
Controlled Hydrogen Loading of Magnesium Thin Films in KOH—Effects on the Hydride Nucleation and Growth Regimes

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Hydrogenography
2.3. Cyclic Voltammetry
3. Results
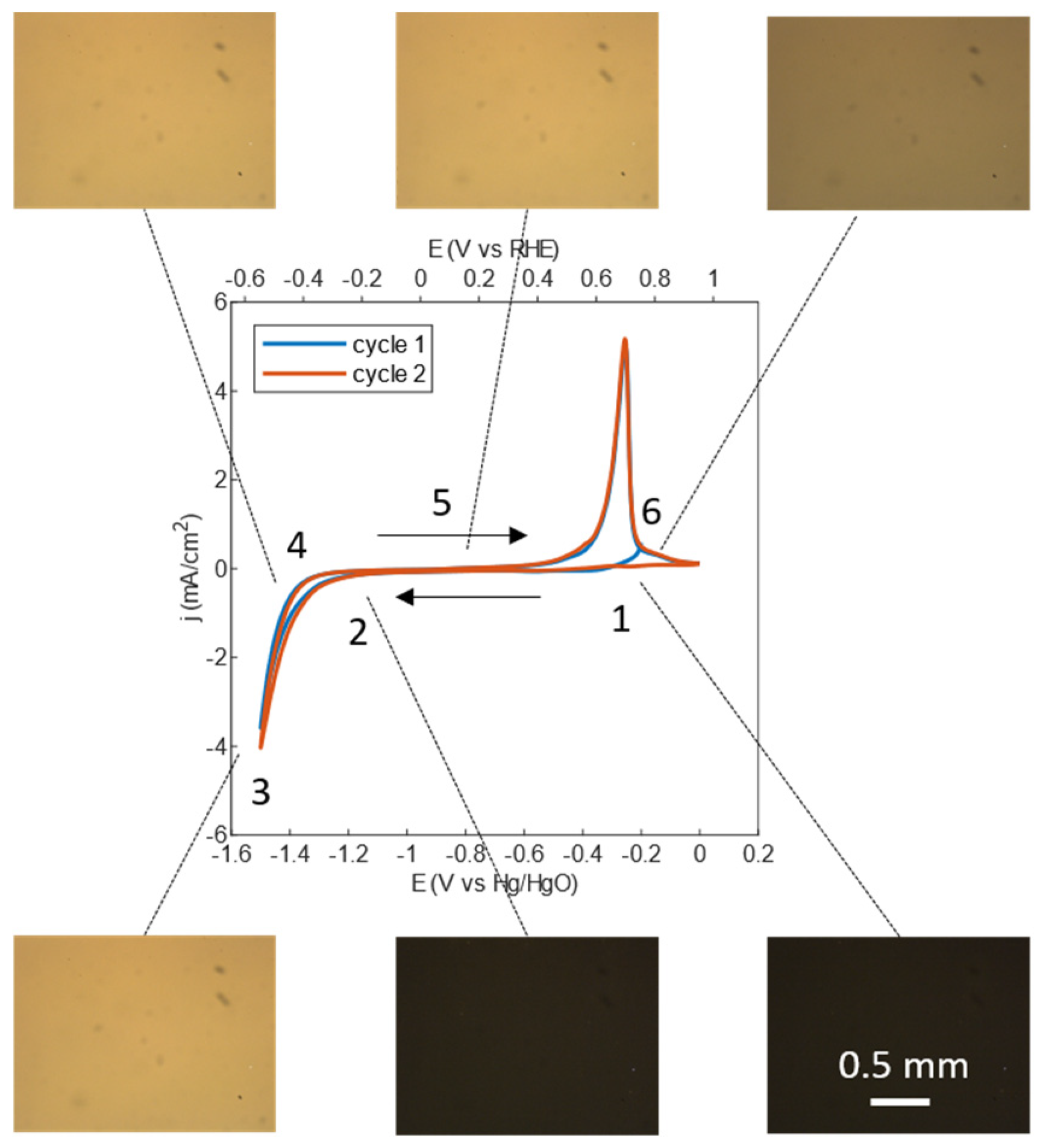
3.1. Cyclic Voltammetry Combined with In Situ Optical Microscopy
3.2. Potential Curves and Hydride Formation
3.3. Dependence of the Potential Curves on the Loading Potential
4. Discussion
5. Conclusions
- -
- The ability to closely follow the time evolution of the chemical potential within each loading step, by measuring the EMF at the thin film sample’s surface. This is impossible to do with gas-phase loading and, as shown in this paper, carries information on the behavior of the system during hydride formation.
