


Apoptosis Regulators Bcl-2 and Caspase-3


Definition
1. Introduction
1.1. Apoptosis
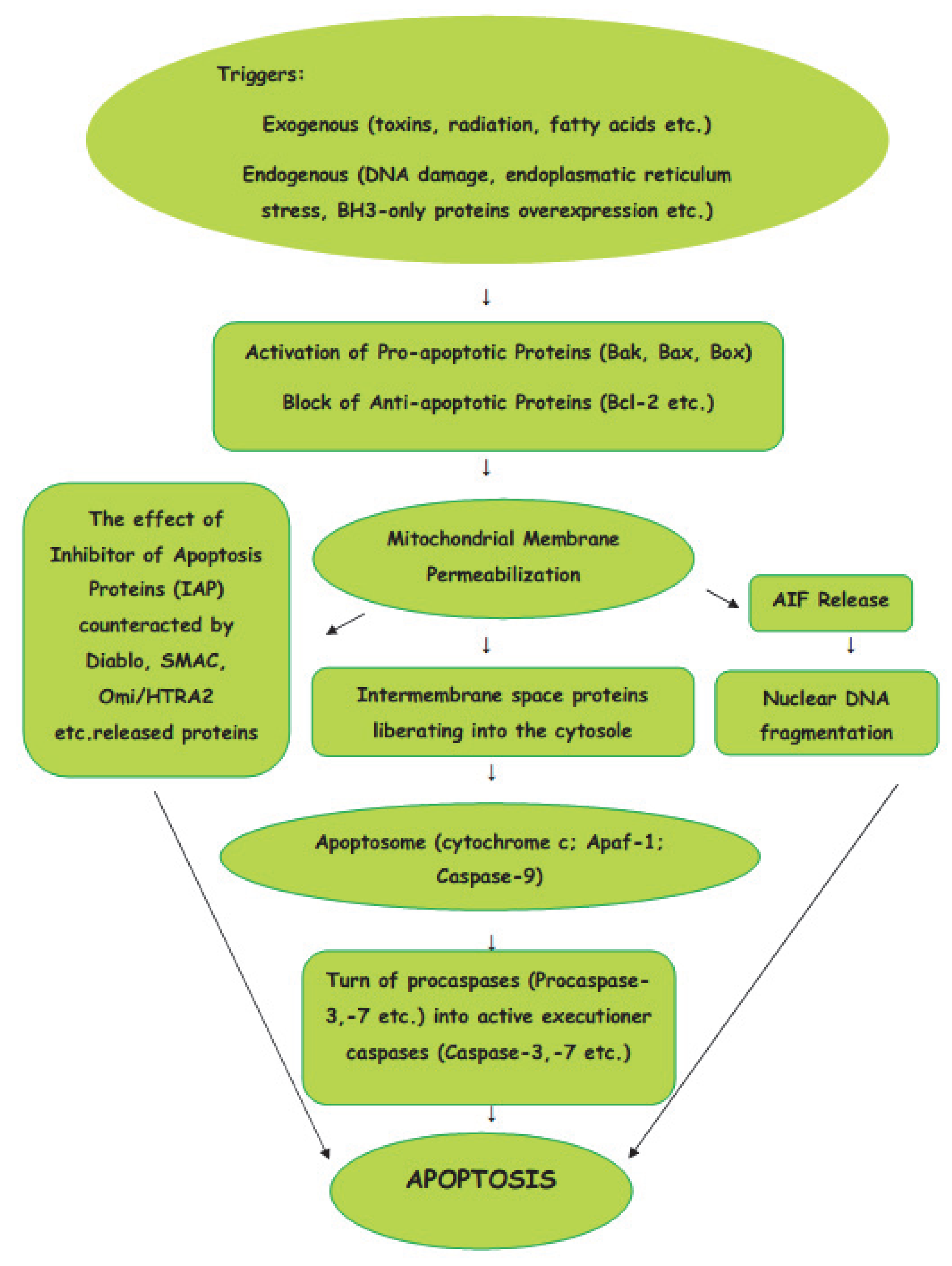
1.2. Intrinsic Pathway
1.3. Extrinsic Pathway
1.3.1. TNF Pathway
1.3.2. Fas Pathway
1.4. Other Pathways of Apoptosis
2. Apoptosis Regulators
2.1. Bcl-2
Apoptosis Regulator Bcl-2
2.2. Caspases
Caspase-3
3. Conclusions
Funding
Conflicts of Interest
References
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Apoptosis: Programmed cell death eliminates unwanted cells. In Molecular Biology of the Cell, 5th ed.; Alberts, B., Ed.; Garland Science: New York, NY, USA, 2008; p. 1115. [Google Scholar]
- Green, D. Means to an End: Apoptosis and Other Cell Death Mechanisms; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2011. [Google Scholar]
- Ucker, P.S. Death by suicide: One way to go in mammalian cellular development. New Biol. 1991, 3, 103–109. [Google Scholar] [PubMed]
- Wyllie, A.H. Cell death: A new classification separating apoptosis from necrosis. In Cell Death in Biology and Pathology; Bowen, I.D., Lockshin, R.A., Eds.; Chapman and Hall: London, UK, 1981; pp. 9–34. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Moujalled, D.; Strasser, A.; Liddell, J.R. Molecular mechanisms of cell death in neurological diseases. Cell Death Differ. 2021, 28, 2029–2044. [Google Scholar] [CrossRef] [PubMed]
- Wyllie, A.H. Apoptosis (the 1992 Frank Rose Memorial Lecture). Br. J. Cancer 1993, 67, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Jan, R.; Chaudhry, G.E. Understanding apoptosis and apoptotic pathways targeted cancer therapeutics. Adv. Pharm. Bull. 2019, 9, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Vaux, D.L. A boom time for necrobiology. Curr. Biol. 1993, 3, 877–878. [Google Scholar] [CrossRef]
- Milliman, C.L.; Korsmeyer, S.J.; Wang, K.; Yin, X.M.; Chao, D.T. BID: A Novel BH3 Domain-only Death Agonist. Genes Dev. 1996, 10, 2859–2869. [Google Scholar] [CrossRef]
- Arvanitis, M.; Li, D.D.; Lee, K.; Mylonakis, E. Apoptosis in C. elegans: Lessons for cancer and immunity. Front. Cell. Infect. Microbiol. 2013, 3, 67. [Google Scholar] [CrossRef]
- Raychaudhuri, S. A minimal model of signalling network elucidates cell-to cell stochastic variability in apoptosis. PLoS ONE 2010, 5, e11930. [Google Scholar] [CrossRef]
- Lossi, L. The concept of intrinsic versus extrinsic apoptosis. Biochem. J. 2022, 479, 357–384. [Google Scholar] [CrossRef]
- Wang, Z.; Figueiredo-Pereira, C.; Oudot, C.; Vieira, H.L.A.; Brenner, C. Mitochondrion: A common organelle for distinct cell deaths? Int. Rev. Cell Mol. Biol. 2017, 331, 245–287. [Google Scholar] [PubMed]
- Urbani, A.; Prosdocimi, E.; Carrer, A.; Checchetto, V.; Szabò, I. Mitochondrial ion channels of the inner membrane and their regulation in cell death signaling. Front. Cell Dev. Biol. 2021, 8, 620081. [Google Scholar] [CrossRef] [PubMed]
