
Vibrational Raman Spectroscopy of the Hydration Shell of Ions


Abstract
1. Introduction
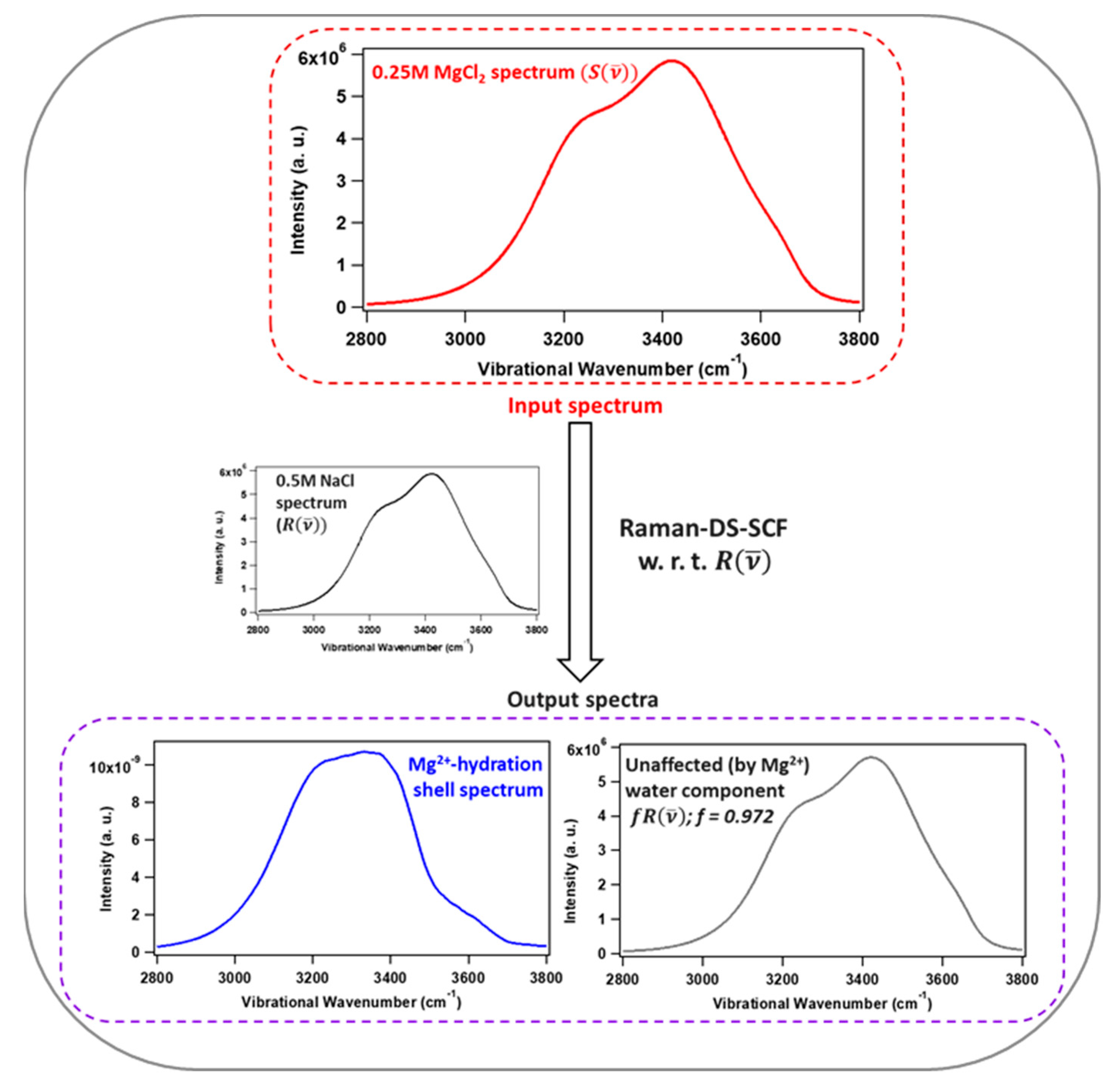
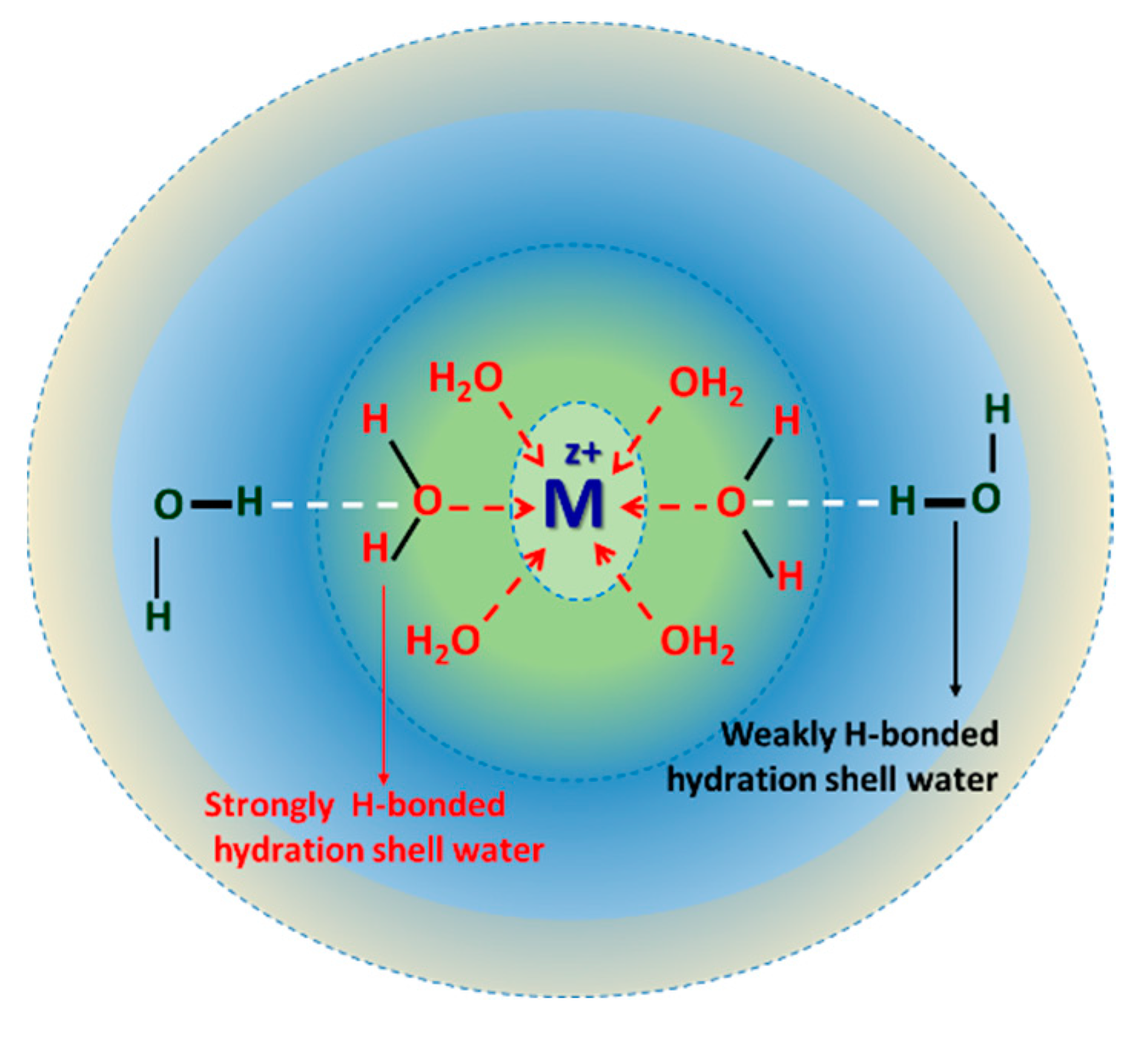
2. Raman-DS-SCF Analysis and Ion Hydration Shell
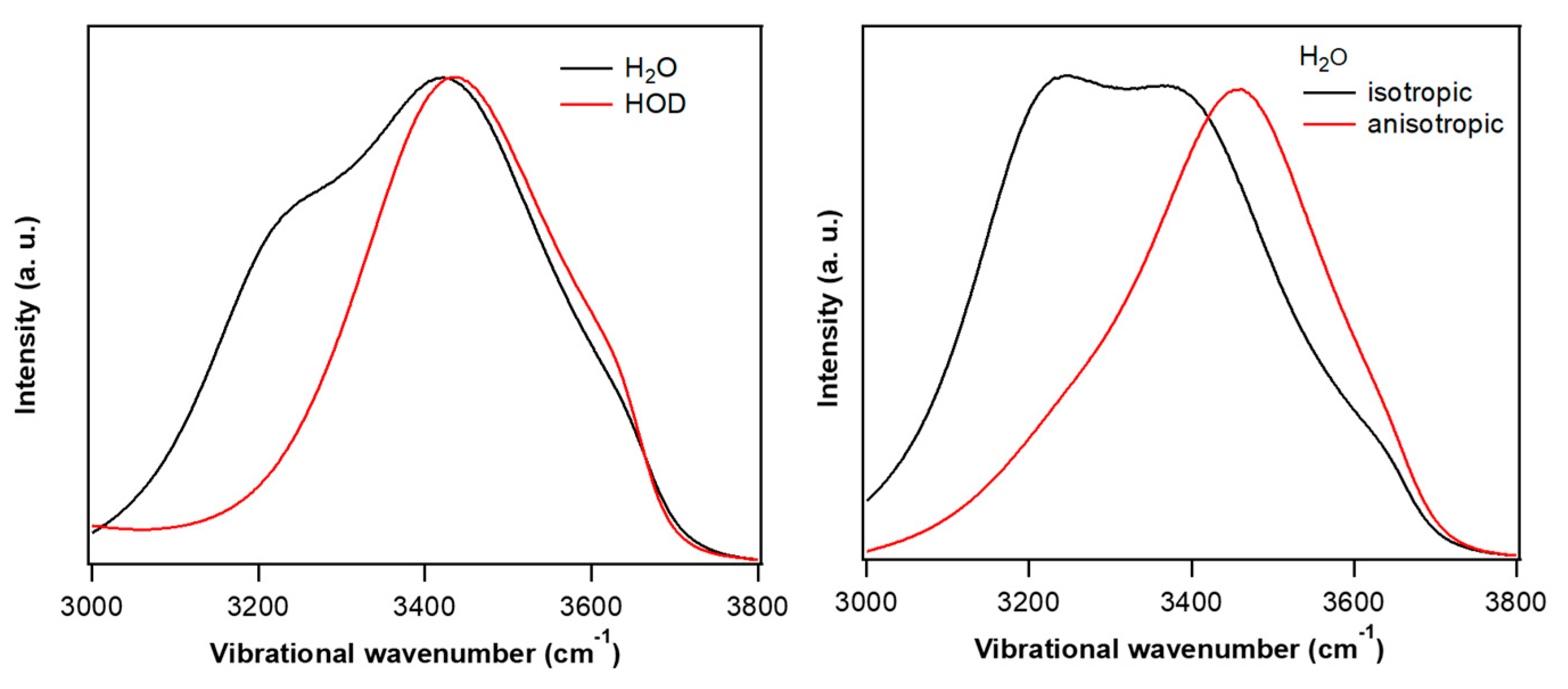
3. OH Stretch Spectra of Neat Water
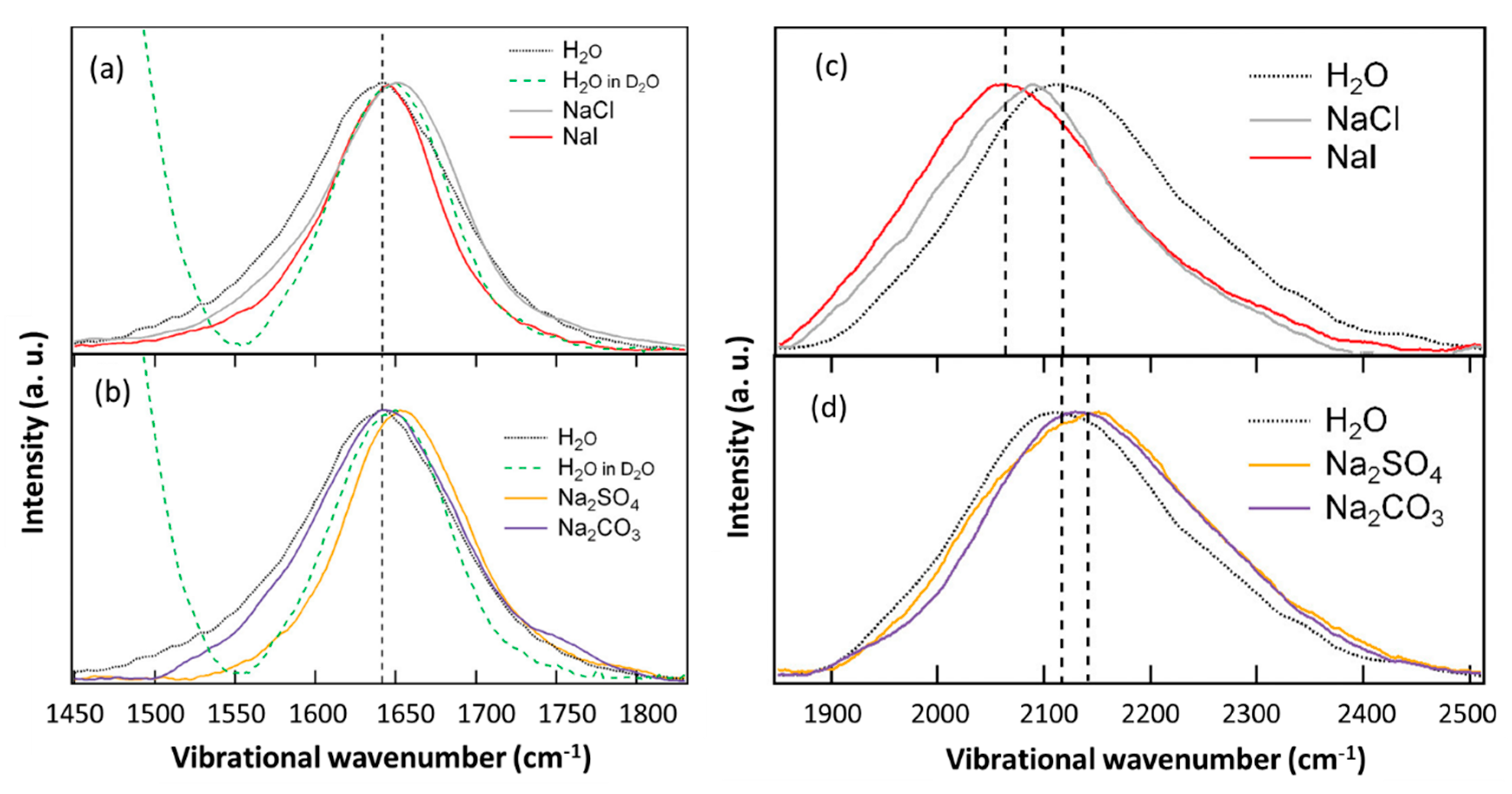
4. Effect of Electrolytes on the OH Stretch Raman Cross-Section and Vibrational Coupling of Water
5. Hydration Shell of Ions
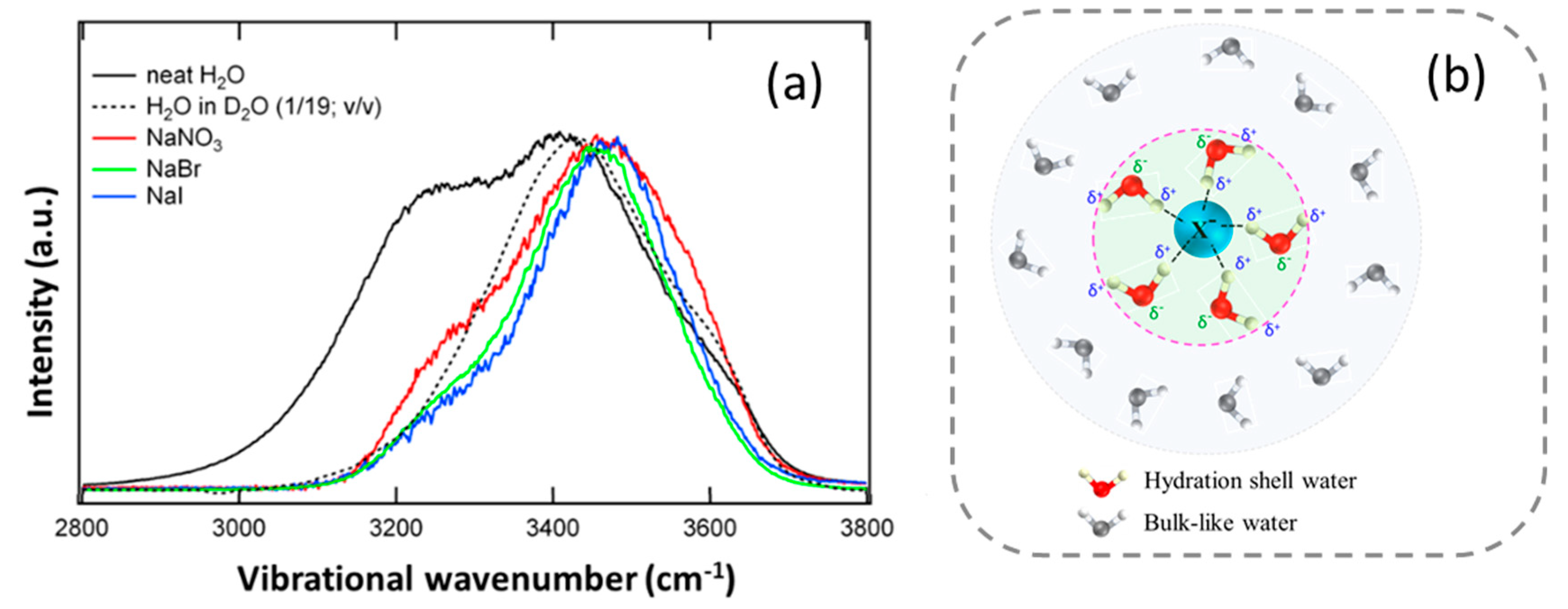
5.1. Water in the Hydration Shell of a Low-Charge-Density Monovalent Anion
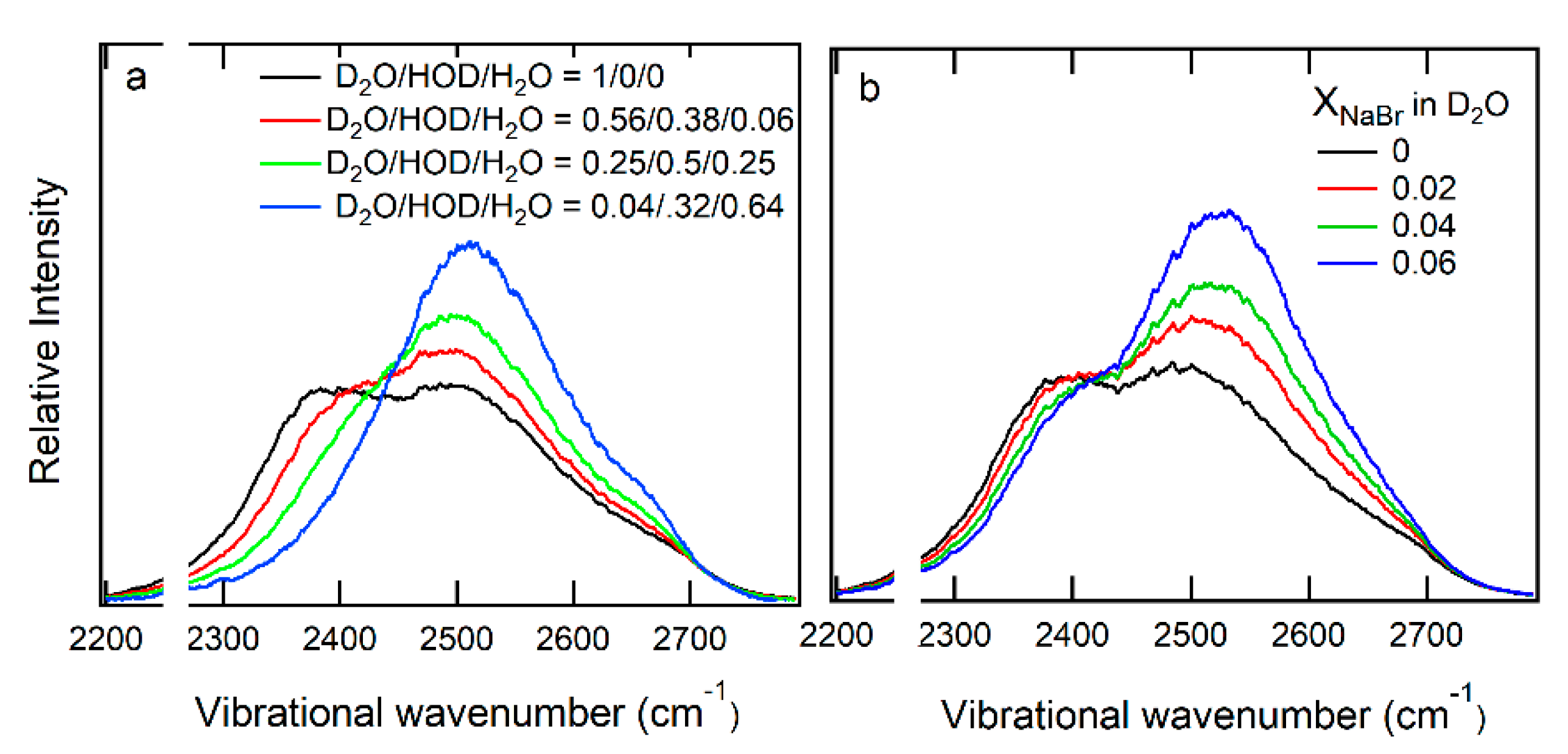
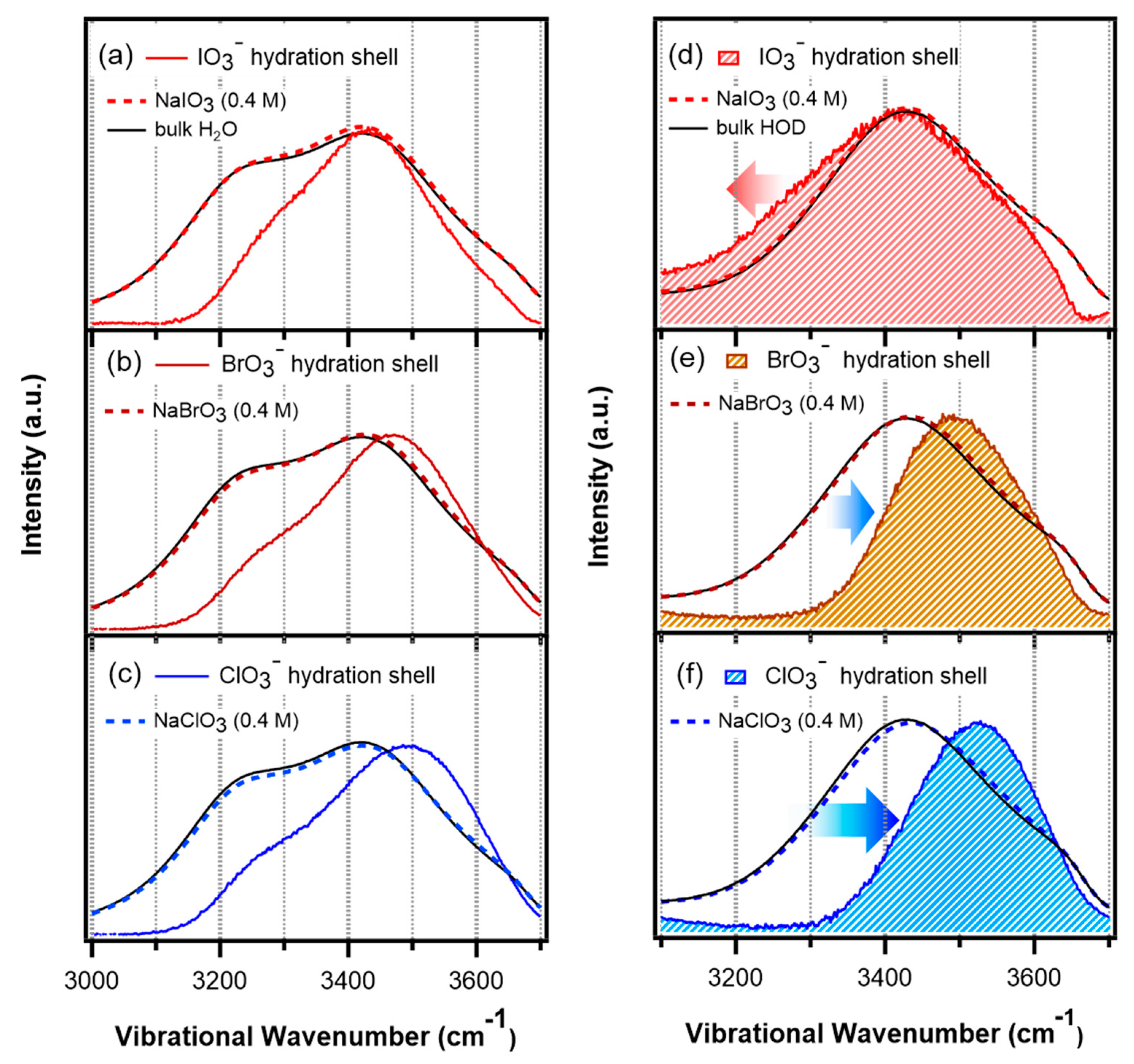
5.2. Hydration Shell of Monovalent Oxyhalide Anion
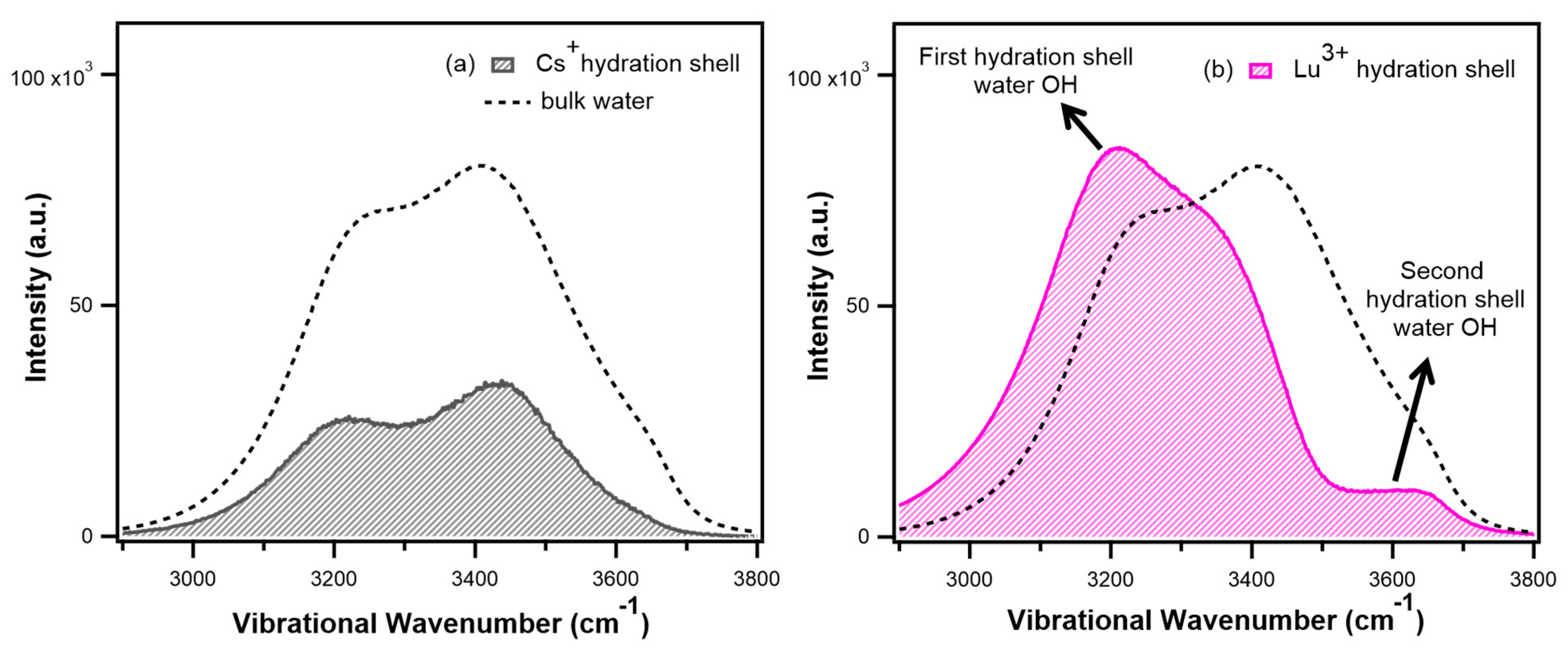
5.3. Hydration Shell of High-Charge-Density Anions
5.4. HOH Bend and Bend+Libration Combination Band of Water in the Anion Hydration Shell
6. Water in the Hydration Shell of a Metal Cation
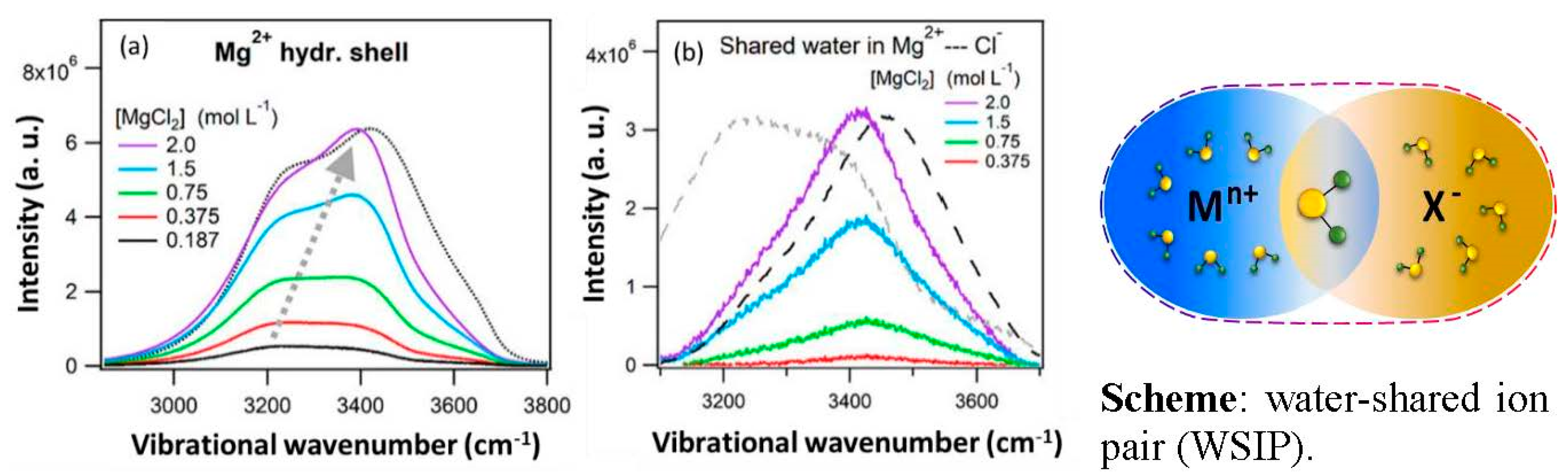
7. Water Shared Ion-Pair Formation and Restructuring of Hydration Shell Water
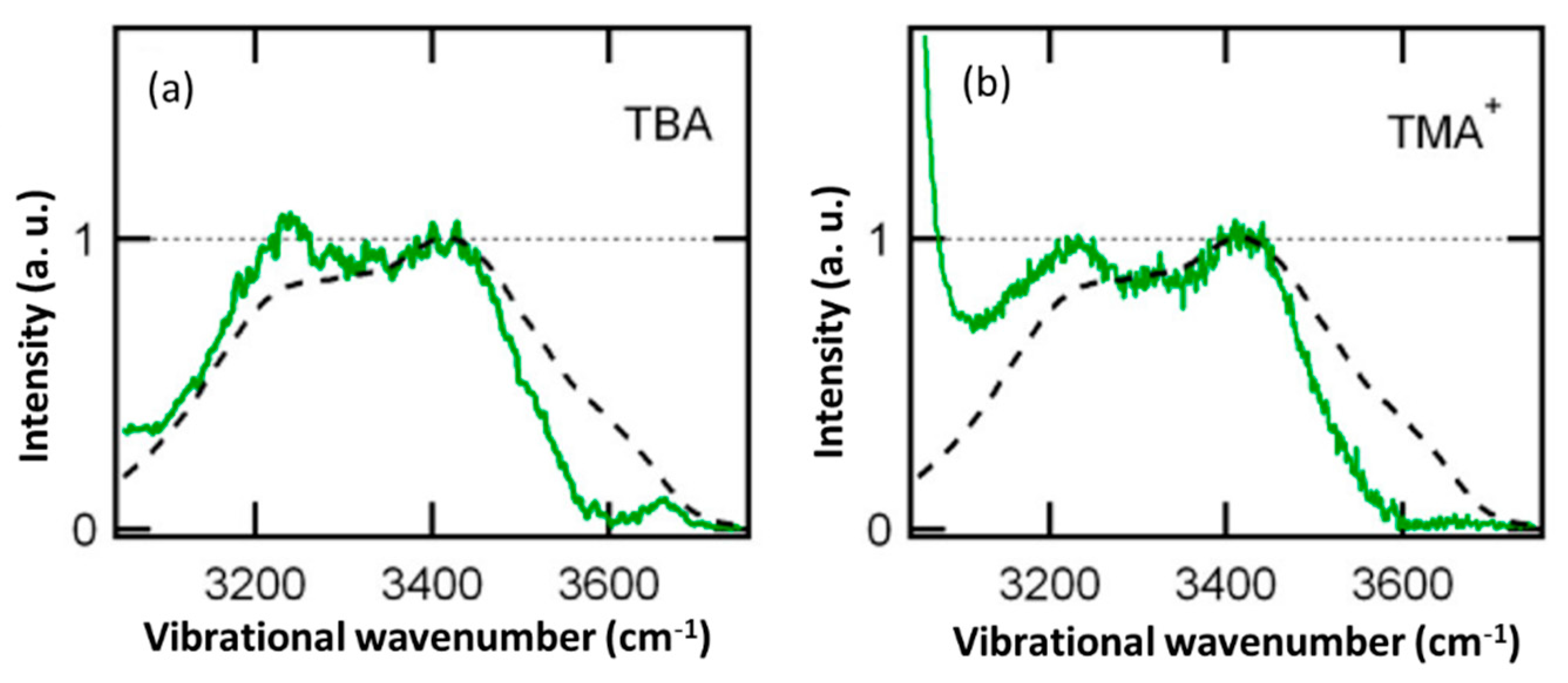
8. Water in the Hydration Shell of Hydrophobic Molecular Cation ((CH3)4N+)
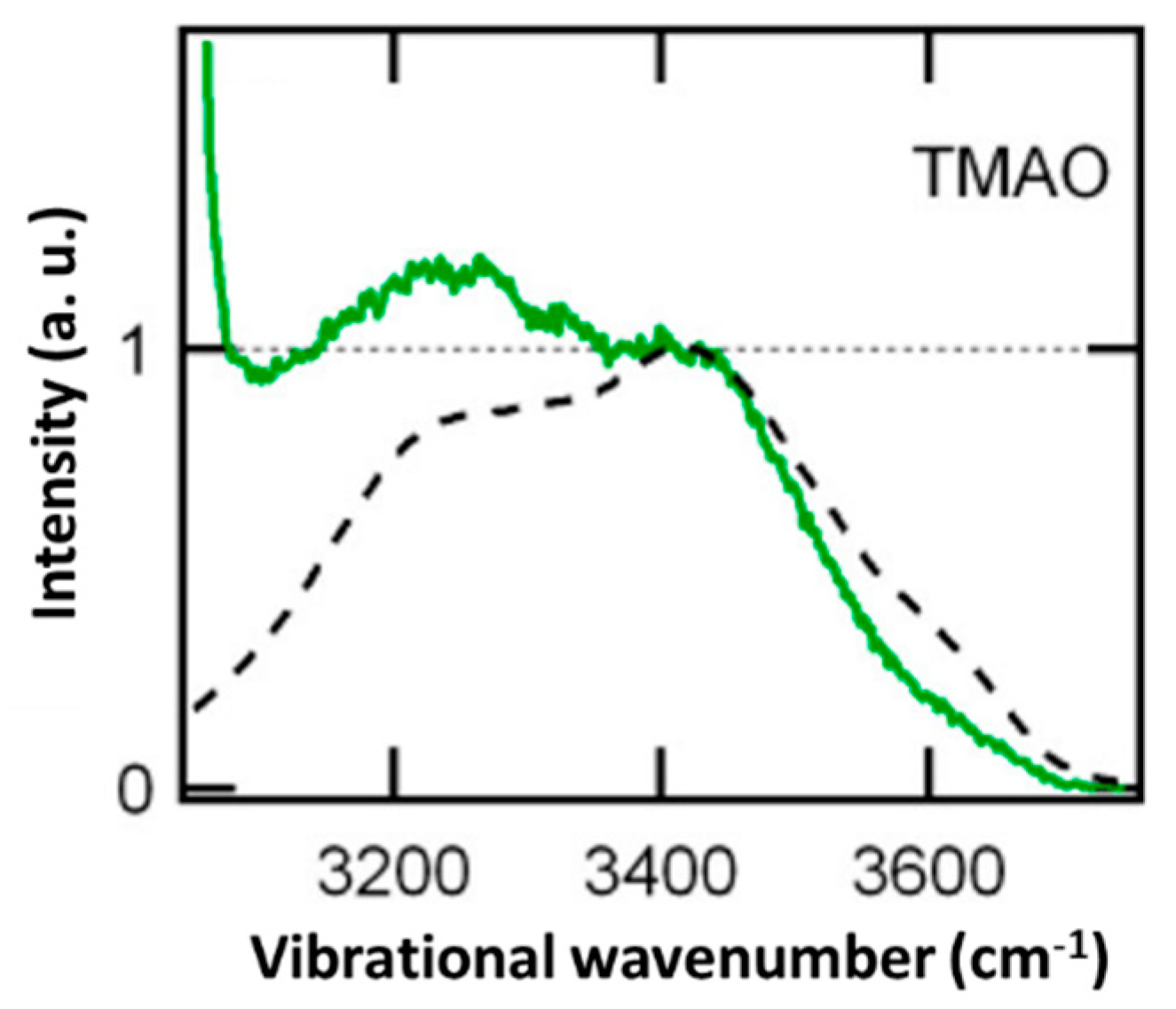
9. Water in the Hydration Shell of Zwitterion, Trimethylamine N-Oxide (CH3)3N+-O−)
10. Conclusions and Future Perspective
Funding
Acknowledgments
Conflicts of Interest
References
- Jacob, D. The role of water vapour in the atmosphere. A short overview from a climate modeller’s point of view. Phys. Chem. Earth Part A Solid Earth Geod. 2001, 26, 523–527. [Google Scholar] [CrossRef]
- Vaida, V. Perspective: Water cluster mediated atmospheric chemistry. J. Chem. Phys. 2011, 135, 020901. [Google Scholar] [CrossRef] [PubMed]
- Wiggins, P.M. Role of water in some biological processes. Microbiol. Rev. 1990, 54, 432–449. [Google Scholar] [CrossRef]
- Pal, S.K.; Zewail, A.H. Dynamics of Water in Biological Recognition. Chem. Rev. 2004, 104, 2099–2124. [Google Scholar] [CrossRef]
- Walrafen, G.E. Raman Spectral Studies of the Effects of Solutes and Pressure on Water Structure. J. Chem. Phys. 1971, 55, 768–792. [Google Scholar] [CrossRef]
- Marcus, Y. Effect of Ions on the Structure of Water: Structure Making and Breaking. Chem. Rev. 2009, 109, 1346–1370. [Google Scholar] [CrossRef]
- Gaballa, G.A.; Neilson, G.W. The effect of pressure on the structure of light and heavy water. Mol. Phys. 1983, 50, 97–111. [Google Scholar] [CrossRef]
- Walrafen, G.E. Raman Spectral Studies of the Effects of Temperature on Water and Electrolyte Solutions. J. Chem. Phys. 1966, 44, 1546–1558. [Google Scholar] [CrossRef]
- Dudev, T.; Lim, C. Importance of Metal Hydration on the Selectivity of Mg2+ versus Ca2+ in Magnesium Ion Channels. J. Am. Chem. Soc. 2013, 135, 17200–17208. [Google Scholar] [CrossRef]
- Cota, R.; Woutersen, S.; Bakker, H.J. Accelerated Vibrational Energy Relaxation of Water in Alkaline Environments. J. Phys. Chem. B 2021, 125, 11980–11986. [Google Scholar] [CrossRef]
- Timmer, R.L.A.; Tielrooij, K.J.; Bakker, H.J. Vibrational Förster transfer to hydrated protons. J. Chem. Phys. 2010, 132, 194504. [Google Scholar] [CrossRef] [PubMed]
