

A Benchmark Test of High-Throughput Atomistic Modeling for Octa-Acid Host–Guest Complexes


Abstract
1. Introduction
2. Computational Setup
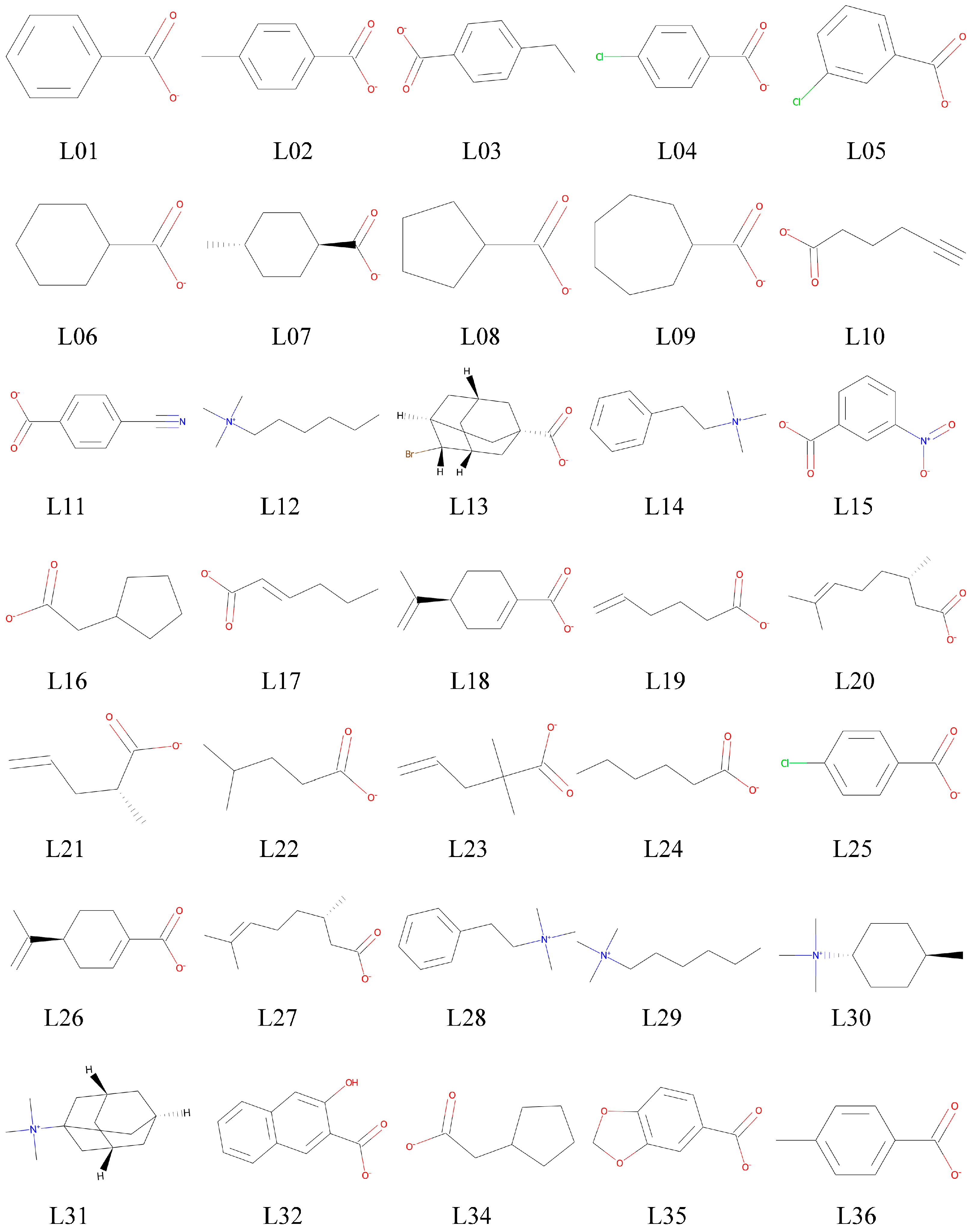
2.1. Docking Screening of Host–Guest Pairs
2.2. Fixed-Charge Parametrization and End-Point Reranking
3. Results and Discussion
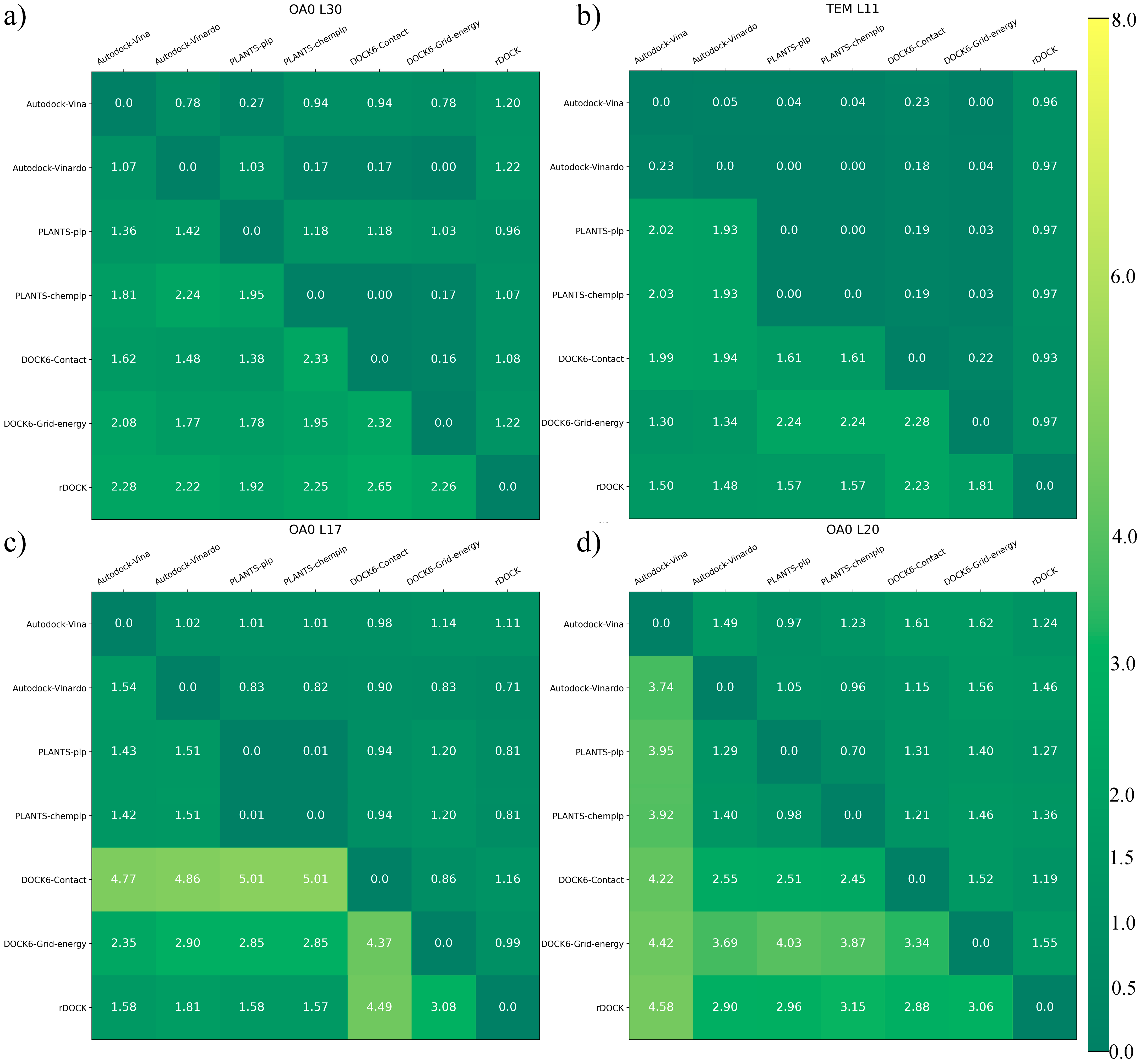
3.1. Performance of Docking Methods
3.1.1. Scoring Power
3.1.2. Computational Cost
3.1.3. Structural Consideration
3.1.4. Take-Home Message
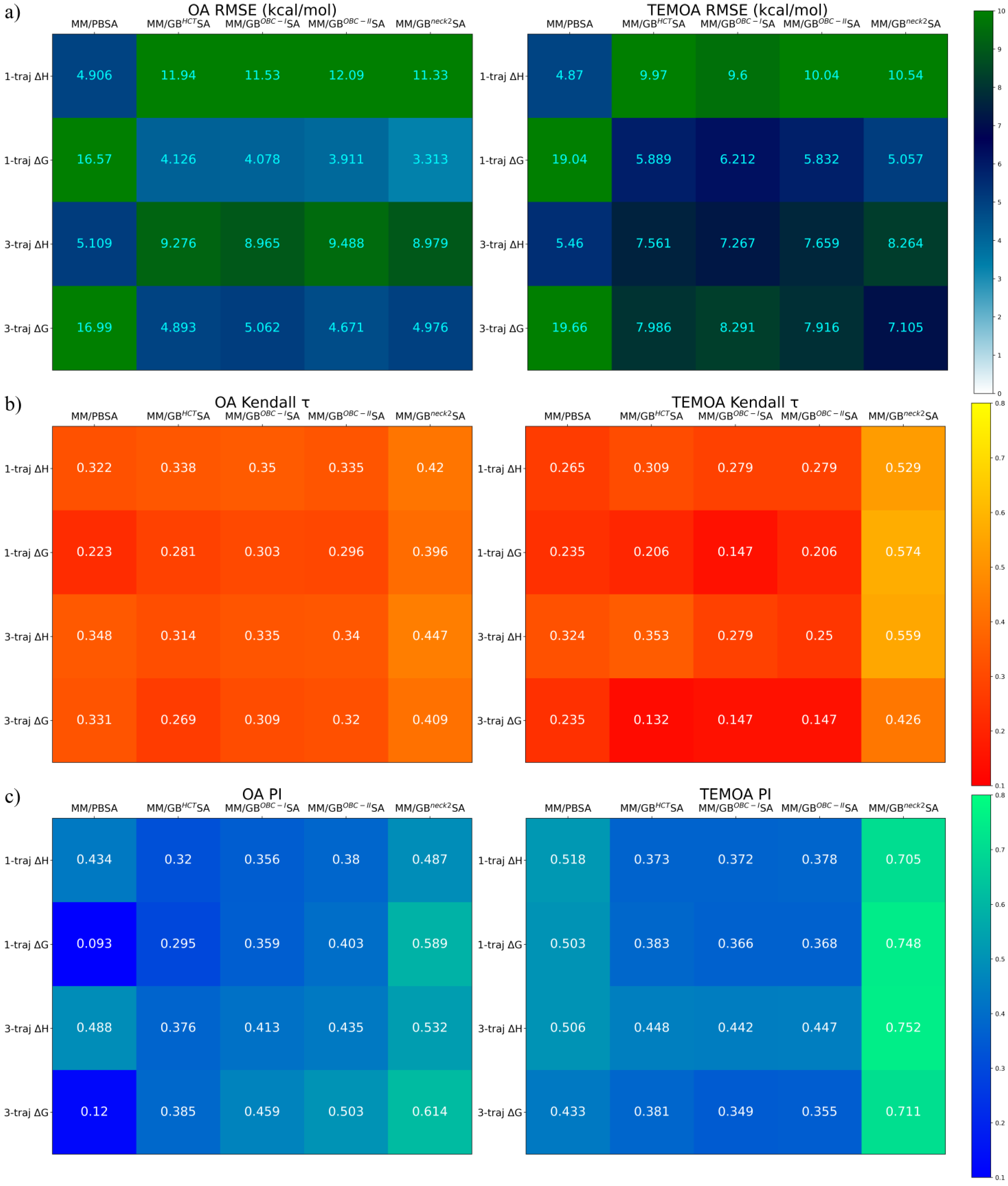
3.2. End-Point Free-Energy Calculations
3.2.1. How Long Do We Need to Converge the Statistics
3.2.2. Screening Power of End-Point Estimators
3.3. The Value of Post-Docking End-Point Rescoring
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Giordano, D.; Biancaniello, C.; Argenio, M.A.; Facchiano, A. Drug Design by Pharmacophore and Virtual Screening Approach. Pharmaceuticals 2022, 15, 646. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, Y.; Yu, Y.; Liu, D.; Zhao, J.; Zhang, L. Deciphering Selectivity Mechanism of BRD9 and TAF1(2) toward Inhibitors Based on Multiple Short Molecular Dynamics Simulations and MM-GBSA Calculations. Molecules 2023, 28, 2583. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Fan, B.; Gao, Y.; Chen, Y.; Han, D.; Lu, J.; Liu, T.; Gao, Q.; Zhang, J.Z.; Wang, M. Design Two Novel Tetrahydroquinoline Derivatives against Anticancer Target LSD1 with 3D-QSAR Model and Molecular Simulation. Molecules 2022, 27, 8358. [Google Scholar] [CrossRef]
- Procacci, P. Reformulating the entropic contribution in molecular docking scoring functions. J. Comput. Chem. 2016, 37, 1819–1827. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.; Hou, T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]
- Jansook, P.; Ogawa, N.; Loftsson, T. Cyclodextrins: Structure, physicochemical properties and pharmaceutical applications. Int. J. Pharm. 2018, 535, 272–284. [Google Scholar] [CrossRef]
- Ahmadian, N.; Mehrnejad, F.; Amininasab, M. Molecular Insight into the Interaction between Camptothecin and Acyclic Cucurbit [4] urils as Efficient Nanocontainers in Comparison with Cucurbit [7] uril: Molecular Docking and Molecular Dynamics Simulation. J. Chem. Inf. Model. 2020, 60, 1791–1803. [Google Scholar] [CrossRef] [PubMed]
- Duan, Q.; Xing, Y.; Guo, K. Carboxylato-Pillar [6] arene-Based Fluorescent Indicator Displacement Assays for Caffeine Sensing. Front. Chem. 2021, 9, 816069. [Google Scholar] [CrossRef] [PubMed]
