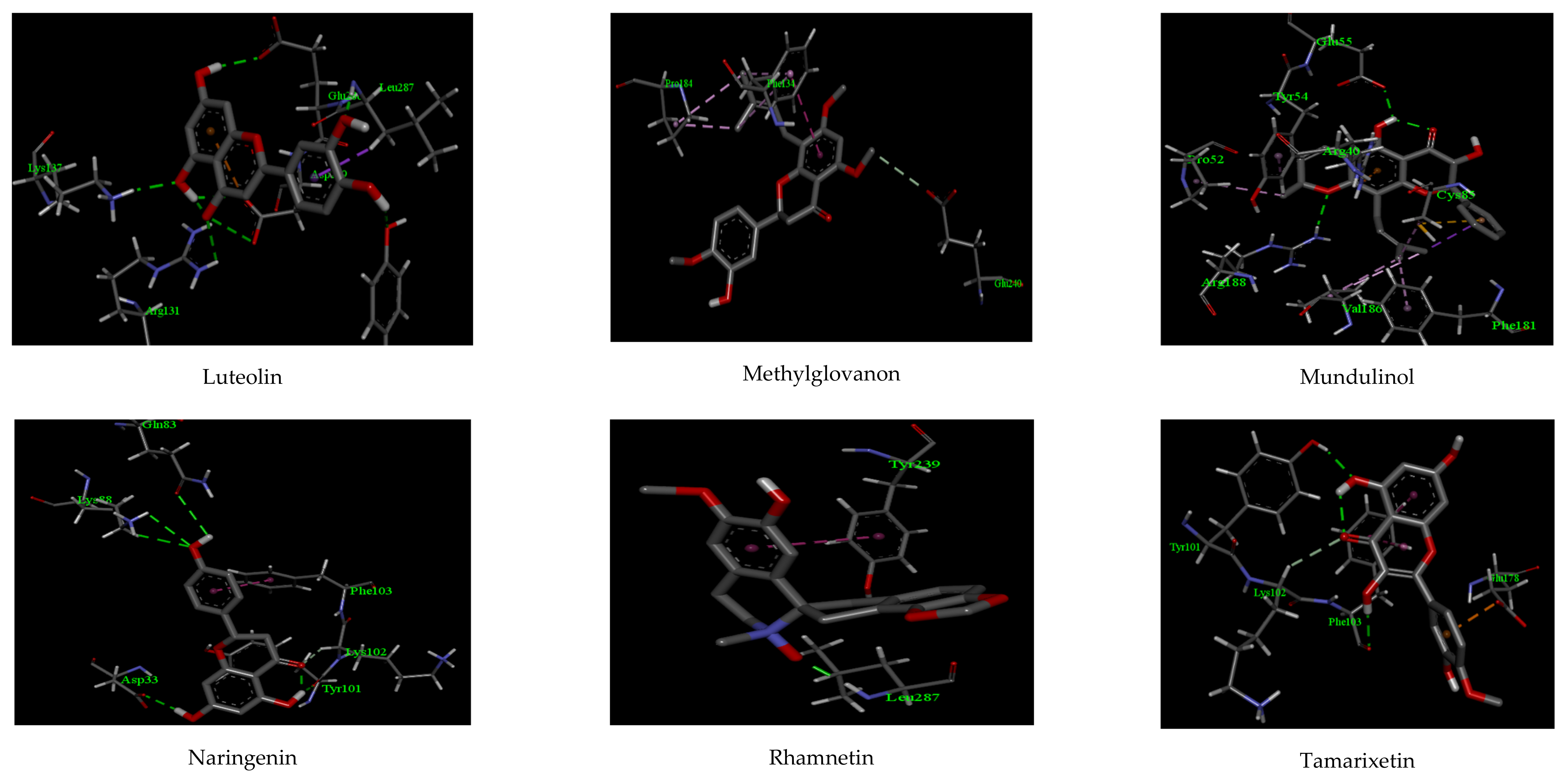
2.1. Molecular Docking
2.2. ADMET Properties and Drug-Likeness Model Score
2.3. Molecular Dynamic Simulation (MD)
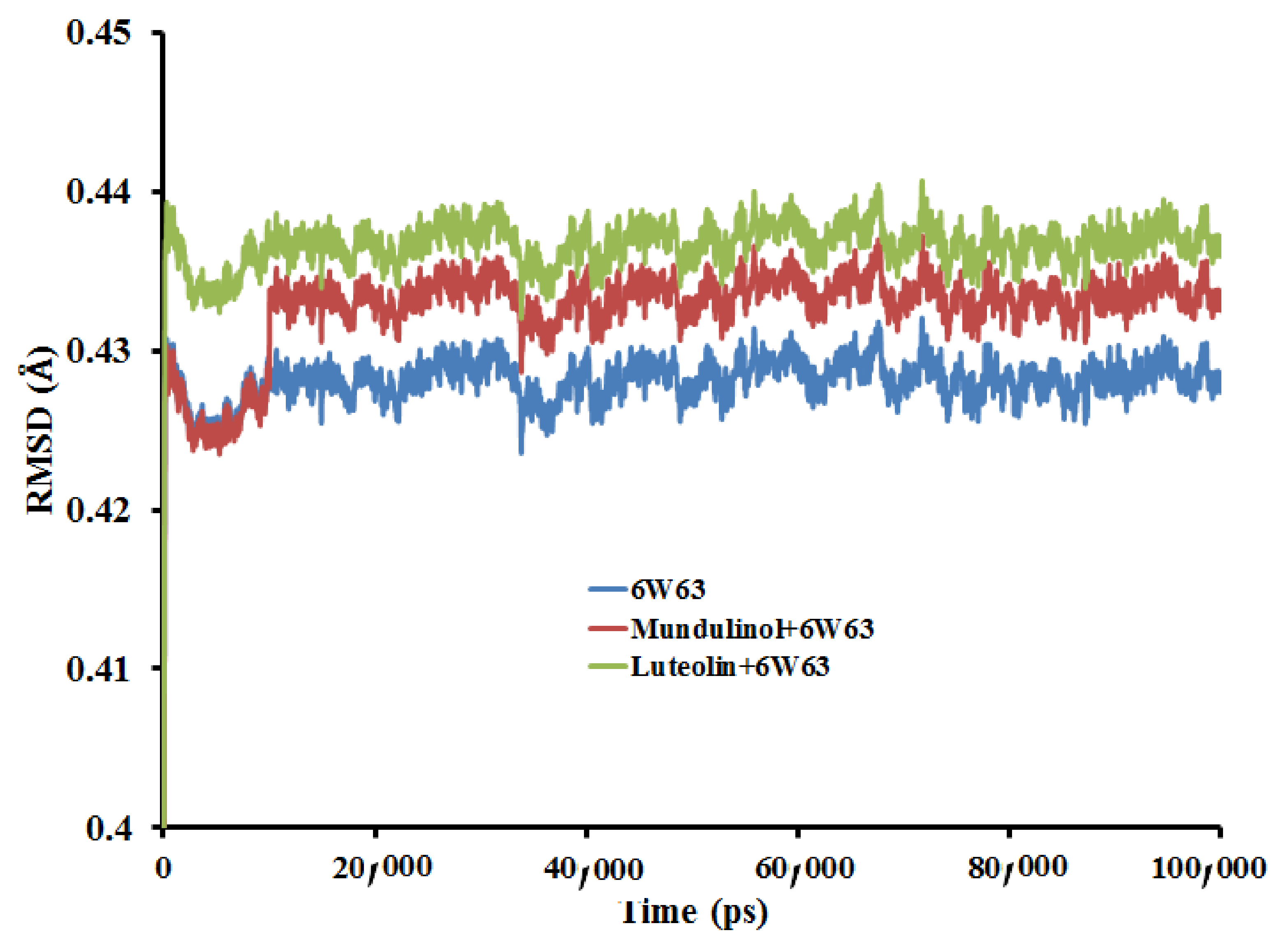
2.3.1. Root Mean Square Deviation (RMSD)
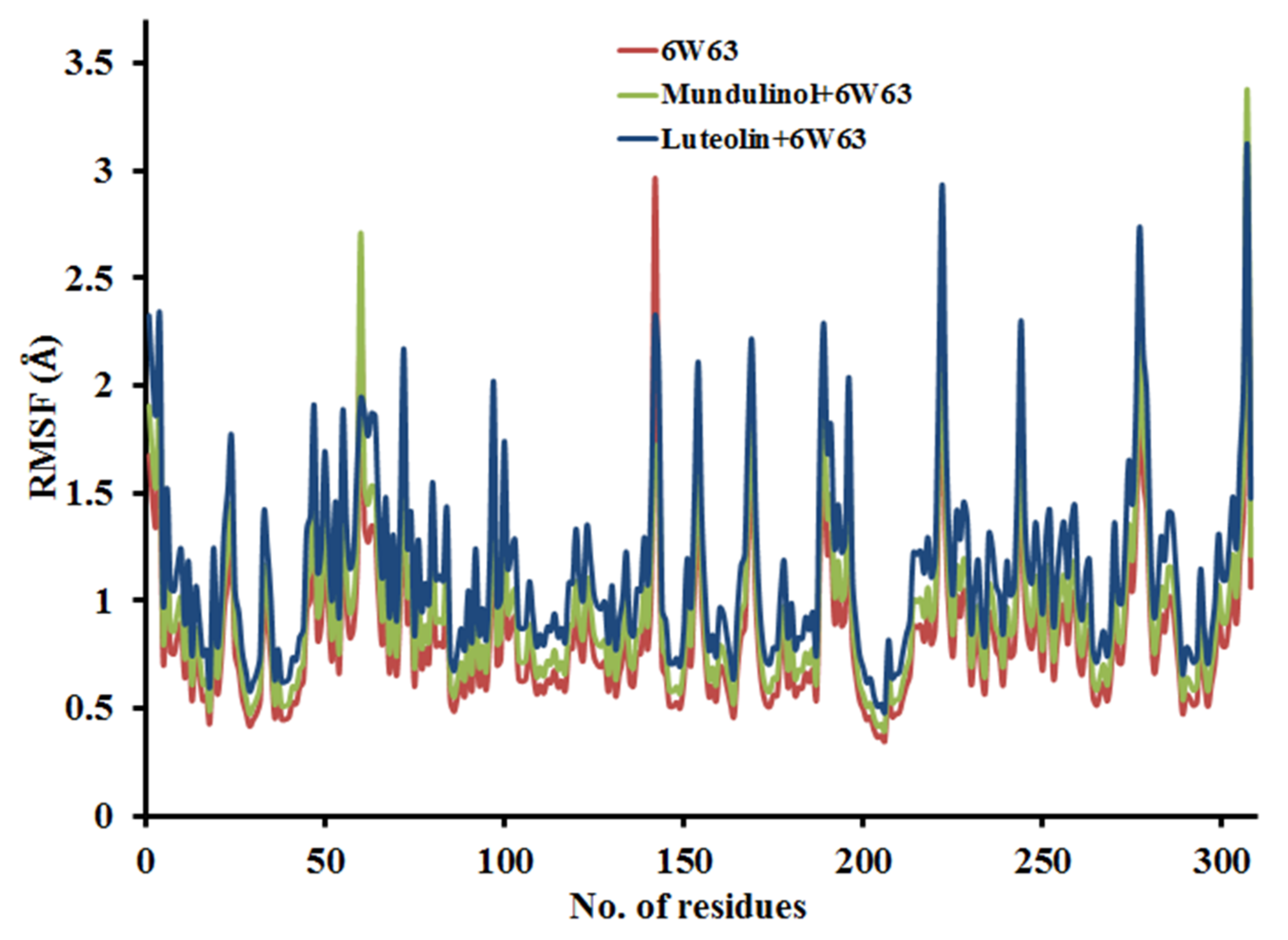
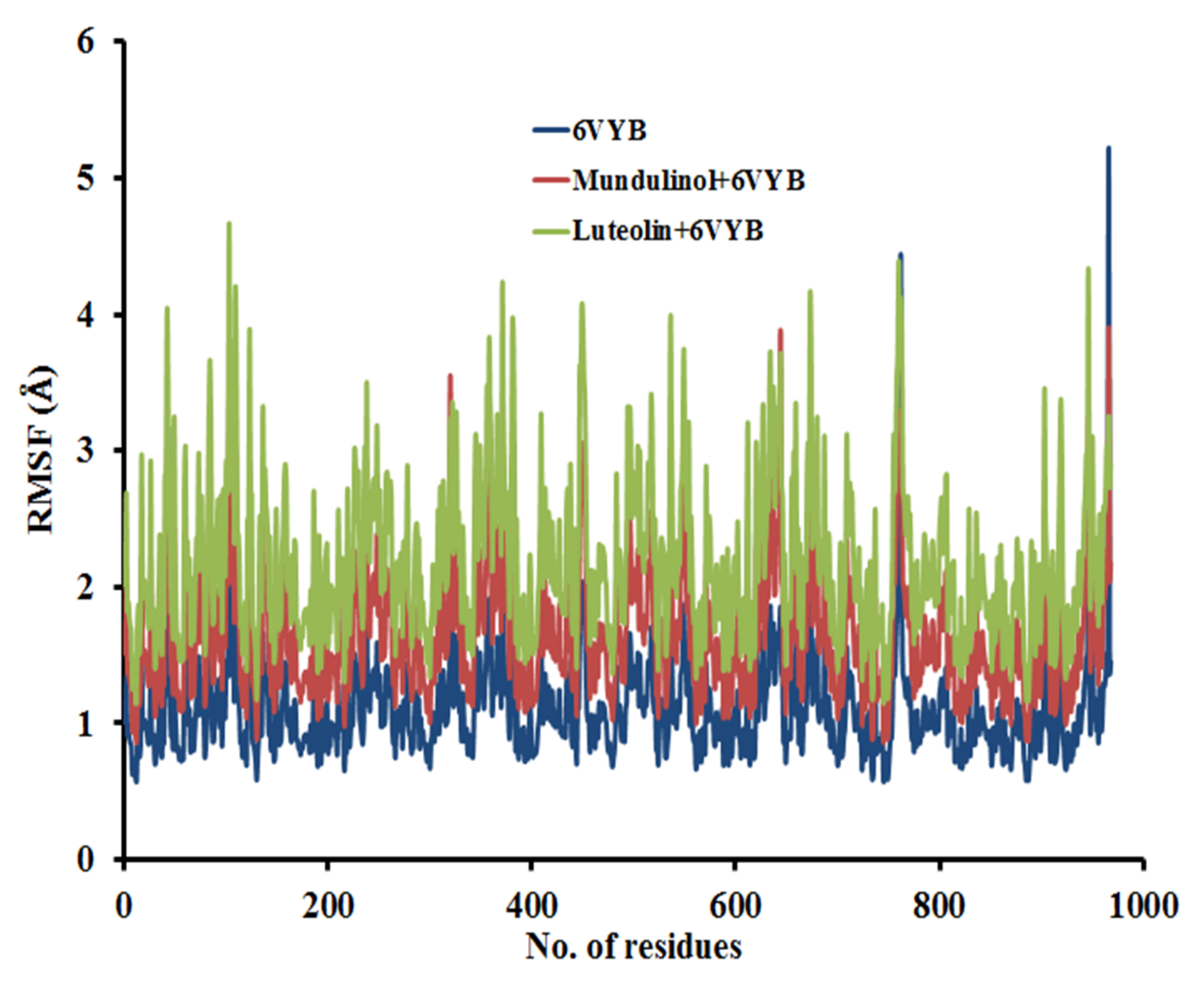
2.3.2. Root Mean Square Fluctuation (RMSF)
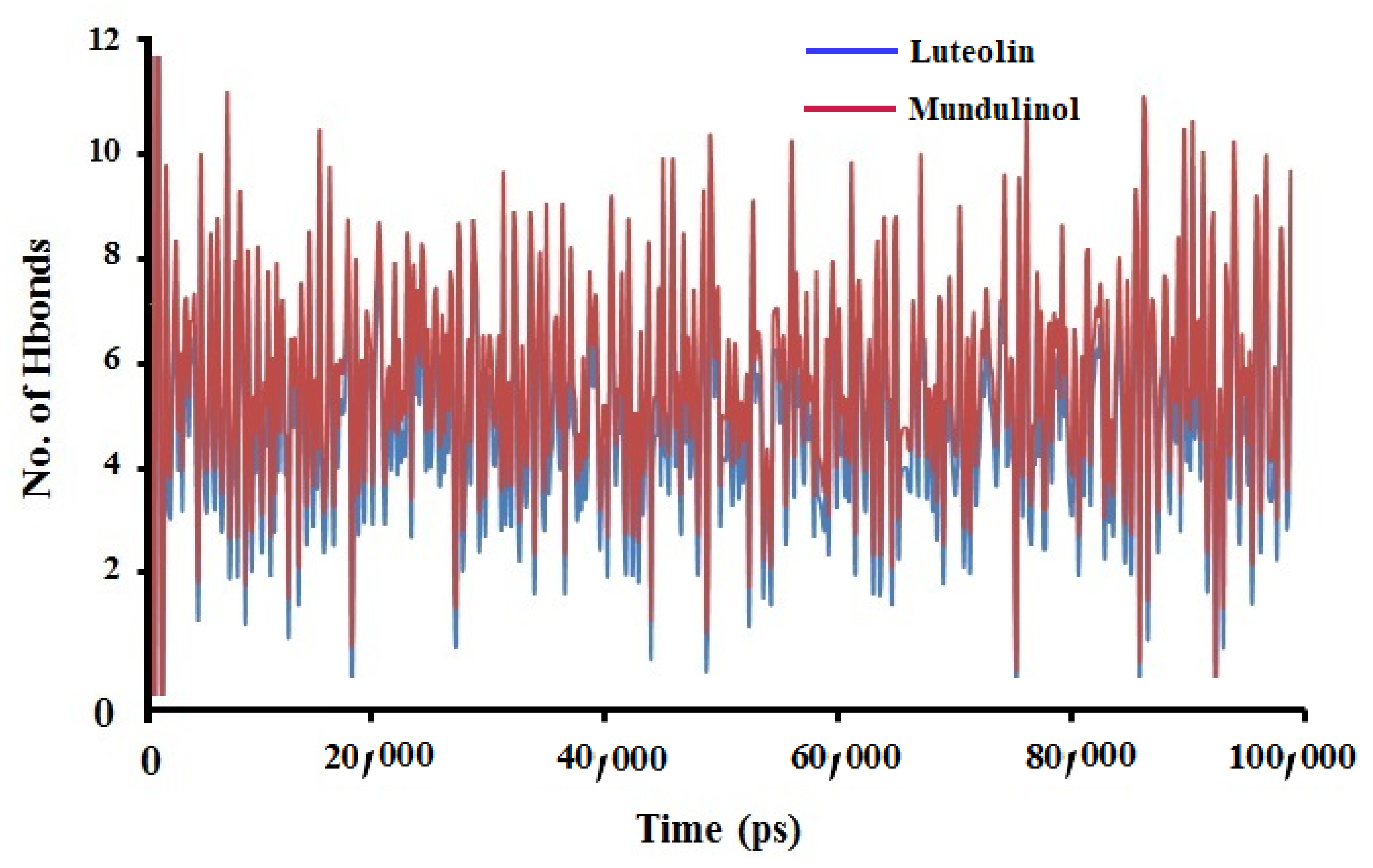
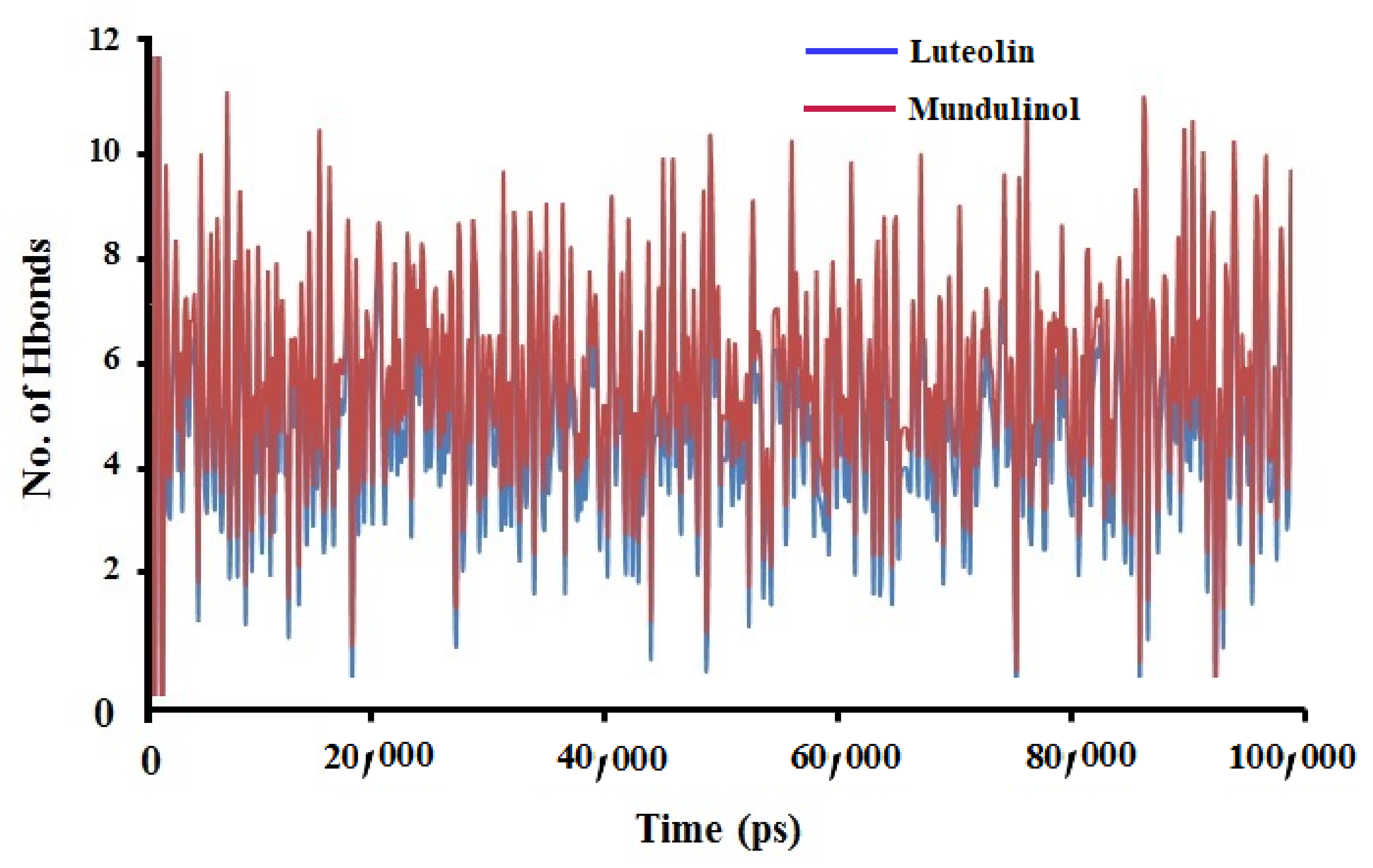
2.3.3. Hydrogen Bonds Analysis
2.4. MM-PBSA Binding Free Energy
3. Materials and Methods
3.1. Ligand and Protein Preparation
3.2. Assessment of Pharmacokinetics and Pharmacological Properties
3.3. Molecular Docking Analysis and Binding Energy Estimation
3.4. Molecular Dynamic (MD) Simulation
3.5. MM-PBSA Calculation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peeri, N.C.; Shrestha, N.; Rahman, M.S.; Zaki, R.; Tan, Z.; Bibi, S.; Baghbanzadeh, M.; Aghamohammadi, N.; Zhang, W.; Haque, U. The SARS, MERS and novel coronavirus (COVID-19) epidemics, the newest and biggest global health threats: What lessons have we learned? Int. J. Epidemiol. 2020, 49, 717–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Wu, T.; Liu, Q.; Yang, Z. The SARS-CoV-2 outbreak: What we know. Int. J. Infect. Dis. IJID Off. Publ. Int. Soc. Infect. Dis. 2020, 94, 44–48. [Google Scholar] [CrossRef]
- Zheng, J. SARS-CoV-2: An Emerging Coronavirus that Causes a Global Threat. Int. J. Biol. Sci. 2020, 16, 1678–1685. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassert, M.; Geerling, E.; Stone, E.T.; Steffen, T.L.; Feldman, M.S.; Dickson, A.L.; Class, J.; Richner, J.M.; Brien, J.D.; Pinto, A.K. mRNA induced expression of human angiotensin-converting enzyme 2 in mice for the study of the adaptive immune response to severe acute respiratory syndrome coronavirus 2. PLoS Pathog. 2020, 16, e1009163. [Google Scholar] [CrossRef]
- Ma, C.; Sacco, M.D.; Hurst, B.; Townsend, J.A.; Hu, Y.; Szeto, T.; Zhang, X.; Tarbet, B.; Marty, M.T.; Chen, Y.; et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692. [Google Scholar] [CrossRef]
- Rut, W.; Lv, Z.; Zmudzinski, M.; Patchett, S.; Nayak, D.; Snipas, S.J.; El Oualid, F.; Huang, T.T.; Bekes, M.; Drag, M.; et al. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: A framework for anti-COVID-19 drug design. Sci. Adv. 2020, 6, eabd4596. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.U.; Froeyen, M. Structural elucidation of SARS-CoV-2 vital proteins: Computational methods reveal potential drug candidates against main protease, Nsp12 polymerase and Nsp13 helicase. J. Pharm. Anal. 2020, 10, 320–328. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Cortegiani, A.; Ingoglia, G.; Ippolito, M.; Giarratano, A.; Einav, S. A systematic review on the efficacy and safety of chloroquine for the treatment of COVID-19. J. Crit. Care 2020, 57, 279–283. [Google Scholar] [CrossRef]
