Abstract
Hyperphosphatemia in dialysis patients is associated with adverse outcomes including bone mineral disease, increased total mortality, and cardiovascular mortality. Therefore, maintaining serum phosphate levels within limits is an important aspect of the clinical care of peritoneal dialysis patients. Unfortunately, hyperphosphatemia is commonly seen in the majority of dialysis patients, at least in the USA, despite apparent optimal dietary and pharmacological intervention and adequate dialysis. Herein, we review major aspects of body phosphate homeostasis in healthy subjects and in dialysis patients in order to provide a good background understanding for a more rational approach to manage serum phosphate. Of note, the phosphate concentration measured in blood by clinical laboratories represents a minute portion of the total body phosphate content but the only one that we can easily access at present; this aspect is discussed in detail in this review. We emphasize the curtailment not only of the total oral phosphate intake but also the intake of highly bioavailable phosphate; this, together with the right use of oral phosphate binders and appropriate dialysis, is an important tool. Emerging therapies with agents that block intestinal absorption of phosphate may offer a promising four-pronged approach to phosphate management.
1. Introduction
Hyperphosphatemia in dialysis patients is associated with adverse outcomes including bone mineral disease, increased total mortality, and cardiovascular mortality [1,2,3]. It has even been suggested that phosphate retention not only increases mortality but also accelerates aging processes in CKD [4]. The KDIGO Clinical Practice Guideline Update for the Diagnosis, Evaluation, Prevention, and Treatment of Chronic Kidney Disease–Mineral and Bone Disorder suggests that serum phosphate levels for ESKD patients should be maintained within the normal range [5]. At any particular time point, serum phosphate levels represent the net balance of phosphate movements between several compartments, i.e., net GI uptake, net movement between extracellular and bone and intracellular compartments, and renal excretion. In dialysis patients, renal clearance is replaced partially or totally with dialysate clearance, depending on residual renal function. Therefore, one would assume that serum phosphate in a dialysis patient should be well controlled by the right combination of decreasing oral intake of phosphate, blocking the availability of phosphate for GI absorption with the use of oral binders, and adequate dialysis. Real life clinical practice, however, reveals that patients on dialysis frequently have elevated serum phosphate levels despite apparent optimal pharmacological intervention, adequate dialysis, and best efforts to instruct them on appropriate dietary restrictions. In fact, a serum phosphate of 4.5 mg/dL (1.45 mmol/L) or higher was found in 72.3% of hemodialysis (HD) patients and 75.1% of PD patients in the US in 2020 [6]. A good understanding of phosphate homeostasis is of paramount importance for nephrologists dealing with dialysis patients, undergoing either hemo- or peritoneal dialysis, in order to devise a rational approach to control serum phosphate.
2. Body Phosphate Homeostasis
The total phosphate content in a 70 kg man is about 700 g. Approximately 90% of this is in the bone and teeth (mostly in the form of calcium phosphate hydroxyapatite, Ca10(PO4)6(OH)2), with 10% in soft tissues and far less than 1% in the extracellular space. Most of intracellular phosphate is present as part of organic molecules such as creatine phosphate, ATP, nucleic acids, phospholipids, and phosphoproteins, with very low concentration of free inorganic phosphate. Therefore, this compartment cannot provide storage for significant accumulation of inorganic phosphate unless there is a simultaneous increase in cell mass [7,8]. The main potential stores for excess body phosphate are bone, teeth, and extraosseous calcifications (vascular and soft tissues). In current clinical practice, the only simple means by which to estimate total body phosphate is via the serum phosphate concentration, although this represents just a minute portion of total body phosphate content and may be markedly affected by shifts of the mineral between different compartments.
Only inorganic serum phosphate is measured by clinical laboratories, with normal values being about 2.5–4.5 mg/dL. The organic phase, although significant, is not included in such clinical determinations (Box 1). One should keep in mind that although the clinical laboratory value may be expressed as “phosphorus” or “phosphate”, only the atomic weight of elemental phosphorus is considered in the result. Therefore, a serum phosphate concentration of 4 mg/dL is 1.29 mmol/L (40 mg/L divided by the molecular weight of elemental phosphorus, i.e., 39). Serum phosphate concentrations are slightly higher in women than men and in children compared to adults, and they show diurnal variations beyond the effect of food intake.
Total intracellular phosphate (including phosphate esters in ATP, nucleic acids, etc.) is much higher (about 30 mmol/L) but is compartmentalized. Free inorganic phosphate comprises much less, around 2 mmol/L. A specific study measured total and free inorganic phosphate by 31P nuclear magnetic resonance spectroscopy and found it to be 28.9 and 2.4 mmol/L in controls subjects and 31.8 and 2.8 mmol/L in hemodialysis patients [9].
Box 1. Basic concepts.
- Elemental phosphorus is a highly reactive compound; the phosphorus we are exposed to in biological systems is combined with oxygen, mostly as orthophosphate.
- At usual body pH, orthophosphate exists in two main forms: H2PO4−1 and HPO4−2.
- Measurements of phosphorus in biological fluids only measure inorganic, not organic phosphate.
- When a clinical laboratory reports a normal serum phosphate level of 4 mg/dL, it is expressed as inorganic phosphorus (MW = 31). Therefore, normal serum Pi= 4 mg/dL =1.29 mmol/L = 2.32 mEq/L (pH 7.4).