- Cova, T.F.; Milne, B.F.; Pais, A.A.C.C. Host flexibility and space filling in supramolecular complexation of cyclodextrins: A free-energy-oriented approach. Carbohydr. Polym. 2019, 205, 42–54. [Google Scholar] [CrossRef]
- Otero-Espinar, F.; Torres-Labandeira, J.; Alvarez-Lorenzo, C.; Blanco-Méndez, J. Cyclodextrins in drug delivery systems. J. Drug Deliv. Sci. Technol. 2010, 20, 289–301. [Google Scholar] [CrossRef]
- Rasheed, A. Cyclodextrins as drug carrier molecule: A review. Sci. Pharm. 2008, 76, 567–598. [Google Scholar] [CrossRef]
- Aksu, H.; Paul, S.K.; Herbert, J.M.; Dunietz, B.D. How Well Does a Solvated Octa-acid Capsule Shield the Embedded Chromophore? A Computational Analysis Based on an Anisotropic Dielectric Continuum Model. J. Phys. Chem. B 2020, 124, 6998–7004. [Google Scholar] [CrossRef]
- Suárez, D.; Díaz, N. Affinity Calculations of Cyclodextrin Host–Guest Complexes: Assessment of Strengths and Weaknesses of End-Point Free Energy Methods. J. Chem. Inf. Model. 2019, 59, 421–440. [Google Scholar] [CrossRef] [PubMed]
- Huai, Z.; Yang, H.; Li, X.; Sun, Z. SAMPL7 TrimerTrip host-guest binding affinities from extensive alchemical and end-point free energy calculations. J. Comput. Aided Mol. Des. 2021, 35, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, M.; Sun, Z. Comprehensive Evaluation of End-Point Free Energy Techniques in Carboxylated-Pillar [6] arene Host-guest Binding: IV. The QM treatment, GB models and the Multi-Trajectory Extension. Liquids 2023, 3, 426–439. [Google Scholar] [CrossRef]
- Zhikol, O.A.; Miasnikova, D.Y.; Vashchenko, O.V.; Pinchukova, N.A.; Zbruyev, O.I.; Shishkina, S.V.; Kyrychenko, A.; Chebanov, V.A. Host-guest complexation of (pyridinyltriazolylthio) acetic acid with cucurbit [n] urils (n = 6, 7, 8): Molecular calculations and thermogravimetric analysis. J. Mol. Struct. 2023, 1294, 136532. [Google Scholar] [CrossRef]
- Liu, X.; Zheng, L.; Qin, C.; Zhang, J.Z.H.; Sun, Z. Comprehensive Evaluation of End-Point Free Energy Techniques in Carboxylated-Pillar [6] arene Host-guest Binding: I. Standard Procedure. J. Comput. Aided Mol. Des. 2022, 36, 735–752. [Google Scholar] [CrossRef] [PubMed]
- Casbarra, L.; Procacci, P. Binding free energy predictions in host-guest systems using Autodock4. A retrospective analysis on SAMPL6, SAMPL7 and SAMPL8 challenges. J. Comput. Aided Mol. Des. 2021, 35, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Gibb, C.L.D.; Gibb, B.C. Binding of cyclic carboxylates to octa-acid deep-cavity cavitand. J. Comput. Aided Mol. Des. 2014, 28, 319–325. [Google Scholar] [CrossRef]