- Hossain, M.J.; Rahman, S.M.A. Repurposing therapeutic agents against SARS-CoV-2 infection: Most promising and neoteric progress. Expert Rev. Anti Infect. Ther. 2021, 19, 1009–1027. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.J.; Jannat, T.; Brishty, S.R.; Roy, U.; Mitra, S.; Rafi, M.O.; Islam, M.R.; Nesa, M.L.; Islam, M.A.; Emran, T. Bin Clinical Efficacy and Safety of Antiviral Drugs in the Extended Use against COVID-19: What We Know So Far. Biologics 2021, 1, 252–284. [Google Scholar] [CrossRef]
- Kumar, V.; Tan, K.-P.; Wang, Y.-M.; Lin, S.-W.; Liang, P.-H. Identification, synthesis and evaluation of SARS-CoV and MERS-CoV 3C-like protease inhibitors. Bioorg. Med. Chem. 2016, 24, 3035–3042. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe Covid-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Chong, Y.P.; Song, J.Y.; Seo, Y.B.; Choi, J.-P.; Shin, H.-S. Antiviral Treatment Guidelines for Middle East Respiratory Syndrome. Infect. Chemother. 2015, 47, 212–222. [Google Scholar] [CrossRef]
- Needle, D.; Lountos, G.T.; Waugh, D.S. Structures of the Middle East respiratory syndrome coronavirus 3C-like protease reveal insights into substrate specificity. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 1102–1111. [Google Scholar] [CrossRef] [Green Version]
- Mirzaie, A.; Halaji, M.; Dehkordi, F.S.; Ranjbar, R.; Noorbazargan, H. A narrative literature review on traditional medicine options for treatment of corona virus disease 2019 (COVID-19). Complement. Ther. Clin. Pract. 2020, 40, 101214. [Google Scholar] [CrossRef]
- Wang, D.; Huang, J.; Yeung, A.W.K.; Tzvetkov, N.T.; Horbańczuk, J.O.; Willschke, H.; Gai, Z.; Atanasov, A.G. The significance of natural product derivatives and traditional medicine for COVID-19. Processes 2020, 8, 937. [Google Scholar] [CrossRef]
- Ren, J.; Zhang, A.-H.; Wang, X.-J. Traditional Chinese medicine for COVID-19 treatment. Pharmacol. Res. 2020, 155, 104743. [Google Scholar] [CrossRef]
- Ipona, E.N.; Inkoto, C.L.; Bongo, G.N.; Mulenga, C.M.; Ilinga, B.L.; Shetonde, O.S.; Mbala, B.M.; Tshilanda, D.D.; Mvingu, B.K.; Kayembe, J.S.; et al. Ethnobotanical survey and ecological study of medicinal plants traditionally used against erectile dysfunction in Democratic Republic of the Congo. Biosci. Bioeng. 2019, 4, 85–91. [Google Scholar]
- Tshilanda, D.D.; Onyamboko, D.N.; Babady-Bila, P.; Ngbolua, K.-T.-N.; Tshibangu, D.S.; Dia Fita Dibwe, E.; Mpiana, P.T. Anti-sickling Activity of Ursolic Acid Isolated from the Leaves of Ocimum gratissimum L. (Lamiaceae). Nat. Prod. Bioprospect. 2015, 5, 215–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orhan, D.D.; Ozçelik, B.; Ozgen, S.; Ergun, F. Antibacterial, antifungal, and antiviral activities of some flavonoids. Microbiol. Res. 2010, 165, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Rathee, P.; Chaudhary, H.; Rathee, S.; Rathee, D.; Kumar, V.; Kohli, K. Mechanism of action of flavonoids as anti-inflammatory agents: A review. Inflamm. Allergy Drug Targets 2009, 8, 229–235. [Google Scholar] [CrossRef]
