Abstract
Viruses that infect bacteria (bacteriophages or phages) have a history of use in both biomedicine and basic molecular biology. Here, I briefly outline the pre-1940 use of phages in biomedicine and then more comprehensively outline the subsequent use of phages in determining the basics of molecular biology. Finally, I outline work that appears to form the foundation for a future, phage-enhanced biomedicine that generally extends medicine in the areas of anti-bacterial therapy (including vaccinology), anti-tumor therapy, and understanding the basic process of amyloid-associated neurodegenerative diseases. The following are general conclusions. (1) In the future, the discipline of phage-based biomedicine will be enhanced by more extensive merging with the discipline of basic phage biology (including molecular biology) and evolution. These two disciplines have been separated post-1940. (2) Biomedicine, in general, will be assisted if the focus is on key problems and key observations, thereby leaving details to later work. (3) Simplicity of strategy is a virtue that can be implemented and should be pursued with phages. (4) Capacity for directed evolution provides phages with generative (artificial intelligence-like) means for increasing biomedical effectiveness without using human design. Two related quotes set the stage (references at the end of the text). “But see that the imagination of nature is far, far greater than the imagination of man” (physicist Richard Feynman). “Nature, in all its variations and seeming paradoxes, speaks to those who pay attention and gives hints and clues to basic facts” (a thought attributed to Felix d’Herelle, a self-trained biologist who developed biological phage isolation and characterization). The integration of natural phenomenon-focused basic science and medical practice is an underlying theme.
1. Introduction
Much current medical research focuses on determining details of genomics, microbiology, and biochemistry. Early studies in these fields include basic science-oriented studies of viruses that infect bacteria. These viruses are called bacteriophages or phages. The phage-based contributions are especially extensive for research that falls in the category of molecular biology (review [1]). I begin by briefly outlining the pre-1940 work on bacteria and phages, which is more heavily weighted toward biomedicine than work of the subsequent era. This text is brief because of the existence of several previous reviews of this material.
Then, I present an outline of the subsequent-era use of phages and bacteria to determine the basics of molecular biology. Some of this topic has a foundation in basic physics. I will present the research philosophy promoted by this foundation.
Finally, I present an outline of recent data that suggest, at least in the near term, that optimal development of medicine includes a significant focus on next-generation, phage-based biomedicine.
2. Early History of Microbial, Including Phage-Related, Biology
The origin of the fundamentals of both modern biochemistry and modern microbiology was in the work of Louis Pasteur on fermentation in the mid-late 1800s. This work was initially not well accepted by chemists (biochemistry being in its infancy) because of the thought that fermentation was a simple chemical (non-biological) process. Pasteur based his conclusion (i.e., that fermentation was a product of living systems), in part, on a key datum: that the products of fermentation, but not laboratory synthesis, were chiral (reviews [2,3,4,5]). The use of light microscopy revealed the relatively small size of the living systems (yeasts and bacteria) involved. Identifying key problems and key data will be a recurrent theme below.
Subsequently, Pasteur developed several vaccines, beginning with an anti-bacterial vaccine for fowl cholera and ending with an anti-virus vaccine for rabies. The viral cause of rabies was too small to be seen by light microscopy, the only option available at the time. Pasteur understood this in general terms; the data were insufficient for specifics (reviews [2,3,4,5]). If Pasteur’s light microscopy had been available to and used by Ignaz Semmelweis (reviews of the work of Semmelweis [6,7]), the bacterial component of pathogenesis might have been recognized and documented 10–20 years earlier than it was.
Post Pasteur, filtration was used to discover that some pathogens were too small to be seen by light microscopy. Viruses were not retained by some filters that retained bacteria; the presence of a virus was detected by disease-causing capacity. A plant virus, tobacco mosaic virus, was the initial subject (review [7,8]). Thus, when both Twort [9] and d’Herelle [10] discovered bacterial filter-passing, bacteria-clearing agents in 1916–1918, a foundation existed for proposing a viral cause. However, again, opposition to this key idea was pervasive (reviews [11,12]).
The counter-idea was that clearing of bacteria was caused by a bacterially produced toxin that activated its own production [11,12]. Even before further characterization of phages, a key observation was that (1) phages formed discontinuous, typically circular regions of reduced turbidity in a bacterial lawn (plaques, or more descriptively, taches vierges [virgin stains] in French) and that (2) plaque number increased linearly with increase in phage concentration [13], as subsequently substantiated by more basic science-oriented investigators [14]. Plaques were subsequently also seen for viruses of eukaryotic cells ([15]; review [16]). Unless only one toxin molecule was needed for the destruction of a bacterial cell, plaque number should increase non-linearly as the concentration of the proposed toxin increased. Thus, even from the simplest basics, phage-as-virus was likely. d’Herelle made the case for this proposition early in the history [13].
That does not imply that the proposed bacteria-generated, bacteria-clearing toxins do not exist. However, observation of propagating, non-plaque-forming bacterial clearing would be unlikely to attract further investigation by current investigators. A causative toxin for such clearing would be distinct from bacteriocins, which are bacterially produced anti-bacterial proteins that are not active on the strain that produces them (review [17]). An intermediate concept is virus-infection activated, but subsequently virus-independent, propagation of change in protein conformation. This concept is the basis of a primitive innate immunity-based explanation of the cause of amyloid-associated neurodegenerative disease ([18] and Section 7).
By the 1920s, the anti-bacterial use of phages was a commercial enterprise that, however, was compromised by a limited understanding of phages, especially by limited understanding and control of (1) phage activity retention during both storage/transport and therapeutic use and (2) development by bacteria of resistance to phages (reviews [11,19]). Activity retention was variable, in part because of variable stability, but probably also because phages stimulated both innate and adaptive immune responses that were variable but not characterized. The field appears to have been stalled by these and possibly other limitations, including the above conceptual conflict, exacerbated by polemics rather than resolved by data. That made easy the decision to abandon phage therapy in the post-antibiotic era, beginning about 1940 [11,19].
The erratic character of phage therapy success, the technical limitations described above, and the polemics were embodied in the question of why phages were so (sometimes: my adaptation of the original question) much more effective in vitro than they were in vivo [11]. Forgetting about the polemics, this is a relevant question. A partial answer will be discussed below.
3. Phages for Investigating the Basics of Molecular Biology
3.1. Early Background: Genetics
Although phage genetic mapping eventually went to the map resolution forefront of molecular genetics, genetic mapping did not start with phages. Fruit fly (Drosophila) genomes were the initial primary subjects. Major advances in fruit fly genetic mapping began with the X-chromosome in 1910–1920. The X-chromosome was chosen because of the haploid (males have only one X-chromosome, rather than two) determination of phenotype (i.e., visible characteristics) in male fruit flies. This simplified the mapping of genotypes (the inherited determinants of phenotypes) by use of phenotypes. The result was a linear co-inheritance (linkage)-based map of genetic determinants of various X-linked phenotypes. Ultimately, four light microscopy-visible chromosomes were found to correspond to the four linkage groups found by genetic mapping (review [20]).
The work on fruit flies was decades ahead of work on phage genetics. Indeed, neither bacteria nor phages had yet been shown to have genes. The finding that bacteria did have genes was, in part, linked to studies of the inter-bacterial transfer of phenotype-determining information by a subcellular entity [21]. The latter work ended with the finding that this entity was DNA [22] (review [23]). Earlier phage research (e.g., [13]) was, in retrospect, observing genetic, DNA-encoded inheritance of phages, although without genetic mapping.
Incidentally, reference [22] also described how to isolate DNA without having the DNA enzymatically degraded. Chelation of divalent cations was a key part of the strategy used. DNA degradation had previously obscured the length and, therefore, the coding capacity of DNA molecules. The extreme lengths of chromosomal DNA molecules were initially found for the largest Drosophila chromosome by use of a physical analysis after release of DNA from cells by extremely gentle (shear breakage-minimizing) procedures [24]. These lengths were subsequently more easily and accurately determined by DNA sequencing, in which a complete chromosomal DNA sequence was assembled from overlaps among sequenced DNA fragments (reviews [25,26]).
The post-Drosophila (1950s–1970s) genetic mapping of phage genes (reviews [27,28,29]) had the advantage of haploid determination of phenotype. Even relatively early in the phage genetic mapping process, phage genetic mapping acquired such high resolution that evidence for the triplet nature of the genetic code was obtained via phage genetics. The data included the reversion of a mutant phenotype by a second, nearby mutation. These mutations were assumed to be deletion–insertion pairs. Deletions or insertions eliminated function if one or two were co-introduced. However, the function was retained if three were co-introduced [30] (three nucleotides, in retrospect). The conclusion was drawn that function was eliminated by altering the reading frame but not by removal of one coding element. The coding element was, therefore, a triplet. The insensitivity of the function studied to the removal of a coding triplet was a lucky aspect.
This work ended with the finding of genetic coding by strings of DNA-associated nucleotide triplets, each triplet coding for one amino acid of a series of amino acids that constitute a protein. This work was at the leading edge of the new discipline of molecular biology, named and heavily supported by the Rockefeller Foundation, led by Warren Weaver [31], and focused on merging biology and physics.
