
Calculation of the Three Partition Coefficients logPow, logKoa and logKaw of Organic Molecules at Standard Conditions at Once by Means of a Generally Applicable Group-Additivity Method



Abstract
1. Introduction
2. Method
2.1. Definition of the Atom and Special Groups
2.2. Calculation of the Atom and Special Group Contributions
2.3. Calculation of Descriptors logPow and logKoa
2.4. Cross-Validation Calculations
3. Sources
3.1. Sources of logPow Values
3.2. Sources of logKoa Values
3.3. Sources of logKaw Values
4. Results
4.1. Partition Coefficient logPow
4.2. Partition Coefficient logKoa
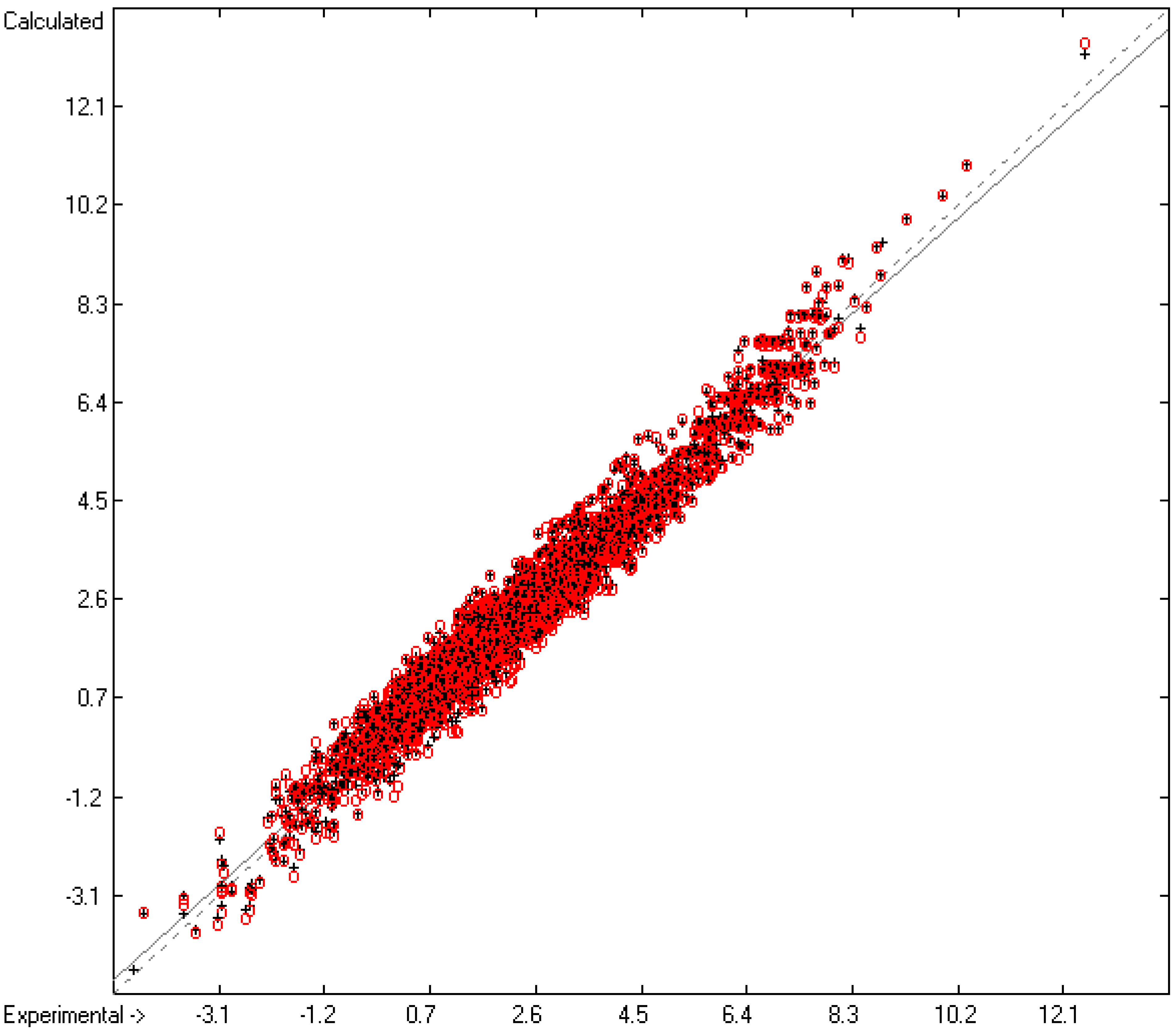
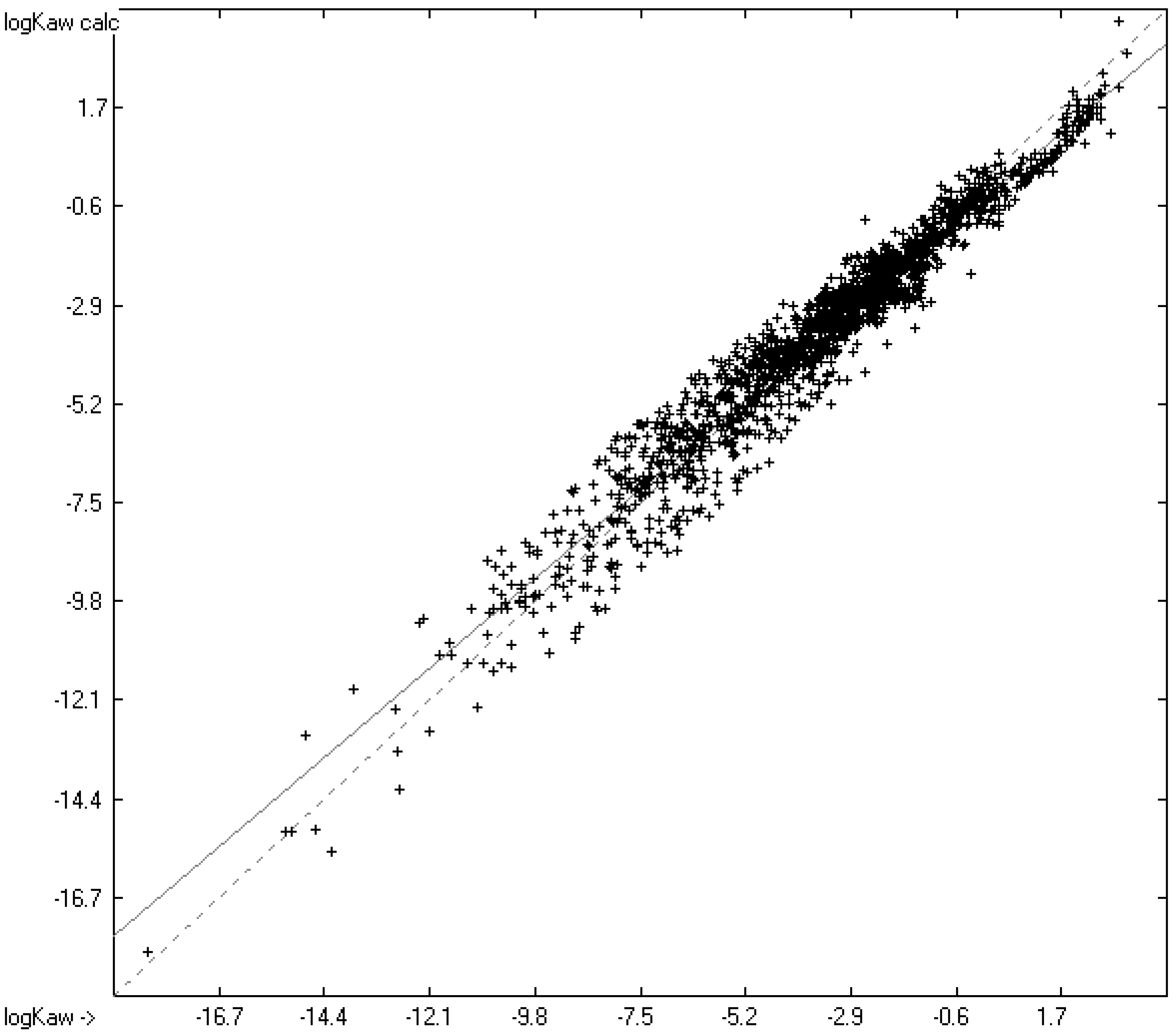
4.3. Partition Coefficient logKaw
4.4. Interpretation of the Special Groups’ Contributions to logPow and logKoa, and Ultimately for logKaw
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Simonich, S.L.; Hites, R.A. Organic Pollutant Accumulation in Vegetation. Environ. Sci. Technol. 1995, 29, 2905–2914. [Google Scholar] [CrossRef]
- McLachlan, M.S. Bioaccumulation of Hydrophobic Chemicals in Agricultural Food Chains. Environ. Sci. Technol. 1996, 30, 252–259. [Google Scholar] [CrossRef]
- Doucette, W.J.; Shunthirsasingham, C.; Dettenmaier, E.M.; Zaleski, R.T.; Fantke, P.; Arnot, J.A. A Review of Measured Bioaccumulation Data on Terrestrial Plants for Organic Chemicals:Metrics, Variability, and the Need for Standardized Measurement Protocols. Environ. Toxicol. Chem. 2018, 37, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Draghi, C.; Fayet, G.; Creton, B.; Rozanska, X.; Rotureau, P.; de Hemptinne, J.-C.; Ungerer, P.; Rousseau, B.; Adamo, C. A General Guidebook for the Theoretical Prediction of Physicochemical Properties of Chemicals for Regulatory Purposes. Chem. Rev. 2015, 115, 13093–13164. [Google Scholar] [CrossRef] [PubMed]
- Cappelli, C.I.; Benfenati, E.; Cester, J. Evaluation of QSAR models for predicting the partition coefficient (log P) of chemicals under the REACH regulation. Environ. Res. 2015, 43, 26–32. [Google Scholar] [CrossRef]
- Ghose, A.K.; Crippen, G.M. Atomic physicochemical parameters for three-dimensional structure-directed quantitative structure-activity relationships I. Partition coefficients as a measure of hydrophobicity. J. Computer. Chem. 1986, 7, 565–577. [Google Scholar] [CrossRef]
- Ghose, A.K.; Pritchett, A.; Crippen, G.M. Atomic physicochemical parameters for three dimensional structure directed quantitative structure-activity relationships III: Modeling hydrophobic interactions. J. Comput. Chem. 1988, 9, 80–90. [Google Scholar] [CrossRef]
- Klopman, G.; Li, J.-Y.; Wang, S.; Dimayuga, M. Computer automated log P calculations based on an extended group contribution approach. J. Chem. Inf. Comput. Sci. 1994, 34, 752–781. [Google Scholar] [CrossRef]
- Visvanadhan, V.N.; Ghose, A.K.; Revankar, G.R.; Robins, R.K. Atomic physicochemical parameters for three dimensional structure directed quantitative structure-activity relationships. 4. Additional parameters for hydrophobic and dispersive interactions and their application for an automated superposition of certain naturally occurring nucleoside antibiotics. J. Chem. Inf. Comput. Sci. 1989, 29, 163–172. [Google Scholar] [CrossRef]
- Leo, A.J. Calculating log Poct from structures. Chem. Rev. 1993, 93, 1281–1306. [Google Scholar] [CrossRef]
- Wang, R.; Fu, Y.; Lai, L. A new atom-additive method for calculating partition coefficients. J. Chem. Inf. Comput. Sci. 1997, 37, 615–621. [Google Scholar] [CrossRef]
- Hou, T.J.; Xu, X.J. ADME evaluation in drug discovery. 2. Prediction of partition coefficient by atom-additive approach based on atom-weighted solvent accessible surface areas. J. Chem. Inf. Comput. Sci. 2003, 43, 1058–1067. [Google Scholar] [CrossRef]
- Naef, R. A Generally Applicable Computer Algorithm Based on the Group Additivity Method for the Calculation of Seven Molecular Descriptors: Heat of Combustion, LogPO/W, LogS, Refractivity, Polarizability, Toxicity and LogBB of Organic Compounds; Scope and Limits of Applicability. Molecules 2015, 20, 18279–18351. [Google Scholar] [CrossRef] [PubMed]