- Elhady, S.S.; Eltamany, E.E.; Shaaban, A.E.; Bagalagel, A.A.; Muhammad, Y.A.; El-Sayed, N.M.; Ayyad, S.N.; Ahmed, A.A.M.; Elgawish, M.S.; Ahmed, S.A. Jaceidin Flavonoid Isolated from Chiliadenus montanus Attenuates Tumor Progression in Mice via VEGF Inhibition: In Vivo and In Silico Studies. Plants 2020, 9, 1031. [Google Scholar] [CrossRef]
- Mpiana, P.T.; Mudogo, V.; Tshibangu, D.S.T.; Kitwa, E.K.; Kanangila, A.B.; Lumbu, J.B.S.; Ngbolua, K.N.; Atibu, E.K.; Kakule, M.K. Antisickling activity of anthocyanins from Bombax pentadrum, Ficus capensis and Ziziphus mucronata: Photodegradation effect. J. Ethnopharmacol. 2008, 120, 413–418. [Google Scholar] [CrossRef]
- Ji, H.-F.; Li, X.-J.; Zhang, H.-Y. Natural products and drug discovery. Can thousands of years of ancient medical knowledge lead us to new and powerful drug combinations in the fight against cancer and dementia? EMBO Rep. 2009, 10, 194–200. [Google Scholar] [CrossRef] [Green Version]
- Mishra, K.P.; Sharma, N.; Diwaker, D.; Ganju, L.; Singh, S.B. Plant derived antivirals: A potential source of drug development. J. Virol. Antivir. Res. 2013, 2, 2. [Google Scholar]
- Delang, L.; Li, C.; Tas, A.; Quérat, G.; Albulescu, I.C.; De Burghgraeve, T.; Guerrero, N.A.S.; Gigante, A.; Piorkowski, G.; Decroly, E.; et al. The viral capping enzyme nsP1: A novel target for the inhibition of chikungunya virus infection. Sci. Rep. 2016, 6, 31819. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, C.; Di Ferdinando, M.; Fini, A.; Pollastri, S.; Tattini, M. Flavonoids as antioxidants and developmental regulators: Relative significance in plants and humans. Int. J. Mol. Sci. 2013, 14, 3540–3555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendra, R.; Ahmad, S.; Sukari, A.; Shukor, M.Y.; Oskoueian, E. Flavonoid analyses and antimicrobial activity of various parts of Phaleria macrocarpa (Scheff.) Boerl fruit. Int. J. Mol. Sci. 2011, 12, 3422–3431. [Google Scholar] [CrossRef] [Green Version]
- Kaul, T.N.; Middleton, E., Jr.; Ogra, P.L. Antiviral effect of flavonoids on human viruses. J. Med. Virol. 1985, 15, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Evers, D.L.; Chao, C.-F.; Wang, X.; Zhang, Z.; Huong, S.-M.; Huang, E.-S. Human cytomegalovirus-inhibitory flavonoids: Studies on antiviral activity and mechanism of action. Antivir. Res. 2005, 68, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Cotin, S.; Calliste, C.-A.; Mazeron, M.-C.; Hantz, S.; Duroux, J.-L.; Rawlinson, W.D.; Ploy, M.-C.; Alain, S. Eight flavonoids and their potential as inhibitors of human cytomegalovirus replication. Antivir. Res. 2012, 96, 181–186. [Google Scholar] [CrossRef]