However, a direct biochemistry-based strategy was used to determine (1) what the genetic code was (e.g., [32]) and, previously, (2) the fact that the code was used to specify the sequence of amino acids in proteins (polypeptides) [33,34]. One feature of the code was the presence of polypeptide chain termination codons that signaled the stopping of the addition of amino acids to a protein.
These discoveries had implications that included understanding of inherited diseases that were caused by miscoding of proteins. These diseases included sickle cell anemia [33,34] and muscular dystrophy (reviews [35,36]). In addition, investigator-introduction of temperature-sensitive and polypeptide chain termination mutations in specific phage genes was used to (1) map phage genes, (2) discover the function of the protein that was mutation-inactivated, and (3) perturb normal intracellular phage-assembly biochemistry in understood ways (reviews [37,38,39,40]). Implications existed for understanding the assembly of subcellular structures of eukaryotes (reviews [39,40]), as also illustrated by specific examples below.
3.2. Early Background: Physiology and Biochemistry
After phage infection, the number of progeny phages was assayed via plaque forming units (PFU) released. In the earliest studies, PFU was found to increase in steps separated by latent phases (work by d’Herelle [13]; also [14,41]). Each increase phase/latent phase was the result of one round of infection. Heritable phenotype variation was observed. The phenotypes observed were plaque morphologies and host ranges (reviews [13,19,31]). Spontaneous (variable in time) mutant generation was discovered via the higher-than-random (higher-than-Poisson) distribution of the number of mutants observed when several different cultures each underwent several rounds of infection. The alternative was phenotype-specific mutant induction by environmental conditions, which would have produced a Poisson distribution of mutant numbers [42]. Induction of mutations had previously been found to be possible by using conditions that altered genomes and were independent of a phenotype that was being selected. These conditions included irradiation with X-rays, as found in experiments with Drosophila (review [20]). Data of this type were also, in retrospect, recognizable in earlier studies of phages [13].
Also, it is very important not to assume that all heritable phage variation has a genomic (DNA-encoded) origin. A case of non-genomic inheritance has been documented in the case of phage T7 [43]. A 1931 summary of anti-bacterial phage therapy optimizations [44] indicates the importance of minimizing pre-therapy laboratory phage propagation. That is more consistent with non-genomic inheritance than genomic inheritance. In any case, discriminating these two possibilities is a key (and easily solvable, given modern DNA sequencing technology) problem for the future of phage therapy of bacterial disease.
Historically, the inactivation of phages by ultraviolet light was observed, as was the exchange of segments of genomes (recombination). Recombination was a mechanism to reconstitute phages damaged by ultraviolet light [45,46]. These data, coupled with evidence of multi-generation-inherited characteristics, led to the assumption, pre-1950, that phages had chromosomes, although obligate parasites. This conclusion was drawn before the final demonstration and acceptance of the fact that genetic encoding was in DNA.
Discussion of whether phages (and other viruses) are living is not productive; sometimes they are living, and sometimes they are not. One of the objectives of phage therapy is to maintain phage-based biomedicines in a reversibly dormant (basically reversibly non-living) storage state for as long as possible.
3.3. Interface with Basic Physics
Early work on phages occurred in the shadow of previous work on the structure of atoms. The latter included electron and proton scattering-based evidence for a central, positively charged nucleus surrounded by negatively charged electrons [47,48]. The initial models of atoms had the electrons orbiting around the nucleus (modern adaptation [49]). This description became problematic enough that a revision was needed. The revision was that atom-associated electrons were delocalized, wavelength-quantized waves of probability (quantum mechanical description of atoms; didactic description [50]). This was part of a major change in physics and chemistry.
Demonstration of the nucleoprotein nature of phages (review [51]) was followed by the introduction of molecular biology to phage research. For some investigators, the basic science-oriented work on phages was motivated by the thought that (1) science should be viewed as a unified discipline, and (2) the study of biology might reveal a missing component of all science, as did the quantum mechanical description of atoms [52,53]. Such a biology-derived, basic component has not yet been observed. A relayed [54] opinion of Richard Feynman, a physicist who spent some time performing phage genetics [55] and knew the advances in biochemistry [56], is that chemistry and accompanying quantum mechanics constitute the foundation of biology. No fundamentally new physics is to be found.
In any case, I think that only limited time should spent on topics like this. The main focus should be on determining what happens empirically. This is especially important in the realm of environmental phages because of the diversity and uncertainty involved. Flexibility of ideas should accompany empiricism. Numerous historical examples exist of the negative consequences of digressing from both empiricism and the accompanying flexibility of ideas [57]. An interesting additional example is telegraphed by the name of the disease, malaria (bad air). Bad air is not the cause of malaria (review [58]).
Phage-based biomedicine, for example, is not an established area of either science or medicine. Among the reasonably anticipated key future discoveries are phages that are significantly different from the phages well known today. The existence of such phages is implicit in work performed over 100 years ago [13]. We do not know enough to forgo flexibility in interpreting data in this area.
3.4. Brief Description of Major Subsequent Developments (Excluding Genetic Engineering)
When phages infect bacterial cells, bacterial protein synthesis is converted to phage protein synthesis. Historically, for phage T2, this change was found to be associated with the appearance of a new RNA, now called messenger RNA, abbreviated mRNA ([59,60]; review [61]). The mRNA carries protein-encoding information from chromosomal DNA to the site of protein synthesis in eukaryotic, as well as prokaryotic, organisms (review [61]). The T2 data are often considered to be the empirical origin of this concept.
Similarly, phages and their bacterial hosts were used in the early studies of the control of the expression of genes. Sites of RNA synthesis initiation, usually known as promoters, were identified in “coliphages”, with a focus on phage lambda (reviews [27,62,63]). Proteins that controlled protein synthesis by interacting with these and neighboring sites on DNA were identified (reviews [27,62,63]). A key procedure was the inactivation of both proteins and protein binding sites by isolation of phage and bacteria that were mutated at these sites. This genetic work was assisted by the relatively rapid propagation of both phages and their bacterial hosts.
Phage DNA not only replicates during a phage infection, it also undergoes recombination: generation of new combinations of genotypes and phenotypes. An early study calculates the number of recombination events [64]. Biotechnological applications of recombination are reviewed for gene modifications/replacements in [65,66]. More recently, an additional proposal is to optimize recombination-based directed phage evolution for the obtaining of desired phenotypes [67].
Early studies revealed recombination-driving proteins of both phages and bacteria. Phage lambda was a focus, apparently, because its replication and recombination were relatively easy to separate from each other, both physically and genetically. Identification of the encoding genes led to one of the first isolations and characterizations of the key proteins involved, an exonuclease and an annealase (reviews [68,69]). In contrast, biochemical analysis revealed that phage T4 had DNA replication and recombination intertwined [70,71], which made more problematic analogous linking of enzymology with DNA structure. This intertwining also appeared to be the case for phage T7 in that most intracellular DNA was isolated as a single, multiply-branched molecule generated by both recombination and DNA replication [72].
In another area, the analysis of intracellular multimolecular assembly was a relatively complex problem that needed simplifying model systems. Two key, assembly-related general concepts arose as spin-offs of the analysis of phages as model systems. The first arose during the development of conditional polypeptide chain termination mutants of phage T4. The phenotypes of these mutants were determined after obtaining stocks of the mutants by propagation in “suppressing” hosts that read the chain termination signal as an amino acid. The general concept developed for assembly of the tail fibers of T4 (Figure 1) was the following. Constituent proteins are assembled in a sequence that was determined by the requirement for an assembly substrate of a limited type before a given protein is added to any partial assembly. Thus, the sub-assemblies and the final assembly observed were relatively independent of the relative concentrations of phage components [39,40,73,74].
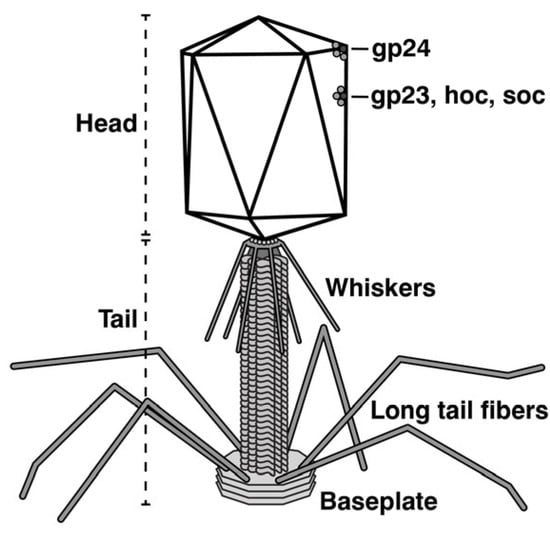
Figure 1.
A sketch of phage T4.