3. Gastrointestinal Phosphate Absorption
Gastrointestinal (GI) phosphate absorption is a linear function of dietary phosphate intake, with a absorption rate of about 60–65%. On a usual mixed American diet of 1400 mg, this would add about 900 mg of phosphate to the extracellular fluid each day (Figure 1). Similar fractional intestinal phosphate absorption has been described for CKD patients using radioisotope tracer methodology [10] and probably would be the same in dialysis patients. Phosphate is absorbed through two main transport systems: one is sodium-dependent, saturable, and transcellular (NaPi-2b), regulated by calcitriol; the other is sodium-independent, non-saturable, and paracellular [11] (Figure 2). The intestinal paracellular phosphate absorption pathway lacks tight regulation and depends on the phosphate concentration gradient across the epithelium, the electrical gradient (lumen negative), and tight junction permeability, controlled by several claudins [12,13]. Although active transport will tend to fall with decreasing levels of calcitriol as CKD progresses, a high oral intake of phosphate will provide enough substrate for passive intercellular transport to continue to occur. This is quite in contrast with the fractional GI absorption of calcium, which is more tightly controlled at about 20–25%, mostly by calcitriol, and falling significantly with worsening kidney function unless calcitriol is provided pharmacologically.
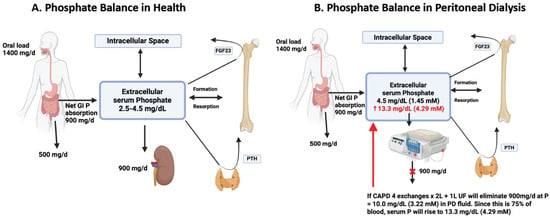
Figure 1.
Panel (A). Phosphate Balance in Healthy Subjects. The figure illustrates an average dietary phosphate intake of about 1400 mg/day, 60% of which (900 mg) will be absorbed into the circulation and exchange with intracellular and bone compartments with 0 net gain in a grown up adult. The net GI absorption of phosphate is excreted in the urine. Extracellular phosphate levels also regulate the levels of two hormones: PTH (produced by the parathyroid glands) and FGF23 (produced by the bone). Panel (B). Phosphate Balance in Peritoneal Dialysis Subjects. In this figure, we assume no residual kidney function and complete replacement of kidney function by dialysis. The same amount of average phosphate intake without the use of oral phosphate binders will provide about 900 mg of phosphate per day to the circulation. A theoretical CAPD patient with four exchanges per day of 2 L each and net ultrafiltration of 1 L/d must eliminate dialysate with a phosphate concentration of 10 mg/dL (3.2 mmol/L) (900 mg divided by 9 L of dialysis output). Since the dialysate phosphate concentration on average only reaches 75% of that in serum, the serum phosphate concentration will keep increasing until reaching a value of about 13.3 mg/dL (4.29 mmol/L). If the dietary intake of phosphate is reduced to about 1000 mg/d and oral phosphate binders are used, the net GI phosphate absorption can be markedly reduced to about 400 mg/d, which could then be eliminated in PD fluid at a concentration of 4.4 mg/dL (1.42 mmol/L) (400 mg divided by 9 L) and a final serum concentration of 5.9 mg/dL (1.89 nmol/L), for example.
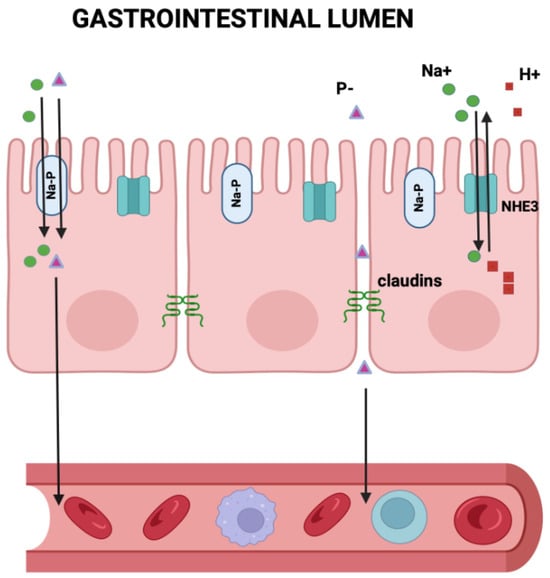
Figure 2.
Gastrointestinal Phosphate absorption. This figure illustrates the two ways to transport phosphate from the GI lumen into the circulation: one is sodium-dependent, saturable, and transcellular (NaPi-2b), regulated by calcitriol; the other is sodium-independent, non-saturable, and paracellular. The intestinal paracellular phosphate absorption pathway lacks tight regulation and depends on the phosphate concentration gradient across the epithelium, the electrical gradient (lumen negative), and tight junction permeability, controlled by several claudins. Notably, tenapanor, which blocks the NHE3 exchanger, also located in the luminal side of the intestinal wall, indirectly decreases the claudin-dependent paracellular absorption of phosphate.
4. Renal Phosphate Handling
The kidneys play a major role in maintaining phosphate homeostasis by matching urinary excretion (output) to GI absorption (input). Obviously, this coupling is lost when dialysis patients lose residual function and become fully dependent on dialytic phosphate removal. Almost 90% of phosphate is filtered at the glomerulus. About 75% of the filtered phosphate is reabsorbed in the proximal tubule, with 10% being filtered in the distal tubule and 15% excreted in the urine. The main phosphate transporter in the apical membrane of the proximal tubule is the Na phosphate cotransporter NaPi-2a, which is electrogenic. In this way, phosphate is transported out of the cell by a phosphate-anion exchanger. The main factors regulating the transport of phosphate in the proximal tubule are PTH and FGF23, with both of them inhibiting it [8].