- Nukui, M.; O’Connor, C.M.; Murphy, E.A. The natural flavonoid compound deguelin inhibits HCMV lytic replication within fibroblasts. Viruses 2018, 10, 614. [Google Scholar] [CrossRef] [Green Version]
- Lyu, S.-Y.; Rhim, J.-Y.; Park, W.-B. Antiherpetic activities of flavonoids against herpes simplex virus type 1 (HSV-1) and type 2 (HSV-2) in vitro. Arch. Pharm. Res. 2005, 28, 1293–1301. [Google Scholar] [CrossRef] [PubMed]
- Keivan, Z.; Teoh, B.-T.; Sam, S.-S.; Wong, P.-F.; Mustafa, M.R.; AbuBakar, S. In vitro antiviral activity of fisetin, rutin and naringenin against dengue virus type-2. J. Med. Plants Res. 2011, 5, 5534–5539. [Google Scholar]
- Lani, R.; Hassandarvish, P.; Shu, M.-H.; Phoon, W.H.; Chu, J.J.H.; Higgs, S.; Vanlandingham, D.; Bakar, S.A.; Zandi, K. Antiviral activity of selected flavonoids against Chikungunya virus. Antivir. Res. 2016, 133, 50–61. [Google Scholar] [CrossRef]
- Khandelwal, N.; Chander, Y.; Kumar, R.; Riyesh, T.; Dedar, R.K.; Kumar, M.; Gulati, B.R.; Sharma, S.; Tripathi, B.N.; Barua, S.; et al. Antiviral activity of Apigenin against buffalopox: Novel mechanistic insights and drug-resistance considerations. Antivir. Res. 2020, 181, 104870. [Google Scholar] [CrossRef]
- Fan, W.; Qian, S.; Qian, P.; Li, X. Antiviral activity of luteolin against Japanese encephalitis virus. Virus Res. 2016, 220, 112–116. [Google Scholar] [CrossRef]
- Den Hartogh, D.J.; Tsiani, E. Antidiabetic Properties of Naringenin: A Citrus Fruit Polyphenol. Biomolecules 2019, 9, 99. [Google Scholar] [CrossRef] [Green Version]
- Qian, S.; Fan, W.; Qian, P.; Zhang, D.; Wei, Y.; Chen, H.; Li, X. Apigenin restricts FMDV infection and inhibits viral IRES driven translational activity. Viruses 2015, 7, 1613–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, M.A.; Kim, S.; Kim, K.H.; Lee, S.-J.; Lee, H. Flavonoid compounds are enriched in lemon balm (Melissa officinalis) leaves by a high level of sucrose and confer increased antioxidant activity. Hortscience 2009, 44, 1907–1913. [Google Scholar] [CrossRef] [Green Version]
- Hameed, I.H.; Cotos, M.R.C.; Hadi, M.Y. A review: Solanum nigrum L. antimicrobial, antioxidant properties, hepatoprotective effects and analysis of bioactive natural compounds. Res. J. Pharm. Technol. 2017, 10, 4063–4068. [Google Scholar] [CrossRef]
- Sala, F.; Thabet, A.M.; Capitani, P.; Bove, F.; Abdelgawad, A.A.; Lovisetti, G. Open Supracondylar-Intercondylar Fractures of the Femur Treatment with Taylor Spatial Frame. J. Orthop. Trauma 2017, 31, 546–553. [Google Scholar] [CrossRef]
- Koteswara Rao, Y.; Vimalamma, G.; Rao, C.V.; Tzeng, Y.-M. Flavonoids and andrographolides from Andrographis paniculata. Phytochemistry 2004, 65, 2317–2321. [Google Scholar] [CrossRef]
- Lin, M.; Zhang, J.; Chen, X. Bioactive flavonoids in Moringa oleifera and their health-promoting properties. J. Funct. Foods 2018, 47, 469–479. [Google Scholar] [CrossRef]