The second concept arose in response to the following key question for both phages and eukaryotes. How is the length of unique-length, polymeric protein filaments determined? This question applied to the length of both polymerized-protein phage tails and some polymerized-protein eukaryotic filaments, such as muscle sarcomere-associated myosin and actin filaments. Phage genetics provided a relatively rapid, empirical, and definitive answer for phage tails and a prototype for other systems. The answer for phage tails was that this length was determined by a ruler (tape measure) protein that, when unraveled to a pre-programmed conformation, signaled the end of the polymerization. The total length of the tape measure protein and its programmed behavior was mRNA determined. The key observation was that, when the length of the phage lambda tape measure protein was genetically changed, the length of the phage tail changed in proportion [75,76]. The tape measure protein gene for a newly isolated phage can be identified from the genomic sequence in part based on the great length and the location of this protein [77,78]. The tape-measure protein for myosin was found to be the protein titin; the tape-measure protein for actin was found to be the protein nebulin [79,80].
4. A Case for Backup Strategies for Therapy of Chronic Diseases
Work on mechanisms of recombination has more recently been extended to eukaryotic systems, sometimes in the context of recombination-driven DNA repair. Deficiency of DNA repair should, in theory (but not always in practice [81]), increase the frequency of cancer (reviews [81,82,83]). Phage/bacteria-analogous mechanisms of eukaryotic recombination have been observed, although without the full spectrum of genetic manipulations used for work on bacteria and phages (reviews [81,82,83]). A rationale for focusing on the details of eukaryotic recombination was that either the genes or the gene products involved would be future targets for anti-cancer chemotherapy. Recombination was known to be used by cancer cells to repair the damage done to them by anti-cancer chemotherapy and radiotherapy.
Without going into the details of the above studies of eukaryotes, I note that cancer cells are notoriously able to evolve resistance to any drug used on them. One might, therefore, want to be flexible enough to have backup strategies for the strategy of the previous paragraph. Flexibility of this type will assist in the avoiding of progress-inhibiting scientific bias (see [57,84]).
For example, in the case of neurodegenerative diseases, the negative effects of inflexibility are becoming obvious via the consequences of using the following to guide research: the hypothesis that both Alzheimer’s disease and other amyloid-associated neurodegenerative diseases are caused by toxicity of the β-sheet structured amyloid proteins that are sometimes, not always, associated with these diseases (amyloid cascade hypothesis [85,86,87]). Assumption of accuracy has led to multiple drug failures. Indeed, the amyloid cascade hypothesis has not been consistent with the data [88,89,90]. The implied inflexibility has become so difficult to understand that this inflexibility risks historical classification as a very expensive scientific bubble. The cost is both human misery-derived and financial.
One cannot project with certainty what will be the future most effective therapies for current life-length and life-quality limiting diseases. The level of “crystal ball” cloudiness does, however, vary. In the next sections, I treat topics in the order of my view of my decreasing ability to project the general outline of what a biomedical, phage-based therapy will be.
5. Projected Future of Phage-Based Biomedicine: Phage Therapy of Bacterial Infections
5.1. Limitations of Antibiotics
Current therapy for bacterial infections almost universally includes the use of antibiotics. In the United States (US), the use of antibiotics was the major factor in reducing bacterial infection-caused deaths per 100,000 people from 216 in 1936 to 59.7 in 1952. These were times during which the major anti-bacterial advance was the introduction of antibiotics. A subsequent reduction was to 38 in 2002 (review [91]). Before 1936, advances in sanitation had caused a reduction [91].
However, from the beginning of the era of antibiotics, bacteria were evolving resistance to antibiotics [92]. Strategy for remedial activity included (1) admonition not to overindulge in the overuse of antibiotics and (2) development of new antibiotics (examples [93,94]). However, the development of new antibiotics was likely to involve internally contradictory aspects of both (1) science, since extreme optimism was required to anticipate an indefinite supply of new antibiotics that were of low enough toxicity, and (2) finances, since great expense was involved for a product for which use was restricted to limit evolving of bacterial resistance [95,96].
Incidentally, warning about the overuse of antibiotics has often arrived in the context of an infection that was thought to be antibiotic-resistant because it was caused by a virus. Inflexibility in warnings of this type had the potential to be problematic because many viral infections have been shown to have bacterial aggravators, for example, influenza virus infections [97,98].
Finally, antibiotics are, in general, not harmless. A list of antibiotic toxicities is in [99]. Interesting examples are the muscle and tendon toxicities of frequently used quinolone antibiotics, which are listed as being under the most active development [100]. These toxicities (1) are apparently most acute for fluoroquinolones, such as ciprofloxacin and levofloxacin, (2) can cause heart damage, and (3) are selectively seen in people who are relatively old and relatively athletic [101,102,103]. This is not a minor problem for people in one or both of these classes.
5.2. Anti-Bacterial Therapy: Phage Therapy
The limitations of antibiotics-based anti-bacterial therapy are, in theory, bypassable by the use of phage therapy, i.e., the use of phages as therapeutic, anti-bacterial agents. Phage therapy, after multiple human trials, has never been found to have serious toxic effects. Typically, no direct toxic effects are detected. The major concerns are (1) indirect effects from the phage-induced release of bacterial lipopolysaccharide (endotoxin) and (2) effects induced by contaminants in phage preparations [104,105,106,107]. In part because of the history of phages being safe, isolation of new, therapy-potential, lytic (in contrast to lysogenic) phages is more rapid than the development of new antibiotics in response to bacterial resistance. In an emergency, if one uses high-speed (e.g., [108,109]), broad-spectrum (e.g., [109]) phage isolation procedures, then less than a week is required. Phage characterization would require an additional 1–2 weeks. This characterization could involve (1) genome sequencing to screen away both lysogenic phages and phages that carry genes for either toxins or antibiotic resistance and (2) whatever is needed to screen for effectiveness since low/no effectiveness is the major limitation (reviews [104,105,106,107,110,111]).
At present, screening for effectiveness is typically performed at a level too low to generate phage therapy cocktails with a high enough success probability to be FDA-approved (reviews [104,105,106,107,110,111]). For this reason, phage therapy is currently used in the US and Western Europe only on a compassionate use basis. In keeping with Pasteur’s focus on key observations, the response should be to both identify and screen for the key effectiveness-promoting phage characteristics.
Effectiveness-promoting characteristics might include the following. (1) High projected in-blood phage lifetime (persistence) is a strong candidate. Typically, the associated screen is performed via in-blood phage persistence in mice. However, we need more efficient (in time and cost) but accurate surrogates. These surrogates might include phage average electrical surface charge density and size, both efficiently determinable by native (infective phage particle) agarose gel electrophoresis [111]. (2) Aggressiveness towards the bacterial host is another strong candidate (see [112]). Screening for aggressiveness might be performed during the original cloning of the phage via both a low number of phage-resistant bacterial colonies and a high speed and long time of phage plaque enlarging. Even phage size screening can be performed during initial isolation via the dependence of plaque size on supporting gel concentration [109].
5.3. Anti-Bacterial Therapy: Vaccines
Vaccination is a technique for preventing infections with all microbes. The original non-natural vaccines were either attenuated or inactivated versions of either the disease-causing microbe or its toxin. Vaccines started with a live, related-virus-derived anti-smallpox vaccine and continued with several live, attenuation-derived anti-bacterial and anti-viral vaccines (review [5]). Vaccination subsequently continued the same way but with the addition of inactivation-derived vaccines (reviews [113,114]). A major contribution of the 20th century was from the industry-associated laboratories of Maurice Hilleman (review [114]). Most vaccines, including all Hilleman vaccines, consisted of an assembled version of the antigen, not a single protein molecule or its mRNA. Some details are the following.
(1) Some FDA-approved, anti-bacterial vaccines consisted of a purified bacterial polysaccharide (to provide the needed vaccine target [antigen]) conjugated to an inactivated toxin, sometimes tetanus toxoid (to boost the immune response [adjuvant]) (e.g., [115]). The final vaccine components were heterogeneous in size and in the range of sizes for some viruses, 20–60 nm [116].
(2) Other vaccines used a phage to present the vaccine antigen (phage display vector). Phages could be used in this way when they had surface proteins that could be modified without inactivating the phage. Figure 1 shows the structure of phage T4. The T4 “head” (Figure 1) includes two surface proteins, hoc and soc, that were found non-essential for T4 propagation in the laboratory. Both proteins could be used to display bacterial and other peptide antigens when phage T4 was used as a vaccine (review [117]).
Other phages can potentially be used in this way, including (1) double-stranded DNA phages lambda and T7, (2) filamentous, single-stranded DNA phages, and (3) RNA phages. Several infectious microbes are targets. However, no phage-based vaccine is currently FDA approved (reviews [117,118]).
One aspect of using whole microbes as vaccines is that the number of antigens and other immunity-triggering signals is maximized. The higher this number is, the more unlikely is the targeted microbe’s escape from an immune response. One signal is the size of the antigen, a signal to which uptake by macrophages and other innate immunity-generating white blood cells [119,120] are sensitive.