5. Exchange of Phosphate Between Extra- and Intracellular Fluids
Since phosphate is a major intracellular anion, the transcellular movement of phosphate has a major impact on extracellular phosphate concentrations. Increased glycolysis by alkaline pH, insulin, and carbohydrate administration [14,15] increases intracellular movement, while acidic pH has the opposite effect.
6. Exchange of Phosphate with Bone
Under steady state conditions, the daily quantities of phosphate deposition in bone or phosphate release from bone are insignificant in relation to the amount reaching the extracellular space through net GI phosphate absorption. Bone mineral is mostly in the form of hydroxyapatite, which has a Ca:P ratio of 400:186 (2.1). Each apatite has 10 molecules of Ca, a molecular weight of 40 [40 × 10 = 400] for six molecules of P, making a molecular weight of 31 [6 × 31 = 186]) [16]. This structural mineral formula determines that whenever bone is dissolved, the release of every 100 mg of Ca would be accompanied by the release of 46 mg of P. Since Ca in bone also exists as calcium carbonate, the total bone Ca:P ratio is somewhat higher, at 2.2 to 2.3. These figures illustrate that the daily contribution of habitual bone loss to urinary phosphate is insignificant in a healthy adult person, and 24-h urinary phosphate excretion is quantitatively almost equal to the net GI absorption of phosphate in the steady state. This relationship is present independent of any renal or hormonal factors influencing the phosphate balance [8,17]. Therefore, discrepancies between intake and output may be found, but only transiently, and a balance has to be reached eventually for the organism to survive. Biologically, any prolonged substantial imbalance of most electrolytes, including phosphate, is incompatible with life (Box 2).
Box 2. Contribution of secondary hyperparathyroidism to serum phosphate in dialysis patients.
Does hyperparathyroidism significantly increase serum P in dialysis patients? Unlikely, because:
- Pi in bone is in the form of hydroxyapatite: Ca10(PO4)6(OH)2
- Therefore, every 46 mg of Pi lost from bone is accompanied by 100 mg of calcium
- For bone release to increase serum Pi by 1 mg/dL, it needs the addition of 160 mg of Pi to the extracellular space (16 L × 10 mg/L) and 348 mg of Ca, which, added to the extracellular space, would raise serum Ca by about 21 mg/L or 2.1 mg/dL (0.5 mmol/L).
7. Oral Intake of Phosphate
Net GI absorption is about 60% with an average diet, but that percentage changes depending on the bioavailability of phosphate in different foods. Phosphate bioavailability is very variable and depends on whether food is of animal or plant origin, whether the food contains calcium or not, and whether phosphate is in liquid form or a solid food matrix. A group of CKD patients were studied in a clinical research center and randomized into two groups. Both received the same amount of protein and total phosphate, but one group got a vegetarian diet while the other got meat-derived protein. The 24-h urine phosphate excretion showed that only about 52% of the dietary phosphate had been absorbed in the vegetarian group, but about 70% of the dietary phosphate had been absorbed in the group on an animal-protein diet [18]. Another study showed that by simply changing the oral intake of calcium from 100 mg/d to about 1600 mg/d, while phosphate intake remained constant at about 1271 mg/d, a 10% decrease in phosphate intestinal absorption was achieved in eight healthy adults [19]. The problem of dietary phosphate bioavailability becomes even more apparent in view of the increasing use of sodium or potassium phosphate salts in modern processed foods, because they are more easily absorbed than the phosphate contained in natural foods. For example, the complete recovery (in the urine) of sodium phosphate added to a liquid diet was demonstrated in two subjects [20]. Hemodialysis patients who had been instructed to avoid processed foods containing inorganic phosphate salts experienced a decrease of their serum phosphate levels by 0.6 mg/dL compared to a control group without any such dietary restriction [21]. In a more extreme case, plant-derived phosphate, mostly in the form of phytate, is generally assumed to be only 50% absorbed [22].
8. Hormonal Regulation of Phosphate
In healthy individuals, the regulation of the serum phosphate concentration within a normal range depends on the interplay of four organs—the kidneys, bone, parathyroid glands, and small intestine—through the action of three hormones—parathyroid hormone (PTH), fibroblast growth factor-23 (FGF23), and calcitriol (1,25-dihydroxy vitamin D [23,24]. The major regulatory impact results from controlling intestinal phosphate absorption and renal tubular phosphate reabsorption. Interestingly, no hormone has been shown to control serum phosphate through action on bone. We assume that the processes of phosphate deposition and release in and out of bone simply emulate that of calcium. PTH is produced by the parathyroid glands and acts directly on bone, increasing its resorption and stimulating FGF23 secretion, and in the kidneys by decreasing expression of NaPi-2a and NaPi-2c, the major Na+-dependent phosphate transporters of the proximal tubule, as well as stimulating renal 1-alpha hydroxylase. PTH indirectly increases phosphate GI absorption through higher levels of calcitriol. FGF23 is a hormone produced in bone in response to excess phosphate; its actions include the suppression of PTH secretion, as well as calcitriol and renal phosphate transport (acting on the same Na-P co-transporters as PTH). Calcitriol upregulates Na-Pi co-transporters in both intestine and proximal renal tubules, leading to increased intestinal absorption of phosphate and increased renal phosphate reabsorption. Calcitriol suppresses PTH secretion and renal 1-alpha hydroxylase but stimulates FGF23 secretion. FGF23 may act through or independently of Klotho [25,26]. Klotho is a beta glucuronidase enzyme present both as a transmembrane protein and a secreted renal protein and can function as a co-receptor of FGF23. In addition to these functions, FGF23 has been implicated in several other metabolic diseases, including cardiovascular disease and obesity-related disorders [25].