- Sullivan, M.R.; Sokkalingam, P.; Nguyen, T.; Donahue, J.P.; Gibb, B.C. Binding of carboxylate and trimethylammonium salts to octa-acid and TEMOA deep-cavity cavitands. J. Comput. Aided Mol. Des. 2017, 31, 21–28. [Google Scholar] [CrossRef]
- Suating, P.; Nguyen, T.T.; Ernst, N.E.; Wang, Y.; Jordan, J.H.; Gibb, C.L.; Ashbaugh, H.S.; Gibb, B.C. Proximal charge effects on guest binding to a non-polar pocket. Chem. Sci. 2020, 11, 3656–3663. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, A.; Murkli, S.; McNeill, J.N.; Yao, W.; Sullivan, M.; Gilson, M.K.; Chiu, M.W.; Isaacs, L.; Gibb, B.C.; Mobley, D.L. Overview of the SAMPL6 host–guest binding affinity prediction challenge. J. Comput.-Aided Mol. Des. 2018, 32, 937–963. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, W.; Tao, Z.; Ni, X.-L. Host–guest interactions in nor-seco-cucurbit [10] uril: Novel guest-dependent molecular recognition and stereoisomerism. Beilstein J. Org. Chem. 2019, 15, 1705–1711. [Google Scholar] [CrossRef]
- Wu, X.; Meng, X.; Cheng, G. A novel 1: 2 cucurbit [8] uril inclusion complex with N-phenylpiperazine hydrochloride. J. Incl. Phenom. Macrocycl. Chem. 2009, 64, 325–329. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Verkhivker, G.M. Computational analysis of ligand binding dynamics at the intermolecular hot spots with the aid of simulated tempering and binding free energy calculations. J. Mol. Graph. Model. 2004, 22, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Verkhivker, G.M.; Bouzida, D.; Gehlhaar, D.K.; Rejto, P.A.; Freer, S.T.; Rose, P.W. Computational detection of the binding-site hot spot at the remodeled human growth hormone–receptor interface. Proteins Struct. Funct. Bioinform. 2003, 53, 201–219. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Bouzida, D.; Gehlhaar, D.K.; Rejto, P.A.; Freer, S.T.; Rose, P.W. Monte Carlo simulations of the peptide recognition at the consensus binding site of the constant fragment of human immunoglobulin G: The energy landscape analysis of a hot spot at the intermolecular interface. Proteins Struct. Funct. Bioinform. 2002, 48, 539–557. [Google Scholar] [CrossRef]
- Gehlhaar, D.K.; Verkhivker, G.M.; Rejto, P.A.; Sherman, C.J.; Fogel, D.R.; Fogel, L.J.; Freer, S.T. Molecular recognition of the inhibitor AG-1343 by HIV-1 protease: Conformationally flexible docking by evolutionary programming. Chem. Biol. 1995, 2, 317–324. [Google Scholar] [CrossRef]