6. Metastatic Cancer
Like antibiotic therapy for bacterial infections, chemotherapy of cancerous tumors has an evolving-of-drug-resistance problem. One response is to raise either drug concentration or drug potency to the point that tumor cells die before they can evolve drug resistance. This must be performed without unacceptable damage to the patient, a constraint that is problematic because cancer is generated by “rogue” cells of the patient. These rogue cells are biochemically similar to healthy cells. Current chemotherapeutic responses are in two general categories: (1) searching for both improved drugs and combinations of drugs that, chemically speaking, have more tumor-specific and/or more numerous targets [121,122,123], and (2) improving the targeting of current anti-cancer drugs (reviews [124,125,126]).
Phage-based responses in category #1 are, for the most part, based on phage display vectors, such as those generated from filamentous phages. A therapeutic protein antigen is (1) cloned in a filamentous phage genome and (2) displayed on the surface of the phage. The phage is selected, during directed evolution, to have either a therapeutic or a diagnostic function, such as specifically binding to a receptor at least partially specific to the surface of cancerous cells (review [127]). The use of directed evolution requires only limited initial knowledge of the details of what will be produced in the end. This is an advantage shared with generative artificial intelligence.
Some phage-based responses in category #2 are (1) also based on the directed evolution of filamentous phages but (2) include covalently conjugating drugs to the external surface of the evolved phage’s capsid; the phage is then used as a drug delivery vehicle (DDV; reviews [128,129]).
Another phage DDV-based strategy is emerging from the use of liposomal DDVs, some FDA-approved [130,131,132], to improve drug tumor-tropism. Even without adding ligands to generate tumor-tropism, liposomal DDVs are tumor-tropic via relatively large pores that are preferentially in tumor-feeding blood vessels in comparison to healthy blood vessels (enhanced permeability and retention, or EPR, effect [133,134,135]). Effectiveness of EPR effect-targeted DDVs is limited by (1) interference-by-stroma to access of DDVs to tumor cells, which is more limiting in humans than in mice [135,136,137], (2) toxicity generated by leakage of drug from the DDV [136,137], and (3) loss of DDVs during DDV circulation [135,136,137] (i.e., low persistence). These are key problems to solve.
In addition to having the high persistence needed for a DDV [138], phage T4 has the following additional characteristics that suggest the potential for the use of T4 and similar phages to solve the problems of DDV leakage and stromal interference. T4 has a natural gate that can be (1) opened to load drugs, and (2) closed to store, transport, inoculate, and circulate drugs in blood [139]. The key remaining characteristic to develop is tumor-specific drug release via either re-opening of the gate or disruption of the phage. This would solve the problem of out-of-tumor drug leakage of the DDV.
The problem of minimizing stromal DDV interference is possibly at least partially solved by the use of the following multi-component strategy with either T4 or similar phages as DDV. (1) Allow more time for the DDV to undergo EPR effect-driven tumor location. This would be possible via both high DDV persistence and closed-gate DDV conformation when not tumor-located. (2) Use directed evolution to solve the stromal interference problem with minimal human participation. This would be performed by serial selection of phages by passage through a tumor-bearing organism, followed by re-propagation of tumor-located phages. Phages propagate rapidly enough so that each selection cycle can be performed in two days. This unexploited, artificial intelligence-like strategy is, thus far, apparently without analog among other DDVs.
7. Amyloid Associated Neurodegenerative Diseases
I will focus here on neurodegenerative diseases that are usually (not always) accompanied by protein aggregates that have starch-like texture; the proteins in the aggregates are called amyloid proteins. The diseases include Alzheimer’s disease, amyotrophic lateral sclerosis, Parkinson’s disease, Huntington’s disease, some ataxias, and prion-associated diseases (reviews [140,141,142,143]). The mechanism (disease pathway) is not rigorously solved for any of these diseases. I begin by noting that phages have the potential to provide a model that helps to solve the disease mechanism puzzle.
In a search of the literature for key data (pieces of the puzzle), one might begin with evidence for the following. (1) Amyloid protein genes are related to genes for viruses. This observation suggests the hypothesis that the healthy function of amyloid proteins includes anti-virus (innate immune) activity [142]. (2) Virus infection is found significantly correlated with subsequent Alzheimer’s disease occurrence, especially for herpesviruses and in the presence of a well-established Alzheimer’s disease-promoting allele [144,145,146,147]; review [148]. These data suggest the following addition to the hypothesis. The disease pathway includes loss of control of the innate immunity that countered the previous infection (review [18]). (3) The association of disease with an amyloid protein suggests that a conformer of this protein is neuro-toxic; this conformer is not necessarily beta-sheet, the conformer found in amyloid plaques, such as those observed by Alzheimer. The data indicate that the toxic conformer is a not-well-known conformer called alpha-sheet (reviews [149,150]). This suggests the further hypothesis-addition that alpha-sheet conformation is involved in innate immunity (review [18]).
Phages assisted in the assembly of the above hypotheses to form a more complete overall hypothesis. The two key observations were the following. (1) Phage DNA packaging in a pre-assembled capsid (called a procapsid) was associated with hyper-expansion and thinning of the outer shell of the capsid. The shell was so thin that a sheet structure was likely (e.g., [18]). (2) Herpesviruses have been found to have phage analogous DNA packaging (review [18]). Thus, the overall hypothesis included the proposal that, in health, alpha-sheet structured amyloid protein inhibited virus propagation by binding to the alpha-sheet structure produced during virus assembly (pathway 1, 3, 4 in Figure 2). In disease, control was lost, and toxic alpha-sheet structured amyloid protein was over-produced (pathway 1, 3, 5 in Figure 2) (review [18]).
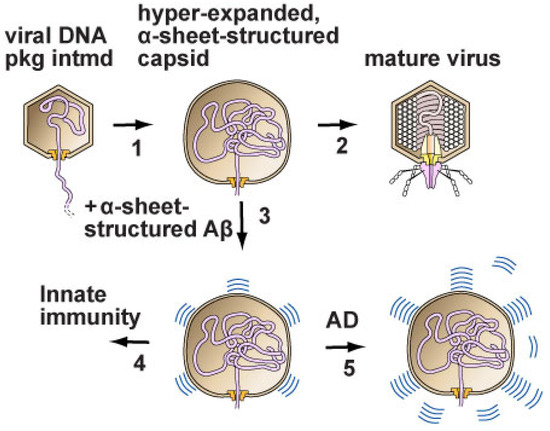
Figure 2.
The disease model. (1) DNA enters the capsid of either a phage (phage T3 is used as an example) or a herpesvirus. Hyper-expansion, with subunit conversion to an alpha-sheet structure, occurs to accelerate DNA packaging when packaging is slowed. (2) In the case of phage, packaging finishes, and a tail is added to the capsid to form a mature phage particle. (3,4) In the case of herpesviruses in humans, amyloid proteins block the progression of the hyper-expanded capsids by converting to an α-sheet structure and then co-assembling with the alpha-sheet of subunits of the capsid. (5) Alzheimer’s disease is initiated by the over-production of amyloid protein α-sheets, which are toxic. Some amyloid protein alpha-sheets are subsequently converted to the beta-sheet amyloid protein of plaques. However, this conversion is not sufficient to avoid toxicity when Alzheimer’s disease is present (Reprinted from [18]).
If the model in Figure 2 is not correct, the exercise involved at least shows that the correct model might be obtained by (1) identifying key data that already exist and (2) assembling the data in a relatively simple and direct way, with the implication that many currently known details would not be needed. If the model of [18] is correct, then the key remaining priority is to use the model to obtain improved therapies (details [18]).
A final model might extend to understanding the origin of some cases of autism. Evidence exists for a microbial infection trigger in some cases (reviews [151,152]). Apparently, the highest probability-viral trigger is the rubella virus. However, a vaccine for rubella has not prevented an increase in autism diagnosis [151]. Although other genetic and environmental factors are involved, I note here that the following three herpesviruses are also on the list of triggers: (1) cytomegalovirus (human herpesvirus 5 or HHV-5), (2) herpes simplex virus (1 and 2), varicella-zoster virus (HHV-3). A vaccine exists for HHV-3, the cause of chickenpox, but not the others [151,152].
Although autism-triggering via immune response is actively considered in the literature [151,152], triggering via an amyloid protein-based innate immune response, like the one in [18] and Figure 2, is not. In any case, one rational anti-autism strategy is the development of additional vaccines, especially for herpes simplex viruses 1 and 2, both of which infect significant percentages of people, 67% and 13%, respectively [151].
I end with a related quote from Maurice Hilleman. “Maybe there are immunities that exist outside of antibodies and cells and cytokines. There may be other mechanisms that we don’t know about.” The congruence of this quote with the quotes in the Abstract is significant. The two quotes in the Abstract are from [153] and [19], respectively. The three individuals quoted are former thought leaders who worked in different areas of science. Before the reader opines about future administrative responses, I recommend reading reference [57].
Funding
This research was funded by the Morrison Trust (2023), San Antonio Medical Foundation (2024), and the US National Science Foundation (2409676).
Acknowledgments
For helpful comments on a draft of this manuscript, I thank Richard F. Ludueña, Anna Malkova, and Alexander Mazin.