- -
- The ability to influence the behavior of the system within each loading step by acting on the applied loading voltage. It is here demonstrated how magnesium hydride formation can be optimized by choosing the appropriate loading voltage.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Schlapbach, L.; Züttel, A. Hydrogen-Storage Materials for Mobile Applications. Nature 2001, 414, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; He, L.; Lin, H.; Li, H.-W. Progress and Trends in Magnesium-Based Materials for Energy-Storage Research: A Review. Energy Technol. 2018, 6, 445–458. [Google Scholar] [CrossRef]
- Leiva, D.R.; Jorge, A.M.; Ishikawa, T.T.; Botta, W.J. Hydrogen Storage in Mg and Mg-Based Alloys and Composites Processed by Severe Plastic Deformation. Mater. Trans. 2019, 60, 1561–1570. [Google Scholar] [CrossRef]
- Zaluska, A.; Zaluski, L.; Ström–Olsen, J.O. Nanocrystalline Magnesium for Hydrogen Storage. J. Alloys Compd. 1999, 288, 217–225. [Google Scholar] [CrossRef]
- Jinzhe, L.; Lider, A.M.; Kudiiarov, V.N. An Overview of Progress in Mg-Based Hydrogen Storage Films*. Chin. Phys. B 2019, 28, 098801. [Google Scholar] [CrossRef]
- Karst, J.; Sterl, F.; Linnenbank, H.; Weiss, T.; Hentschel, M.; Giessen, H. Watching in Situ the Hydrogen Diffusion Dynamics in Magnesium on the Nanoscale. Sci. Adv. 2020, 6, eaaz0566. [Google Scholar] [CrossRef]
- Hadjixenophontos, E.; Michalek, L.; Weigel, A.; Schmitz, G. Hydrogen Sorption Kinetics in MgH2 and TiH2 Thin Films. Defect Diffus. Forum 2018, 383, 127–132. [Google Scholar] [CrossRef]
- Hadjixenophontos, E.; Zhang, K.; Weigel, A.; Stender, P.; Schmitz, G. Hydrogenation of Pd/Mg Films: A Quantitative Assessment of Transport Coefficients. Int. J. Hydrogen Energy 2019, 44, 27862–27875. [Google Scholar] [CrossRef]
- Teichmann, N.; Hamm, M.; Pundt, A. Fast Lateral Hydrogen Diffusion in Magnesium-Hydride Films on Sapphire Substrates Studied by Electrochemical Hydrogenography. Int. J. Hydrogen Energy 2018, 43, 1634–1642. [Google Scholar] [CrossRef]
- Drechsler, V.; Krauth, J.; Karst, J.; Giessen, H.; Hentschel, M. Switchable Optical Nonlinearity at the Metal to Insulator Transition in Magnesium Thin Films. ACS Photonics 2020, 7, 1560–1568. [Google Scholar] [CrossRef]
- Hamm, M.; Bongers, M.D.; Roddatis, V.; Dietrich, S.; Lang, K.-H.; Pundt, A. In Situ Observation of Hydride Nucleation and Selective Growth in Magnesium Thin-Films with Environmental Transmission Electron Microscopy. Int. J. Hydrogen Energy 2019, 44, 32112–32123. [Google Scholar] [CrossRef]
- Uchida, H.T.; Wagner, S.; Hamm, M.; Kürschner, J.; Kirchheim, R.; Hjörvarsson, B.; Pundt, A. Absorption Kinetics and Hydride Formation in Magnesium Films: Effect of Driving Force Revisited. Acta Mater. 2015, 85, 279–289. [Google Scholar] [CrossRef]