- Cagnol, S.; Mansour, A.; Van Obberghen-Schilling, E.; Chambard, J.C. Raf-1 activation prevents caspase 9 processing downstream of apoptosome formation. J. Signal Transduct. 2011, 2011, 834948. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H. The Fas signaling pathway: More than a paradigm. Science 2002, 296, 1635–1636. [Google Scholar] [CrossRef] [PubMed]
- Azzwali, A.A.A.; Azab, A.E. Apoptosis: Insight into Stages, Extrinsic and Intrinsic Pathways. Clin. Med. 2019, 7, 80–82. [Google Scholar]
- Muntané, J. Harnessing tumor necrosis factor receptors to enhance antitumor activities of drugs. Chem. Res. Toxicol. 2011, 24, 1610–1616. [Google Scholar] [CrossRef] [PubMed]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [CrossRef]
- Aral, K.; Aral, C.A.; Kapila, Y. The role of caspase-8, caspase-9, and apoptosis inducing factor in periodontal disease. J. Periodontol. 2019, 90, 288–294. [Google Scholar] [CrossRef]
- Slee, E.A.; Adrain, C.; Martin, S.J. Executioner caspase-3, -6, and-7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J. Biol. Chem. 2001, 276, 7320–7326. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, J.; Guo, Z.; Shi, X.; Zhang, Y.; Zhang, L.; Yu, Q.; Han, L. Effect of Oxidative Stress on AIF-mediated Apoptosis and Bovine Muscle Tenderness during Postmortem Aging. J. Food Sci. 2020, 85, 77–85. [Google Scholar] [CrossRef]
- Talanian, R.V.; Allen, H. Roles of caspases in inflammation and apoptosis: Prospects as drug discovery targets. Annu. Rep. Med. Chem. 1998, 33, 273–282. [Google Scholar]
- Bojarska-Junak, A.; Sieklucka, M.; Hus, I.; Wąsik-Szczepanek, E.; Kusz, M.L.; Surdacka, A.; Chocholska, S.; Dmoszyńska, A.; Roliński, J. Assessment of the pathway of apoptosis involving PAR-4, DAXX and ZIPK proteins in CLL patients and its relationship with the principal prognostic factors. Folia Histochem. Cytobiol. 2011, 49, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Dey, D.K.; Chang, S.N.; Vadlamudi, Y.; Park, J.G.; Kang, S.C. Synergistic therapy with tangeretin and 5-fluorouracil accelerates the ROS/JNK mediated apoptotic pathway in human colorectal cancer cell. Food Chem. Toxicol. 2020, 143, 111529. [Google Scholar] [CrossRef] [PubMed]
- Zamzami, N.; Brenner, C.; Marzo, I.; Susin, S.A.; Kroemer, G. Subcellular and submitochondrial mode of action of Bcl-2 like oncoproteins. Oncogene 1998, 16, 2265–2282. [Google Scholar] [CrossRef] [PubMed]
- Nalepa, G.; Zukowska-Szczechowska, E. Caspases and apoptosis: Die and let live. Wiad. Lec. 2002, 55, 100–106. [Google Scholar]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Hussar, P.; Žuravskaja, M.; Kärner, M. Apoptosis regulator BCL-2. Pap. Anthropol. 2013, XXII, 63–67. [Google Scholar] [CrossRef]