- Cottiglia, F.; Casu, L.; Bonsignore, L.; Casu, M.; Floris, C.; Sosa, S.; Altinier, G.; Della Loggia, R. Topical anti-inflammatory activity of flavonoids and a new xanthone from Santolina insularis. Z. Nat. C 2005, 60, 63–66. [Google Scholar] [CrossRef]
- Rafi, M.O.; Al-Khafaji, K.; Tok, T.T.; Rahman, M.S. Computer-based identification of potential compounds from Salviae miltiorrhizae against Neirisaral adhesion A regulatory protein. J. Biomol. Struct. Dyn. 2020, 2020, 1–13. [Google Scholar] [CrossRef]
- Vijayakumar, B.G.; Ramesh, D.; Joji, A.; Kannan, T. In silico pharmacokinetic and molecular docking studies of natural flavonoids and synthetic indole chalcones against essential proteins of SARS-CoV-2. Eur. J. Pharmacol. 2020, 886, 173448. [Google Scholar] [CrossRef]
- Ahmed, S.A.; Abdelrheem, D.A.; El-Mageed, H.R.A. Destabilizing the structural integrity of COVID-19 by caulerpin and its derivatives along with some antiviral drugs: An in silico approaches for a combination therapy. Struct. Chem. 2020, 31, 2391–2412. [Google Scholar] [CrossRef]
- Abdelrheem, D.A.; Ahmed, S.A.; El-Mageed, H.R.A.; Mohamed, H.S.; Rahman, A.A.; Elsayed, K.N.M.; Ahmed, S.A. The inhibitory effect of some natural bioactive compounds against SARS-CoV-2 main protease: Insights from molecular docking analysis and molecular dynamic simulation. J. Environ. Sci. Health Part A 2020, 55, 1373–1386. [Google Scholar] [CrossRef]
- Hamilton, W. Protein Data Bank; Brookhaven National Laboratory: Upton, NY, USA, 1971. [Google Scholar]
- Rasool, N.; Akhtar, A.; Hussain, W. Insights into the inhibitory potential of selective phytochemicals against Mpro of 2019-nCoV: A computer-aided study. Struct. Chem. 2020, 31, 1777–1783. [Google Scholar] [CrossRef] [PubMed]
- Biovia, D.S. Discovery Studio Visualizer; Discover: San Diego, CA, USA, 2017; p. 936. Available online: https://discover.3ds.com/discovery-studio-visualizer-download (accessed on 10 June 2021).
- Bikadi, Z.; Hazai, E. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J. Cheminform. 2009, 1, 15. [Google Scholar] [CrossRef] [Green Version]
- Aldahham, B.J.M.; Al-Khafaji, K.; Saleh, M.Y.; Abdelhakem, A.M.; Alanazi, A.M.; Islam, M.A. Identification of naphthyridine and quinoline derivatives as potential Nsp16-Nsp10 inhibitors: A pharmacoinformatics study. J. Biomol. Struct. Dyn. 2020, 2020, 1–8. [Google Scholar] [CrossRef]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef]
- Qaddir, I.; Rasool, N.; Hussain, W.; Mahmood, S. Computer-aided analysis of phytochemicals as potential dengue virus inhibitors based on molecular docking, ADMET and DFT studies. J. Vector Borne Dis. 2017, 54, 255–262. [Google Scholar] [CrossRef]
- Hussain, W.; Amir, A.; Rasool, N. Computer-aided study of selective flavonoids against chikungunya virus replication using molecular docking and DFT-based approach. Struct. Chem. 2020, 31, 1363–1374. [Google Scholar] [CrossRef]