- Xin, G.; Wang, Y.; Fu, H.; Li, G.; Zheng, J.; Li, X. Promising Electrochemical Hydrogen Storage Properties of Thick Mg-Pd Films Obtained by Insertion of Thin Ti Interlayers. Phys. Chem. Chem. Phys. 2014, 16, 3001–3006. [Google Scholar] [CrossRef] [PubMed]
- Lyu, J.; Elman, R.R.; Svyatkin, L.A.; Kudiiarov, V.N. Theoretical and Experimental Research of Hydrogen Solid Solution in Mg and Mg-Al System. Materials 2022, 15, 1667. [Google Scholar] [CrossRef]
- Pacanowski, S.; Wachowiak, M.; Jabłoński, B.; Szymański, B.; Smardz, L. Interface Mixing and Hydrogen Absorption in Pd/Mg and Pd/Al/Mg Thin Films. Int. J. Hydrogen Energy 2021, 46, 806–813. [Google Scholar] [CrossRef]
- Pivak, Y.; Schreuders, H.; Dam, B. Thermodynamic Properties, Hysteresis Behavior and Stress-Strain Analysis of MgH2 Thin Films, Studied over a Wide Temperature Range. Crystals 2012, 2, 710–729. [Google Scholar] [CrossRef]
- Mooij, L.P.A.; Baldi, A.; Boelsma, C.; Shen, K.; Wagemaker, M.; Pivak, Y.; Schreuders, H.; Griessen, R.; Dam, B. Interface Energy Controlled Thermodynamics of Nanoscale Metal Hydrides. Adv. Energy Mater. 2011, 1, 754–758. [Google Scholar] [CrossRef]
- Mounkachi, O.; Akrouchi, A.; Tiouitchi, G.; Lakhal, M.; Salmani, E.; Benyoussef, A.; Kara, A.; El Kenz, A.; Ez-Zahraouy, H.; El Moutaouakil, A. Stability, Electronic Structure and Thermodynamic Properties of Nanostructured MgH2 Thin Films. Energies 2021, 14, 7737. [Google Scholar] [CrossRef]
- Crivello, J.-C.; Dam, B.; Denys, R.V.; Dornheim, M.; Grant, D.M.; Huot, J.; Jensen, T.R.; Jongh, P.; Latroche, M.; Milanese, C.; et al. Review of Magnesium Hydride-Based Materials: Development and Optimisation. Appl. Phys. A 2016, 122, 97. [Google Scholar] [CrossRef]
- Zeng, K. Critical Assessment and Thermodynamic Modeling of the Mg–H System. Int. J. Hydrogen Energy 1999, 24, 989–1004. [Google Scholar] [CrossRef]
- Stampfer, J.F.; Holley, C.E.; Suttle, J.F. The Magnesium-Hydrogen System 1–3. J. Am. Chem. Soc. 1960, 82, 3504–3508. [Google Scholar] [CrossRef]
- San-Martin, A.; Manchester, F.D. The H−Mg (Hydrogen-Magnesium) System. J. Phase Equilibria 1987, 8, 431–437. [Google Scholar] [CrossRef]
- Kirchheim, R.; Pundt, A. Hydrogen in Metals. In Physical Metallurgy; Elsevier: Amsterdam, The Netherlands, 2014; pp. 2597–2705. ISBN 978-0-444-53770-6. [Google Scholar]
- Borgschulte, A.; Lohstroh, W.; Westerwaal, R.J.; Schreuders, H.; Rector, J.H.; Dam, B.; Griessen, R. Combinatorial Method for the Development of a Catalyst Promoting Hydrogen Uptake. J. Alloys Compd. 2005, 404–406, 699–705. [Google Scholar] [CrossRef]
- Gremaud, R.; Broedersz, C.P.; Borsa, D.M.; Borgschulte, A.; Mauron, P.; Schreuders, H.; Rector, J.H.; Dam, B.; Griessen, R. Hydrogenography: An Optical Combinatorial Method to Find New Light-Weight Hydrogen-Storage Materials. Adv. Mater. 2007, 19, 2813–2817. [Google Scholar] [CrossRef]