- Plante, J.; Werner, S. JPlogP: An improved logP predictor trained using predicted data. J. Cheminform. 2018, 10, 61. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, N.; Goss, K.-U.; Ebert, A. Exploring the octanol–water partition coefficient dataset using deep learning techniques and data augmentation. Com. Chem. 2021, 4, 90. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Hou, T.; He, X.; Man, V.H.; Wang, J. Development and test of highly accurate endpoint free energy methods. 2: Prediction of logarithm of n-octanol–water partition coefficient (logP) for druglike molecules using MM-PBSA method. J. Comput. Chem. 2023, 44, 1300–1311. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Harner, T.; Schramm, K.W.; Quan, X.; Xue, X.; Wu, W.; Kettrup, A. Quantitative relationships between molecular structures, environmental temperatures and octanol/air partition coefficients of PCDD/Fs. Sci. Total Environ. 2002, 300, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Harner, T.; Yang, P.; Quan, X.; Chen, S.; Schramm, K.W.; Kettrup, A. Quantitative predictive models for octanol/air partition coefficients of polybrominated diphenyl ethers at different temperatures. Chemosphere 2003, 51, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Harner, T.; Schramm, K.W.; Quan, X.; Xue, X.; Kettrup, A. Quantitative relationships between molecular structures, environmental temperatures and octanol/air partition coefficients of polychlorinated biphenyls. Comput. Biol. Chem. 2003, 27, 405–421. [Google Scholar] [CrossRef]
- Zhao, H.; Chen, J.; Quan, X.; Qu, B.; Liang, X. Octanol/air partition coefficients of polybrominated biphenyls. Chemosphere 2009, 74, 1490–1494. [Google Scholar] [CrossRef]
- Staikova, M.; Wania, F.; Donaldson, D. Molecular polarizability as a single parameter predictor of vapour pressures and octanoleair partitioning coefficients of non-polar compounds: A priori approach and results. Atmos. Environ. 2004, 38, 213–225. [Google Scholar] [CrossRef]
- Zhao, H.; Zhang, Q.; Chen, J.; Xue, X.; Liang, X. Prediction of octanol/air partition coefficients of semivolatile organic compounds based on molecular connectivity index. Chemosphere 2005, 59, 1421–1426. [Google Scholar] [CrossRef]
- Zeng, X.L.; Zhang, X.L.; Wang, Y. Qspr modeling of n-octanol/air partition coefficients and liquid vapor pressures of polychlorinated dibenzo-p-dioxins. Chemosphere 2013, 91, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Shi, J.; Liu, H.; Wang, Z. Improved 3D-QSPR analysis of the predictive octanol/air partition coefficients of hydroxylated and methoxylated polybrominated diphenyl ethers. Atmos. Environ. 2013, 77, 840–845. [Google Scholar] [CrossRef]
- Jiao, L.; Gao, M.; Wang, X.; Li, H. QSPR study on the octanol/air partition coefficient of polybrominated diphenyl ethers by using molecular distance-edge vector index. Chem. Cent. J. 2014, 8, 36. [Google Scholar] [CrossRef][Green Version]
- Chen, Y.; Cai, X.; Jiang, L.; Li, Y. Prediction of octanol-air partition coefficients for polychlorinated biphenyls (PCBs) using 3D-SQAR models. Ecotoxicol. Environ. Saf. 2016, 124, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Chen, J.; Li, X.; Wang, Y.; Yu, H. Comparison of prediction methods for octanol-air partition coefficients of diverse organic compounds. Chemosphere 2016, 148, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Fu, Z.; Li, X.; Chen, J. Development of polyparameter linear free energy relationship models for octanol/air partition coefficients of diverse chemicals. Environ. Sci. Process. Impact. 2017, 19, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, J.; Zhang, L.; Qiao, X.; Huang, L. The fragment constant method for predicting octanol/air partition coefficients of persistent organic pollutants at different temperatures. J. Phys. Chem. Ref. Data 2006, 35, 1365–1384. [Google Scholar] [CrossRef]
- Ebert, R.-U.; Kühne, R.; Schüürmann, G. Henry’s Law Constant—A General-Purpose Fragment Model to Predict Log Kaw from Molecular Structure. Environ. Sci. Technol. 2023, 57, 160–167. [Google Scholar] [CrossRef]
- Hardtwig, E. Fehler—Und Ausgleichsrechnung; Bibliographisches Institut AG: Mannheim, Germany, 1968. [Google Scholar]
- Sangster, J. Octanol-water partition coefficients of simple organic compounds. J. Phys. Chem. Ref. Data 1989, 18, 1111–1229. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Tewari, Y.B.; Miller, M.M.; St. Wasik, P.; Martire, D.E. Aqueous Solubility and Octanol/Water Partition Coefficient of Organic Compounds at 25.0 °C. J. Chem. Eng. Data 1982, 27, 451–454. [Google Scholar] [CrossRef]
- Abraham, M.H.; Chadha, H.S.; Whiting, G.S.; Mitchell, R.C. Hydrogen Bonding. 32. An Analysis of Water-Octanol and Water-Alkane Partitioning and the Δlog P Parameter of Seiler. J. Pharm. Sci. 1994, 83, 1085–1100. [Google Scholar] [CrossRef] [PubMed]
- St. Czerwinski, E.; Skvorak, J.P.; Maxwell, D.M.; Lenz, D.E.; St. Baskin, I. Organophosphorus Compounds on Biodistribution and Percutaneous Toxicity. J. Biochem. Mol. Tox. 2006, 20, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Boddu, V.M.; Abburi, K.; St. Maloney, W.; Damavarapu, R. Thermophysical Properties of an Insensitive Munitions Compound, 2,4-Dinitroanisole. J. Chem. Eng. Data 2008, 53, 1120–1125. [Google Scholar] [CrossRef]
- Li, X.-J.; Shan, G.; Liu, H.; Wang, Z.-Y. Determination of lgKow and QSPR Study on Some Fluorobenzene Derivatives. Chin. J. Struct. Chem. 2009, 28, 1236–1241. [Google Scholar]