Conflicts of Interest
P.S. is a founding member of a company, Phage Refinery LLC, that is working on the implementation of some of the ideas discussed here. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| DDV | Drug delivery vehicle |
| EPR | Enhanced permeability and retention |
| HHV | Human herpesvirus |
| mRNA | Messenger RNA |
| US | United States |
References
- Keen, E.C. A century of phage research: Bacteriophages and the shaping of modern biology. Bioessays 2015, 37, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Dubos, R. Louis Pasteur: Free Lance of Science, revised ed.; Da Capo PR Inc.: Boston, MA, USA, 1986. [Google Scholar]
- Institute Pasteur. Our History: The Early Years 1847–1862. Available online: https://www.pasteur.fr/en/institut-pasteur/history/early-years-1847-1862 (accessed on 17 March 2025).
- Berche, P. Louis Pasteur, from crystals of life to vaccination. Clin. Microbiol. Infect. 2012, 18, 1–6. [Google Scholar] [CrossRef]
- Smith, K.A. Louis Pasteur, the father of immunology? Front. Immunol. 2012, 3, 68. [Google Scholar] [CrossRef]
- Kadar, N. Rediscovering Ignaz Philipp Semmelweis (1818–1865). Am. J. Obstet. Gynecol. 2019, 220, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Pittet, D.; Allegranzi, B. Preventing sepsis in healthcare—200 years after the birth of Ignaz Semmelweis. Euro Surveill. 2018, 23, 18-00222. [Google Scholar] [CrossRef] [PubMed]
- Lecoq, H. Découverte du premier virus, le virus de la mosaïque du tabac: 1892 ou 1898? Comptes Rendus Acad. Sci. III 2001, 324, 929–933. [Google Scholar] [CrossRef]
- Twort, F.W. An investigation on the nature of ultra-microscopic viruses. Lancet 1915, 186, 1241–1243. [Google Scholar] [CrossRef]
- d’Herelle, F. Sur un microbe invisible antagoniste des bacillus dysentérique. Comptes Rendus Acad. Sci. Paris 1917, 165, 373–375. [Google Scholar]
- Summers, W.C. The strange history of phage therapy. Bacteriophage 2012, 2, 130–133. [Google Scholar] [CrossRef]
- Emrich, J.; Richter, C. Bacteria Eaters: The “Twort-d’Hérelle Phenomenon”. The American Association of Immunologists. 2021. Available online: https://www.aai.org/About/History/History-Articles-Keep-for-Hierarchy/Bacteria-Eaters-The-Twort-d’Herelle-Phenomenon”#:~:text=The%20“Twort%2Dd'Hérelle%20Phenomenon%2C”%20also%20known,know%20it%20now%20as%20bacteriophage (accessed on 18 March 2025).
- d’Herelle, F. Le Bactériophage; Son Rôle dans l’Immunité; Masson et cie: Paris, France, 1921. [Google Scholar]
- Ellis, E.L.; Delbrück, M. The growth of bacteriophage. J. Gen. Physiol. 1939, 22, 365–384. [Google Scholar] [CrossRef]
- Dulbecco, R. Production of plaques in monolayer tissue cultures by single particles of an animal virus. Proc. Natl. Acad. Sci. USA 1952, 38, 747–752. [Google Scholar] [CrossRef]
- Baer, A.; Kehn-Hall, K. Viral concentration determination through plaque assays: Using traditional and novel overlay systems. J. Vis. Exp. 2014, 93, e52065. [Google Scholar] [CrossRef]
- Solis-Balandra, M.A.; Sanchez-Salas, J.L. Classification and multi-functional use of bacteriocins in health, biotechnology, and food industry. Antibiotics 2024, 13, 666. [Google Scholar] [CrossRef]
- Weaver-Rosen, M.S.; Serwer, P. Alzheimer’s Disease: A molecular model and implied path to improved therapy. Int. J. Mol. Sci. 2024, 25, 3479. [Google Scholar] [CrossRef]
- Summers, W.C. Félix d’Herelle and the Origins of Molecular Biology; Yale University Press: New Haven, CT, USA, 1999. [Google Scholar]
- Carlson, E.A. Genes, Radiation, and Society: The Life and Work of H.J. Muller; Cornell University Press: Ithaca, NY, USA, 1981. [Google Scholar]
- Griffith, F. The significance of pneumococcal types. J. Hyg. 1928, 27, 113–159. [Google Scholar] [CrossRef] [PubMed]
- Avery, O.T.; MacLeod, C.M.; McCarty, M. Studies on the chemical nature of the substance inducing transformation of pneumococcal types. Induction of transformation by a desoxyribonucleic acid fraction isolated from pneumococcus type III. J. Exp. Med. 1944, 89, 137–158. [Google Scholar] [CrossRef] [PubMed]
- Petsko, G.A. Transformation. Genome Biol. 2006, 7, 117. [Google Scholar] [CrossRef][Green Version]
- Kavenoff, R.; Zimm, B.H. Chromosome-sized DNA molecules from Drosophila. Chromosoma 1973, 41, 1–27. [Google Scholar] [CrossRef]
- Brlek, P.; Bulić, L.; Bračić, M.; Projić, P.; Škaro, V.; Shah, N.; Shah, P.; Primorac, D. Implementing whole genome sequencing (WGS) in clinical practice: Advantages, challenges, and future perspectives. Cells 2024, 13, 504. [Google Scholar] [CrossRef]
- Scarano, C.; Veneruso, I.; De Simone, R.R.; Di Bonito, G.; Secondino, A.; D’Argenio, V. The third- generation sequencing challenge: Novel insights for the omic sciences. Biomolecules 2024, 14, 568. [Google Scholar] [CrossRef] [PubMed]
- Casjens, S.; Hendrix, R.W. Bacteriophage lambda: Early pioneer and still relevant. Virology 2015, 479–480, 310–330. [Google Scholar] [CrossRef]
- Stahl, F.W.; Edgar, R.S.; Steinberg, J. The linkage map of bacteriophage T4. Genetics 1964, 50, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Studier, F.W. The genetics and physiology of bacteriophage T7. Virology 1969, 39, 562–574. [Google Scholar] [CrossRef]
- Crick, F.H.C.; Barnett, L.; Brenner, S.; Watts-Tobin, R.J. General nature of the genetic code for proteins. Nature 1961, 192, 1227–1232. [Google Scholar] [CrossRef]
- Summers, W.C. The American Phage Group: Founders of Molecular Biology; Yale University Press: New Haven, CT, USA, 2023. [Google Scholar]
- Nirenberg, M.; Leder, P.; Bernfield, M.; Brimacombe, R.; Trupin, J.; Rottman, F.; O’Neal, C. RNA codewords and protein synthesis, VII. On the general nature of the RNA code. Proc. Natl. Acad. Sci. USA 1965, 53, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L.; Itano, H.A.; Singer, S.J.; Wells, I.C. Sickle cell anemia, a molecular disease. Science 1949, 110, 543–548. [Google Scholar] [CrossRef]
- Ingram, V.M. A specific chemical difference between the globins of normal human and sickle-cell anaemia haemoglobin. Nature 1956, 178, 792–794. [Google Scholar] [CrossRef]
- Heydemann, A.; Siemionow, M. A brief review of Duchenne muscular dystrophy treatment options, with an emphasis on two novel strategies. Biomedicines 2023, 11, 830. [Google Scholar] [CrossRef]
- Krishna, L.; Prashant, A.; Kumar, Y.H.; Paneyala, S.; Patil, S.J.; Ramachandra, S.C.; Vishwanath, P. Molecular and biochemical therapeutic strategies for Duchenne muscular dystrophy. Neurol. Int. 2024, 16, 731–760. [Google Scholar] [CrossRef]
- Kuhn, A.; Thomas, J.A. The beauty of bacteriophage T4 research: Lindsay W. Black and the T4 head assembly. Viruses 2022, 14, 700. [Google Scholar] [CrossRef] [PubMed]
- Kutter, E.; Bryan, D.; Ray, G.; Brewster, E.; Blasdel, B.; Guttman, B. From host to phage metabolism: Hot tales of phage T4’s takeover of E. coli. Viruses 2018, 10, 387. [Google Scholar] [CrossRef] [PubMed]
- Edgar, B. The genome of bacteriophage T4: An archeological dig. Genetics 2004, 168, 575–582. [Google Scholar] [CrossRef]
- Wood, W.B. Bacteriophage T4 morphogenesis as a model for assembly of subcellular structure. Q. Rev. Biol. 1980, 55, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Hyman, P.; Abedon, S.T. Practical methods for determining phage growth parameters. Meth. Mol Biol. 2009, 501, 175–202. [Google Scholar] [CrossRef]
- Luria, S.E.; Delbrück, M. Mutations of bacteria from virus sensitivity to virus resistance. Genetics 1943, 28, 491–511. [Google Scholar] [CrossRef]
- Gabashvili, I.S.; Khan, S.A.; Hayes, S.J.; Serwer, P. Polymorphism of bacteriophage T7. J. Mol. Biol. 1997, 273, 658–667. [Google Scholar] [CrossRef]
- d’Herelle, F. Bacteriophage as a treatment in acute medical and surgical infections. Bull. N. Y. Acad. Med. 1931, 7, 329–348. [Google Scholar] [PubMed]
- Luria, S.E.; Latarjet, R. Ultraviolet irradiation of bacteriophage during intracellular growth. J. Bacteriol. 1947, 53, 149–163. [Google Scholar] [CrossRef]
- Hyman, P. The genetics of the Luria-Latarjet effect in bacteriophage T4: Evidence for the involvement of multiple DNA repair pathways. Genet. Res. 1993, 62, 1–9. [Google Scholar] [CrossRef]
- Thomson, J.J. On the scattering of rapidly moving electrified particles. Proc. Cambridge Phil. Soc. 1910, 15, 465–471. [Google Scholar]
- Rutherford, E. The structure of the atom. Nature 1913, 92, 423. [Google Scholar] [CrossRef]
- Svidzinsky, A.A.; Scully, M.O.; Herschbach, D.R. Bohr’s 1913 molecular model revisited. Proc. Natl. Acad. Sci. USA 2005, 102, 11985–11988. [Google Scholar] [CrossRef]
- Available online: https://uh.edu/~chem1p/c7/c7F99.pdf (accessed on 17 February 2025).