- Wagner, S.; Pundt, A. Quasi-Thermodynamic Model on Hydride Formation in Palladium–Hydrogen Thin Films: Impact of Elastic and Microstructural Constraints. Int. J. Hydrogen Energy 2016, 41, 2727–2738. [Google Scholar] [CrossRef]
- Vermeulen, P.; Ledovskikh, A.; Danilov, D.; Notten, P.H.L. Thermodynamics and Kinetics of the Thin Film Magnesium–Hydrogen System. Acta Mater. 2009, 57, 4967–4973. [Google Scholar] [CrossRef]
- Vermeulen, P.; van Thiel, E.F.M.J.; Notten, P.H.L. Ternary MgTiX-Alloys: A Promising Route towards Low-Temperature, High-Capacity, Hydrogen-Storage Materials. Chemistry 2007, 13, 9892–9898. [Google Scholar] [CrossRef]
- Gao, J.; Shen, Y.; Sun, Y.; Feng, Z.; Shi, P.; Xie, K.; Lin, L.; Guo, X.; Zhang, S. CrSe2 Based Single-Cluster Catalysts with Controllable Charge States for the Oxygen Reduction and Hydrogen Evolution Reactions. J. Colloid Interface Sci. 2025, 678, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Nivedhitha, K.S.; Umarfarooq, M.A.; Banapurmath, N.R.; Venkatesh, R.; Khan, T.; Shreeshail, M.L. Hydrogen Storage Properties of Magnesium Based Alloys Mg67Ni(32−x)Nb1Alx (x = 1, 3, and 5) by Using Response Surface Methodology. Phys. Scr. 2024, 99, 055013. [Google Scholar] [CrossRef]
- Nivedhitha, K.S.; Venkatesh, R.; Banapurmath, N.R.; Ramesh, K.; Sajjan, A.M.; Chikkatti, P.B.S.; Yogesh; Jain, A.; Nithya, C. Exploring the Influence of Single-Walled Carbon Nanotubes Substituted Mg–Ti Alloy for Hydriding and Dehydriding Properties. Int. J. Hydrogen Energy 2024, 59, 272–281. [Google Scholar] [CrossRef]
- Nivedhitha, K.S.; Venkatesh, R.; Banapurmath, N.R. Activation Energy Study on Nanostructured Niobium Substituted Mg2Ni Intermetallic Alloy for Hydrogen Storage Application. Phys. Scr. 2022, 98, 015706. [Google Scholar] [CrossRef]
- Verbovytskyy, Y.; Vlad, K.; Zavaliy, I.; Rożdżyńska-Kiełbik, B.; Pavlyuk, V. Crystal Structure and Hydrogen Sorption Properties of Nd0.5Y0.5MgNi4−xCox Alloys (x = 0–3). Solid State Sci. 2024, 156, 107674. [Google Scholar] [CrossRef]
- Verbovytskyy, Y.; Oprysk, V.; Zavaliy, I.; Vlad, K.; Berezovets, V.; Kosarchyn, Y. The Impact of La/Y and Ni/Co Substitutions on the Gas-Phase and Electrochemical Hydrogenation Properties of the La3−xMgxNi9 Alloys. J. Alloys Compd. 2024, 977, 173247. [Google Scholar] [CrossRef]
- Verbovytskyy, Y.V.; Oprysk, V.V.; Shtender, V.V.; Zavaliy, I.Y. Hydrogen Sorption Properties of Materials Based on Alloys and Compounds with High Contents of Magnesium. Mater. Sci. 2021, 57, 366–376. [Google Scholar] [CrossRef]