- Saranjampour, P.; Vebrosky, E.N.; Armbrust, K.L. Salinity Impacts on Water Solubility and n-Octanol/Water Partition Coefficients of Selected Pesticides and Oil Constituents. Environ. Toxicol. Chem. 2017, 36, 2274–2280. [Google Scholar] [CrossRef] [PubMed]
- Ebert, R.-U.; Kühne, R.; Schüürmann, G. Octanol/Air Partition Coefficient. A General-Purpose Fragment Model to Predict Log Koa from Molecular Structure. Environ. Sci. Technol. 2023, 57, 976–984. [Google Scholar] [CrossRef] [PubMed]
- Puzyn, T.; Falandysz, J.; Rostkowski, P.; Piliszek, S.; Wilczynska, A. Computational estimation of logarithm of octanol/air partition coefficients and subcooled vapour pressures for each of 75 chloronaphtalene congeners. Phys.-Chem. Prop. Distr. Model. Organohal. Compds. 2004, 66, 2354–2360. [Google Scholar] [CrossRef]
- Odabasi, M.; Cetin, E.; Sofuoglu, A. Determination of octanol–air partition coefficients and supercooled liquid vapor pressures of PAHs as a function of temperature: Application to gas–particle partitioning in an urban atmosphere. Atm. Environ. 2006, 40, 6615–6625. [Google Scholar] [CrossRef]
- Xu, S.; Kropscott, B. Method for Simultaneous Determination of Partition Coefficients for Cyclic Volatile Methylsiloxanes and Dimethylsilanediol. Anal. Chem. 2012, 84, 1948–1955. [Google Scholar] [CrossRef] [PubMed]
- Easterbrook, K.D.; Vona, M.A.; Osthoff, H.D. Measurement of Henry’s law constants of ethyl nitrate in deionized water, synthetic sea salt solutions, and n-octanol. Chemosphere 2024, 346, 140482. [Google Scholar] [CrossRef] [PubMed]
- Sander, R. Compilation of Henry’s law constants (version 5.0.0) for water as solvent. Atmos. Chem. Phys. 2023, 23, 10901–12440. [Google Scholar] [CrossRef]
- Allen, G.; Dwek, R.A. An n.m.r. study of keto-enol tautomerism in β-diketones. J. Chem. Soc. B 1966, 1966, 161–163. [Google Scholar] [CrossRef]
- COSMOlogic GmbH Co. KG. A Dassault Systemes Company, Version 19.0.4, COSMOthermX. 2019. Available online: www.cosmologic.de (accessed on 4 December 2023).
- US EPA. Estimation Programs Interface Suite for Microsoft Windows; Version 4.11, Module KOAWIN v. 1.11; United States Environmental Protection Agency: Washington, DC, USA, 2015. [Google Scholar]
- Naef, R.; Acree, W.E., Jr. Calculation of Five Thermodynamic Molecular Descriptors by Means of a General Computer Algorithm Based on the Group-Additivity Method: Standard Enthalpies of Vaporization, Sublimation and Solvation, and Entropy of Fusion of Ordinary Organic Molecules and Total Phase-Change Entropy of Liquid Crystals. Molecules 2017, 22, 1059. [Google Scholar] [CrossRef]
- Naef, R.; Acree, W.E. Application of a General Computer Algorithm Based on the Group-Additivity Method for the Calculation of Two Molecular Descriptors at Both Ends of Dilution: Liquid Viscosity and Activity Coefficient inWater at Infinite Dilution. Molecules 2018, 23, 5. [Google Scholar] [CrossRef]
- Naef, R.; Acree, W.E., Jr. Calculation of the Surface Tension of Ordinary Organic and Ionic Liquids by Means of a Generally Applicable Computer Algorithm Based on the Group-Additivity Method. Molecules 2018, 23, 1224. [Google Scholar] [CrossRef] [PubMed]
- Naef, R. Calculation of the Isobaric Heat Capacities of the Liquid and Solid Phase of Organic Compounds at 298.15K by Means of the Group-Additivity Method. Molecules 2020, 25, 1147. [Google Scholar] [CrossRef]
- Naef, R.; Acree, W.E., Jr. Calculation of the Vapour Pressure of Organic Molecules by Means of a Group-Additivity Method and Their Resultant Gibbs Free Energy and Entropy of Vaporization at 298.15 K. Molecules 2021, 26, 1045. [Google Scholar] [CrossRef] [PubMed]
- Naef, R.; Acree, W.E., Jr. Revision and Extension of a Generally Applicable Group-Additivity Method for the Calculation of the Standard Heat of Combustion and Formation of Organic Molecules. Molecules 2021, 26, 6101. [Google Scholar] [CrossRef] [PubMed]
- Naef, R.; Acree, W.E., Jr. Revision and Extension of a Generally Applicable Group Additivity Method for the Calculation of the Refractivity and Polarizability of Organic Molecules at 298.15 K. Liquids 2022, 2, 327–377. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atom Type | Neighbours | Meaning |
|---|---|---|
| H | H Acceptor | Correction value for intramolecular H bridge between acidic H (on O, N or S) and basic acceptor (O, N or F) |
| (COH)n | n > 1 | Correction value for each additional hydroxy group |
| (COOH)n | n > 1 | Correction value for each additional carboxylic acid group |
| Alkane | No. of C atoms | Correction value for each C atom in a pure alkane |
| Unsaturated HC | No. of C atoms | Correction value for each C atom in an aromatic hydrocarbon |
| Endocyclic bonds | No. of single bonds | Correction value for each single endocyclic bond |
| Entry | Atom Type | Neighbours | Contribution | Occurrences | Molecules |
|---|---|---|---|---|---|
| 1 | Const | 0.73 | 3332 | 3332 | |
| 2 | B(−) | F4 | 2.71 | 10 | 10 |