- Wachter, W.; Kunz, W.; Buchner, R.; Hefter, G. Is There an Anionic Hofmeister Effect on Water Dynamics? Dielectric Spectroscopy of Aqueous Solutions of NaBr, NaI, NaNO3, NaClO4, and NaSCN. J. Phys. Chem. A 2005, 109, 8675–8683. [Google Scholar] [CrossRef] [PubMed]
- Tielrooij, K.J.; Garcia-Araez, N.; Bonn, M.; Bakker, H.J. Cooperativity in Ion Hydration. Science 2010, 328, 1006–1009. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Wu, Z.; Morrow, A.R.; Yethiraj, A.; Yethiraj, A. Self-Diffusion and Viscosity in Electrolyte Solutions. J. Phys. Chem. B 2012, 116, 12007–12013. [Google Scholar] [CrossRef]
- Cota, R.; van Dam, E.P.; Woutersen, S.; Bakker, H.J. Slowing Down of the Molecular Reorientation of Water in Concentrated Alkaline Solutions. J. Phys. Chem. B 2020, 124, 8309–8316. [Google Scholar] [CrossRef]
- Collins, K.D.; Washabaugh, M.W. The Hofmeister effect and the behaviour of water at interfaces. Q. Rev. Biophys. 1985, 18, 323–422. [Google Scholar] [CrossRef]
- Collins, K.D. Charge Density-Dependent Strength of Hydration and Biological Structure. Biophys. J. 1997, 72, 65–76. [Google Scholar] [CrossRef]
- Collins, K.D. Why continuum electrostatics theories cannot explain biological structure, polyelectrolytes or ionic strength effects in ion–protein interactions. Biophys. Chem. 2012, 167, 43–59. [Google Scholar] [CrossRef]
- Gurney, R.W. Ionic Processes in Solution; McGraw-Hill: New York, NY, USA, 1953. [Google Scholar]
- Jones, G.; Dole, M. The viscosity of aqueous solutions of strong electrolytes with special reference to barium chloride. J. Am. Chem. Soc. 1929, 51, 2950–2964. [Google Scholar] [CrossRef]
- Näslund, L.-Å.; Edwards, D.C.; Wernet, P.; Bergmann, U.; Ogasawara, H.; Pettersson, L.G.M.; Myneni, S.; Nilsson, A. X-ray Absorption Spectroscopy Study of the Hydrogen Bond Network in the Bulk Water of Aqueous Solutions. J. Phys. Chem. A 2005, 109, 5995–6002. [Google Scholar] [CrossRef]
- Hofmesiter, F. About the science of the effects of salts: About the water withdrawing effect of the salts. Arch. Exp. Pathol. Pharmacol. 1888, 24, 247–260. [Google Scholar]
- Mason, P.E.; Ansell, S.; Neilson, G.W.; Rempe, S.B. Neutron Scattering Studies of the Hydration Structure of Li+. J. Phys. Chem. B 2015, 119, 2003–2009. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, G.; Bowron, D.T.; Zhu, F.; Hannon, A.C.; Zhou, Y.; Liu, X.; Shi, G. Unveiling the structure of aqueous magnesium nitrate solutions by combining X-ray diffraction and theoretical calculations. Phys. Chem. Chem. Phys. 2022, 24, 22939–22949. [Google Scholar] [CrossRef]
- van der Vegt, N.F.A.; Haldrup, K.; Roke, S.; Zheng, J.; Lund, M.; Bakker, H.J. Water-Mediated Ion Pairing: Occurrence and Relevance. Chem. Rev. 2016, 116, 7626–7641. [Google Scholar] [CrossRef]
- Ohtaki, H.; Radnai, T. Structure and dynamics of hydrated ions. Chem. Rev. 1993, 93, 1157–1204. [Google Scholar] [CrossRef]
- Jalilehvand, F.; Spångberg, D.; Lindqvist-Reis, P.; Hermansson, K.; Persson, I.; Sandström, M. Hydration of the Calcium Ion. An EXAFS, Large-Angle X-ray Scattering, and Molecular Dynamics Simulation Study. J. Am. Chem. Soc. 2001, 123, 431–441. [Google Scholar] [CrossRef]