9. Body Phosphate Balance
In the presence of normal kidney function, phosphate balance is achieved over a relatively short period of weeks or months; however, anuric dialysis patients may be in a state of constant phosphate retention, leading to high prevalence of vascular and valvular calcifications, tumoral calcinosis, and even calciphylaxis [27,28,29]. CKD patients not on dialysis may or may not be in phosphate balance, depending on how much GI phosphate absorption takes place in relation to residual renal function. A CKD Stage IV (estimated GFR = 20 mL/min = 29 L/day) patient, not on oral phosphate binders, eating an average mixed diet with 1400 mg P/day, would have a GI absorption of about 900 mg/d that needs to be excreted in the urine to achieve phosphate balance. At a serum phosphate concentration of 4 mg/dL (40 mg/L, 1 mmol/L), his/her daily filtered load of phosphorus would be 1044 mg (40 mg/L, but only 90% filterable and GFR of 29 L/d). If we assume a fractional excretion of phosphate of about 40% at that level of GFR [30], his/her daily urinary phosphate excretion would be about 418 mg. Since this patient gets 900 mg phosphate from GI uptake, he/she patient would have to retain about 482 mg/d of phosphate. As phosphate is retained, the serum phosphate level will increase while simultaneously precipitating calcium and inducing secondary hyperparathyroidism. This large daily retention of phosphate requires calcium to precipitate, i.e., as much as 930 mg if deposited as calcium hydroxyapatite, and slightly less if deposited in other crystal forms. Since decreased intestinal absorption of calcium is an early manifestation of CKD [31,32,33,34], one would not expect this amount of calcium to be absorbed in a stage 4 CKD patient. Since blood has a very limited capacity for the retention of phosphate, excess phosphate retention would only be possible with a large supply of calcium from bone. Consequently, substantial daily phosphate retention is only possible in subjects who have large amounts of calcium available to deposit with phosphate, which would require huge intake and calcium and active vitamin D or a constant supply of calcium from bone. Secondary hyperparathyroidism could supply calcium from the bone but at the expense of decreased bone density over time.
A progressive decline in urinary phosphate excretion has been associated with worsening kidney function. A study of 1836 subjects with different degrees of renal function found a mean difference in 24 h urine phosphate excretion of 400 mg/day between CKD stage 1 and 4 [35]. In another study involving 3879 CKD patients, the mean difference was 200 mg/day [36]. Therefore, when CKD progresses, one of two things happen: either patients decrease their intake of phosphate (no data on dietary intake are provided) or they develop a positive phosphate balance (retaining an amount determined by the difference between their net GI phosphate absorption and daily urinary excretion of phosphate). If the 400 mg/day change in urinary phosphate excretion as CKD advances reflects actual phosphate retention, one would expect a positive balance of 146 g per year! If retention were to occur as hydroxyapatite, a simultaneous retention of 860 mg calcium/day (314 g per year) would be required. A positive phosphate balance of this magnitude would therefore be detected by an increase bone density—not a regular feature in CKD. The only other place for calcium phosphate deposits would be soft tissues, where a tumoral calcinosis [27] can accommodate large amounts of calcium phosphate, or in heart valves or the arterial walls [28]. A theoretical quantitative analysis suggested that a huge amount of positive phosphate can be present in the vascular tree [37,38]. The main issue here is where to get the calcium needed to retain this much phosphate. Since there is a limited amount of Ca absorbed from the GI tract, the needed Ca could only come from bone in the form of calcium carbonate (removing calcium phosphate will add to the already high degree of phosphate retention), but there is also a limit to the supply of calcium carbonate [39,40]. This theoretical analysis supports the idea that the only reasonable explanation for the above findings is the existence of a neutral phosphorus balance, i.e., as CKD progresses, there is a progressive decrease in dietary food intake, including phosphate. As CKD progresses, each step of GFR decline would produce a minimal increase in serum phosphate that would increase Ca x P enough to induce extra skeletal calcification and therefore phosphate retention. As mentioned above, a phosphate balance eventually has to be reached, but at the expense of some amount of phosphate being retained in soft tissues and/or by increasing the serum phosphate level. This situation becomes more compounded in dialysis patients, particularly when all residual function is lost.
Another interesting point, not always considered, is that phosphate deposition in the form of hydroxyapatite would always result in sequestering alkali in bone and therefore adding a significant acidic load to the extraosseous space [41].
10. Clinical Approach to Hyperphosphatemia in PD
As mentioned in the introduction to this chapter, hyperphosphatemia remains a significant challenge in the management of ESRD patients, contributing to increased morbidity and mortality. The presence of hyperphosphatemia implies problems with excessive dietary intake of phosphate, problems with the use of oral phosphate binders, or insufficient dialysis (Box 3).