| 3 | C sp3 | H3C | 0.27 | 2614 | 1498 |
| 4 | C sp3 | H3N | 0.14 | 457 | 320 |
| 5 | C sp3 | H3N(+) | −1.35 | 2 | 2 |
| 6 | C sp3 | H3O | −0.26 | 375 | 285 |
| 7 | C sp3 | H3P | −0.3 | 4 | 4 |
| 8 | C sp3 | H3S | −0.34 | 61 | 53 |
| 9 | C sp3 | H3Si | 0.76 | 44 | 5 |
| 10 | C sp3 | H2C2 | 0.44 | 3262 | 1046 |
| 11 | C sp3 | H2CN | 0.42 | 741 | 429 |
| 12 | C sp3 | H2CN(+) | −0.86 | 32 | 25 |
| 13 | C sp3 | H2CO | −0.1 | 799 | 604 |
| 14 | C sp3 | H2CS | −0.33 | 97 | 69 |
| 15 | C sp3 | H2CF | −0.29 | 5 | 5 |
| 16 | C sp3 | H2CCl | 0.33 | 84 | 67 |
| 17 | C sp3 | H2CBr | 0.41 | 54 | 48 |
| 18 | C sp3 | H2CJ | 1.08 | 6 | 6 |
| 19 | C sp3 | H2CP | 2.77 | 1 | 1 |
| 20 | C sp3 | H2N2 | 2.05 | 3 | 3 |
| 21 | C sp3 | H2NO | 0.46 | 4 | 4 |
| 22 | C sp3 | H2NS | 0.72 | 3 | 3 |
| 23 | C sp3 | H2O2 | −0.17 | 6 | 6 |
| 24 | C sp3 | H2S2 | −0.86 | 6 | 6 |
| 25 | C sp3 | HC3 | 0.45 | 417 | 269 |
| 26 | C sp3 | HC2N | 0.58 | 200 | 157 |
| 27 | C sp3 | HC2N(+) | −0.73 | 25 | 24 |
| 28 | C sp3 | HC2O | 0.1 | 383 | 241 |
| 29 | C sp3 | HC2S | −0.21 | 8 | 8 |
| 30 | C sp3 | HC2F | −0.36 | 2 | 2 |
| 31 | C sp3 | HC2Cl | 0.69 | 64 | 22 |
| 32 | C sp3 | HC2Br | 0.81 | 26 | 22 |
| 33 | C sp3 | HCN2 | 1.2 | 6 | 5 |
| 34 | C sp3 | HCNO | 1.15 | 17 | 17 |
| 35 | C sp3 | HCNS | 0.9 | 25 | 25 |
| 36 | C sp3 | HCO2 | −0.02 | 31 | 22 |
| 37 | C sp3 | HCOS | 0.6 | 3 | 3 |
| 38 | C sp3 | HCOCl | 0.19 | 3 | 1 |
| 39 | C sp3 | HCOBr | 1.03 | 1 | 1 |
| 40 | C sp3 | HCOP | 0.31 | 1 | 1 |
| 41 | C sp3 | HCF2 | −0.02 | 2 | 2 |
| 42 | C sp3 | HCCl2 | 0.93 | 13 | 12 |
| 43 | C sp3 | HOF2 | −0.04 | 1 | 1 |
| 44 | C sp3 | C4 | 0.54 | 144 | 111 |
| 45 | C sp3 | C3N | 0.71 | 37 | 36 |
| 46 | C sp3 | C3N(+) | −0.43 | 6 | 6 |
| 47 | C sp3 | C3O | 0.04 | 54 | 52 |
| 48 | C sp3 | C3S | −0.1 | 17 | 17 |
| 49 | C sp3 | C3F | 0.94 | 4 | 4 |
| 50 | C sp3 | C3Cl | 0.8 | 21 | 8 |
| 51 | C sp3 | C3Br | 0.59 | 5 | 4 |
| 52 | C sp3 | C2N2 | −1.17 | 1 | 1 |
| 53 | C sp3 | C2NO | 0.52 | 5 | 5 |
| 54 | C sp3 | C2O2 | 1.65 | 5 | 5 |
| 55 | C sp3 | C2F2 | 0.67 | 2 | 2 |
| 56 | C sp3 | C2Cl2 | 0.84 | 9 | 9 |
| 57 | C sp3 | CNO2 | 1.46 | 1 | 1 |
| 58 | C sp3 | CF3 | 0.86 | 80 | 76 |
| 59 | C sp3 | CF2Cl | 1.1 | 3 | 2 |
| 60 | C sp3 | CFCl2 | 1.1 | 3 | 2 |
| 61 | C sp3 | CCl3 | 1.6 | 23 | 21 |
| 62 | C sp3 | CCl2Br | 0 | 1 | 1 |
| 63 | C sp3 | CBr3 | 2.44 | 1 | 1 |
| 64 | C sp3 | OF3 | 0.8 | 2 | 2 |
| 65 | C sp3 | SF3 | 1.04 | 8 | 8 |
| 66 | C sp3 | SFCl2 | 1.9 | 1 | 1 |
| 67 | C sp3 | SCl3 | 0.76 | 3 | 3 |
| 68 | C sp2 | H2=C | 0.25 | 97 | 87 |
| 69 | C sp2 | H2=N | −0.62 | 1 | 1 |
| 70 | C sp2 | HC=C | 0.24 | 449 | 285 |
| 71 | C sp2 | HC=N | −1.98 | 18 | 18 |
| 72 | C sp2 | HC=N(+) | 0.94 | 10 | 10 |
| 73 | C sp2 | H=CN | −0.08 | 146 | 109 |
| 74 | C sp2 | H=CN(+) | −0.6 | 18 | 18 |
| 75 | C sp2 | HC=O | −0.73 | 45 | 45 |
| 76 | C sp2 | H=CO | 0.32 | 14 | 13 |
| 77 | C sp2 | H=CS | 0.02 | 17 | 16 |
| 78 | C sp2 | H=CCl | 0.51 | 8 | 6 |
| 79 | C sp2 | H=CBr | 0.59 | 1 | 1 |
| 80 | C sp2 | HN=N | −0.06 | 65 | 52 |
| 81 | C sp2 | HN=O | −0.63 | 16 | 15 |
| 82 | C sp2 | HO=O | −0.4 | 10 | 10 |
| 83 | C sp2 | H=NS | −0.51 | 4 | 4 |
| 84 | C sp2 | C2=C | 0.38 | 160 | 133 |
| 85 | C sp2 | C2=N | −0.25 | 105 | 102 |
| 86 | C sp2 | C2=N(+) | 2.45 | 1 | 1 |
| 87 | C sp2 | C2=O | −0.86 | 242 | 194 |
| 88 | C sp2 | C=CN | 0.76 | 76 | 64 |
| 89 | C sp2 | C=CN(+) | −0.56 | 3 | 3 |
| 90 | C sp2 | C=CO | 0.64 | 41 | 36 |
| 91 | C sp2 | C=CS | −0.16 | 17 | 15 |
| 92 | C sp2 | C=CF | −0.01 | 3 | 3 |
| 93 | C sp2 | C=CCl | 0.81 | 31 | 21 |
| 94 | C sp2 | C=CBr | 0.94 | 4 | 4 |
| 95 | C sp2 | C=CJ | 0.89 | 1 | 1 |
| 96 | C sp2 | C=CP | 0 | 1 | 1 |
| 97 | C sp2 | =CN2 | 1.36 | 19 | 19 |
| 98 | C sp2 | =CN2(+) | 0.74 | 11 | 11 |
| 99 | C sp2 | CN=N | 0.24 | 67 | 63 |
| 100 | C sp2 | CN=N(+) | −0.67 | 1 | 1 |
| 101 | C sp2 | CN=O | −0.69 | 449 | 364 |
| 102 | C sp2 | C=NO | −0.76 | 1 | 1 |
| 103 | C sp2 | =CNO | −0.01 | 4 | 4 |
| 104 | C sp2 | =CNO(+) | −0.37 | 2 | 2 |
| 105 | C sp2 | CN=S | −0.36 | 8 | 8 |
| 106 | C sp2 | C=NS | 0.07 | 5 | 4 |
| 107 | C sp2 | =CNS | 0.37 | 4 | 4 |
| 108 | C sp2 | =CNCl | 1.94 | 1 | 1 |
| 109 | C sp2 | =CNBr | 0.7 | 5 | 3 |
| 110 | C sp2 | C=NCl | 1.75 | 1 | 1 |
| 111 | C sp2 | CO=O | −0.13 | 700 | 613 |
| 112 | C sp2 | CO=O(-) | −2.16 | 35 | 35 |
| 113 | C sp2 | C=OS | −0.99 | 4 | 4 |
| 114 | C sp2 | C=OCl | 0.28 | 4 | 4 |
| 115 | C sp2 | =COCl | 1.27 | 1 | 1 |
| 116 | C sp2 | =CS2 | 0 | 3 | 3 |
| 117 | C sp2 | =CSBr | −2.41 | 1 | 1 |
| 118 | C sp2 | =CF2 | 0.26 | 1 | 1 |
| 119 | C sp2 | =CCl2 | 1.21 | 12 | 10 |
| 120 | C sp2 | =CBr2 | 1.36 | 1 | 1 |
| 121 | C sp2 | N2=N | 0.79 | 26 | 25 |
| 122 | C sp2 | N2=N(+) | 0.74 | 1 | 1 |
| 123 | C sp2 | N2=O | 0.07 | 135 | 134 |
| 124 | C sp2 | N=NO | 0.11 | 1 | 1 |
| 125 | C sp2 | N2=S | 0.11 | 9 | 8 |
| 126 | C sp2 | N=NS | 0.24 | 25 | 24 |
| 127 | C sp2 | N=NCl | 1.13 | 3 | 3 |