- Reed, J.C.; Zha, H.; Aime-Sempe, C.; Takayama, S.; Wang, H.G. Structure-function analysis of Bcl-2 family proteins. Regulators of programmed cell death. Adv. Exp. Med. Biol. 1996, 406, 99–112. [Google Scholar] [CrossRef]
- Banjara, S.; Suraweera, C.D.; Hinds, M.G.; Kvansakul, M. The Bcl-2 Family: Ancient Origins, Conserved Structures, and Divergent Mechanisms. Biomolecules 2020, 10, 128. [Google Scholar] [CrossRef]
- Kale, J.; Osterlund, E.; Andrews, D. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef]
- Boise, L.H.; González-García, M.; Postema, C.E.; Ding, L.; Lindsten, T.; Turka, L.A.; Mao, X.; Nuñez, G.; Thompson, C.B. Bcl-x, a bcl-2 related gene that functions as a dominant regulator of apoptotic cell death. Cell 1993, 74, 597–608. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Shimizu, S. Bcl-2 family: Life-or-death switch. FEBS Lett. 2000, 466, 6–10. [Google Scholar] [CrossRef]
- Green, D. Apoptotic pathways: Paper wraps stone blunts scissors. Cell 2000, 102, 1–4. [Google Scholar] [CrossRef]
- Martinou, J.-C.; Green, R.D. Breaking the mitochondrial barrier. Nat. Rev. Mol. Cell Biol. 2001, 2, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y.; Shimizu, S. VDAC regulation by the Bcl-2 family of proteins. Cell Death Differ. 2000, 7, 1174–1181. [Google Scholar] [CrossRef] [PubMed]
- Zamzami, N.; Kroemer, G. The mitochondrion in apoptosis: How Pandora’s box opens. Nat. Rev. Mol. Cell Biol. 2001, 2, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibtion. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef]
- Samali, A.; Cai, J.; Zhivotovsky, B.; Jones, D.P.; Orrenius, S. Presence of a pre-apoptotic complex of pro- caspase -3, Hsp60 and Hsp10 in the mitochondrial fraction of jurkat cells. EMBO J. 1999, 18, 2040–2048. [Google Scholar] [CrossRef]
- Tsujimoto, Y. Bcl-2 Family of Proteins: Life-or-Death Switch in Mitochondria. Biosci. Rep. 2002, 22, 47–58. [Google Scholar] [CrossRef]
- Hippenstiel, S.; Schmeck, B.; N’Guessan, P.D.; Seybold, J.; Krüll, M.; Preissner, K.; Eichel-Streiber, C.V.; Suttorp, N. Rho protein inactivation induced apoptosis of cultured human endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2002, 283, L830–L838. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Bouchier-Hayes, L.; Green, D.R. Mitochondrial outer membrane permeabilization during apoptosis: The innocent bystander scenario. Cell Death Differ. 2006, 13, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Vo, T.T.; Letai, A. BH3-only proteins and their effects on cancer. Adv. Exp. Med. Biol 2010, 687, 49–63. [Google Scholar] [CrossRef]
- MacManus, J.P.; Matthew, D. Gene expression induced by cerebral ischemia: An apoptotic perspective. J. Cereb. Blood Flow Metab. 1997, 17, 815–832. [Google Scholar] [CrossRef] [PubMed]