Box 3. Factors influencing phosphate removal in peritoneal dialysis patients.
- Patient’s size may affect dwell fluid volume
- Peritoneal membrane transport characteristics (slow or fast transporters)
- Presence of residual renal function
- Dwelling time not matching with peritoneal membrane characteristics (for example, short dwell times in a slow transporter)
- Automatic peritoneal dialysis versus CAPD; consider that more than 90% of PD patients in the USA are currently on APD (cycler) [42]
- PD vintage is important, since membrane transport characteristics also affecting phosphoate dialytic removal develop as time on PD progresses
Beyond total dietary phosphate intake, phosphate bioavailability in food is a major consideration. Dietary phosphate intake includes not only the phosphate contained naturally in foods but also that which is added during processing. Generally, organic natural phosphate is less well absorbed than inorganic phosphate in additives [43]. In fact, the phosphate content of the American diet has been increasing, mostly due to the consumption of foods processed with phosphate additives [43].
Lower socioeconomic status has been associated with higher serum phosphate in the United States [44]. Individuals living in low-income neighborhoods have limited access to healthy foods, leading to the overconsumption of processed and fast foods, which are rich in highly absorbable phosphate additives [45]. The situation may be quite different in other societies, where poverty is linked to reduced food intake rather than the overconsumption of low-quality foods [46,47].
Another source of oral phosphate, usually not considered, comes from the use of phosphate-containing dietary supplements and OTC or prescription medications. The latter source of phosphate may easily amount to over 100 mg/d in patients taking multiple medications [43].
The association between dietary intake and serum phosphate levels is additionally affected by oral phosphate binders, the use of which varies substantially among the dialysis population [48]. Data from the USRDS reported about 61.6% PD patients were taking one or more phosphate binders in the United States in 2020 [6]. The effectiveness of currently commercially available phosphate binders is rather modest, with average intestinal binding of only about 200 mg of phosphorus per day; this may amount to the same amount of phosphate additives consumed daily in food [49,50].
Furthermore, there is some controversy about choosing between calcium-containing and non-calcium-containing phosphate binders. Calcium-based binders, although effective, are associated with risks of hypercalcemia and vascular calcification, which can exacerbate cardiovascular morbidity. Non-calcium-based binders, such as Sevelamer and Lanthanum, have a lower risk of hypercalcemia and have been shown to reduce vascular calcification, but their superior clinical outcome remains to be determined. The economic and pill burdens associated with phosphate binders are important factors to consider, since they may lead to poor compliance [51]. Emerging therapies, such as Tenapanor [52,53], offer a novel, four pronged approach to phosphate management through a distinct mechanism of action (See Box 4).
Box 4. Four-pronged approach to control hyperphosphatemia.
- Dietary Phosphate control
- Limit total phosphate intake
- Limit more bioavailable phosphate intake (e.g., avoid processed foods with easily absorbable phosphates, prefer plant-derived foods with less absorbable phytate)
- 2.
- Use of oral phosphate binders
- Calcium carbonate
- Calcium acetate
- Sevelamer carbonate
- Lanthanum carbonate
- Ferric citrate
- Sucroferric oxyhydroxide
- 3.
- Decrease GI phosphate absorption
- Niacin (blocking intestinal Na-P cotransporter)
- Tenapanor (blocking NHE3 exchanger)
- 4.
- Increase dialysis phosphate removal
Serum phosphate levels are inversely correlated with peritoneal phosphate clearance, and several studies have shown that dialysis phosphate clearance is comparable to dialysis creatinine clearance, suggesting that creatinine clearance may be an important marker of dialysis adequacy in this population beyond the usual urea Kt/V [54,55,56]. This may be an important consideration [57]. However, phosphate removal by PD is limited, and anuric patients relying only on dialytic removal may develop severe hyperphosphatemia if attention is not paid to phosphate oral intake or the use of phosphate binders or GI absorption blockers (Figure 1B).
Within this context, it is important to emphasize that the relationship between serum phosphate and outcomes in patients with kidney failure is only an epidemiologic association and not a proven cause-and-effect relationship. Currently, there is one randomized controlled trial trying to elucidate whether changing serum phosphate levels in ESRD patients on dialysis indeed changes the clinical outcomes, but no results are available yet [58].
In the above discussion, we assumed that PD patients do not have significant residual kidney function to contribute to phosphate clearance; this assumption may not apply in predominantly incident dialysis patients, particularly peritoneal dialysis patients, who may have some significant degree of residual kidney function. Another important consideration is that as PD vintage increases, changes may take place in the peritoneal membrane transport characteristics, which will secondarily affect urea, creatinine, and phosphate dialytic removal.
11. Conclusions
Serum phosphate is a major determinant of outcome in ESRD patients on dialysis. Unfortunately, keeping normal serum phosphate levels during PD is extremely difficult, particularly after losing residual renal function. In view of the limited dialytic removal of phosphate, detailed attention to dietary phosphorus is of paramount importance in this population. Current oral phosphorus binders are relatively inefficient. It remains to be seen whether new agents that control the GI absorption of phosphate will be a game changer in this area.