- Korb, O.; Stutzle, T.; Exner, T.E. Empirical scoring functions for advanced protein—Ligand docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, T. Evaluation of AutoDock and AutoDock Vina on the CASF-2013 Benchmark. J. Chem. Inf. Model. 2018, 58, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Nguyen, T.H.; Pham, T.N.H.; Huy, N.T.; Bay, M.V.; Pham, M.Q.; Nam, P.C.; Vu, V.V.; Ngo, S.T. Autodock Vina Adopts More Accurate Binding Poses but Autodock4 Forms Better Binding Affinity. J. Chem. Inf. Model. 2020, 60, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Quiroga, R.; Villarreal, M.A. Vinardo: A Scoring Function Based on Autodock Vina Improves Scoring, Docking, and Virtual Screening. PLoS ONE 2016, 11, e0155183. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Stützle, T.; Exner, T.E. PLANTS: Application of ant colony optimization to structure-based drug design. In International Workshop on ant Colony Optimization and Swarm Intelligence; Springer: Berlin/Heidelberg, Germany, 2006; pp. 247–258. [Google Scholar]
- Deneubourg, J.-L.; Aron, S.; Goss, S.; Pasteels, J.M. The self-organizing exploratory pattern of the argentine ant. J. Insect Behav. 1990, 3, 159–168. [Google Scholar] [CrossRef]
- Goss, S.; Aron, S.; Deneubourg, J.-L.; Pasteels, J.M. Self-organized shortcuts in the Argentine ant. Naturwissenschaften 1989, 76, 579–581. [Google Scholar] [CrossRef]
- Stützle, T.; Hoos, H.H. MAX–MIN ant system. Future Gener. Comput. Syst. 2000, 16, 889–914. [Google Scholar] [CrossRef]
- Clark, M.; Cramer III, R.D.; Van Opdenbosch, N. Validation of the general purpose tripos 5.2 force field. J. Comput. Chem. 1989, 10, 982–1012. [Google Scholar] [CrossRef]
- Murray, C.W.; Auton, T.R.; Eldridge, M.D. Empirical scoring functions. II. The testing of an empirical scoring function for the prediction of ligand-receptor binding affinities and the use of Bayesian regression to improve the quality of the model. J. Comput.-Aided Mol. Des. 1998, 12, 503–519. [Google Scholar] [CrossRef]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput.-Aided Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef] [PubMed]
- Allen, W.J.; Balius, T.E.; Mukherjee, S.; Brozell, S.R.; Moustakas, D.T.; Lang, P.T.; Case, D.A.; Kuntz, I.D.; Rizzo, R.C. DOCK 6: Impact of new features and current docking performance. J. Comput. Chem. 2015, 36, 1132–1156. [Google Scholar] [CrossRef] [PubMed]
- Shoichet, B.K.; Kuntz, I.D.; Bodian, D.L. Molecular docking using shape descriptors. J. Comput. Chem. 1992, 13, 380–397. [Google Scholar] [CrossRef]