- Muchmore, S.W.; Sattler, M.; Liang, H.; Meadows, R.P.; Harlan, J.E.; Yoon, H.S.; Nettesheim, D.; Chang, B.S.; Thompson, C.B.; Wong, S.-L.; et al. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature 1996, 381, 335–341. [Google Scholar] [CrossRef]
- Minn, A.J.; Velez, P.; Schendel, S.L.; Liang, H.; Muchmore, S.W.; Fesik, S.W.; Fill, M.; Thompson, C.B. Bcl-x(L) forms an ion channel in synthetic lipid membranes. Nature 1997, 385, 353–357. [Google Scholar] [CrossRef]
- Horvitz, H.R. Genetic control of programmed cell death in the nematode Caenorhabditis elegans. Cancer Res. 1999, 59, 1701s–1706s. [Google Scholar]
- Tsujimoto, Y.; Finger, L.R.; Yunis, J.; Nowell, P.C.; Croce, C.M. Cloning of the chromosome breakpoint of neoplastic B cells with the t (14;18) chromosome translocation. Science 1984, 226, 1097–1099. [Google Scholar] [CrossRef]
- Cleary, M.L.; Smith, S.D.; Sklar, J. Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl-2/immunoglobulin transcript resulting from the (14;18) translocation. Cell 1986, 47, 19–28. [Google Scholar] [CrossRef]
- Willis, S.; Day, C.L.; Hinds, M.G.; Huang, D.C.S. The Bcl-2-regulated apoptotic pathway. J Cell Sci. 2003, 116, 4053–4056. [Google Scholar] [CrossRef]
- Otake, Y.; Soundararajan, S.; Sengupta, T.K.; Kio, E.A.; Smith, J.C.; Pineda-Roman, M.; Stuart, R.K.; Spicer, E.K.; Fernandes, D.J. Overexpression of nucleolin in chronic lymphocytic leukemia cells induces stabilization of bcl2 mRNA. Blood 2007, 109, 3069–3075. [Google Scholar] [CrossRef] [PubMed]
- Handschuh, L.; Wojciechowski, P.; Kazmierczak, M.; Lewandowski, K. Transcript-Level Dysregulation of BCL2 Family Genes in Acute Myeloblastic Leukemia. Cancers 2021, 13, 3175. [Google Scholar] [CrossRef] [PubMed]
- Glantz, L.A.; Gilmore, J.H.; Lieberman, J.A.; Jarskog, L.F. Apoptotic mechanisms and the synaptic pathology of schizophrenia. Schizophr. Res. 2006, 81, 47–63. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Finger, L.R.; Richardson, A.; Kaye, S.B. Pharmacological inhibition of the Bcl-2 family of apoptosis regulators as cancer therapy. Curr. Mol. Pharmacol. 2008, 1, 244–254. [Google Scholar] [CrossRef]
- Charlotte, F.; L’Herminé, A.; Martin, N.; Geleyn, Y.; Nollet, M.; Gaulard, P.; Zafrani, E.S. Immunohistochemical detection of bcl-2 protein in normal and pathological human liver. Am. J. Pathol. 1994, 144, 460–465. [Google Scholar]
- Patel, T.; Gores, G.J. Apoptosis and hepatobiliary disease. Hepatology 1995, 21, 1725–1741. [Google Scholar] [CrossRef]
- Shojaie, L.; Iorga, A.; Dara, L. Cell Death in Liver Diseases: A Review. Int. J. Mol. Sci. 2020, 21, 9682. [Google Scholar] [CrossRef]