- Schwaab, G.; Sebastiani, F.; Havenith, M. Ion Hydration and Ion Pairing as Probed by THz Spectroscopy. Angew. Chem. Int. Ed. 2019, 58, 3000–3013. [Google Scholar] [CrossRef]
- Sebastiani, F.; Verde, A.V.; Heyden, M.; Schwaab, G.; Havenith, M. Cooperativity and ion pairing in magnesium sulfate aqueous solutions from the dilute regime to the solubility limit. Phys. Chem. Chem. Phys. 2020, 22, 12140–12153. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, Y.-H.; Zhao, L.-J. ATR-FTIR spectroscopic studies on aqueous LiClO4, NaClO4, and Mg(ClO4)2 solutions. Phys. Chem. Chem. Phys. 2004, 6, 537–542. [Google Scholar] [CrossRef]
- Max, J.-J.; Blois, S.d.; Veilleux, A.; Chapados, C. IR Spectroscopy of aqueous alkali halides. Factor analysis. Can. J. Chem. 2001, 79, 13–21. [Google Scholar] [CrossRef]
- Yonehama, K.; Yoshimura, Y.; Takekiyo, T.; Kanno, H. Variation of the Uncoupled OD Stretching Frequency with Electrolyte Concentration in Aqueous Electrolyte Solutions. Bull. Chem. Soc. Jpn. 2009, 82, 563–569. [Google Scholar] [CrossRef]
- Perera, P.; Wyche, M.; Loethen, Y.; Ben-Amotz, D. Solute-Induced Perturbations of Solvent-Shell Molecules Observed Using Multivariate Raman Curve Resolution. J. Am. Chem. Soc. 2008, 130, 4576–4577. [Google Scholar] [CrossRef] [PubMed]
- Fega, K.R.; Wilcox, A.S.; Ben-Amotz, D. Application of Raman Multivariate Curve Resolution to Solvation-Shell Spectroscopy. Appl. Spectrosc. 2012, 66, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Rankin, B.M.; Ben-Amotz, D. Expulsion of Ions from Hydrophobic Hydration Shells. J. Am. Chem. Soc. 2013, 135, 8818–8821. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Namboodiri, V.; Singh, A.K.; Mondal, J.A.; Sarkar, S.K. How Ions Affect the Structure of Water: A Combined Raman Spectroscopy and Multivariate Curve Resolution Study. J. Phys. Chem. B 2013, 117, 16479–16485. [Google Scholar] [CrossRef]
- Ahmed, M.; Singh, A.K.; Mondal, J.A.; Sarkar, S.K. Water in the Hydration Shell of Halide Ions Has Significantly Reduced Fermi Resonance and Moderately Enhanced Raman Cross Section in the OH Stretch Regions. J. Phys. Chem. B 2013, 117, 9728–9733. [Google Scholar] [CrossRef]
- Ahmed, M.; Singh, A.K.; Mondal, J.A. Hydrogen-bonding and vibrational coupling of water in a hydrophobic hydration shell as observed by Raman-MCR and isotopic dilution spectroscopy. Phys. Chem. Chem. Phys. 2016, 18, 2767–2775. [Google Scholar] [CrossRef]
- Ahmed, M.; Namboodiri, V.; Singh, A.K.; Mondal, J.A. On the Intermolecular Vibrational Coupling, Hydrogen Bonding, and Librational Freedom of Water in the Hydration Shell of Mono- and Bivalent Anions. J. Chem. Phys. 2014, 141, 164708. [Google Scholar] [CrossRef]
- Ben-Amotz, D. Hydration-Shell Vibrational Spectroscopy. J. Am. Chem. Soc. 2019, 141, 10569–10580. [Google Scholar] [CrossRef]
- Roy, S.; Biswas, B.; Ghosh, N.; Singh, P.C.; Mondal, J.A. Hydrophobic Hydration of Fluoroalkyl (C-F) is Distinctly Different from that of Its Hydrogenated Counterpart (C-H) as Observed by Raman Difference with Simultaneous Curve Fitting Analysis. J. Phys. Chem. C 2019, 123, 27012–27019. [Google Scholar] [CrossRef]