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Carmona, S.; Alvarez-Garcia, D.; Foloppe, N.; Garmendia-Doval, A.B.; Juhos, S.; Schmidtke, P.; Barril, X.; Hubbard, R.E.; Morley, S.D. rDock: A Fast, Versatile and Open Source Program for Docking Ligands to Proteins and Nucleic Acids. PLoS Comp. Biol. 2014, 10, e1003571. [Google Scholar] [CrossRef] [PubMed]
- Morley, S.D.; Afshar, M. Validation of an empirical RNA-ligand scoring function for fast flexible docking using RiboDock®. J. Comput.-Aided Mol. Des. 2004, 18, 189–208. [Google Scholar] [CrossRef] [PubMed]
- Hasel, W.; Hendrickson, T.F.; Still, W.C. A rapid approximation to the solvent accessible surface areas of atoms. Tetrahedron Comput. Methodol. 1988, 1, 103–116. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1992, 97, 10269–10280. [Google Scholar] [CrossRef]
- Hertwig, R.H.; Koch, W. On the parameterization of the local correlation functional. What is Becke-3-LYP? Chem. Phys. Lett. 1997, 268, 345–351. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. IV. A new dynamical correlation functional and implications for exact-exchange mixing. J. Chem. Phys. 1996, 104, 1040–1046. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Mcweeny, R.; Diercksen, G. Self-Consistent Perturbation Theory. II. Extension to Open Shells. J. Chem. Phys. 1968, 49, 4852–4856. [Google Scholar] [CrossRef]
- Pople, J.A.; Nesbet, R.K. Self-Consistent Orbitals for Radicals. J. Chem. Phys. 1954, 22, 571–572. [Google Scholar] [CrossRef]
- Roothaan, C.C.J. New Developments in Molecular Orbital Theory. Rev. Mod. Phys. 1951, 23, 69–89. [Google Scholar] [CrossRef]
- Sun, Z.; Zheng, L.; Kai, W.; Huai, Z.; Liu, Z. Primary vs Secondary: Directionalized Guest Coordination in β-Cyclodextrin Derivatives. Carbohydr. Polym. 2022, 297, 120050. [Google Scholar] [CrossRef]
- Sun, Z.; Huai, Z.; He, Q.; Liu, Z. A General Picture of Cucurbit [8] uril Host–Guest Binding. J. Chem. Inf. Model. 2021, 61, 6107–6134. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; He, Q.; Li, X.; Zhu, Z. SAMPL6 host–guest binding affinities and binding poses from spherical-coordinates-biased simulations. J. Comput.-Aided Mol. Des. 2020, 34, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Zheng, L.; Zhang, Z.-y.; Cong, Y.; Yang, J.; Wang, X. Molecular Modelling of Ionic Liquids: Perfluorinated Anionic Species with Enlarged Halogen Substitutions. J. Mol. Liq. 2023, 378, 121599. [Google Scholar] [CrossRef]