- Frommel, T.O.; Yong, S.; Zarling, E.J. Immunohistochemical evaluation of Bcl-2 gene family expression in liver of hepatitis C and cirrhotic patients: A novel mechanism to explain the high incidence of hepatocarcinoma in cirrhotics. Am. J. Gastroenterol. 1999, 94, 178–182. [Google Scholar] [CrossRef]
- Tsamandas, A.C.; Thomopoulos, K.; Zolota, V.; Kourelis, T.; Karatzas, T.; Ravazoula, P.; Tepetes, K.; Petsas, T.; Karavias, D.; Karatza, C.; et al. Potential Role of Bcl-2 and Bax mRNA and Protein Expression in Chronic Hepatitis Type B and C: A Clinicopathologic Study. Mod. Pathol. 2003, 16, 1273–1288. [Google Scholar] [CrossRef]
- Tokin, I.I.; Hussar, P.; Tokin, I.B.; Filimonova, G.; Zuravaskaja, M.; Tisler, A.; Järveots, T. Immunohiostochemical evaluation of expression of Bcl-2 in chronic viral hepatitis B. BJVM 2012, 15, 66. [Google Scholar]
- Arbel, N.; Shoshan-Barmatz, V. Voltage-dependent Anion Channel 1-based Peptides Interact with Bcl-2 to Prevent Antiapoptotic Activity. J. Biol. Chem. 2010, 285, 6053–6062. [Google Scholar] [CrossRef] [PubMed]
- Antonsson, B.; Montessuit, S.; Lauper, S.; Eskes, R.; Martinou, J.C. Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem J. 2000, 345 Pt 2, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Fesik, S.W.; Shi, Y. Structural biology. Controlling the caspases. Science 2001, 294, 1477–1478. [Google Scholar] [CrossRef] [PubMed]
- Thornberry, N.A.; Lazebnik, Y. Caspases: Enemies Within. Science 1998, 281, 1312–1326. [Google Scholar] [CrossRef]
- Galluzzi, L.; López-Soto, A.; Kumar, S.; Kroemer, G. Caspases Connect Cell-Death Signaling to Organismal Homeostasis. Immunity 2016, 44, 221–231. [Google Scholar] [CrossRef]
- Ahmad, M.; Srinivasula, S.M.; Hegde, R.; Mukattash, R.; Fernandes-Alnemri, T.; Alnemri, E.S. Identification and characterization of murine caspase-14, a new member of the caspase family. Cancer Res. 1998, 58, 5201–5205. [Google Scholar]
- Markiewicz, A.; Sigorski, D.; Markiewicz, M.; Owczarczyk-Saczonek, A.; Placek, W. Caspase-14—From Biomolecular Basics to Clinical Approach. A Review of Available Data. Int. J. Mol. Sci. 2021, 22, 5575. [Google Scholar] [CrossRef]
- Brenner, C.; Grimm, S. The permeability transition pore complex in cancer cell death. Oncogene 2006, 25, 4744–4756. [Google Scholar] [CrossRef]
- Fuentes-Prior, P.; Salvesen, G.S. The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem. J. 2004, 384 Pt 2, 201–232. [Google Scholar] [CrossRef]
- Shalini, S.; Dorstyn, L.; Dawar, S.; Kumar, S. Old, new and emerging functions of caspases. Cell Death Differ. 2015, 22, 526–539. [Google Scholar] [CrossRef]
- Goodsell, D.S. The molecular perspective: Caspases. Oncologist 2000, 5, 435–436. [Google Scholar] [CrossRef] [PubMed]
- Bantel, H.; Ruck, P.; Schulze-Osthoff, K. In situ monitoring of caspase activation in hepatobiliary diseases. Cell Death Differ. 2000, 7, 504–505. [Google Scholar] [CrossRef] [PubMed]