- Stent, G.S. A short epistemology of bacteriophage multiplication. Biophys. J. 1962, 2, 13–23. [Google Scholar] [CrossRef]
- Delbrück, M. A physicist looks at biology. In Phage and the Origins of Molecular Biology; Cairns, J., Stent, G.S., Watson, J.D., Eds.; Cold Spring Laboratory of Quantitative Biology: Cold Spring Harbor, NY, USA, 1966; pp. 9–22. [Google Scholar]
- Strauss, B.S. A physicist’s quest in biology: Max Delbrück and “complementarity”. Genetics 2017, 206, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Bridges, W.B. Archives California Institute of Technology, Pasadena, California, Electrical Engineering, Applied Physics. Available online: https://oralhistories.library.caltech.edu/117/1/OH_Bridges_W.pdf (accessed on 17 February 2025).
- Edgar, R.S.; Feynman, R.P.; Klein, S.; Lielausis, I.; Steinberg, C.M. Mapping experiments with r mutants of bacteriophage T4D. Genetics 1962, 47, 179–186. [Google Scholar] [CrossRef]
- Feynman, R.P. (Notes Taken and Transcribed by John T. Neer). Feynman Hughes Lectures, Volume 4, Biology, Organic Chemistry and Microbiology 1966. Available online: http://www.thehugheslectures.info/wp-content/uploads/lectures/FeynmanHughesLectures_Vol4.pdf (accessed on 17 February 2025).
- Barrio, J.R. Consensus science and the peer review. Mol. Imaging Biol. 2009, 11, 293. [Google Scholar] [CrossRef] [PubMed]
- Boualam, M.A.; Pradines, B.; Drancourt, M.; Barbieri, R. Malaria in Europe: A historical perspective. Front. Med. 2021, 8, 691095. [Google Scholar] [CrossRef]
- Astrachan, L.; Volkin, E. Properties of ribonucleic acid turnover in T2-infected Escherichia coli. Biochim. Biophys. Acta. 1958, 29, 536–544. [Google Scholar] [CrossRef]
- Nomura, M.; Hall, B.D.; Spiegelman, S. Characterization of RNA synthesized in Escherichia coli after bacteriophage T2 infection. J. Mol. Biol. 1960, 2, 306–326. [Google Scholar] [CrossRef]
- Cobb, M. Who discovered messenger RNA? Curr. Biol. 2015, 25, R526–R532. [Google Scholar] [CrossRef] [PubMed]
- Murray, N.E. The impact of phage lambda: From restriction to recombineering. Biochem. Soc. Trans. 2006, 34 (Pt 2), 203–207. [Google Scholar] [CrossRef]
- Gottesman, M. Bacteriophage lambda: The untold story. J. Mol. Biol. 1999, 293, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Visconti, N.; Delbrück, M. The mechanism of genetic recombination in phage. Genetics 1953, 38, 5–33. [Google Scholar] [CrossRef] [PubMed]
- Nafissi, N.; Slavcev, R. Bacteriophage recombination systems and biotechnical applications. Appl. Microbiol. Biotechnol. 2014, 98, 2841–2851. [Google Scholar] [CrossRef]
- Hillyar, C.R.T. Genetic recombination in bacteriophage lambda. Biosci. Horizons Int. J. Student Res. 2012, 5, hzs001. [Google Scholar] [CrossRef]
- Bull, J.J.; Wichman, H.A.; Krone, S.M.; Molineux, I.J. Controlling recombination to evolve bacteriophages. Cells 2024, 13, 585. [Google Scholar] [CrossRef]
- Muniyappa, K.; Radding, C.M. The homologous recombination system of phage lambda. Pairing activities of beta protein. J. Biol. Chem. 1986, 261, 7472–7478. [Google Scholar] [CrossRef] [PubMed]
- Brewster, J.L.; Tolun, G. Half a century of bacteriophage lambda recombinase: In vitro studies of lambda exonuclease and Red-beta annealase. IUBMB Life 2020, 72, 1622–1633. [Google Scholar] [CrossRef]
- Mosig, G.; Gewin, J.; Luder, A.; Colowick, N.; Vo, D. Two recombination-dependent DNA replication pathways of bacteriophage T4, and their roles in mutagenesis and horizontal gene transfer. Proc. Natl. Acad. Sci. USA 2001, 98, 8306–8311. [Google Scholar] [CrossRef]
- Kreuzer, K.N. Recombination-dependent DNA replication in phage T4. Trends Biochem. Sci. 2000, 25, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Serwer, P. Fast sedimenting bacteriophage T7 DNA from T7-infected Escherichia coli. Virology 1974, 59, 70–88. [Google Scholar] [CrossRef]
- Leiman, P.G.; Arisaka, F.; van Raaij, M.J.; Kostyuchenko, V.A.; Aksyuk, A.A.; Kanamaru, S.; Rossmann, M.G. Morphogenesis of the T4 tail and tail fibers. Virol. J. 2010, 7, 355. [Google Scholar] [CrossRef] [PubMed]
- Wood, W.B.; Eiserling, F.A.; Crowther, R.A. Long tail fibers: Genes, proteins, structure, and assembly. In Molecular Biology of Bacteriophage T4; Karam, J.D., Ed.; American Society for Microbiology: Washington, DC, USA, 1994; pp. 282–290. [Google Scholar]
- Katsura, I.; Hendrix, R.W. Length determination in bacteriophage lambda tails. Cell 1984, 3 Pt 2, 691–698. [Google Scholar] [CrossRef]
- Katsura, I. Determination of bacteriophage lambda tail length by a protein ruler. Nature 1987, 327, 73–75. [Google Scholar] [CrossRef]
- Levin, M.E.; Hendrix, R.W.; Casjens, S.R. A programmed translational frameshift is required for the synthesis of a bacteriophage lambda tail assembly protein. J. Mol. Biol. 1993, 234, 124–139. [Google Scholar] [CrossRef] [PubMed]
- Pedulla, M.L.; Ford, M.E.; Houtz, J.M.; Karthikeyan, T.; Wadsworth, C.; Lewis, J.A.; Jacobs-Sera, D.; Falbo, J.; Gross, J.; Pannunzio, N.R.; et al. Origins of highly mosaic mycobacteriophage genomes. Cell 2003, 113, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Wang, K. Titin/connectin and nebulin: Giant protein rulers of muscle structure and function. Adv. Biophys. 1996, 33, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Tskhovrebova, L.; Trinick, J. Titin and nebulin in thick and thin filament length regulation. Subcell. Biochem. 2017, 82, 285–318. [Google Scholar] [CrossRef]
- Matos-Rodrigues, G.; Guirouilh-Barbat, J.; Martini, E.; Lopez, B.S. Homologous recombination, cancer and the ‘RAD51 paradox’. NAR Cancer 2021, 3, zcab016. [Google Scholar] [CrossRef]
- Waters, K.L.; Spratt, D.E. New discoveries on protein recruitment and regulation during the early stages of the DNA damage response pathways. Int. J. Mol. Sci. 2024, 25, 1676. [Google Scholar] [CrossRef]
- San Filippo, J.; Sung, P.; Klein, H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008, 77, 229–257. [Google Scholar] [CrossRef] [PubMed]
- Socol, Y.; Shaki, Y.Y.; Yanovskiy, M. Interests, bias, and consensus in science and regulation. Dose-Response 2019, 17, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, H.; Li, R.; Sterling, K.; Song, W. Amyloid β-based therapy for Alzheimer’s disease: Challenges, successes and future. Sig. Transduct. Target Ther. 2023, 8, 248. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Hong, F.; Yang, S. Amyloidosis in Alzheimer’s disease: Pathogeny, etiology, and related therapeutic directions. Molecules 2022, 27, 1210. [Google Scholar] [CrossRef]
- Herrup, K. The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 2015, 18, 794–799. [Google Scholar] [CrossRef]
- Mullane, K.; Williams, M. Alzheimer’s disease (AD) therapeutics—1: Repeated clinical failures continue to question the amyloid hypothesis of AD and the current understanding of AD causality. Biochem. Pharmacol. 2018, 158, 359–375. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer Disease and aducanumab: Adjusting our approach. Nat. Rev. Neurol. 2019, 15, 365–366. [Google Scholar] [CrossRef]
- Gottfried, J. History Repeating? Avoiding a Return to the Pre-Antibiotic Age. Available online: http://nrs.harvard.edu/urn-3:HUL.InstRepos:8889467 (accessed on 22 February 2025).