- Sun, Z.; Zheng, L.; Zhang, Z.-Y.; Cong, Y.; Wang, M.; Wang, X.; Yang, J.; Liu, Z.; Huai, Z. Molecular Modelling of Ionic Liquids: Situations When Charge Scaling Seems Insufficient. Molecules 2023, 28, 800. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, M.; He, Q.; Liu, Z. Molecular Modelling of Ionic Liquids: Force-Field Validation and Thermodynamic Perspective from Large-Scale Fast-Growth Solvation Free Energy Calculations. Adv. Theory Simul. 2022, 5, 2200274. [Google Scholar] [CrossRef]
- Wang, X.; Huai, Z.; Sun, Z. Host Dynamics under General-Purpose Force Fields. Molecules 2023, 28, 5940. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; He, Q.; Gong, Z.; Kalhor, P.; Huai, Z.; Liu, Z. A General Picture of Cucurbit [8] uril Host-Guest Binding: Recalibrating Bonded Interactions. Molecules 2023, 28, 3124. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1173. [Google Scholar] [CrossRef]
- Price, D.J.; Brooks III, C.L. A Modified TIP3P Water Potential for Simulation with Ewald Summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef] [PubMed]
- Joung, I.S.; Cheatham III, T.E. Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef] [PubMed]
- Joung, I.S.; Cheatham, T.E. Molecular Dynamics Simulations of the Dynamic and Energetic Properties of Alkali and Halide Ions Using Water-Model-Specific Ion Parameters. J. Phys. Chem. B 2009, 113, 13279–13290. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E.; Tom, D.; Holger, G.; Luo, R.; Merz, K.M.; Alexey, O.; Carlos, S.; Bing, W.; Woods, R.J. The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of The Cartesian Equations of Motion of A System with Constraints: Molecular Dynamics of n -alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An Analytical Version of The SHAKE and RATTLE Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Case, D.A. Normal mode analysis of protein dynamics. Curr. Opin. Struct. Biol. 2010, 4, 285–290. [Google Scholar] [CrossRef]
- Liu, X.; Zheng, L.; Qin, C.; Yalong, C.; Zhang, J.Z.; Sun, Z. Comprehensive Evaluation of End-Point Free Energy Techniques in Carboxylated-Pillar [6] arene Host-Guest Binding: III. Force-Field Comparison, Three-Trajectory Realization and Further Dielectric Augmentation. Molecules 2023, 28, 2767. [Google Scholar] [CrossRef]
- Hawkins, G.D.; Cramer, C.J.; Truhlar, D.G. Parametrized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. J. Phys. Chem. 1996, 100, 19824–19839. [Google Scholar] [CrossRef]
- Hawkins, G.D.; Cramer, C.J.; Truhlar, D.G. Pairwise solute descreening of solute charges from a dielectric medium. Chem. Phys. Lett. 1995, 246, 122–129. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins Struct. Funct. Bioinform. 2004, 55, 383–394. [Google Scholar] [CrossRef]