- Verbovytskyy, Y.; Oprysk, V.; Paul-Boncour, V.; Zavaliy, I.; Berezovets, V.; Lyutyy, P.; Kosarchyn, Y. Solid Gas and Electrochemical Hydrogenation of the Selected Alloys (R′,R″)2−xMgxNi4−yCoy (R′, R″ = Pr, Nd; x = 0.8–1.2; y = 0–2). J. Alloys Compd. 2021, 876, 160155. [Google Scholar] [CrossRef]
- Jiang, W.; Chen, Y.; Hu, M.; Zeng, C.; Liang, C. Rare Earth-Mg-Ni-Based Alloys with Superlattice Structure for Electrochemical Hydrogen Storage. J. Alloys Compd. 2021, 887, 161381. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Qi, Y.; Zhang, W.; Gao, J.-L.; Zhang, Y.-H. Highly Improved Hydrogen Storage Dynamics of Nanocrystalline and Amorphous NdMg12-Type Alloys by Mechanical Milling. J. Iron Steel Res. Int. 2020, 27, 1236–1246. [Google Scholar] [CrossRef]
- Liu, J.; Zhu, S.; Cheng, H.; Zheng, Z.; Zhu, Z.; Yan, K.; Han, S. Enhanced Cycling Stability and High Rate Dischargeability of A2B7-Type La–Mg–Ni-Based Alloys by in-Situ Formed (La,Mg)5Ni19 Superlattice Phase. J. Alloys Compd. 2019, 777, 1087–1097. [Google Scholar] [CrossRef]
- Muthu, P.; Sinnaeruvadi, K. Facilitation of Quasi-Reversible Effect with Rapid Diffusion Kinetics on Mg0.9−xTi0.1Nix High Energy Ball Milled Powders for Ni-MH Batteries. J. Alloys Compd. 2019, 793, 155–169. [Google Scholar] [CrossRef]
- Venkateswari, A.; Nithya, C.; Kumaran, S. Electrochemical Behaviour of Mg67Ni(33−x)Nbx (X = 0, 1, 2 and 4) Alloy Synthesized by High Energy Ball Milling. Procedia Mater. Sci. 2014, 5, 679–687. [Google Scholar] [CrossRef]
- Wan, C.; Denys, R.V.; Yartys, V.A. Effect of Yttrium Content in the La2−xYxMgNi9 Battery Anode Alloys on the Structural, Hydrogen Storage and Electrochemical Properties. Dalton Trans. 2022, 51, 12986–12995. [Google Scholar] [CrossRef] [PubMed]
- Mütschele, T.; Kirchheim, R. Hydrogen as a Probe for the Average Thickness of a Grain Boundary. Scr. Metall. 1987, 21, 1101–1104. [Google Scholar] [CrossRef]
- Borsa, D.M.; Gremaud, R.; Baldi, A.; Schreuders, H.; Rector, J.H.; Kooi, B.; Vermeulen, P.; Notten, P.H.L.; Dam, B.; Griessen, R. Structural, Optical, and Electrical Properties of MgyTi1−yHx Thin Films. Phys. Rev. B 2007, 75, 205408. [Google Scholar] [CrossRef]
- Isidorsson, J.; Giebels, I.A.M.E.; Arwin, H.; Griessen, R. Optical Properties of MgH2 Measured in Situ by Ellipsometry and Spectrophotometry. Phys. Rev. B 2003, 68, 115112. [Google Scholar] [CrossRef]
- Kirchheim, R.; McLellan, R.B. Electrochemical Methods for Measuring Diffusivities of Hydrogen in Palladium and Palladium Alloys. J. Electrochem. Soc. 1980, 127, 2419–2425. [Google Scholar] [CrossRef]
- Mütschele, T.; Kirchheim, R. Segregation and Diffusion of Hydrogen in Grain Boundaries of Palladium. Scr. Metall. 1987, 21, 135–140. [Google Scholar] [CrossRef]
- Kirchheim, R.; Mütschele, T.; Kieninger, W.; Gleiter, H.; Birringer, R.; Koblé, T.D. Hydrogen in Amorphous and Nanocrystalline Metals. Mater. Sci. Eng. 1988, 99, 457–462. [Google Scholar] [CrossRef]