- Briskin, D.P. Medicinal plants and phytomedicines. Linking plant biochemistry and physiology to human health. Plant Physiol. 2000, 124, 507–514. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Khafaji, K.; Taskin Tok, T. Understanding the mechanism of amygdalin’s multifunctional anti-cancer action using computational approach. J. Biomol. Struct. Dyn. 2021, 39, 1600–1610. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, S.; Rafi, M.O.; Paul, G.K.; Promi, M.M.; Shimu, M.S.S.; Biswas, S.; Emran, T.B.; Dhama, K.; Alyami, S.A.; Moni, M.A.; et al. Designing a multi-epitope vaccine candidate to combat MERS-CoV by employing an immunoinformatics approach. Sci. Rep. 2021, 11, 15431. [Google Scholar] [CrossRef] [PubMed]
- Rafi, M.O.; Bhattacharje, G.; Al-Khafaji, K.; Taskin-Tok, T.; Alfasane, M.A.; Das, A.K.; Parvez, M.A.K.; Rahman, M.S. Combination of QSAR, molecular docking, molecular dynamic simulation and MM-PBSA: Analogues of lopinavir and favipiravir as potential drug candidates against COVID-19. J. Biomol. Struct. Dyn. 2020, 2020, 1–20. [Google Scholar] [CrossRef]
- Kalé, L.; Skeel, R.; Bhandarkar, M.; Brunner, R.; Gursoy, A.; Krawetz, N.; Phillips, J.; Shinozaki, A.; Varadarajan, K.; Schulten, K. NAMD2: Greater scalability for parallel molecular dynamics. J. Comput. Phys. 1999, 151, 283–312. [Google Scholar] [CrossRef]
- Al-Anazi, M.; Al-Najjar, B.O.; Khairuddean, M. Structure-Based Drug Design Studies toward the Discovery of Novel Chalcone Derivatives as Potential Epidermal Growth Factor Receptor (EGFR) Inhibitors. Molecules 2018, 23, 3203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Refaei, M.A.; Makki, R.M.; Ali, H.M. Structure prediction of transferrin receptor protein 1 (TfR1) by homology modelling, docking, and molecular dynamics simulation studies. Heliyon 2020, 6, e03221. [Google Scholar] [CrossRef]
- Balaji, B.; Ramanathan, M. Prediction of estrogen receptor β ligands potency and selectivity by docking and MM-GBSA scoring methods using three different scaffolds. J. Enzym. Inhib. Med. Chem. 2012, 27, 832–844. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
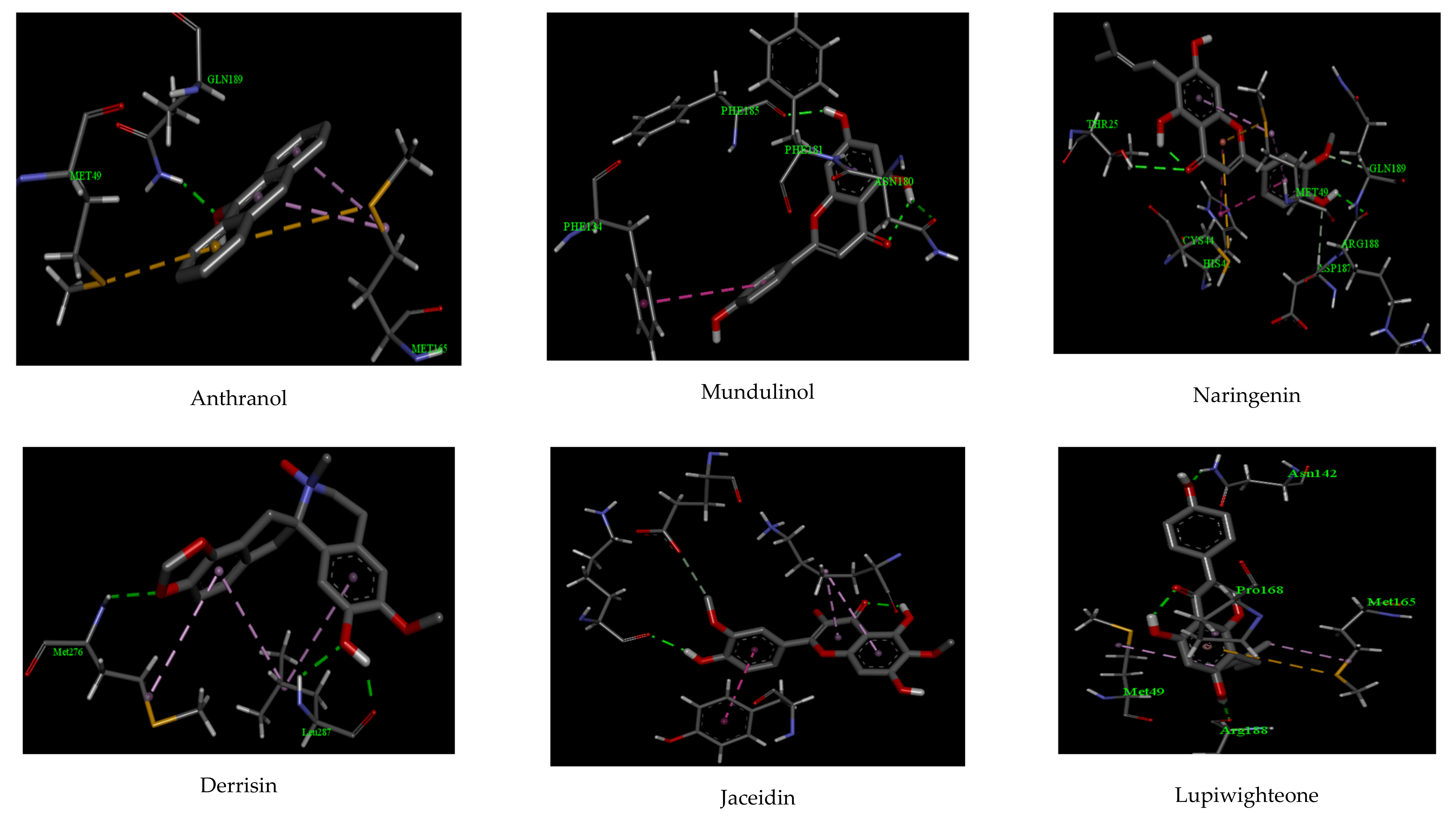
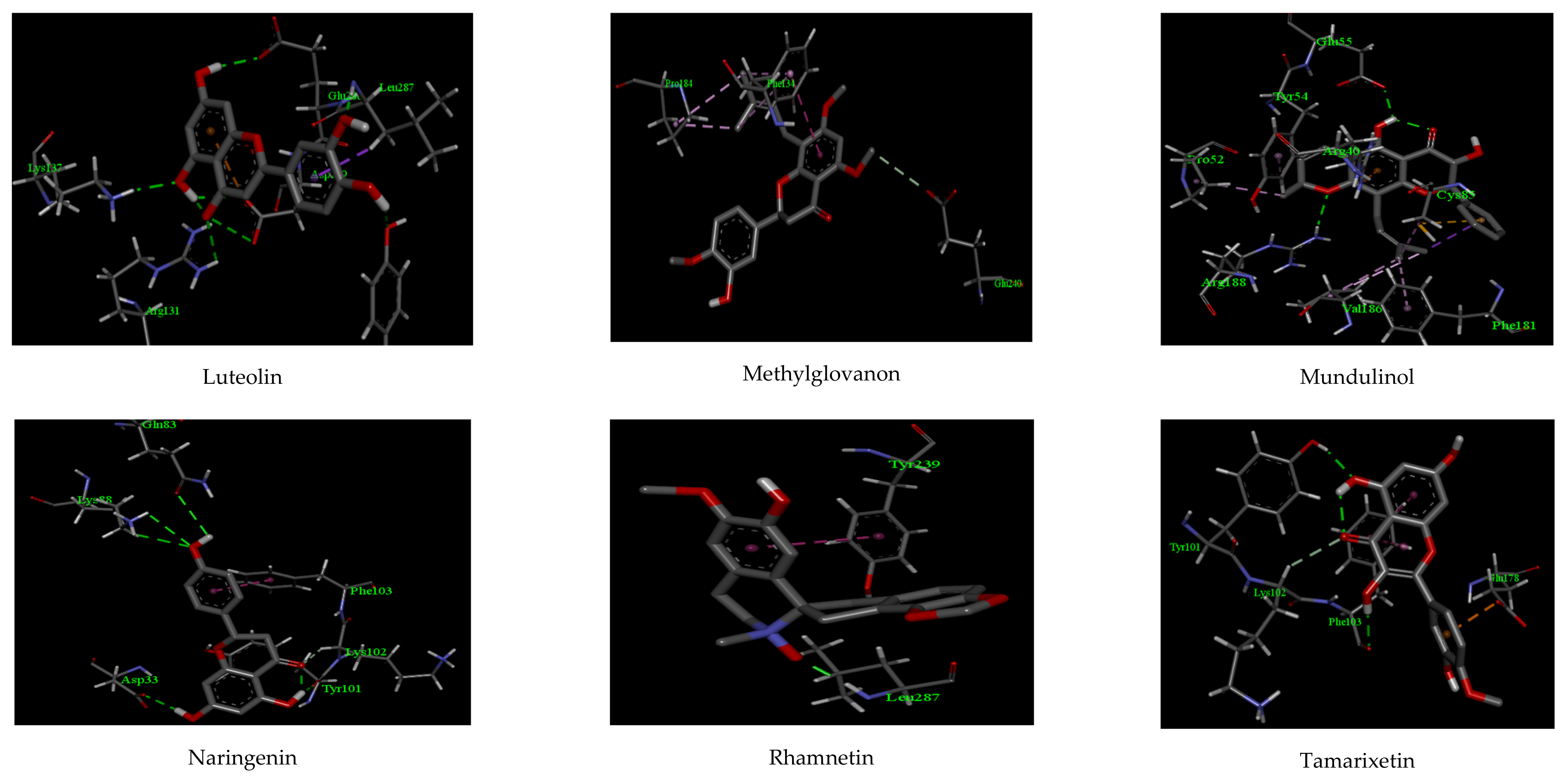
| Compounds | Interactions | |||||
|---|---|---|---|---|---|---|
| H-Bonds | Hydrophobic | |||||
| Binding Energy | Number | Residue | Distance (Å) | Number | Residue | |
| Anthranol | −10.22 | 1 | GLU 189 | 2.11 | 2 | MET 165, MET 49 |
| Apigenin | −12.32 | 2 | PHE 185, ASN 180 | 2.07, 2.22 | 2 | PHE 181, PHE 134 |
| Cannflavin | −10.88 | 2 | ARG 188, THR 25 | 2.27, 2.58 | 2 | CYS 44, MET 49 |
| Derrisin | −11.44 | 2 | ARG 188, GLU 166 | 2.44, 2.55 | 3 | MET 165, HIS 41, CYS 145 |
| Jaceidin | −9.66 | 1 | LYS 137 | 2.06 | 2 | TYR 126, GLU 290 |
| Lupiwighteone | −11.02 | 2 | ARG 188, ASN 142 | 2.07, 2.18 | 2 | PRO 168, MET 49 |