- Bantel, H.; Lügering, A.; Poremba, C.; Lügering, N.; Held, J.; Domschke, W.; Schulze-Osthoff, K. Caspase activation correlates with the degree of inflammatory liver injury in chronic hepatitis C virus infection. Hepatology 2001, 34 Pt 1, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Bantel, H.; Ruck, P.; Gregor, M.; Schulze-Osthoff, K. Detection of elevated caspase activation and early apoptosis in liver diseases. Eur. J. Cell Biol. 2001, 80, 230–239. [Google Scholar] [CrossRef]
- Bantel, H.; Lügering, A.; Heidemann, J.; Volkmann, X.; Poremba, C.; Strassburg, C.P.; Manns, M.P.; Schulze-Osthoff, K. Detection of apoptotic caspase activation in sera from patients with chronic HCV infection is associated with fibrotic liver injury. Hepatology 2004, 40, 1078–1087. [Google Scholar] [CrossRef]
- Wieckowska, A.; Zein, N.N.; Yerian, L.M.; Lopez, A.R.; McCullough, A.J.; Feldstein, A.E. In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease. Hepatology 2006, 44, 27–33. [Google Scholar] [CrossRef]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef]
- Lahm, A.; Paradisi, A.; Green, D.R.; Melino, G. Death fold domain interaction in apoptosis. Cell Death Differ. 2003, 10, 10–12. [Google Scholar] [CrossRef]
- Gu, J.; Zhan, A.J.; Jiang, J.L.; Chen, Y.; Xu, J.; Ye, L.; Mao, M.G. Conserved function of Pacific cod Caspase-3 in apoptosis. Gene 2020, 732, 144370. [Google Scholar] [CrossRef]
- Salvesen, G.S. Caspases: Opening the boxes and interpreting the arrows. Cell Death Differ. 2002, 9, 3–5. [Google Scholar] [CrossRef]
- Ghavami, S.; Hashemi, M.; Ande, S.R.; Yeganeh, B.; Xiao, W.; Eshraghi, M.; Bus, C.J.; Kadkhoda, K.; Wiechec, E.; Halayko, A.J.; et al. Apoptosis and cancer: Mutations within caspase genes. J. Med. Genet. 2009, 46, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Walters, J.; Pop, C.; Scott, F.L.; Drag, M.; Swartz, P.; Mattos, C.; Salvesen, G.S.; Clark, A.C. A constitutively active and uninhibitable caspase-3 zymogen efficiently induces apoptosis. Biochem. J. 2009, 424, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Perry, D.K.; Smyth, M.J.; Stennicke, H.R.; Salvesen, G.S.; Duriez, P.; Poirier, G.G.; Hannun, Y.A. Zinc is a potent inhibitor of the apoptotic protease, caspase -3. A novel target for zinc in the inhibition of apoptosis. J. Biol. Chem. 1997, 272, 18530–18533. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Porter, A.G.; Jänicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef]
- Katunuma, N.; Matsui, A.; Le, Q.; Utsumi, K.; Salvesen, G.; Ohashi, A. Novel procaspase-3 activating cascade mediated by lysoapoptases and its biological significances in apoptosis. Adv. Enzym. Regul. 2001, 41, 237–250. [Google Scholar] [CrossRef]