- Patra, A.; Roy, S.; Saha, S.; Palit, D.K.; Mondal, J.A. Observation of Extremely Weakly Interacting OH (~3600 cm−1) in the Vicinity of High Charge Density Metal Ions (Mz+; z = 1, 2, 3): A Structural Heterogeneity in the Extended Hydration Shell. J. Phys. Chem. C 2020, 124, 3028–3036. [Google Scholar] [CrossRef]
- Daly, C.A.; Streacker, L.M.; Sun, Y.; Pattenaude, S.R.; Hassanali, A.A.; Petersen, P.B.; Corcelli, S.A.; Ben-Amotz, D. Decomposition of the Experimental Raman and Infrared Spectra of Acidic Water into Proton, Special Pair, and Counterion Contributions. J. Phys. Chem. Lett. 2017, 8, 5246–5252. [Google Scholar] [CrossRef] [PubMed]
- Stangret, J.; Gampe, T. Ionic Hydration Behavior Derived from Infrared Spectra in HDO. J. Phys. Chem. A 2002, 106, 5393–5402. [Google Scholar] [CrossRef]
- Perera, P.N.; Browder, B.; Ben-Amotz, D. Perturbations of Water by Alkali Halide Ions Measured using Multivariate Raman Curve Resolution. J. Phys. Chem. B 2009, 113, 1805–1809. [Google Scholar] [CrossRef]
- Lindgren, J.; Hermansson, K.; Wojcik, M.J. Theoretical Simulation and Experimental-Determination of OH And OD Stretching Bands of Isotopically Diluted Hdo Molecules in Aqueous-Electrolyte Solutions. J. Phys. Chem. 1993, 97, 5254–5259. [Google Scholar] [CrossRef]
- Kristiansson, O.; Lindgren, J.; De Villepin, J. A quantitative infrared spectroscopic method for the study of the hydration of ions in aqueous solutions. J. Phys. Chem. 1988, 92, 2680–2685. [Google Scholar] [CrossRef]
- Śmiechowski, M.; Stangret, J. Vibrational spectroscopy of semiheavy water (HDO) as a probe of solute hydration. Pure Appl. Chem. 2010, 82, 1869–1887. [Google Scholar] [CrossRef]
- Sokołowska, A.; Kęcki, Z. Inter- and intra-molecular coupling and Fermi resonance in the Raman spectra of liquid water. J. Raman Spectrosc. 1986, 17, 29–33. [Google Scholar] [CrossRef]
- Auer, B.M.; Skinner, J.L. IR and Raman Spectra of Liquid Water: Theory and Interpretation. J. Chem. Phys. 2008, 128, 224511. [Google Scholar] [CrossRef]
- Hare, D.E.; Sorensen, C.M. Interoscillator coupling effects on the OH stretching band of liquid water. J. Chem. Phys. 1992, 96, 13–22. [Google Scholar] [CrossRef]
- Sovago, M.; Campen, R.K.; Wurpel, G.W.H.; Müller, M.; Bakker, H.J.; Bonn, M. Vibrational Response of Hydrogen-Bonded Interfacial Water is Dominated by Intramolecular Coupling. Phys. Rev. Lett. 2008, 100, 173901. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Pakoulev, A.; Pang, Y.; Dlott, D.D. Vibrational Substructure in the OH Stretching Transition of Water and HOD. J. Phys. Chem. A 2004, 108, 9054–9063. [Google Scholar] [CrossRef]
- Eaves, J.D.; Loparo, J.J.; Fecko, C.J.; Roberts, S.T.; Tokmakoff, A.; Geissler, P.L. Hydrogen bonds in liquid water are broken only fleetingly. Proc. Natl. Acad. Sci. USA 2005, 102, 13019–13022. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Skinner, J.L. Signatures of Coherent Vibrational Energy Transfer in IR and Raman Line Shapes for Liquid Water. Phys. Chem. Chem. Phys. 2010, 12, 982–991. [Google Scholar] [CrossRef]
- Saha, S.; Roy, S.; Mathi, P.; Mondal, J.A. Polyatomic Iodine Species at the Air–Water Interface and Its Relevance to Atmospheric Iodine Chemistry: An HD-VSFG and Raman-MCR Study. J. Phys. Chem. A 2019, 123, 2924–2934. [Google Scholar] [CrossRef]