A good understanding of the phosphate balance provides a solid, necessary background for a rational approach to control serum phosphate levels in dialysis patients; at present, this aspect remains substandard. The lack of a simple clinical tool to assess body phosphate balance forces us to rely on serum phosphate, which represents only a minute portion of the total body phosphate content. This may therefore yield misleading results, particularly when isolated serum phosphate values are used. Since the bone contents of calcium and phosphate are not increased in CKD, a sustained positive balance of these minerals can occur only with the development of extra skeletal calcifications. Phosphate precipitation in soft tissue requires a supply of a large amount of calcium that may not be easily available, given the limited GI absorption of calcium in CKD; this initiates compensatory calcium removal from bone, mediated by secondary hyperparathyroidism. Adequate control of CKD secondary hyperparathyroidism is only possible by preventing phosphate retention, i.e., by either decreasing the amount of GI absorption or increasing dialysis removal.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Hiyamuta, H.; Yamada, S.; Taniguchi, M.; Tokumoto, M.; Tsuruya, K.; Nakano, T.; Kitazono, T. Association of hyperphosphatemia with an increased risk of sudden death in patients on hemodialysis: Ten-year outcomes of the Q-Cohort Study. Atherosclerosis 2021, 316, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Block, G.A.; Hulbert-Shearon, T.E.; Levin, N.W.; Port, F.K. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: A national study. Am. J. Kidney Dis. 1998, 31, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Li, X.; Sun, L.; Qu, Z.; Jiang, L.; Du, Y. Phosphorus and mortality risk in end-stage renal disease: A meta-analysis. Clin. Chim. Acta 2017, 474, 108–113. [Google Scholar] [CrossRef]
- Kuro-o, M. A potential link between phosphate and aging--lessons from Klotho-deficient mice. Mech. Ageing Dev. 2010, 131, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Stevens, P.E.; Ahmed, S.B.; Carrero, J.J.; Foster, B.; Francis, A.; Hall, R.K.; Herrington, W.G.; Hill, G.; Inker, L.A.; Kazancıoğlu, R.; et al. KDIGO 2024 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. 2024, 105, S117–S314. [Google Scholar] [CrossRef]
- NIH. 2022 USRDS Annual Data Report: Epidemiology of Kidney Disease in the United States; NIH: Bethesda, MD, USA, 2022.
- Nadkarni, G.N.; Uribarri, J. Phosphorus and the kidney: What is known and what is needed. Adv. Nutr. 2014, 5, 98–103. [Google Scholar] [CrossRef]
- Wagner, C.A. The basics of phosphate metabolism. Nephrol. Dial. Transplant. 2024, 39, 190–201. [Google Scholar] [CrossRef]
- Durozard, D.; Pimmel, P.; Baretto, S.; Caillette, A.; Labeeuw, M.; Baverel, G.; Zech, P. 31P NMR spectroscopy investigation of muscle metabolism in hemodialysis patients. Kidney Int. 1993, 43, 885–892. [Google Scholar] [CrossRef]
- Stremke, E.R.; Wiese, G.N.; Moe, S.M.; Wastney, M.E.; Moorthi, R.N.; Hill Gallant, K.M. Intestinal Phosphorus Absorption in Moderate CKD and Healthy Adults Determined Using a Radioisotopic Tracer. J. Am. Soc. Nephrol. 2021, 32, 2057–2069. [Google Scholar] [CrossRef]
- Fishbane, S.N.; Nigwekar, S. Phosphate Absorption and Hyperphosphatemia Management in Kidney Disease: A Physiology-Based Review. Kidney Med. 2021, 3, 1057–1064. [Google Scholar] [CrossRef]
- Marks, J. The role of SLC34A2 in intestinal phosphate absorption and phosphate homeostasis. Pflug. Arch. 2019, 471, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Saurette, M.; Alexander, R.T. Intestinal phosphate absorption: The paracellular pathway predominates? Exp. Biol. Med. 2019, 244, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Mehanna, H.M.; Moledina, J.; Travis, J. Refeeding syndrome: What it is, and how to prevent and treat it. BMJ 2008, 336, 1495–1498. [Google Scholar] [CrossRef] [PubMed]
- Megapanou, E.; Florentin, M.; Milionis, H.; Elisaf, M.; Liamis, G. Drug-Induced Hypophosphatemia: Current Insights. Drug Saf. 2020, 43, 197–210. [Google Scholar] [CrossRef]
- Michigami, T.; Ozono, K. Roles of Phosphate in Skeleton. Front. Endocrinol. 2019, 10, 180. [Google Scholar] [CrossRef]
- Hughes, E.A.B.; Robinson, T.E.; Bassett, D.B.; Cox, S.C.; Grover, L.M. Critical and diverse roles of phosphates in human bone formation. J. Mater. Chem. B 2019, 7, 7460–7470. [Google Scholar] [CrossRef]