- Li, P.; Nijhawan, D.; Wang, X. Mitochondrial activation of apoptosis. Cell 2004, 116 (Suppl. S2), S57–S59. [Google Scholar] [CrossRef]
- Lavrik, I.N.; Golks, A.; Krammer, P.H. Caspases: Pharmacological manipulation of cell death. J. Clin. Investig. 2005, 115, 2665–2672. [Google Scholar] [CrossRef]
- Denault, J.-B.; Eckelman, B.P.; Shin, H.; Pop, C.; Salvesen, G.S. Caspase 3 attenuates XIAP (X-linked inhibitor of apoptosis protein)–mediated inhibition of caspase 9. Biochem. J. 2007, 405, 11–19. [Google Scholar] [CrossRef]
- Agosto, M.; Azrin, M.; Singh, K.; Jaffe, A.S.; Liang, B.T. Serum Caspase-3 p17 Fragment Is Elevated in Patients With ST-Segment Elevation Myocardial Infarction: A Novel Observation. J. Am. Coll. Cardiol. 2011, 57, 220–221. [Google Scholar] [CrossRef]
- Abdul-Ghani, M.; Megeney, L.A. Rehabilitation of a Contract Killer: Caspase-3 Directs Stem Cell Differentiation. Cell Stem Cell 2008, 2, 515–516. [Google Scholar] [CrossRef] [PubMed]
- Asadi, M.; Taghizadeh, S.; Kaviani, E.; Vakili, O.; Taheri-Anganeh, M.; Tahamtan, M.; Savardashtaki, A. Caspase-3: Structure, function, and biotechnological aspects. Biotechnol. Appl. Biochem. 2021, 69, 1633–1645. [Google Scholar] [CrossRef] [PubMed]
- Jänicke, R.U.; Sprengart, M.L.; Wati, M.R.; Porter, A.G. Caspase-3 Is Required for DNA Fragmentation and Morphological Changes Associated with Apoptosis. J. Biol. Chem. 1998, 273, 9357–9360. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef]
- Tokin, I.I.; Tokin, I.B.; Filimonova, G.F.; Hussar, P. Comparative Immunohistochemical Detection of Caspase-3 Activation in Liver Biopsy Specimens of Patients with Mono and Mixed Infection. AJPNM 2013, 1, 92–97. [Google Scholar]
- Rusin, P.; Jabłońska, K. Disturbances in the Mechanism of Apoptosis as One of the Causes of the Development of Cancer Diseases. Studia Ecol. Bioethicae 2020, 18, 63–73. [Google Scholar] [CrossRef]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Skommer, J.; Brittain, T.; Raychaudhuri, S. Bcl-2 inhibits apoptosis by increasing the time-to-death and intrinsic cell-to-cell variations in the mitochondrial pathway of cell death. Apoptosis 2010, 15, 1223–1233. [Google Scholar] [CrossRef]
- Hatok, J.; Racay, P. Bcl-2 family proteins: Master regulators of cell survival. Biomol. Concepts 2016, 7, 259–270. [Google Scholar] [CrossRef]
- Suraweera, C.D.; Banjara, S.; Hinds, M.G.; Kvansakul, M. Metazoans and Intrinsic Apoptosis: An Evolutionary Analysis of the Bcl-2 Family. Int. J. Mol. Sci. 2022, 23, 3691. [Google Scholar] [CrossRef]
- Letai, A.; Bassik, M.C.; Walensky, L.D.; Sorcinelli, M.D.; Weiler, S.; Korsmeyer, S.J. Distinct BH3 domaineither sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002, 2, 183–192. [Google Scholar] [CrossRef]