| Luteolin | −13.97 | 6 | TYR 239, LEU 287 GLU 288, ASP 289 LYS 137, ARG 131 | 2.56, 2.02 2.17, 2.44 2.11, 2.27 | 1 | ASP 289 |
| Methylglovanon | −9.66 | - | - | - | 2 | PHE 134, GLU 240 |
| Mundulinol | −13.12 | 2 | ARG 188, GLU 55 | 2.29, 2.01 | 4 | PHE 181, VAL 186, PRO 52, TYR 54 |
| Naringenin | −12.72 | 4 | ASP 33, TYR 101 GLN 83, LYS 88 | 2.36, 2.18 2.19, 2.26 | 1 | PHE 103 |
| Rhamnetin | −10.15 | 1 | LEU 287 | 1.88 | 1 | TYR 239 |
| Tamarixetin | −11.13 | 2 | PHE 103, TYR 101 | 2.11, 2.03 | 2 | LYS 102, GLU 178 |
| Favipiravir | −13.02 | 3 | ASP 33, TYR 101 LYS 88 | 2.38, 2.16 2.24 | 2 | PHE 103, TYR 239 |
| Lopinavir | −12.98 | 2 | GLU 288, ASP 289 | 2.11, 2.32 | 2 | PHE 181, VAL 186 |
| Hydroxychloroquine | −12.45 | 2 | ARG 188, GLU 166 | 2.25, 2.34 | 3 | VAL 186, PRO 52, TYR 54 |
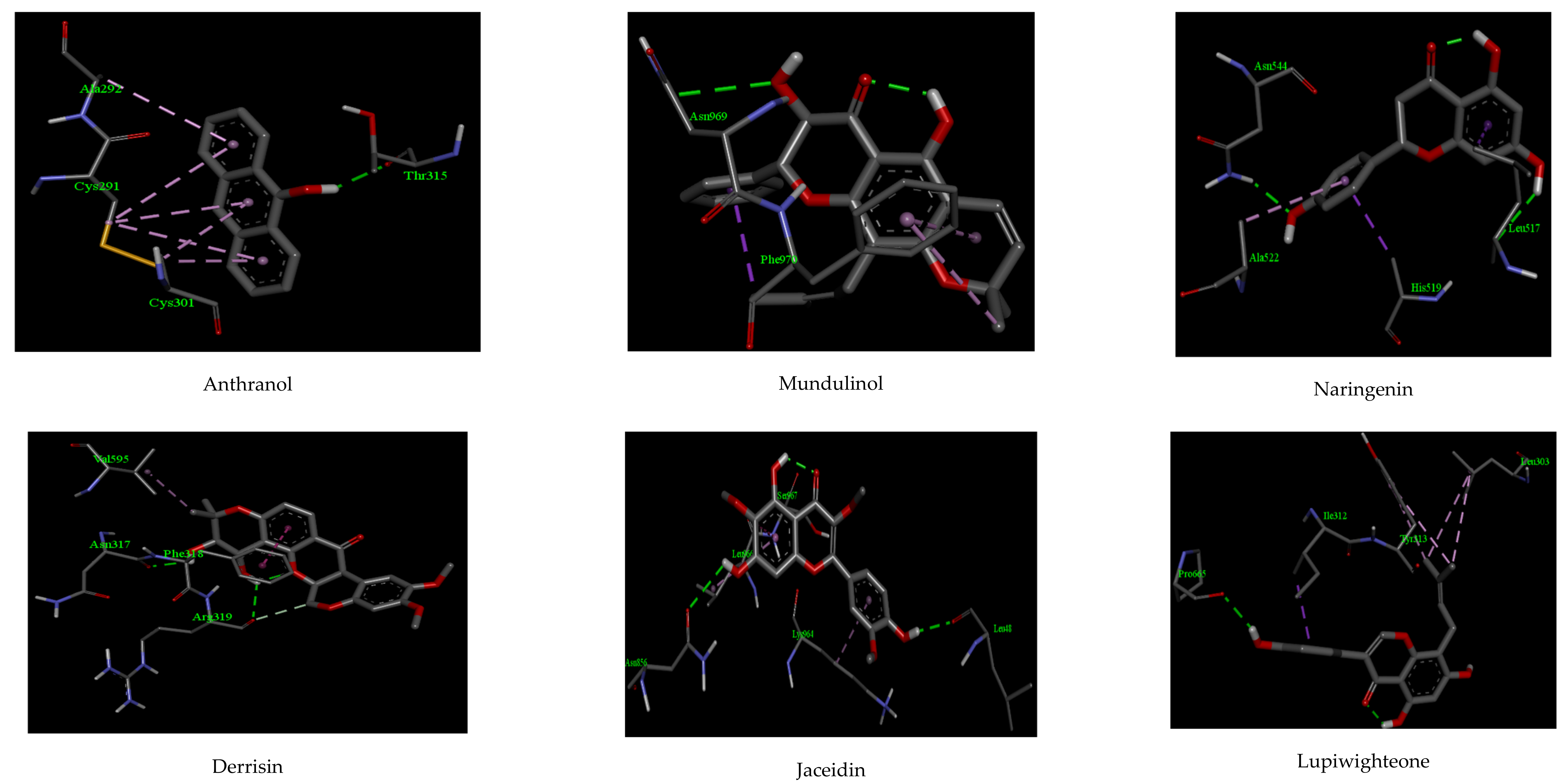
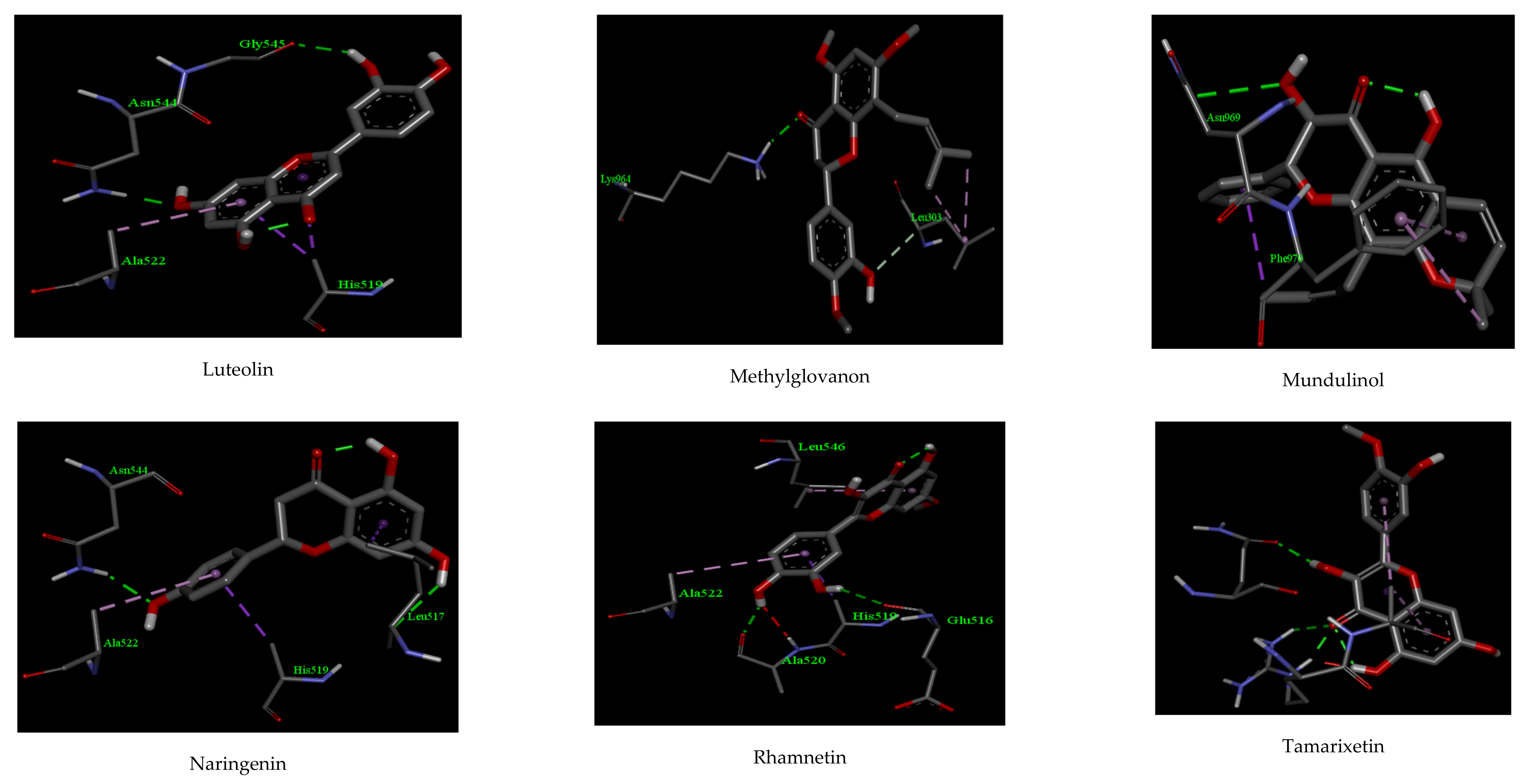
| Compounds | Interactions | |||||
|---|---|---|---|---|---|---|
| H-Bonds | Hydrophobic Bonds | |||||
| Binding Energy | Number | Residue | Distance (Å) | Number | Residue | |
| Anthranol | −9.08 | 1 | THR 315 | 2.11 | 2 | CYS 301, ALA 292 |
| Apigenin | −10.09 | 5 | MET 731, LYS 733 SER 730, HIS 1058 ASP 867 | 2.15, 2.26 2.27, 2.31 2.40 | 2 | VAL 860, PRO 863 |
| Cannflavin | −9.11 | 1 | THR 315 | 2.46 | 2 | CYS 301, ALA 292 |
| Derrisin | −11.04 | 2 | ARG 319, ASN 317 | 2.38, 2.49 | 2 | PHE 318, VAL 595 |
| Jaceidin | −10.54 | 2 | LEU 48, ASN 856 | 2.14, 2.28 | 2 | LEU 966, SER 967 |
| Lupiwighteone | −9.92 | 1 | PRO 665 | 2.46 | 3 | ILE 312, LEU 303, TYR 313 |
| Luteolin | −10.92 | 2 | ASN 544, GLY 545 | 2.32, 2.16 | 3 | ALA 522, HIS 519 |
| Methylglovanon | −9.43 | 1 | LYS 964 | 2.56 | 1 | LEU 303 |
| Mundulinol | −11.08 | 2 | ASN 969, ASP 867 | 2.33, 2.39 | 1 | PHE 970 |
| Naringenin | −10.12 | 2 | LEU 517 ASN 544 | 2.31 2.26 | 2 | ALA 522, ASN 544 |
| Rhamnetin | −10.15 | 2 | GLN 516 ALA 520 | 2.28 2.19 | 2 | LEU 546, HIS 519 |
| Tamarixetin | −10.33 | 2 | ARG 765, GLN 762 | 2.15, 2.23 | 2 | ALA 766 |
| Favipiravir | −10.76 | 2 | ASN 969, THR 315 | 2.24, 2.56 | 2 | PHE 318, ALA 292 |
| Lopinavir | −10.72 | 2 | MET 731, LYS 733 | 2.19, 2.38 | 1 | SER 967 |
| Hydroxychloroquine | −10.35 | 1 | GLY 545 | 2.33 | 1 | ASN 544 |
| Compounds | Estimated Solubility Log Score | Estimated Solubility Class | GIT Absorption | Caco-2 Permeability | BBB Penetration | Lipinski Violations | Toxicity | Carcinogenicity |
|---|---|---|---|---|---|---|---|---|
| Anthranol | −2.63 | High soluble | High | 1.14 | No | 0 | Non-toxic | Non-carcinogenic |
| Apigenin | −2.74 | High Soluble | High | 1.01 | No | 0 | Non-toxic | Non-carcinogenic |
| Cannflavin | −5.47 | Moderately soluble | High | 1.21 | No | 0 | Non-toxic | Non-carcinogenic |
| Derrisin | −2.72 | Hight Soluble | High | 1.03 | No | 0 | Non-toxic | Non-carcinogenic |
| Jaceidin | −5.11 | Moderately soluble | High | 1.28 | No | 0 | Non-toxic | Non-carcinogenic |