- Moe, S.M.; Zidehsarai, M.P.; Chambers, M.A.; Jackman, L.A.; Radcliffe, J.S.; Trevino, L.L.; Donahue, S.E.; Asplin, J.R. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 257–264. [Google Scholar] [CrossRef]
- Lewis, N.M.; Marcus, M.S.; Behling, A.R.; Greger, J.L. Calcium supplements and milk: Effects on acid-base balance and on retention of calcium, magnesium, and phosphorus. Am. J. Clin. Nutr. 1989, 49, 527–533. [Google Scholar] [CrossRef]
- Relman, A.S.; Lennon, E.J.; Lemann, J., Jr. Endogenous production of fixed acid and the measurement of the net balance of acid in normal subjects. J. Clin. Investig. 1961, 40, 1621–1630. [Google Scholar] [CrossRef]
- Sullivan, C.; Sayre, S.S.; Leon, J.B.; Machekano, R.; Love, T.E.; Porter, D.; Marbury, M.; Sehgal, A.R. Effect of food additives on hyperphosphatemia among patients with end-stage renal disease: A randomized controlled trial. JAMA 2009, 301, 629–635. [Google Scholar] [CrossRef]
- Calvo, M.S.; Uribarri, J. Perspective: Plant-based Whole-Grain Foods for Chronic Kidney Disease: The Phytate-Phosphorus Conundrum. Adv. Nutr. 2021, 12, 2056–2067. [Google Scholar] [CrossRef] [PubMed]
- Jacquillet, G.; Unwin, R.J. Physiological regulation of phosphate by vitamin D, parathyroid hormone (PTH) and phosphate (Pi). Pflug. Arch. 2019, 471, 83–98. [Google Scholar] [CrossRef]
- Bergwitz, C.; Jüppner, H. Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu. Rev. Med. 2010, 61, 91–104. [Google Scholar] [CrossRef]
- Kawai, M. The FGF23/Klotho axis in the regulation of mineral and metabolic homeostasis. Horm. Mol. Biol. Clin. Investig. 2016, 28, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Kuro, O.M. The Klotho proteins in health and disease. Nat. Rev. Nephrol. 2019, 15, 27–44. [Google Scholar] [CrossRef]
- Cofan, F.; García, S.; Combalia, A.; Campistol, J.M.; Oppenheimer, F.; Ramón, R. Uremic tumoral calcinosis in patients receiving longterm hemodialysis therapy. J. Rheumatol. 1999, 26, 379–385. [Google Scholar]
- Smith, E.R. Vascular Calcification in Uremia: New-Age Concepts about an Old-Age Problem. Methods Mol. Biol. 2016, 1397, 175–208. [Google Scholar] [CrossRef] [PubMed]
- Nigwekar, S.U.; Thadhani, R.; Brandenburg, V.M. Calciphylaxis. N. Engl. J. Med. 2018, 378, 1704–1714. [Google Scholar] [CrossRef]
- Gutierrez, O.; Isakova, T.; Rhee, E.; Shah, A.; Holmes, J.; Collerone, G.; Jüppner, H.; Wolf, M. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J. Am. Soc. Nephrol. 2005, 16, 2205–2215. [Google Scholar] [CrossRef]
- Hill, K.M.; Martin, B.R.; Wastney, M.E.; McCabe, G.P.; Moe, S.M.; Weaver, C.M.; Peacock, M. Oral calcium carbonate affects calcium but not phosphorus balance in stage 3–4 chronic kidney disease. Kidney Int. 2013, 83, 959–966. [Google Scholar] [CrossRef]
- Spiegel, D.M.; Brady, K. Calcium balance in normal individuals and in patients with chronic kidney disease on low- and high-calcium diets. Kidney Int. 2012, 81, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Coburn, J.W.; Koppel, M.H.; Brickman, A.S.; Massry, S.G. Study of intestinal absorption of calcium in patients with renal failure. Kidney Int. 1973, 3, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.H. Are we mismanaging calcium and phosphate metabolism in renal failure? Am. J. Kidney Dis. 1997, 29, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Craver, L.; Marco, M.P.; Martínez, I.; Rue, M.; Borràs, M.; Martín, M.L.; Sarró, F.; Valdivielso, J.M.; Fernández, E. Mineral metabolism parameters throughout chronic kidney disease stages 1-5--achievement of K/DOQI target ranges. Nephrol. Dial. Transplant. 2007, 22, 1171–1176. [Google Scholar] [CrossRef]
- Isakova, T.; Wahl, P.; Vargas, G.S.; Gutiérrez, O.M.; Scialla, J.; Xie, H.; Appleby, D.; Nessel, L.; Bellovich, K.; Chen, J.; et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011, 79, 1370–1378. [Google Scholar] [CrossRef]
- Uribarri, J.; Oh, M.S. Phosphorus Intake and Whole-Body Phosphorus Homeostasis. In Dietary Phosphorus: Health, Nutrition and Regulatory Aspects; Uribarri, J., Calvo, M.S., Eds.; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Artunc, F. Widespread calcifications delineating an arterial vessel tree in a peritoneal dialysis patient. Turk. J. Nephrol. 2022, 31, 391–392. [Google Scholar] [CrossRef]
- Pellegrino, E.D.; Biltz, R.M. The composition of human bone in uremia. Observations on the reservoir functions of bone and demonstration of a labile fraction of bone carbonate. Medicine 1965, 44, 397–418. [Google Scholar] [CrossRef]