| 128 | C sp2 | N=NBr | 0.24 | 3 | 2 |
| 129 | C sp2 | NO=O | 0.2 | 117 | 114 |
| 130 | C sp2 | =NOS | −0.19 | 1 | 1 |
| 131 | C sp2 | N=OS | 0.05 | 7 | 7 |
| 132 | C sp2 | NO=S | 0.97 | 1 | 1 |
| 133 | C sp2 | =NS2 | −1.65 | 2 | 2 |
| 134 | C sp2 | NS=S | −1.02 | 5 | 3 |
| 135 | C sp2 | =NSCl | 1.17 | 1 | 1 |
| 136 | C sp2 | O2=O | 0 | 3 | 3 |
| 137 | C sp2 | O=OCl | −0.13 | 3 | 3 |
| 138 | C aromatic | H:C2 | 0.25 | 9963 | 2133 |
| 139 | C aromatic | H:C:N | −0.49 | 283 | 193 |
| 140 | C aromatic | H:C:N(+) | 0.22 | 33 | 27 |
| 141 | C aromatic | H:N2 | −0.91 | 9 | 9 |
| 142 | C aromatic | :C3 | 0.25 | 389 | 170 |
| 143 | C aromatic | C:C2 | 0.32 | 2023 | 1351 |
| 144 | C aromatic | C:C:N | −0.38 | 74 | 62 |
| 145 | C aromatic | C:C:N(+) | −3.29 | 4 | 3 |
| 146 | C aromatic | :C2N | 0.39 | 653 | 534 |
| 147 | C aromatic | :C2N(+) | −0.15 | 194 | 161 |
| 148 | C aromatic | :C2:N | −0.09 | 93 | 72 |
| 149 | C aromatic | :C2:N(+) | −3.54 | 19 | 19 |
| 150 | C aromatic | :C2O | 0.57 | 1076 | 742 |
| 151 | C aromatic | :C2S | 0.08 | 208 | 170 |
| 152 | C aromatic | :C2F | 0.27 | 126 | 86 |
| 153 | C aromatic | :C2Cl | 0.78 | 1718 | 565 |
| 154 | C aromatic | :C2Br | 0.9 | 248 | 111 |
| 155 | C aromatic | :C2J | 1.26 | 50 | 34 |
| 156 | C aromatic | :C2P | 1.08 | 1 | 1 |
| 157 | C aromatic | C:N2 | −1.81 | 9 | 9 |
| 158 | C aromatic | :C:N2 | −0.13 | 1 | 1 |
| 159 | C aromatic | :CN:N | 0.49 | 38 | 34 |
| 160 | C aromatic | :CN:N(+) | −0.83 | 1 | 1 |
| 161 | C aromatic | :C:NO | 0.97 | 21 | 15 |
| 162 | C aromatic | :C:NS | −0.16 | 5 | 5 |
| 163 | C aromatic | :C:NF | −0.23 | 4 | 3 |
| 164 | C aromatic | :C:NCl | 0.16 | 18 | 16 |
| 165 | C aromatic | :C:NBr | 0.06 | 1 | 1 |
| 166 | C aromatic | N:N2 | −0.05 | 51 | 41 |
| 167 | C aromatic | N:N2(+) | 0 | 1 | 1 |
| 168 | C aromatic | :N2O | 1.53 | 8 | 8 |
| 169 | C aromatic | :N2S | 0.8 | 3 | 3 |
| 170 | C aromatic | :N2Cl | 0.89 | 6 | 6 |
| 171 | C(+) aromatic | H:N2 | 0.21 | 25 | 25 |
| 172 | C sp | H#C | −0.27 | 28 | 28 |
| 173 | C sp | C#C | 0.2 | 86 | 57 |
| 174 | C sp | C#N | −0.7 | 136 | 130 |
| 175 | C sp | N#N | 0.04 | 3 | 3 |
| 176 | C sp | #NS | −0.59 | 5 | 5 |
| 177 | C sp | =N=O | 0.64 | 4 | 4 |
| 178 | C sp | =N=S | 1.53 | 27 | 26 |
| 179 | N sp3 | H2C | −1.57 | 86 | 84 |
| 180 | N sp3 | H2C(pi) | −1.05 | 326 | 292 |
| 181 | N sp3 | H2N | −0.85 | 20 | 20 |
| 182 | N sp3 | H2S | −1.55 | 34 | 34 |
| 183 | N sp3 | HC2 | −1.3 | 74 | 73 |
| 184 | N sp3 | HC2(pi) | −0.93 | 225 | 203 |
| 185 | N sp3 | HC2(2pi) | −0.47 | 311 | 272 |
| 186 | N sp3 | HCN | −1.1 | 4 | 3 |
| 187 | N sp3 | HCN(pi) | −0.49 | 14 | 13 |
| 188 | N sp3 | HCN(2pi) | 1.65 | 42 | 42 |
| 189 | N sp3 | HCO(pi) | −1.32 | 9 | 9 |
| 190 | N sp3 | HCS | −1.69 | 4 | 4 |
| 191 | N sp3 | HCS(pi) | −0.98 | 47 | 47 |
| 192 | N sp3 | HCP | −1.78 | 3 | 3 |
| 193 | N sp3 | HCP(pi) | −0.41 | 1 | 1 |
| 194 | N sp3 | C3 | −1.03 | 122 | 108 |
| 195 | N sp3 | C3(pi) | −0.73 | 153 | 138 |
| 196 | N sp3 | C3(2pi) | −0.72 | 149 | 136 |
| 197 | N sp3 | C3(3pi) | −0.75 | 23 | 23 |
| 198 | N sp3 | C2N | −1.57 | 1 | 1 |
| 199 | N sp3 | C2N(pi) | −1.41 | 31 | 28 |
| 200 | N sp3 | C2N(2pi) | −0.67 | 51 | 47 |
| 201 | N sp3 | C2N(3pi) | −0.44 | 10 | 10 |
| 202 | N sp3 | C2O(pi) | −0.31 | 5 | 5 |
| 203 | N sp3 | C2S | −1.42 | 5 | 5 |
| 204 | N sp3 | C2S(pi) | 0.03 | 7 | 6 |
| 205 | N sp3 | C2S(2pi) | 0.76 | 2 | 2 |
| 206 | N sp3 | C2P | −0.33 | 5 | 3 |
| 207 | N sp3 | CN2(2pi) | 1.36 | 1 | 1 |
| 208 | N sp3 | CS2 | 0.27 | 1 | 1 |
| 209 | N sp3 | CS2(pi) | −0.29 | 1 | 1 |
| 210 | N sp2 | H=C | −0.67 | 12 | 11 |
| 211 | N sp2 | C=C | −0.72 | 200 | 180 |
| 212 | N sp2 | C=N | 0.01 | 13 | 12 |
| 213 | N sp2 | =CN | 0.49 | 96 | 78 |
| 214 | N sp2 | C=N(+) | −6.61 | 1 | 1 |
| 215 | N sp2 | =CN(+) | −1.02 | 2 | 2 |
| 216 | N sp2 | =CO | −0.64 | 47 | 41 |
| 217 | N sp2 | C=O | −1.05 | 2 | 2 |
| 218 | N sp2 | =CS | −1.44 | 5 | 4 |
| 219 | N sp2 | N=N | −0.78 | 25 | 18 |
| 220 | N sp2 | N=O | 0.16 | 40 | 37 |
| 221 | N aromatic | C2:C(+) | 0 | 50 | 25 |
| 222 | N aromatic | :C2 | 0.38 | 354 | 258 |
| 223 | N aromatic | :C:N | −0.35 | 4 | 2 |
| 224 | N(+) sp3 | H3C | −1.03 | 26 | 26 |
| 225 | N(+) sp3 | H2C2 | 1.2 | 5 | 5 |
| 226 | N(+) sp3 | HC3 | 2.68 | 1 | 1 |
| 227 | N(+) sp3 | C4 | 3.03 | 1 | 1 |
| 228 | N(+) sp2 | C=CO(−) | −2.3 | 10 | 10 |
| 229 | N(+) sp2 | CO=O(−) | 0.27 | 235 | 198 |
| 230 | N(+) sp2 | NO=O(−) | −0.19 | 2 | 2 |
| 231 | N(+) sp2 | O2=O(−) | 0.44 | 55 | 29 |
| 232 | N(+) aromatic | H:C2 | 2.5 | 3 | 3 |
| 233 | N(+) aromatic | C:C2 | −0.48 | 7 | 6 |
| 234 | N(+) aromatic | :C2O(−) | 1.73 | 19 | 19 |
| 235 | N(+) sp | =C=N(−) | 1.8 | 1 | 1 |
| 236 | N(+) sp | =N2(−) | 0 | 1 | 1 |
| 237 | O | HC | −0.96 | 481 | 344 |
| 238 | O | HC(pi) | −0.72 | 627 | 557 |
| 239 | O | HN | −0.15 | 11 | 11 |
| 240 | O | HN(pi) | −0.24 | 6 | 6 |