- Baldry, P. The Battle Against Bacteria: A Fresh Look, 2nd ed.; Cambridge University Press: Cambridge, UK, 1976. [Google Scholar]
- Hays, J.P.; Ruiz-Alvarez, M.J.; Roson-Calero, N.; Amin, R.; Murugaiyan, J.; van Dongen, M.B.M. Global AMR insights ambassador network. Perspectives on the ethics of antibiotic overuse and on the implementation of (new) antibiotics. Infect. Dis. Ther. 2022, 11, 1315–1326. [Google Scholar] [CrossRef]
- Capuozzo, M.; Zovi, A.; Langella, R.; Ottaiano, A.; Cascella, M.; Scognamiglio, M.; Ferrara, F.O. Optimizing antibiotic use: Addressing resistance through effective strategies and health policies. Antibiotics 2024, 13, 1112. [Google Scholar] [CrossRef] [PubMed]
- Dutescu, I.A.; Hillier, S.A. Encouraging the development of new antibiotics: Are financial incentives the right way forward? A systematic review and case study. Infect. Drug Resist. 2021, 14, 415–434. [Google Scholar] [CrossRef]
- Pfizer. 2022 Annual Review: Breakthroughs Changing More than 1.3 Billion Lives. Available online: https://www.pfizer.com/sites/default/files/investors/financial_reports/annual_reports/2022/files/Pfizer_Annual_Review.pdf (accessed on 23 February 2025).
- Azevedo, M.; Mullis, L.; Agnihothram, S. Viral and bacterial co-infection and its implications. SciFed. Virol. Res. J. 2017, 1. [Google Scholar] [CrossRef]
- Liu, Y.; Ling, L.; Wong, S.H.; Wang, M.H.; Fitzgerald, J.R.; Zou, X.; Fang, S.; Liu, X.; Wang, X.; Hu, W.; et al. Outcomes of respiratory viral-bacterial co-infection in adult hospitalized patients. EClinicalMedicine 2021, 37, 100955. [Google Scholar] [CrossRef]
- Centers for Diseases Control and Prevention, US. Department of Health and Human Services. Antibiotic Resistance Threats in the United States 2019, p.40. Available online: https://www.cdc.gov/antimicrobial-resistance/media/pdfs/2019-ar-threats-report-508.pdf (accessed on 24 February 2025).
- Shi, Z.; Zhang, J.; Tian, L.; Xin, L.; Liang, C.; Ren, X.; Li, M. A Comprehensive overview of the antibiotics approved in the last two decades: Retrospects and prospects. Molecules 2023, 28, 1762. [Google Scholar] [CrossRef]
- Rusu, A.; Munteanu, A.-C.; Arbănași, E.-M.; Uivarosi, V. Overview of side-effects of antibacterial fluoroquinolones: New drugs versus old drugs, a step forward in the safety profile? Pharmaceutics 2023, 15, 804. [Google Scholar] [CrossRef] [PubMed]
- Scavone, C.; Mascolo, A.; Ruggiero, R.; Sportiello, L.; Rafaniello, C.; Berrino, L.; Capuano, A. Quinolones-induced musculoskeletal, neurological, and psychiatric ADRs: A pharmacovigilance study based on data from the Italian spontaneous reporting system. Front. Pharmacol. 2020, 11, 428. [Google Scholar] [CrossRef]
- Stahlmann, R.; Lode, H.M. Risks associated with the therapeutic use of fluoroquinolones. Expert Opin. Drug Saf. 2013, 12, 497–505. [Google Scholar] [CrossRef]
- Sulakvelidze, A.; Alavidze, Z.; Morris, J.G., Jr. Bacteriophage therapy. Antimicrob. Agents Chemother. 2001, 45, 649–659. [Google Scholar] [CrossRef]
- Liu, D.; Van Belleghem, J.D.; de Vries, C.R.; Burgener, E.; Chen, Q.; Manasherob, R.; Aronson, J.R.; Amanatullah, D.F.; Tamma, P.D.; Suh, G.A. The safety and toxicity of phage therapy: A review of animal and clinical studies. Viruses 2021, 13, 1268. [Google Scholar] [CrossRef]
- Stacey, H.J.; De Soir, S.; Jones, J.D. The safety and efficacy of phage therapy: A systematic review of clinical and safety trials. Antibiotics 2022, 11, 1340. [Google Scholar] [CrossRef] [PubMed]
- Sawa, T.; Moriyama, K.; Kinoshita, M. Current status of bacteriophage therapy for severe bacterial infections. J. Intensive Care 2024, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Olsen, N.S.; Hendriksen, N.B.; Hansen, L.H.; Kot, W. A new high-throughput screening method for phages: Enabling crude isolation and fast identification of diverse phages with therapeutic potential. Phage 2020, 1, 137–148. [Google Scholar] [CrossRef]
- Serwer, P.; Hayes, S.J.; Zaman, S.; Lieman, K.; Rolando, M.; Hardies, S.C. Improved isolation of under sampled bacteriophages: Finding of distant terminase genes. Virology 2004, 329, 412–424. [Google Scholar] [CrossRef]
- Dedrick, R.M.; E. Smith, B.; Cristinziano, M.; Freeman, K.G.; Jacobs-Sera, D.; Belessis, Y.; Brown, A.W.; Cohen, K.; Davidson, R.M.; van Duin, D.; et al. Phage therapy of Mycobacterium infections: Compassionate use of phages in 20 patients with drug-resistant mycobacterial disease. Clin. Infect. Dis. 2023, 76, 103–112. [Google Scholar] [CrossRef]
- Chambers, J.P.; Aldis, M.; Thomas, J.A.; Gonzales, C.B.; White, R.A., III; Serwer, P. Biophysical breakthroughs projected for the phage therapy of bacterial disease. Biophysica 2024, 4, 195–206. [Google Scholar] [CrossRef]
- Rojero, M.; Weaver-Rosen, M.; Serwer, P. Bypassing evolution of bacterial resistance to phages: The example of hyper-aggressive phage 0524phi7-1. Int. J. Mol. Sci. 2025, 26, 2914. [Google Scholar] [CrossRef] [PubMed]
- Montero, D.A.; Vidal, R.M.; Velasco, J.; Carreño, L.J.; Torres, J.P.; Benachi O., M.A.; Tovar-Rosero, Y.Y.; Oñate, A.A.; O’Ryan, M. Two centuries of vaccination: Historical and conceptual approach and future perspectives. Front. Public Health 2024, 11, 1326154. [Google Scholar] [CrossRef] [PubMed]
- Offit, P.A. Vaccinated: One Man’s Quest to Defeat the World’s Deadliest Diseases; HarperCollins: New York, NY, USA, 2007. [Google Scholar]
- Claesson, B.A.; Trollfors, B.; Lagergard, T.; Taranger, J.; Bryla, D.; Otterman, G.; Cramton, T.; Yang, Y.; Reimer, C.B.; Robbins, J.B.; et al. Clinical and immunologic responses to the capsular polysaccharide of Haemophilus influenzae type b alone or conjugated to tetanus toxoid in 18- to 23-month-old children. J. Pediatr. 1988, 112, 695–702. [Google Scholar] [CrossRef]
- Tietz, D.; Aldroubi, A.; Schneerson, R.; Unser, M.; Chrambach, A. The distribution of particles characterized by size and free mobility within polydisperse populations of protein-polysaccharide conjugates, determined from two-dimensional agarose electropherograms. Electrophoresis 1991, 12, 46–54. [Google Scholar] [CrossRef]
- Hess, K.L.; Jewell, C.M. Phage display as a tool for vaccine and immunotherapy development. Bioeng. Transl. Med. 2019, 5, e10142. [Google Scholar] [CrossRef] [PubMed]
- Palma, M. Aspects of phage-based vaccines for protein and epitope immunization. Vaccines 2023, 11, 436. [Google Scholar] [CrossRef] [PubMed]
- Peterson, E.; Söderström, B.; Prins, N.; Le, G.H.B.; Hartley-Tassell, L.E.; Evenhuis, C.; Grønnemose, R.; Andersen, T.E.; Møller-Jensen, J.; Iosifidis, G.; et al. The role of bacterial size, shape and surface in macrophage engulfment of uropathogenic E. coli cells. PLoS Pathog. 2024, 20, e1012458. [Google Scholar] [CrossRef] [PubMed]
- Baranov, M.V.; Kumar, M.; Sacanna, S.; Thutupalli, S.; van den Bogaart, G. Modulation of immune responses by particle size and shape. Front. Immunol. 2021, 11, 607945. [Google Scholar] [CrossRef]
- NIH, National Cancer Institute. Study Identifies Hundreds of Potential Targets for Cancer Drugs. Available online: https://www.cancer.gov/news-events/cancer-currents-blog/2024/new-cancer-drug-targets-from-proteogenomics-data (accessed on 26 February 2025).