- Jena, N.K.; Josefsson, I.; Eriksson, S.K.; Hagfeldt, A.; Siegbahn, H.; Björneholm, O.; Rensmo, H.; Odelius, M. Solvent-Dependent Structure of the I3− Ion Derived from Photoelectron Spectroscopy and Ab Initio Molecular Dynamics Simulations. Chem.—Eur. J. 2015, 21, 4049–4055. [Google Scholar] [CrossRef]
- Josefsson, I.; Eriksson, S.K.; Ottosson, N.; Öhrwall, G.; Siegbahn, H.; Hagfeldt, A.; Rensmo, H.; Björneholm, O.; Odelius, M. Collective hydrogen-bond dynamics dictates the electronic structure of aqueous I3−. Phys. Chem. Chem. Phys. 2013, 15, 20189–20196. [Google Scholar] [CrossRef]
- Bergstroem, P.A.; Lindgren, J.; Kristiansson, O. An IR Study of the Hydration of Perchlorate, Nitrate, Iodide, Bromide, Chloride and Sulfate Anions in Aqueous Solution. J. Phys. Chem. 1991, 95, 8575–8580. [Google Scholar] [CrossRef]
- Roy, S.; Mondal, J.A. “Breaking” and “Making” of Water Structure at the Air/Water–Electrolyte (NaXO3; X = Cl, Br, I) Interface. J. Phys. Chem. Lett. 2021, 12, 1955–1960. [Google Scholar] [CrossRef]
- Baer, M.D.; Pham, V.-T.; Fulton, J.L.; Schenter, G.K.; Balasubramanian, M.; Mundy, C.J. Is Iodate a Strongly Hydrated Cation? J. Phys. Chem. Lett. 2011, 2, 2650–2654. [Google Scholar] [CrossRef]
- Sharma, B.; Chandra, A. Nature of hydration shells of a polyoxy-anion with a large cationic centre: The case of iodate ion in water. J. Comput. Chem. 2018, 39, 1226–1235. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Chandra, A. Born–Oppenheimer Molecular Dynamics Simulations of a Bromate Ion in Water Reveal Its Dual Kosmotropic and Chaotropic Behavior. J. Phys. Chem. B 2018, 122, 2090–2101. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Mondal, J.A. Kosmotropic Electrolyte (Na2CO3, NaF) Perturbs the Air/Water Interface through Anion Hydration Shell without Forming a Well-Defined Electric Double Layer. J. Phys. Chem. B 2021, 125, 3977–3985. [Google Scholar] [CrossRef]
- Boisson, J.; Stirnemann, G.; Laage, D.; Hynes, J.T. Water reorientation dynamics in the first hydration shells of F− and I−. Phys. Chem. Chem. Phys. 2011, 13, 19895–19901. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Chandra, A. Structural and Dynamical Nature of Hydration Shells of the Carbonate Ion in Water: An Ab Initio Molecular Dynamics Study. J. Phys. Chem. B 2018, 122, 1495–1504. [Google Scholar] [CrossRef] [PubMed]
- Kameda, Y.; Hosoya, K.; Sakamoto, S.; Suzuki, H.; Usuki, T.; Uemura, O. Hydrogen-bonded structure in aqueous sulfuric acid solutions. J. Mol. Liq. 1995, 65–66, 305–308. [Google Scholar] [CrossRef]
- Rudolph, W.W.; Irmer, G. Raman and Infrared Spectroscopic Investigations on Aqueous Alkali Metal Phosphate Solutions and Density Functional Theory Calculations of Phosphate—Water Clusters. Appl. Spectrosc. 2007, 61, 1312–1327. [Google Scholar] [CrossRef]
- Sharma, B.; Chandra, A. Ab Initio Molecular Dynamics Simulation of the Phosphate Ion in Water: Insights into Solvation Shell Structure, Dynamics, and Kosmotropic Activity. J. Phys. Chem. B 2017, 121, 10519–10529. [Google Scholar] [CrossRef]
- Yu, C.-C.; Chiang, K.-Y.; Okuno, M.; Seki, T.; Ohto, T.; Yu, X.; Korepanov, V.; Hamaguchi, H.-O.; Bonn, M.; Hunger, J.; et al. Vibrational couplings and energy transfer pathways of water’s bending mode. Nat. Commun. 2020, 11, 5977. [Google Scholar] [CrossRef]