- Feig, M.; Onufriev, A.; Lee, M.S.; Im, W.; Case, D.A. Performance comparison of generalized born and Poisson methods in the calculation of electrostatic solvation energies for protein structures. J. Comput. Chem. 2004, 25, 265–284. [Google Scholar] [CrossRef] [PubMed]
- Hai, N.; Pérez, A.; Bermeo, S.; Simmerling, C. Refinement of Generalized Born Implicit Solvation Parameters for Nucleic Acids and their Complexes with Proteins. J. Chem. Theory Comput. 2015, 11, 3714. [Google Scholar]
- Weiser, J.; Shenkin, P.S.; Still, W.C. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J. Comput. Chem. 1999, 20, 217–230. [Google Scholar] [CrossRef]
- Kendall, M.G. A New Measure of Rank Correlation. Biometrika 1938, 30, 81–93. [Google Scholar] [CrossRef]
- Pearlman, D.A.; Charifson, P.S. Are Free Energy Calculations Useful in Practice? A Comparison with Rapid Scoring Functions for the p38 MAP Kinase Protein System. J. Med. Chem. 2001, 44, 3417–3423. [Google Scholar] [CrossRef]
- Nutho, B.; Khuntawee, W.; Rungnim, C.; Pongsawasdi, P.; Wolschann, P.; Karpfen, A.; Kungwan, N.; Rungrotmongkol, T. Binding mode and free energy prediction of fisetin/β-cyclodextrin inclusion complexes. Beilstein J. Org. Chem. 2014, 10, 2789–2799. [Google Scholar] [CrossRef]
- Rungrotmongkol, T.; Chakcharoensap, T.; Pongsawasdi, P.; Kungwan, N.; Wolschann, P. The inclusion complexation of daidzein with β-cyclodextrin and 2,6-dimethyl-β-cyclodextrin: A theoretical and experimental study. Monatsh. Chem. 2018, 149, 1739–1747. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Docking Program | Sampling/Scoring Protocol | Docking Protocol |
|---|---|---|
| AutoDock Vina | Vina | Autodock Vina |
| AutoDock Vina | Vinardo | Autodock Vinardo |
| PLANTS | plp | PLANTS-plp |
| PLANTS | chemplp | PLANTS-chemplp |
| DOCK6 | Contact | DOCK6-Contact |
| DOCK6 | Grid Energy | DOCK6-Grid-energy |
| rDock | rDock | rDOCK |
| Sampling Protocol | With/Without Normal-Mode Entropy | Energy Evaluation | ||||
|---|---|---|---|---|---|---|
| MM/PBSA | MM/GBHCTSA | MM/GBOBC-ISA | MM/GBOBC-IISA | MM/GBneck2SA | ||
| Single-trajectory sampling | enthalpy-only ΔH | single-trajectory MM/PBSA ΔH | single-trajectory MM/GBHCTSA ΔH | single-trajectory MM/GBOBC-ISA ΔH | single-trajectory MM/GBOBC-IISA ΔH | single-trajectory MM/GBneck2SA ΔH |
| with entropy ΔG | single-trajectory MM/PBSA ΔG | single-trajectory MM/GBHCTSA ΔG | single-trajectory MM/GBOBC-ISA ΔG | single-trajectory MM/GBOBC-IISA ΔG | single-trajectory MM/GBneck2SA ΔG | |