- Garg, P.; Malhotra, J.; Kulkarni, P.; Horne, D.; Salgia, R.; Singhal, S.S. Emerging therapeutic strategies to overcome drug resistance in cancer cells. Cancers 2024, 16, 2478. [Google Scholar] [CrossRef]
- Eslami, M.; Memarsadeghi, O.; Davarpanah, A.; Arti, A.; Nayernia, K.; Behnam, B. Overcoming chemotherapy resistance in metastatic cancer: A comprehensive review. Biomedicines 2024, 12, 183. [Google Scholar] [CrossRef]
- Li, J.; Wang, Q.; Xia, G.; Adilijiang, N.; Li, Y.; Hou, Z.; Fan, Z.; Li, J. Recent advances in targeted drug delivery strategy for enhancing oncotherapy. Pharmaceutics 2023, 15, 2233. [Google Scholar] [CrossRef]
- Doostmohammadi, A.; Jooya, H.; Ghorbanian, K.; Gohari, S.; Dadashpou, M. Potentials and future perspectives of multi-target drugs in cancer treatment: The next generation anti-cancer agents. Cell Commun. Signal. 2024, 22, 228. [Google Scholar] [CrossRef]
- Hao, Y.; Li, B.; Huang, D.; Wu, S.; Wang, T.; Fu, L.; Liu, X. Developing a semi-supervised approach using a PU-learning-based data augmentation strategy for multitarget drug discovery. Int. J. Mol. Sci. 2024, 25, 8239. [Google Scholar] [CrossRef]
- Li, Y.; Yang, K.D.; Duan, H.Y.; Du, Y.N.; Ye, J.F. Phage-based peptides for pancreatic cancer diagnosis and treatment: Alternative approach. Front. Microbiol. 2023, 14, 1231503. [Google Scholar] [CrossRef]
- Ju, Z.; Sun, W. Drug delivery vectors based on filamentous bacteriophages and phage-mimetic nanoparticles. Drug Deliv. 2017, 24, 1898–1908. [Google Scholar] [CrossRef]
- Emencheta, S.C.; Onugwu, A.L.; Kalu, C.F.; Ezinkwo, P.N.; Eze, O.C.; Vila, M.M.D.C.; Balcão, V.M.; Attama, A.A.; Onuigbo, E.B. Bacteriophages as nanocarriers for targeted drug delivery and enhanced therapeutic effects. Mater. Adv. 2024, 5, 986–1016. [Google Scholar] [CrossRef]
- Gatto, M.S.; Johnson, M.P.; Najahi-Missaoui, W. Targeted liposomal drug delivery: Overview of the current applications and challenges. Life 2024, 14, 672. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, H.; Kouchak, M.; Mirveis, Z.; Hajipour, F.; Khodarahmi, M.; Rahbar, N.; Handali, S. What we need to know about liposomes as drug nanocarriers: An updated review. Adv. Pharm. Bull. 2023, 13, 7–23. [Google Scholar] [CrossRef]
- Barenholz, Y. Doxil®—The first FDA-approved nano-drug: From basics via CMC, cell culture and animal studies to clinical use. In Nanomedicines: Design, Delivery and Detection; Braddock, M., Ed.; The Royal Society of Chemistry: Cambridge, UK, 2016; Chapter 13; pp. 315–345. [Google Scholar]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46 Pt 1, 6387–6392. [Google Scholar] [PubMed]
- Ikeda-Imafuku, M.; Wang, L.L.; Rodrigues, D.; Shaha, S.; Zhao, Z.; Mitragotri, S. Strategies to improve the EPR effect: A mechanistic perspective and clinical translation. J. Control. Release 2022, 345, 512–536. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Cho, H.; Lim, D.K.; Joo, M.K.; Kim, K. Perspectives for improving the tumor targeting of nanomedicine via the EPR effect in clinical tumors. Int. J. Mol. Sci. 2023, 24, 10082. [Google Scholar] [CrossRef]
- Kakkar, A.; Traverso, G.; Farokhzad, O.C.; Weissleder, R.; Langer, R. Evolution of macromolecular complexity in drug delivery systems. Nat. Rev. Chem. 2017, 1, 0063. [Google Scholar] [CrossRef]
- Li, Y.; Ji, T.; Torre, M.; Shao, R.; Zheng, Y.; Wang, D.; Li, X.; Liu, A.; Zhang, W.; Deng, X.; et al. Aromatized liposomes for sustained drug delivery. Nat. Commun. 2023, 14, 6659. [Google Scholar] [CrossRef]
- Serwer, P.; Wright, E.T.; De La Chapa, J.; Gonzales, C.B. Basics for improved use of phages for therapy. Antibiotics 2021, 10, 723. [Google Scholar] [CrossRef]
- Serwer, P.; Wright, E.T. Gated ethidium- and bleomycin-loading in phage T4 that is subsequently purified leak-free. Biophysica 2022, 2, 366–380. [Google Scholar] [CrossRef]
- Koo, E.H.; Lansbury, P.T., Jr.; Kelly, J.W. Amyloid diseases: Abnormal protein aggregation in neurodegeneration. Proc. Natl. Acad. Sci. USA 1999, 96, 9989–9990. [Google Scholar] [CrossRef] [PubMed]
- Candelise, N.; Scaricamazza, S.; Salvatori, I.; Ferri, A.; Valle, C.; Manganelli, V.; Garofalo, T.; Sorice, M.; Misasi, R. Protein aggregation landscape in neurodegenerative diseases: Clinical relevance and future applications. Int. J. Mol. Sci. 2021, 22, 6016. [Google Scholar] [CrossRef] [PubMed]
- Bandea, C.I. Aβ, tau, α-synuclein, huntingtin, TDP-43, PrP and AA are members of the innate immune system: A unifying hypothesis on the etiology of AD, PD, HD, ALS, CJD and RSA as innate immunity disorders. bioRxiv 2013. Available online: http://biorxiv.org/content/early/2013/11/18/000604 (accessed on 27 February 2025).
- Ciurea, A.V.; Mohan, A.G.; Covache-Busuioc, R.-A.; Costin, H.-P.; Glavan, L.-A.; Corlatescu, A.-D.; Saceleanu, V.M. Unraveling molecular and genetic insights into neurodegenerative diseases: Advances in understanding Alzheimer’s, Parkinson’s, and Huntington’s diseases and amyotrophic lateral sclerosis. Int. J. Mol. Sci. 2023, 24, 10809. [Google Scholar] [CrossRef]
- Cairns, D.M.; Itzhaki, R.F.; Kaplan, D.L. Potential involvement of Varicella Zoster Virus in Alzheimer’s Disease via reactivation of quiescent Herpes Simplex Virus Type 1. J. Alzheimer’s Dis. 2022, 88, 1189–1200. [Google Scholar] [CrossRef]
- Itzhaki, R.F. Overwhelming evidence for a major role for Herpes Simplex Virus Type 1 (HSV1) in Alzheimer’s Disease (AD); Underwhelming evidence against. Vaccines 2021, 9, 679. [Google Scholar] [CrossRef]
- Itzhaki, R.F. Hypothesis: Does the apparent protective action of Green Valley’s drug GV971 against cognitive decline result from antiviral action against Herpes Simplex Virus type 1 in brain? J. Alzheimer’s Dis. 2020, 76, 85–87. [Google Scholar] [CrossRef]
- Eyting, M.; Xie, M.; Michalik, F.; Heß, S.; Chung, S.; Geldsetzer, P. A natural experiment on the effect of herpes zoster vaccination on dementia. Nature 2025. [Google Scholar] [CrossRef]
- Hammarström, P.; Nyström, S. Viruses and amyloids—A vicious liaison. Prion 2023, 17, 82–104. [Google Scholar] [CrossRef]
- Prosswimmer, T.; Heng, A.; Daggett, V. Mechanistic insights into the role of amyloid-β in innate immunity. Sci. Rep. 2024, 14, 5376. [Google Scholar] [CrossRef] [PubMed]
- Bi, T.M.; Daggett, V. The Role of α-sheet in amyloid oligomer aggregation and toxicity. Yale J. Biol. Med. 2018, 91, 247–255. [Google Scholar] [PubMed]
- Al-Beltagi, M.; Saeed, N.K.; Elbeltagi, R.; Bediwy, A.S.; Aftab, S.A.S.; Alhawamdeh, R. Viruses and autism: A bi-mutual cause and effect. World J. Virol. 2023, 12, 172–192. [Google Scholar] [CrossRef] [PubMed]
- Maltsev, D.; Solonko, I.; Sydorenko, O. The assessment of microbial infection in children with autism spectrum disorders and genetic folate cycle deficiency. BMC Pediatr. 2024, 24, 200. [Google Scholar] [CrossRef]
- Feynman, R.P. The Meaning of It All: Thoughts of a Citizen Scientist; Perseus Books: New York, NY, USA, 1998. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).