- Seki, T.; Chiang, K.-Y.; Yu, C.-C.; Yu, X.; Okuno, M.; Hunger, J.; Nagata, Y.; Bonn, M. The Bending Mode of Water: A Powerful Probe for Hydrogen Bond Structure of Aqueous Systems. J. Phys. Chem. Lett. 2020, 11, 8459–8469. [Google Scholar] [CrossRef]
- Walrafen, G.E. Raman Spectral Studies of the Effects of Temperature on Water Structure. J. Chem. Phys. 1967, 47, 114–126. [Google Scholar] [CrossRef]
- Cho, M.; Fleming, G.R.; Saito, S.; Ohmine, I.; Stratt, R.M. Instantaneous normal mode analysis of liquid water. J. Chem. Phys. 1994, 100, 6672–6683. [Google Scholar] [CrossRef]
- Nash, C.P.; Donnelly, T.C.; Rock, P.A. Raman spectra in the libration region of concentrated aqueous solutions of lithium-6 and lithium-7 halides. Evidence for tetrahedral Li(OH2)4+. J. Solut. Chem. 1977, 6, 663–670. [Google Scholar] [CrossRef]
- Carey, D.M.; Korenowski, G.M. Measurement of the Raman spectrum of liquid water. J. Chem. Phys. 1998, 108, 2669–2675. [Google Scholar] [CrossRef]
- Cappa, C.D.; Smith, J.D.; Messer, B.M.; Cohen, R.C.; Saykally, R.J. Effects of Cations on the Hydrogen Bond Network of Liquid Water: New Results from X-ray Absorption Spectroscopy of Liquid Microjets. J. Phys. Chem. B 2006, 110, 5301–5309. [Google Scholar] [CrossRef] [PubMed]
- Bergstroem, P.A.; Lindgren, J. Infrared study on the hydration of manganese (2+), iron (2+), cobalt (2+), lanthanum (3+), neodymium (3+), dysprosium (3+), and ytterbium (3+) ions in dilute aqueous solution. Inorg. Chem. 1992, 31, 1529–1533. [Google Scholar] [CrossRef]
- Rudolph, W.W.; Irmer, G. Raman spectroscopic characterization of light rare earth ions: La3+, Ce3+, Pr3+, Nd3+ and Sm3+–hydration and ion pair formation. Dalton Trans. 2017, 46, 4235–4244. [Google Scholar] [CrossRef]
- Rudolph, W.; Irmer, G. On the Hydration of Heavy Rare Earth Ions: Ho3+, Er3+, Tm3+, Yb3+ and Lu3+—A Raman Study. Molecules 2019, 24, 1953. [Google Scholar] [CrossRef]
- Das, B.; Mondal, S.; Chandra, A. Two-Dimensional Infrared Spectroscopy of Aqueous Solutions of Metal Nitrates: Slowdown of Spectral Diffusion in the Presence of Divalent Cations. J. Phys. Chem. B 2020, 124, 7391–7404. [Google Scholar] [CrossRef]
- Zeng, Y.; Jia, Y.; Yan, T.; Zhuang, W. Binary structure and dynamics of the hydrogen bonds in the hydration shells of ions. Phys. Chem. Chem. Phys. 2021, 23, 11400–11410. [Google Scholar] [CrossRef]
- Habenschuss, A.; Spedding, F.H. The coordination (hydration) of rare earth ions in aqueous chloride solutions from x-ray diffraction. I.TbCl3, DyCl3, ErCl3, TmCl3, and LuCl3. J. Chem. Phys. 1979, 70, 2797–2806. [Google Scholar] [CrossRef]
- Habenschuss, A.; Spedding, F.H. The coordination (hydration) of rare earth ions in aqueous chloride solutions from x-ray diffraction. II. LaCl3, PrCl3, and NdCl3a). J. Chem. Phys. 1979, 70, 3758–3763. [Google Scholar] [CrossRef]
- Duvail, M.; Vitorge, P.; Spezia, R. Building a polarizable pair interaction potential for lanthanoids(III) in liquid water: A molecular dynamics study of structure and dynamics of the whole series. J. Chem. Phys. 2009, 130, 104501. [Google Scholar] [CrossRef] [PubMed]