| Three-trajectory sampling | enthalpy-only ΔH | three-trajectory MM/PBSA ΔH | three-trajectory MM/GBHCTSA ΔH | three-trajectory MM/GBOBC-ISA ΔH | three-trajectory MM/GBOBC-IISA ΔH | three-trajectory MM/GBneck2SA ΔH |
| with entropy ΔG | three-trajectory MM/PBSA ΔG | three-trajectory MM/GBHCTSA ΔG | three-trajectory MM/GBOBC-ISA ΔG | three-trajectory MM/GBOBC-IISA ΔG | three-trajectory MM/GBneck2SA ΔG | |
| Host | OA Top-1 | OA Top-2 | TEMOA Top-1 | TEMOA Top-2 | |
|---|---|---|---|---|---|
| Metrics | |||||
| RMSE | Single-trajectory MM/GBneck2SA ΔG | Single-trajectory MM/GBOBC-IISA ΔG | Single-trajectory MM/PBSA ΔH | Single-trajectory MM/GBneck2SA ΔG | |
| Kendall τ | Three-trajectory MM/GBneck2SA ΔH | Single-trajectory MM/GBneck2SA ΔH | Single-trajectory MM/GBneck2SA ΔG | Three-trajectory MM/GBneck2SA ΔH | |
| PI | Three-trajectory MM/GBneck2SA ΔG | Single-trajectory MM/GBneck2SA ΔG | Three-trajectory MM/GBneck2SA ΔH | Single-trajectory MM/GBneck2SA ΔG | |
| Pearson r | Three-trajectory MM/GBneck2SA ΔH | Single-trajectory MM/GBneck2SA ΔH | Three-trajectory MM/GBneck2SA ΔH | Single-trajectory MM/GBneck2SA ΔG | |
| Metrics | Robust Selection |
|---|---|
| RMSE | Single-trajectory MM/GBneck2SA ΔG |
| Kendall τ | Three-trajectory MM/GBneck2SA ΔH |
| PI | Single-trajectory MM/GBneck2SA ΔG |
| Pearson r | Three-trajectory MM/GBneck2SA ΔH |
| End-Point Protocol | Number of Observations |
|---|---|
| Single-trajectory MM/GBneck2SA ΔG | 6 |
| Single-trajectory MM/GBneck2SA ΔH | 2 |
| Single-trajectory MM/GBOBC-IISA ΔG | 1 |
| Single-trajectory MM/PBSA ΔH | 1 |
| Three-trajectory MM/GBneck2SA ΔH | 5 |
| Three-trajectory MM/GBneck2SA ΔG | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Huai, Z.; Zheng, L.; Liu, M.; Sun, Z. A Benchmark Test of High-Throughput Atomistic Modeling for Octa-Acid Host–Guest Complexes. Liquids 2024, 4, 485-504. https://doi.org/10.3390/liquids4030027
Wang X, Huai Z, Zheng L, Liu M, Sun Z. A Benchmark Test of High-Throughput Atomistic Modeling for Octa-Acid Host–Guest Complexes. Liquids. 2024; 4(3):485-504. https://doi.org/10.3390/liquids4030027
Chicago/Turabian StyleWang, Xiaohui, Zhe Huai, Lei Zheng, Meili Liu, and Zhaoxi Sun. 2024. "A Benchmark Test of High-Throughput Atomistic Modeling for Octa-Acid Host–Guest Complexes" Liquids 4, no. 3: 485-504. https://doi.org/10.3390/liquids4030027
APA StyleWang, X., Huai, Z., Zheng, L., Liu, M., & Sun, Z. (2024). A Benchmark Test of High-Throughput Atomistic Modeling for Octa-Acid Host–Guest Complexes. Liquids, 4(3), 485-504. https://doi.org/10.3390/liquids4030027