- Kaye, M.; Frueh, A.J.; Silverman, M.; Henderson, J.; Thibault, T. A study of vertebral bone powder from patients with chronic renal failure. J. Clin. Investig. 1970, 49, 442–453. [Google Scholar] [CrossRef]
- Oh, M.S. Irrelevance of bone buffering to acid-base homeostasis in chronic metabolic acidosis. Nephron 1991, 59, 7–10. [Google Scholar] [CrossRef]
- Moor, V.; Wagner, R.; Sayer, M.; Petsch, M.; Rueb, S.; Häring, H.U.; Heyne, N.; Artunc, F. Routine Monitoring of Sodium and Phosphorus Removal in Peritoneal Dialysis (PD) Patients Treated with Continuous Ambulatory PD (CAPD), Automated PD (APD) or Combined CAPD+APD. Kidney Blood Press. Res. 2017, 42, 257–266. [Google Scholar] [CrossRef]
- Calvo, M.S.; Uribarri, J. Contributions to total phosphorus intake: All sources considered. Semin. Dial. 2013, 26, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, O.M.; Anderson, C.; Isakova, T.; Scialla, J.; Negrea, L.; Anderson, A.H.; Bellovich, K.; Chen, J.; Robinson, N.; Ojo, A.; et al. Low socioeconomic status associates with higher serum phosphate irrespective of race. J. Am. Soc. Nephrol. 2010, 21, 1953–1960. [Google Scholar] [CrossRef] [PubMed]
- Butt, S.; Leon, J.B.; David, C.L.; Chang, H.; Sidhu, S.; Sehgal, A.R. The prevalence and nutritional implications of fast food consumption among patients receiving hemodialysis. J. Ren. Nutr. 2007, 17, 264–268. [Google Scholar] [CrossRef][Green Version]
- Correia, M.I.; Hegazi, R.A.; Diaz-Pizarro Graf, J.I.; Gomez-Morales, G.; Fuentes Gutiérrez, C.; Goldin, M.F.; Navas, A.; Pinzón Espitia, O.L.; Tavares, G.M. Addressing Disease-Related Malnutrition in Healthcare: A Latin American Perspective. J. Parenter. Enteral Nutr. 2016, 40, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Paniagua, R.; Ramos, A.; Fabian, R.; Lagunas, J.; Amato, D. Chronic kidney disease and dialysis in Mexico. Perit. Dial. Int. 2007, 27, 405–409. [Google Scholar] [CrossRef]
- Scialla, J.J.; Kendrick, J.; Uribarri, J.; Kovesdy, C.P.; Gutiérrez, O.M.; Jimenez, E.Y.; Kramer, H.J. State-of-the-Art Management of Hyperphosphatemia in Patients With CKD: An NKF-KDOQI Controversies Perspective. Am. J. Kidney Dis. 2021, 77, 132–141. [Google Scholar] [CrossRef]
- Kestenbaum, B.R.; de Boer, I.H. Comparative Safety of Phosphate Binders Without Proven Efficacy-How Did We Get Here? JAMA Intern. Med. 2019, 179, 749–750. [Google Scholar] [CrossRef]
- Calvo, M.S.; Sherman, R.A.; Uribarri, J. Dietary Phosphate and the Forgotten Kidney Patient: A Critical Need for FDA Regulatory Action. Am. J. Kidney Dis. 2019, 73, 542–551. [Google Scholar] [CrossRef]
- Marcuccilli, M.; Chonchol, M.; Jovanovich, A. Phosphate Binders and Targets Over Decades: Do We have it Right Now? Semin. Dial. 2017, 30, 134–141. [Google Scholar] [CrossRef]
- King, A.J.; Siegel, M.; He, Y.; Nie, B.; Wang, J.; Koo-McCoy, S.; Minassian, N.A.; Jafri, Q.; Pan, D.; Kohler, J.; et al. Inhibition of sodium/hydrogen exchanger 3 in the gastrointestinal tract by tenapanor reduces paracellular phosphate permeability. Sci. Transl. Med. 2018, 10, eaam6474. [Google Scholar] [CrossRef]
- Pergola, P.E.; Rosenbaum, D.P.; Yang, Y.; Chertow, G.M. A Randomized Trial of Tenapanor and Phosphate Binders as a Dual-Mechanism Treatment for Hyperphosphatemia in Patients on Maintenance Dialysis (AMPLIFY). J. Am. Soc. Nephrol. 2021, 32, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Debowska, M.; Gomez, R.; Pinto, J.; Waniewski, J.; Lindholm, B. Phosphate clearance in peritoneal dialysis. Sci. Rep. 2020, 10, 17504. [Google Scholar] [CrossRef] [PubMed]
- Peruzzo, D.; Guedes, M.; Larkin, J.W.; Yokoyama, G.; Dos Santos, T.L.; Pecoits-Filho, R.; Ribeiro, S.C.; Ramos, A.; Barretti, P.; de Moraes, T.P. Peritoneal dialysis modality transition and impact on phosphate and potassium serum levels. PLoS ONE 2021, 16, e0257140. [Google Scholar] [CrossRef] [PubMed]
- Sedlacek, M.; Dimaano, F.; Uribarri, J. Relationship between phosphorus and creatinine clearance in peritoneal dialysis: Clinical implications. Am. J. Kidney Dis. 2000, 36, 1020–1024. [Google Scholar] [CrossRef]
- Uribarri, J.; Guedes, M.; Diaz Bessone, M.I.; Chan, L.; De La Torre, A.; Mermelstein, A.; Garcia-Garcia, G.; Raimann, J.; Moraes, T.; Peters, V.; et al. The Role of Kt/V and Creatinine clearance on Assisting Optimization of Serum Phosphorus Levels among Patients on PD. Kidney360 2024, 6, 105–111. [Google Scholar] [CrossRef]
- AKTN. PHOSPHATE. Available online: https://aktn.org.au (accessed on 18 March 2025).


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).