- Verde, A.V.; Santer, M.; Lipowsky, R. Solvent-shared pairs of densely charged ions induce intense but short-range supra-additive slowdown of water rotation. Phys. Chem. Chem. Phys. 2016, 18, 1918–1930. [Google Scholar] [CrossRef]
- Yadav, S.; Chandra, A. Preferential solvation, ion pairing, and dynamics of concentrated aqueous solutions of divalent metal nitrate salts. J. Chem. Phys. 2017, 147, 244503. [Google Scholar] [CrossRef]
- Marcus, Y.; Hefter, G. Ion Pairing. Chem. Rev. 2006, 106, 4585–4621. [Google Scholar] [CrossRef]
- Roy, S.; Patra, A.; Saha, S.; Palit, D.K.; Mondal, J.A. Restructuring of Hydration Shell Water due to Solvent-Shared Ion Pairing (SSIP): A Case Study of Aqueous MgCl2 and LaCl3 Solutions. J. Phys. Chem. B 2020, 124, 8141–8148. [Google Scholar] [CrossRef]
- Baumler, S.M.; Hartt, V.W.H.; Allen, H.C. Hydration of ferric chloride and nitrate in aqueous solutions: Water-mediated ion pairing revealed by Raman spectroscopy. Phys. Chem. Chem. Phys. 2019, 21, 19172–19180. [Google Scholar] [CrossRef]
- Friesen, S.; Hefter, G.; Buchner, R. Cation Hydration and Ion Pairing in Aqueous Solutions of MgCl2 and CaCl2. J. Phys. Chem. B 2019, 123, 891–900. [Google Scholar] [CrossRef]
- Fulton, J.L.; Heald, S.M.; Badyal, Y.S.; Simonson, J.M. Understanding the Effects of Concentration on the Solvation Structure of Ca2+ in Aqueous Solution. I: The Perspective on Local Structure from EXAFS and XANES. J. Phys. Chem. A 2003, 107, 4688–4696. [Google Scholar] [CrossRef]
- Badyal, Y.S.; Barnes, A.C.; Cuello, G.J.; Simonson, J.M. Understanding the Effects of Concentration on the Solvation Structure of Ca2+ in Aqueous Solution. II: Insights into Longer Range Order from Neutron Diffraction Isotope Substitution. J. Phys. Chem. A 2004, 108, 11819–11827. [Google Scholar] [CrossRef]
- de Oliveira, D.M.; Bredt, A.J.; Miller, T.C.; Corcelli, S.A.; Ben-Amotz, D. Spectroscopic and Structural Characterization of Water-Shared Ion-Pairs in Aqueous Sodium and Lithium Hydroxide. J. Phys. Chem. B 2021, 125, 1439–1446. [Google Scholar] [CrossRef] [PubMed]
- Drexler, C.I.; Miller, T.C.; Rogers, B.A.; Li, Y.C.; Daly, C.A.; Yang, T.; Corcelli, S.A.; Cremer, P.S. Counter Cations Affect Transport in Aqueous Hydroxide Solutions with Ion Specificity. J. Am. Chem. Soc. 2019, 141, 6930–6936. [Google Scholar] [CrossRef] [PubMed]
- Perera, P.N.; Fega, K.R.; Lawrence, C.; Sundstrom, E.J.; Tomlinson-Phillips, J.; Ben-Amotz, D. Observation of water dangling OH bonds around dissolved nonpolar groups. Proc. Natl. Acad. Sci. USA 2009, 106, 12230–12234. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.G.; Gierszal, K.P.; Wang, P.; Ben-Amotz, D. Water Structural Transformation at Molecular Hydrophobic Interfaces. Nature 2012, 491, 582–585. [Google Scholar] [CrossRef]
- Ahmed, M.; Namboodiri, V.; Mathi, P.; Singh, A.K.; Mondal, J.A. How Osmolyte and Denaturant Affect Water at the Air–Water Interface and in Bulk: A Heterodyne-Detected Vibrational Sum Frequency Generation (HD-VSFG) and Hydration Shell Spectroscopic Study. J. Phys. Chem. C 2016, 120, 10252–10260. [Google Scholar] [CrossRef]
- Mondal, J.A. Effect of Trimethylamine N-Oxide on Interfacial Electrostatics at Phospholipid Monolayer–Water Interfaces and Its Relevance to Cardiovascular Disease. J. Phys. Chem. Lett. 2016, 7, 1704–1708. [Google Scholar] [CrossRef]