| 241 | O | C2 | 0.06 | 156 | 115 |
| 242 | O | C2(pi) | −0.13 | 726 | 588 |
| 243 | O | C2(2pi) | −0.51 | 301 | 280 |
| 244 | O | CN | 0.4 | 3 | 3 |
| 245 | O | CN(pi) | 0.82 | 4 | 4 |
| 246 | O | CN(+)(pi) | 0.01 | 55 | 29 |
| 247 | O | CN(2pi) | 0.53 | 13 | 12 |
| 248 | O | CS | −0.13 | 13 | 8 |
| 249 | O | CS(pi) | −0.1 | 3 | 3 |
| 250 | O | CP | 0.23 | 132 | 68 |
| 251 | O | CP(pi) | −0.49 | 36 | 26 |
| 252 | O | CSi | −0.15 | 8 | 2 |
| 253 | O | N2(2pi) | 1.91 | 5 | 5 |
| 254 | O | NP(pi) | −1.95 | 14 | 14 |
| 255 | O | Si2 | 0.09 | 18 | 4 |
| 256 | S2 | HC | 0.65 | 14 | 12 |
| 257 | S2 | HC(pi) | 0.14 | 31 | 31 |
| 258 | S2 | C2 | 1.39 | 48 | 45 |
| 259 | S2 | C2(pi) | 0.98 | 68 | 63 |
| 260 | S2 | C2(2pi) | 0.98 | 55 | 54 |
| 261 | S2 | CN | 0 | 3 | 3 |
| 262 | S2 | CN(2pi) | 2.3 | 1 | 1 |
| 263 | S2 | CS | 0.87 | 2 | 1 |
| 264 | S2 | CS(pi) | 1.97 | 4 | 2 |
| 265 | S2 | CP | 1.12 | 17 | 15 |
| 266 | S2 | CP(pi) | 0.48 | 3 | 2 |
| 267 | S2 | N2 | −2.2 | 2 | 2 |
| 268 | S2 | N2(2pi) | 5.96 | 1 | 1 |
| 269 | S4 | C2=O | −1.13 | 11 | 11 |
| 270 | S4 | C2=O2 | −0.5 | 16 | 16 |
| 271 | S4 | CO=O2 | −0.48 | 2 | 1 |
| 272 | S4 | CN=O2 | −0.05 | 85 | 80 |
| 273 | S4 | C=O2F | 0.24 | 2 | 2 |
| 274 | S4 | NO=O2 | 0 | 3 | 3 |
| 275 | S4 | N2=O2 | 0.77 | 5 | 5 |
| 276 | S4 | O2=O | 0.83 | 2 | 2 |
| 277 | S4 | O2=O2 | 0.5 | 2 | 2 |
| 278 | S4 | O2=O2(−) | −1.14 | 3 | 3 |
| 279 | P4 | CO2=O | −1.11 | 2 | 2 |
| 280 | P4 | CO2=S | 0.26 | 1 | 1 |
| 281 | P4 | CO=OS | −2.58 | 1 | 1 |
| 282 | P4 | CO=OF | −0.88 | 3 | 3 |
| 283 | P4 | COS=S | −2.04 | 1 | 1 |
| 284 | P4 | O3=O | −0.56 | 29 | 29 |
| 285 | P4 | O3=S | 1.12 | 18 | 18 |
| 286 | P4 | O2S=S | 0.7 | 12 | 11 |
| 287 | P4 | O=OS2 | −0.54 | 2 | 2 |
| 288 | P4 | N3=O | −0.31 | 1 | 1 |
| 289 | P4 | N2O=O | 0.24 | 2 | 2 |
| 290 | P4 | NO=OS | −1.5 | 2 | 2 |
| 291 | Si | C4 | −0.51 | 1 | 1 |
| 292 | Si | C3O | −1.7 | 2 | 1 |
| 293 | Si | C2O2 | 0.13 | 17 | 4 |
| 294 | Si | O4 | 0 | 2 | 2 |
| 295 | Halide | 1.1 | 20 | 19 | |
| 296 | H | H Acceptor | 0.51 | 164 | 154 |
| 297 | (COH)n | n > 1 | 0.26 | 137 | 74 |
| 298 | (COOH)n | n > 1 | −0.15 | 26 | 25 |
| 299 | Alkane | No. of C atoms | 0.09 | 290 | 32 |
| 300 | Unsaturated HC | No. of C atoms | 0.02 | 1584 | 135 |
| 301 | Endocyclic bonds | No. of single bds | −0.14 | 2338 | 384 |
| A | Based on | Valid groups | 214 | 3332 | |
| B | Goodness of fit | R2 | 0.9648 | 3246 | |
| C | Deviation | Average | 0.31 | 3246 | |
| D | Deviation | Standard | 0.39 | 3246 | |
| E | K-fold cv | K | 10 | 3164 | |
| F | Goodness of fit | Q2 | 0.9599 | 3164 | |
| G | Deviation | Average (cv) | 0.33 | 3164 | |
| H | Deviation | Standard (cv) | 0.42 | 3164 |
| Entry | Atom Type | Neighbours | Contribution | Occurrences | Molecules |
|---|---|---|---|---|---|
| 1 | Const | 1.46 | 1900 | 1900 | |
| 2 | C sp3 | H3C | −0.07 | 1800 | 875 |
| 3 | C sp3 | H3N | 3.42 | 131 | 87 |
| 4 | C sp3 | H3N(+) | 1.42 | 1 | 1 |
| 5 | C sp3 | H3O | 2.24 | 292 | 219 |
| 6 | C sp3 | H3S | 1.51 | 30 | 26 |
| 7 | C sp3 | H3P | −0.42 | 3 | 3 |
| 8 | C sp3 | H3Si | 0.42 | 68 | 11 |
| 9 | C sp3 | H2C2 | 0.43 | 1732 | 538 |
| 10 | C sp3 | H2CN | 3.91 | 191 | 129 |
| 11 | C sp3 | H2CN(+) | 1.64 | 6 | 5 |
| 12 | C sp3 | H2CO | 2.61 | 535 | 342 |
| 13 | C sp3 | H2CS | 1.76 | 57 | 44 |
| 14 | C sp3 | H2CP | 2.58 | 3 | 3 |
| 15 | C sp3 | H2CF | −0.77 | 3 | 3 |
| 16 | C sp3 | H2CCl | 0.71 | 75 | 56 |
| 17 | C sp3 | H2CBr | 1.05 | 23 | 18 |
| 18 | C sp3 | H2CJ | 1.13 | 5 | 5 |
| 19 | C sp3 | H2CSi | 2.91 | 4 | 4 |
| 20 | C sp3 | H2N2 | 4.89 | 8 | 3 |
| 21 | C sp3 | H2NO | 5.65 | 9 | 8 |
| 22 | C sp3 | H2NS | 4.67 | 5 | 5 |
| 23 | C sp3 | H2O2 | 4.78 | 6 | 4 |
| 24 | C sp3 | H2S2 | 3.74 | 4 | 4 |
| 25 | C sp3 | HC3 | 0.64 | 268 | 180 |
| 26 | C sp3 | HC2N | 4.08 | 64 | 53 |
| 27 | C sp3 | HC2N(+) | 2.05 | 1 | 1 |
| 28 | C sp3 | HC2O | 2.86 | 169 | 135 |
| 29 | C sp3 | HC2S | 1.76 | 9 | 7 |
| 30 | C sp3 | HC2F | −1.66 | 1 | 1 |
| 31 | C sp3 | HC2Cl | 1.21 | 43 | 17 |
| 32 | C sp3 | HC2Br | 1.31 | 14 | 9 |
| 33 | C sp3 | HC2J | 1.95 | 1 | 1 |
| 34 | C sp3 | HCNO | 8.18 | 3 | 3 |
| 35 | C sp3 | HCNS | 2.08 | 1 | 1 |
| 36 | C sp3 | HCO2 | 5.73 | 6 | 6 |
| 37 | C sp3 | HCF2 | −0.18 | 7 | 7 |
| 38 | C sp3 | HCFCl | 0.02 | 2 | 2 |
| 39 | C sp3 | HCCl2 | 1.18 | 15 | 14 |
| 40 | C sp3 | HCClBr | 0.77 | 1 | 1 |
| 41 | C sp3 | HOF2 | 1.79 | 3 | 3 |
| 42 | C sp3 | C4 | 0.73 | 98 | 84 |
| 43 | C sp3 | C3N | 4.1 | 13 | 13 |
| 44 | C sp3 | C3O | 3.11 | 40 | 37 |
| 45 | C sp3 | C3S | 2.6 | 3 | 3 |
| 46 | C sp3 | C3Cl | 0.87 | 37 | 15 |
| 47 | C sp3 | C2NO | 5.94 | 1 | 1 |
| 48 | C sp3 | C2O2 | 5.94 | 6 | 6 |
| 49 | C sp3 | C2F2 | 0.23 | 58 | 10 |
| 50 | C sp3 | C2Cl2 | 1.24 | 18 | 17 |
| 51 | C sp3 | CNO2 | 9.56 | 1 | 1 |
| 52 | C sp3 | COF2 | 3.06 | 3 | 3 |
| 53 | C sp3 | CF3 | −0.06 | 55 | 51 |
| 54 | C sp3 | CF2Cl | −0.02 | 4 | 3 |
| 55 | C sp3 | CFCl2 | 0.37 | 3 | 2 |