- Guedes, J.P.; Baptista, V.; Santos-Pereira, C.; Sousa, M.J.; Manon, S.; Chaves, S.R.; Côrte-Real, M. Acetic acid triggers cytochrome c release in yeast heterologously expressing human Bax. Apoptosis 2022, 27, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Scaini, G.; Mason, B.L.; Diaz, A.P.; Jha, M.K.; Soares, J.C.; Trivedi, M.H.; Quevedo, J. Dysregulation of mitochondrial dynamics, mitophagy and apoptosis in major depressive disorder: Does inflammation play a role? Mol. Psychiatry 2021, 27, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Sarić, N.; Hashimoto-Torii, K.; Jevtović-Todorović, V.; Ishibashi, N. Nonapoptotic caspases in neural development and in anesthesia-induced neurotoxicity. Trends Neurosci. 2022, 45, 446–458. [Google Scholar] [CrossRef]
- Christgen, S.; Tweedell, R.E.; Kanneganti, T.-D. Programming inflammatory cell death for therapy. Pharmacol. Ther. 2021, 232, 108010. [Google Scholar] [CrossRef]
- Ersoy, T.; Ozmen, O. Immunohistochemical detection of caspase 3 and proliferating cell nuclear antigen in the intestines of dogs naturally infected with parvovirus. Vet. Res. Forum 2022, 1, 127–131. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Pro-apoptotic members | Pro-apoptotic proteins (pore-formers): Bax Bcl-2-like protein 4 Bak Bcl-2 homologous antagonist killer Bok Bcl-2-related ovarian killer Mtd Matador |
| Pro-apoptotic (BH3-only) proteins: Bad Bcl-2-associated death promoter Bid BH3 interacting domain death agonist Bik Bcl-2-interacting killer Bim Bcl-2-like protein 11 Bmf Bcl-2-modifying factor Egl-1 Caenorhabditis elegans gene egl-1 Hrk Activator of apoptosis harakiri Puma p53 upregulated modulator of apoptosis etc. | |
| Anti-apoptotic members | Bcl-2 B-cell CLL/lymphoma 2 Bcl-xL B-cell lymphoma-extra large Mcl-1 MCL1, myeloid cell leukemia 1 Bcl-w Bcl-2-like protein 2, B-cell lymphoma-w CED-9 C. elegans gene ced-9 Bfl-1/A1 Burkholderia lethal factor 1 etc. |
| Inflammatory type of caspases | Caspase-1, -4, -5, -11, -12, -13 |
| Initiator type of caspases | Caspase-2, -8, -9, -10 |
| Executioner type of caspases | Caspase-3, -6, -7 |
| Other type of caspases | Caspase-14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussar, P. Apoptosis Regulators Bcl-2 and Caspase-3. Encyclopedia 2022, 2, 1624-1636. https://doi.org/10.3390/encyclopedia2040111
Hussar P. Apoptosis Regulators Bcl-2 and Caspase-3. Encyclopedia. 2022; 2(4):1624-1636. https://doi.org/10.3390/encyclopedia2040111
Chicago/Turabian StyleHussar, Piret. 2022. "Apoptosis Regulators Bcl-2 and Caspase-3" Encyclopedia 2, no. 4: 1624-1636. https://doi.org/10.3390/encyclopedia2040111
APA StyleHussar, P. (2022). Apoptosis Regulators Bcl-2 and Caspase-3. Encyclopedia, 2(4), 1624-1636. https://doi.org/10.3390/encyclopedia2040111