| Lupiwighteone | −3.71 | High Soluble | High | 0.79 | No | 0 | Non-toxic | Non-carcinogenic |
| Luteolin | −3.04 | high soluble | High | 0.69 | No | 0 | Non-toxic | Non-carcinogenic |
| Methylglovanon | −5.86 | Moderately soluble | High | 1.25 | No | 0 | Non-toxic | Non-carcinogenic |
| Mundulinol | −1.49 | High Soluble | High | 0.46 | No | 0 | Non-toxic | Non-carcinogenic |
| Naringenin | −2.88 | High Soluble | High | 1.07 | No | 0 | Non-toxic | Non-carcinogenic |
| Rhamnetin | −4.54 | Moderately soluble | High | 1.12 | No | 0 | Non-toxic | Non-carcinogenic |
| Tamarixetin | −4.63 | Moderately soluble | High | 1.15 | No | 0 | Non-toxic | Non-carcinogenic |
| Compounds | Drug-Likeness Model Score |
|---|---|
| Anthranol | 0.60 |
| Apigenin | 0.98 |
| Cannflavin | 0.67 |
| Derrisin | −1.08 |
| Jaceidin | 0.25 |
| Lupiwighteone | 0.36 |
| Luteolin | 0.95 |
| Methylglovanon | 0.18 |
| Mundulinol | 0.92 |
| Naringenin | 0.90 |
| Rhamnetin | −0.50 |
| Tamarixetin | 0.27 |
| Complexes | ΔEElectrostatic (kJ/mol) | ΔEVan derWaal (kJ/mol) | ΔEpolar (kJ/mol) | SASA (kJ/mol) | ΔEbinding (kJ/mol) |
|---|---|---|---|---|---|
| Mundulinol + 6W63 | −114.54 ± 1.33 | −33.65 ± 2.18 | 35.55 ± 2.16 | −36.32 ± 2.18 | −148.96 ± 3.55 |
| Luteolin + 6W63 | −117.54 ± 2.06 | −33.65 ± 2.36 | 35.55 ± 2.65 | −35.32 ± 3.34 | −150.96 ± 2.64 |
| Mundulinol + 6VYB | −115.54 ± 2.32 | −37.65 ± 2.98 | 38.55 ± 2.39 | −37.32 ± 4.05 | −151.96 ± 3.55 |
| Luteolin + 6VYB | −125.21 ± 2.64 | −36.23 ± 3.49 | 35.15 ± 2.18 | −37.65 ± 3.54 | −163.94 ± 2.63 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Mageed, H.R.A.; Abdelrheem, D.A.; Rafi, M.O.; Sarker, M.T.; Al-Khafaji, K.; Hossain, M.J.; Capasso, R.; Emran, T.B. In Silico Evaluation of Different Flavonoids from Medicinal Plants for Their Potency against SARS-CoV-2. Biologics 2021, 1, 416-434. https://doi.org/10.3390/biologics1030024
El-Mageed HRA, Abdelrheem DA, Rafi MO, Sarker MT, Al-Khafaji K, Hossain MJ, Capasso R, Emran TB. In Silico Evaluation of Different Flavonoids from Medicinal Plants for Their Potency against SARS-CoV-2. Biologics. 2021; 1(3):416-434. https://doi.org/10.3390/biologics1030024
Chicago/Turabian StyleEl-Mageed, H. R. Abd, Doaa A. Abdelrheem, Md. Oliullah Rafi, Md. Takim Sarker, Khattab Al-Khafaji, Md. Jamal Hossain, Raffaele Capasso, and Talha Bin Emran. 2021. "In Silico Evaluation of Different Flavonoids from Medicinal Plants for Their Potency against SARS-CoV-2" Biologics 1, no. 3: 416-434. https://doi.org/10.3390/biologics1030024
APA StyleEl-Mageed, H. R. A., Abdelrheem, D. A., Rafi, M. O., Sarker, M. T., Al-Khafaji, K., Hossain, M. J., Capasso, R., & Emran, T. B. (2021). In Silico Evaluation of Different Flavonoids from Medicinal Plants for Their Potency against SARS-CoV-2. Biologics, 1(3), 416-434. https://doi.org/10.3390/biologics1030024