- Niessen, R.A.H.; Notten, P.H.L. The Influence of O2 on the Electrochemistry of Thin Film, Hydrogen Storage, Electrodes. Electrochim. Acta 2005, 50, 2959–2965. [Google Scholar] [CrossRef]
- Kumar, S.; Pavloudis, T.; Singh, V.; Nguyen, H.; Steinhauer, S.; Pursell, C.; Clemens, B.; Kioseoglou, J.; Grammatikopoulos, P.; Sowwan, M. Hydrogen Flux through Size Selected Pd Nanoparticles into Underlying Mg Nanofilms. Adv. Energy Mater. 2018, 8, 1701326. [Google Scholar] [CrossRef]
- Spatz, P.; Aebischer, H.A.; Krozer, A.; Schlapbach, L. The Diffusion of H in Mg and the Nucleation and Growth of MgH2 in Thin Films*. Z. Phys. Chem. 1993, 181, 393–397. [Google Scholar] [CrossRef]
- Klyukin, K.; Shelyapina, M.G.; Fruchart, D. DFT Calculations of Hydrogen Diffusion and Phase Transformations in Magnesium. J. Alloys Compd. 2015, 644, 371–377. [Google Scholar] [CrossRef]
- Vegge, T. Locating the Rate-Limiting Step for the Interaction of Hydrogen with Mg(0001) Using Density-Functional Theory Calculations and Rate Theory. Phys. Rev. B 2004, 70, 035412. [Google Scholar] [CrossRef]
- Nishimura, C.; Komaki, M.; Amano, M. Hydrogen Permeation through Magnesium. J. Alloys Compd. 1999, 293–295, 329–333. [Google Scholar] [CrossRef]
- Huiberts, J.N.; Griessen, R.; Rector, J.H.; Wijngaarden, R.J.; Dekker, J.P.; Groot, D.G.; Koeman, N.J. Yttrium and Lanthanum Hydride Films with Switchable Optical Properties. Nature 1996, 380, 231–234. [Google Scholar] [CrossRef]
- van der Sluis, P.; Ouwerkerk, M.; Duine, P.A. Optical Switches Based on Magnesium Lanthanide Alloy Hydrides. Appl. Phys. Lett. 1997, 70, 3356–3358. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guardi, G.; Sarapulova, A.; Dsoke, S.; Wagner, S.; Pasquini, L.; Pundt, A. Controlled Hydrogen Loading of Magnesium Thin Films in KOH—Effects on the Hydride Nucleation and Growth Regimes. Micro 2024, 4, 765-777. https://doi.org/10.3390/micro4040047
Guardi G, Sarapulova A, Dsoke S, Wagner S, Pasquini L, Pundt A. Controlled Hydrogen Loading of Magnesium Thin Films in KOH—Effects on the Hydride Nucleation and Growth Regimes. Micro. 2024; 4(4):765-777. https://doi.org/10.3390/micro4040047
Chicago/Turabian StyleGuardi, Giorgia, Angelina Sarapulova, Sonia Dsoke, Stefan Wagner, Luca Pasquini, and Astrid Pundt. 2024. "Controlled Hydrogen Loading of Magnesium Thin Films in KOH—Effects on the Hydride Nucleation and Growth Regimes" Micro 4, no. 4: 765-777. https://doi.org/10.3390/micro4040047
APA StyleGuardi, G., Sarapulova, A., Dsoke, S., Wagner, S., Pasquini, L., & Pundt, A. (2024). Controlled Hydrogen Loading of Magnesium Thin Films in KOH—Effects on the Hydride Nucleation and Growth Regimes. Micro, 4(4), 765-777. https://doi.org/10.3390/micro4040047