- Mondal, J.A. Metabolite-Affected Interfacial Electrostatics and Its Role in the Pathogenesis of Cardiovascular Disease: An Interface-Selective Vibrational Spectroscopic Study. J. Phys. Chem. C 2016, 120, 21642–21651. [Google Scholar] [CrossRef]
- Roy, S.; Patra, A.; Palit, D.K.; Mondal, J.A. Interaction of Zwitterionic Osmolyte Trimethylamine N-oxide (TMAO) with Molecular Hydrophobes: An Interplay of Hydrophobic and Electrostatic Interactions. J. Phys. Chem. B 2021, 125, 10939–10946. [Google Scholar] [CrossRef]
- Salazar-Salinas, K.; Baldera-Aguayo, P.A.; Encomendero-Risco, J.J.; Orihuela, M.; Sheen, P.; Seminario, J.M.; Zimic, M. Metal-Ion Effects on the Polarization of Metal-Bound Water and Infrared Vibrational Modes of the Coordinated Metal Center of Mycobacterium tuberculosis Pyrazinamidase via Quantum Mechanical Calculations. J. Phys. Chem. B 2014, 118, 10065–10075. [Google Scholar] [CrossRef]
- Vergauwe, R.M.A.; Thomas, A.; Nagarajan, K.; Shalabney, A.; George, J.; Chervy, T.; Seidel, M.; Devaux, E.; Torbeev, V.; Ebbesen, T.W. Modification of Enzyme Activity by Vibrational Strong Coupling of Water. Angew. Chem. Int. Ed. 2019, 58, 15324–15328. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.-H.; Shen, Y.; Chen, H.; Chen, Y.; Jiang, B.; Wu, G.; Yang, X.; Yuan, K.; Zheng, J. Ordered-to-Disordered Transformation of Enhanced Water Structure on Hydrophobic Surfaces in Concentrated Alcohol–Water Solutions. J. Phys. Chem. Lett. 2019, 10, 7922–7928. [Google Scholar] [CrossRef] [PubMed]
- Verma, P.K.; Kundu, A.; Puretz, M.S.; Dhoonmoon, C.; Chegwidden, O.S.; Londergan, C.H.; Cho, M. The Bend+Libration Combination Band Is an Intrinsic, Collective, and Strongly Solute-Dependent Reporter on the Hydrogen Bonding Network of Liquid Water. J. Phys. Chem. B 2018, 122, 2587–2599. [Google Scholar] [CrossRef] [PubMed]
- Shattuck, J.; Shah, P.; Erramilli, S.; Ziegler, L.D. Structure Making and Breaking Effects of Cations in Aqueous Solution: Nitrous Oxide Pump–Probe Measurements. J. Phys. Chem. B 2016, 120, 10569–10580. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coefficients | Peak 1 | Peak 2 | Peak 3 | Peak 4 |
|---|---|---|---|---|
| Amplitude, (×10−4) | 2.2 | 17.2 | 9.9 | 3.0 |
| Peak center (cm−1), | 3048 | 3234 | 3399 | 3561 |
| fwhm (cm−1), | 155 | 113 | 65 | 92 |
| , (×10−4) | −0.27 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghosh, N.; Roy, S.; Bandyopadhyay, A.; Mondal, J.A. Vibrational Raman Spectroscopy of the Hydration Shell of Ions. Liquids 2023, 3, 19-39. https://doi.org/10.3390/liquids3010003
Ghosh N, Roy S, Bandyopadhyay A, Mondal JA. Vibrational Raman Spectroscopy of the Hydration Shell of Ions. Liquids. 2023; 3(1):19-39. https://doi.org/10.3390/liquids3010003
Chicago/Turabian StyleGhosh, Nishith, Subhadip Roy, Anisha Bandyopadhyay, and Jahur Alam Mondal. 2023. "Vibrational Raman Spectroscopy of the Hydration Shell of Ions" Liquids 3, no. 1: 19-39. https://doi.org/10.3390/liquids3010003
APA StyleGhosh, N., Roy, S., Bandyopadhyay, A., & Mondal, J. A. (2023). Vibrational Raman Spectroscopy of the Hydration Shell of Ions. Liquids, 3(1), 19-39. https://doi.org/10.3390/liquids3010003