| 56 | C sp3 | CCl3 | 1.62 | 17 | 16 |
| 57 | C sp3 | CBr3 | 0.57 | 1 | 1 |
| 58 | C sp3 | O2F2 | 6.85 | 1 | 1 |
| 59 | C sp3 | OF3 | 1.86 | 3 | 3 |
| 60 | C sp2 | H2=C | −0.19 | 88 | 76 |
| 61 | C sp2 | HC=C | 0.34 | 233 | 141 |
| 62 | C sp2 | HC=N | 0.85 | 8 | 8 |
| 63 | C sp2 | HC=O | 1 | 27 | 27 |
| 64 | C sp2 | H=CN | 1.1 | 19 | 13 |
| 65 | C sp2 | H=CO | 0.48 | 15 | 14 |
| 66 | C sp2 | H=CS | −1.08 | 9 | 7 |
| 67 | C sp2 | H=CCl | 0.44 | 12 | 10 |
| 68 | C sp2 | H=CBr | 0.6 | 3 | 2 |
| 69 | C sp2 | H=CSi | 2.17 | 1 | 1 |
| 70 | C sp2 | HN=N | 1.73 | 53 | 30 |
| 71 | C sp2 | HN=O | 2.27 | 3 | 3 |
| 72 | C sp2 | HO=O | 0.92 | 4 | 4 |
| 73 | C sp2 | H=NS | 2.88 | 1 | 1 |
| 74 | C sp2 | C2=C | 0.8 | 103 | 79 |
| 75 | C sp2 | C2=N | 1.62 | 34 | 30 |
| 76 | C sp2 | C=CN | 1.6 | 19 | 16 |
| 77 | C sp2 | C2=O | 1.08 | 87 | 75 |
| 78 | C sp2 | C=CO | 1.22 | 27 | 26 |
| 79 | C sp2 | C=CP | −0.09 | 1 | 1 |
| 80 | C sp2 | C=CS | −0.41 | 14 | 10 |
| 81 | C sp2 | C=CCl | 0.62 | 39 | 24 |
| 82 | C sp2 | C=CBr | 1.01 | 12 | 5 |
| 83 | C sp2 | =CN2 | 2.98 | 2 | 2 |
| 84 | C sp2 | CN=N | 2.75 | 7 | 7 |
| 85 | C sp2 | CN=O | 2.64 | 93 | 88 |
| 86 | C sp2 | C=NO | 1.26 | 5 | 5 |
| 87 | C sp2 | =CNO | −1.24 | 3 | 3 |
| 88 | C sp2 | C=NS | 0.46 | 6 | 6 |
| 89 | C sp2 | =CNCl | 3.35 | 6 | 3 |
| 90 | C sp2 | CO=O | 1.73 | 244 | 210 |
| 91 | C sp2 | C=OS | −0.61 | 3 | 2 |
| 92 | C sp2 | =CS2 | −0.66 | 1 | 1 |
| 93 | C sp2 | =CF2 | −1.14 | 1 | 1 |
| 94 | C sp2 | =CCl2 | 1.14 | 16 | 14 |
| 95 | C sp2 | N2=N | 3.36 | 9 | 9 |
| 96 | C sp2 | N2=O | 3.65 | 43 | 40 |
| 97 | C sp2 | N=NO | 2.64 | 4 | 4 |
| 98 | C sp2 | N=NS | 0.71 | 7 | 7 |
| 99 | C sp2 | NO=O | 2.81 | 38 | 36 |
| 100 | C sp2 | N=OS | 0.93 | 17 | 17 |
| 101 | C sp2 | NO=S | 4.26 | 1 | 1 |
| 102 | C sp2 | =NOS | 0.68 | 3 | 3 |
| 103 | C sp2 | NS=S | 6.03 | 3 | 2 |
| 104 | C sp2 | =NSCl | −5.44 | 2 | 2 |
| 105 | C sp2 | O2=O | 2.56 | 3 | 3 |
| 106 | C aromatic | H:C2 | 0.31 | 5436 | 1136 |
| 107 | C aromatic | H:C:N | 0.53 | 81 | 49 |
| 108 | C aromatic | H:N2 | 0.17 | 6 | 6 |
| 109 | C aromatic | :C3 | 0.89 | 441 | 148 |
| 110 | C aromatic | C:C2 | 0.79 | 1163 | 657 |
| 111 | C aromatic | C:C:N | 0.68 | 42 | 30 |
| 112 | C aromatic | :C2N | 1.35 | 164 | 146 |
| 113 | C aromatic | :C2N(+) | 2.09 | 96 | 69 |
| 114 | C aromatic | :C2:N | 1.01 | 13 | 10 |
| 115 | C aromatic | :C2O | 1.27 | 769 | 453 |
| 116 | C aromatic | :C2P | 3.53 | 5 | 3 |
| 117 | C aromatic | :C2S | −0.19 | 38 | 33 |
| 118 | C aromatic | :C2Si | −0.25 | 1 | 1 |
| 119 | C aromatic | :C2F | 0.13 | 99 | 41 |
| 120 | C aromatic | :C2Cl | 0.91 | 1844 | 550 |
| 121 | C aromatic | :C2Br | 1.24 | 391 | 143 |
| 122 | C aromatic | :C2J | 2.14 | 10 | 9 |
| 123 | C aromatic | C:N2 | 0.77 | 11 | 10 |
| 124 | C aromatic | :CN:N | 0.8 | 4 | 4 |
| 125 | C aromatic | :C:NO | 1.2 | 28 | 24 |
| 126 | C aromatic | :C:NCl | 0.9 | 14 | 12 |
| 127 | C aromatic | N:N2 | 1.18 | 60 | 36 |
| 128 | C aromatic | :N2O | 1.15 | 11 | 11 |
| 129 | C aromatic | :N2S | −0.6 | 8 | 8 |
| 130 | C aromatic | :N2Cl | 0.43 | 9 | 8 |
| 131 | C sp | H#C | −0.45 | 18 | 17 |
| 132 | C sp | C#C | 0.67 | 18 | 17 |
| 133 | C sp | C#N | 0.73 | 46 | 43 |
| 134 | C sp | N#N | 5.32 | 1 | 1 |
| 135 | C sp | #NP | −5.58 | 1 | 1 |
| 136 | C sp | =N=S | −0.13 | 2 | 2 |
| 137 | N sp3 | H2C | −2.18 | 17 | 16 |
| 138 | N sp3 | H2C(pi) | 1.02 | 57 | 53 |
| 139 | N sp3 | H2N | 3.57 | 5 | 5 |
| 140 | N sp3 | H2S | 1.81 | 1 | 1 |
| 141 | N sp3 | HC2 | −5.94 | 12 | 11 |
| 142 | N sp3 | HC2(pi) | −2.38 | 93 | 70 |
| 143 | N sp3 | HC2(2pi) | 0.08 | 65 | 56 |
| 144 | N sp3 | HCN(pi) | 0.02 | 5 | 4 |
| 145 | N sp3 | HCN(2pi) | 1.2 | 4 | 4 |
| 146 | N sp3 | HCO(pi) | 1.13 | 1 | 1 |
| 147 | N sp3 | HCP | −4.1 | 3 | 3 |
| 148 | N sp3 | HCP(pi) | 1.51 | 1 | 1 |
| 149 | N sp3 | HCS(pi) | −1.54 | 8 | 8 |
| 150 | N sp3 | C3 | −9.44 | 17 | 17 |
| 151 | N sp3 | C3(pi) | −6.39 | 58 | 55 |
| 152 | N sp3 | C3(2pi) | −4.82 | 49 | 45 |
| 153 | N sp3 | C3(3pi) | −3.61 | 9 | 9 |
| 154 | N sp3 | C2N | −5.12 | 1 | 1 |
| 155 | N sp3 | C2N(pi) | −2.54 | 15 | 14 |
| 156 | N sp3 | C2N(+)(pi) | −1.93 | 7 | 2 |
| 157 | N sp3 | C2N(2pi) | −3.84 | 36 | 36 |
| 158 | N sp3 | C2N(3pi) | −0.65 | 13 | 12 |
| 159 | N sp3 | C2P | 0 | 1 | 1 |
| 160 | N sp3 | C2P(pi) | −2.97 | 1 | 1 |
| 161 | N sp3 | C2P(2pi) | −4.07 | 1 | 1 |
| 162 | N sp2 | H=C | 0.51 | 1 | 1 |
| 163 | N sp2 | C=C | −0.97 | 54 | 48 |
| 164 | N sp2 | C=N | 0.61 | 6 | 4 |
| 165 | N sp2 | =CN | 0.03 | 54 | 49 |
| 166 | N sp2 | =CN(+) | 9.74 | 2 | 2 |
| 167 | N sp2 | =CO | −3.65 | 30 | 26 |
| 168 | N sp2 | N=N | −1.3 | 4 | 3 |
| 169 | N sp2 | N=O | −2.02 | 13 | 13 |
| 170 | N aromatic | :C2 | 0.54 | 194 | 109 |
| 171 | N aromatic | :C:N | 0.47 | 4 | 1 |
| 172 | N(+) sp2 | CO=O(−) | −0.36 | 104 | 76 |
| 173 | N(+) sp2 | NO=O(−) | 0 | 9 | 4 |
| 174 | N(+) sp2 | O2=O(−) | −1.09 | 63 | 35 |
| 175 | O | HC | −0.66 | 143 | 121 |
| 176 | O | HC(pi) | 1.39 | 175 | 159 |
| 177 | O | HN(pi) | 4.18 | 2 | 2 |
| 178 | O | HP | 2.11 | 4 | 2 |
| 179 | O | HSi | 1.91 | 3 | 2 |
| 180 | O | C2 | −4.17 | 139 | 105 |
| 181 | O | C2(pi) | −2.68 | 392 | 317 |
| 182 | O | C2(2pi) | −0.92 | 255 | 228 |
| 183 | O | CN(pi) | 0.51 | 20 | 16 |
| 184 | O | CN(+)(pi) | 0.1 | 63 | 35 |
| 185 | O | CN(2pi) | 3.07 | 8 | 8 |
| 186 | O | CO(pi) | −1.03 | 2 | 1 |
| 187 | O | CS | −0.88 | 11 | 6 |
| 188 | O | CP | −1.2 | 183 | 93 |
| 189 | O | CP(pi) | −0.01 | 70 | 54 |
| 190 | O | CSi | −2.38 | 9 | 3 |
| 191 | O | NP(pi) | 4.65 | 1 | 1 |
| 192 | O | P2 | 1.7 | 1 | 1 |
| 193 | O | Si2 | 0 | 21 | 6 |
| 194 | P4 | C3=O | −5.7 | 1 | 1 |
| 195 | P4 | CNO=O | 1.2 | 1 | 1 |
| 196 | P4 | CO2=O | 1.47 | 3 | 3 |
| 197 | P4 | CO2=S | −1.5 | 3 | 3 |
| 198 | P4 | CO=OS | 1.99 | 1 | 1 |
| 199 | P4 | CO=OF | 1.94 | 1 | 1 |
| 200 | P4 | COS=S | −0.86 | 1 | 1 |
| 201 | P4 | NO2=O | 3.42 | 1 | 1 |
| 202 | P4 | NO2=S | 1.88 | 3 | 3 |
| 203 | P4 | NO=OS | 1.2 | 2 | 2 |
| 204 | P4 | O3=O | 0.09 | 29 | 29 |
| 205 | P4 | O3=S | −0.4 | 32 | 30 |
| 206 | P4 | O2=OS | 0.43 | 5 | 5 |
| 207 | P4 | O2=OF | −0.17 | 1 | 1 |
| 208 | P4 | O=OS2 | 1.58 | 3 | 3 |
| 209 | P4 | O2S=S | −0.27 | 18 | 17 |
| 210 | P4 | =OS3 | 1.46 | 1 | 1 |
| 211 | S2 | HC | −1.08 | 2 | 2 |
| 212 | S2 | HC(pi) | 1.54 | 1 | 1 |
| 213 | S2 | C2 | −1.5 | 14 | 14 |
| 214 | S2 | C2(pi) | 0.4 | 41 | 39 |
| 215 | S2 | C2(2pi) | 2.82 | 24 | 23 |
| 216 | S2 | CS | −0.58 | 4 | 2 |
| 217 | S2 | CS(pi) | −2.98 | 2 | 1 |
| 218 | S2 | CP | −0.12 | 33 | 28 |
| 219 | S2 | CP(pi) | 1.78 | 3 | 2 |
| 220 | S4 | C2=O | 0.6 | 2 | 2 |
| 221 | S4 | C2=O2 | 2.13 | 3 | 3 |
| 222 | S4 | CN=O2 | 3.26 | 9 | 9 |
| 223 | S4 | CO=O2 | −0.05 | 1 | 1 |
| 224 | S4 | O2=O | −0.35 | 2 | 2 |
| 225 | S4 | O2=O2 | 0.24 | 3 | 3 |
| 226 | S6 | CF5 | 1.92 | 3 | 3 |
| 227 | Si | C4 | −1.33 | 3 | 3 |
| 228 | Si | C3O | −0.65 | 7 | 4 |
| 229 | Si | C2O2 | 0.1 | 19 | 6 |
| 230 | Si | CO3 | 0 | 3 | 3 |
| 231 | H | H Acceptor | −1.51 | 47 | 45 |
| 232 | (COH)n | n > 1 | 0.06 | 22 | 15 |
| 233 | (COOH)n | n > 1 | 1.2 | 6 | 6 |
| 234 | Alkane | No. of C atoms | −0.05 | 268 | 34 |
| 235 | Unsaturated HC | No. of C atoms | −0.03 | 1512 | 140 |
| 236 | Endocyclic bonds | No. of single bds | −0.11 | 1109 | 210 |
| A | Based on | Valid groups | 167 | 1900 | |
| B | Goodness of fit | R2 | 0.9765 | 1829 | |
| C | Deviation | Average | 0.34 | 1829 | |
| D | Deviation | Standard | 0.44 | 1829 | |
| E | K-fold cv | K | 10 | 1765 | |
| F | Goodness of fit | Q2 | 0.9717 | 1765 | |
| G | Deviation | Average (cv) | 0.37 | 1765 | |
| H | Deviation | Standard (cv) | 0.48 | 1765 |
| Atom Type | C sp3 | C sp3 | C sp3 | C sp3 | C sp2 | O | S4 | Endocycl. Bonds | Const | Sum |
|---|---|---|---|---|---|---|---|---|---|---|
| Neighbors | H2CO | HC3 | C3Cl | C2Cl2 | C=CCl | CS | O2=O2 | n C-C | ||
| Contribution | 2.61 | 0.64 | 0.87 | 1.24 | 0.62 | −0.88 | 0.24 | −0.11 | 1.46 | |
| n Groups | 2 | 2 | 2 | 1 | 2 | 2 | 1 | 9 | ||
| n × Contribution | 5.22 | 1.28 | 1.74 | 1.24 | 1.24 | −1.76 | 0.24 | −0.99 | 1.46 | 9.67 |
| Descriptor | 2-Nitroaniline | 3-Nitroaniline |
|---|---|---|
| logPow | 1.85 (1.70) | 1.37 (1.19) |
| logKoa | 6.46 (5.29) | 7.62 (6.80) |
| Descriptor | Hexanoic Acid | 1,6-Hexanedioic Acid |
|---|---|---|
| logPow | 1.92 (1.91) | 0.08 (0.64) |
| logKoa | 6.31 (6.23) | 10.74 (10.62) |
| Compound | logKaw Exp | logKaw Calc |
|---|---|---|
| 2-Nitroaniline | −4.77 | −3.59 |
| 3-Nitroaniline | −6.49 | −5.61 |
| Hexanoic Acid | −4.531 | −4.32 |
| 1,6-Hexanedioic Acid | −11.15 | −9.98 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naef, R.; Acree, W.E., Jr. Calculation of the Three Partition Coefficients logPow, logKoa and logKaw of Organic Molecules at Standard Conditions at Once by Means of a Generally Applicable Group-Additivity Method. Liquids 2024, 4, 231-260. https://doi.org/10.3390/liquids4010011
Naef R, Acree WE Jr. Calculation of the Three Partition Coefficients logPow, logKoa and logKaw of Organic Molecules at Standard Conditions at Once by Means of a Generally Applicable Group-Additivity Method. Liquids. 2024; 4(1):231-260. https://doi.org/10.3390/liquids4010011
Chicago/Turabian StyleNaef, Rudolf, and William E. Acree, Jr. 2024. "Calculation of the Three Partition Coefficients logPow, logKoa and logKaw of Organic Molecules at Standard Conditions at Once by Means of a Generally Applicable Group-Additivity Method" Liquids 4, no. 1: 231-260. https://doi.org/10.3390/liquids4010011
APA StyleNaef, R., & Acree, W. E., Jr. (2024). Calculation of the Three Partition Coefficients logPow, logKoa and logKaw of Organic Molecules at Standard Conditions at Once by Means of a Generally Applicable Group-Additivity Method. Liquids, 4(1), 231-260. https://doi.org/10.3390/liquids4010011