

Thermodynamic Modeling of Mineral Scaling in High-Temperature and High-Pressure Aqueous Environments



Abstract
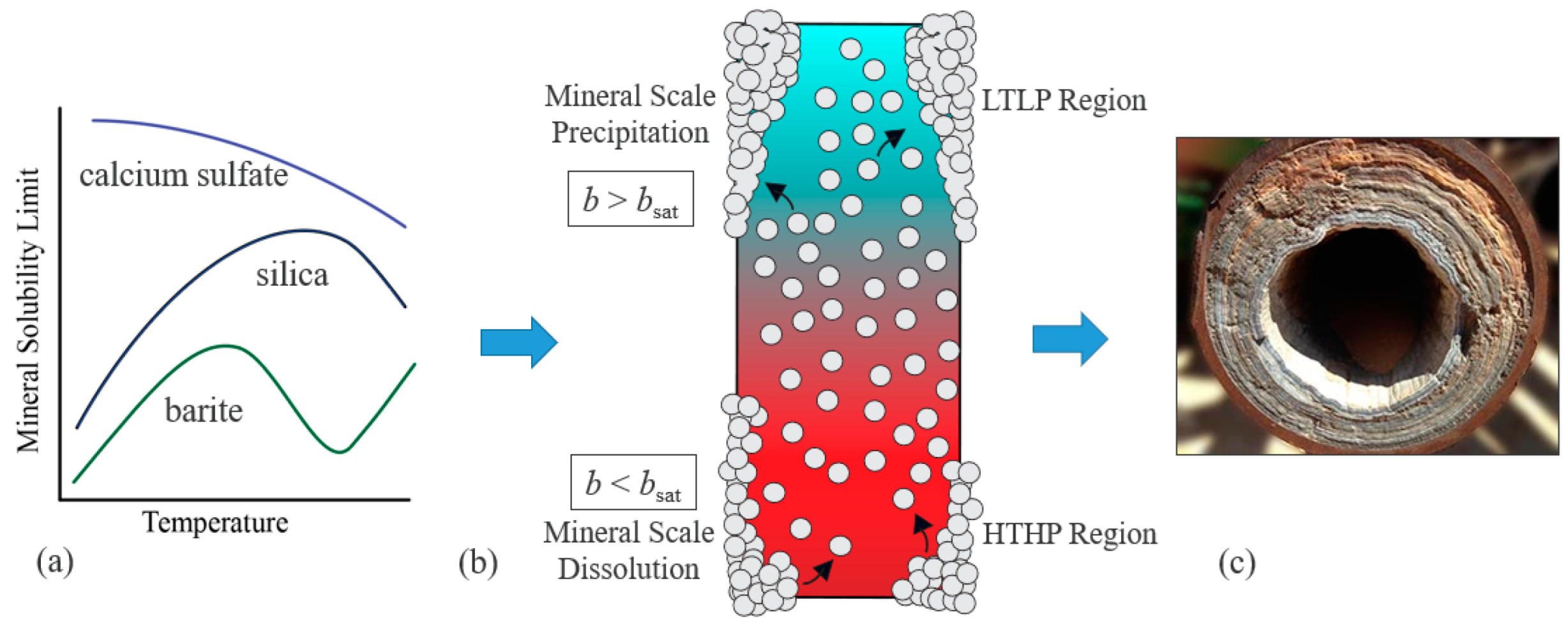
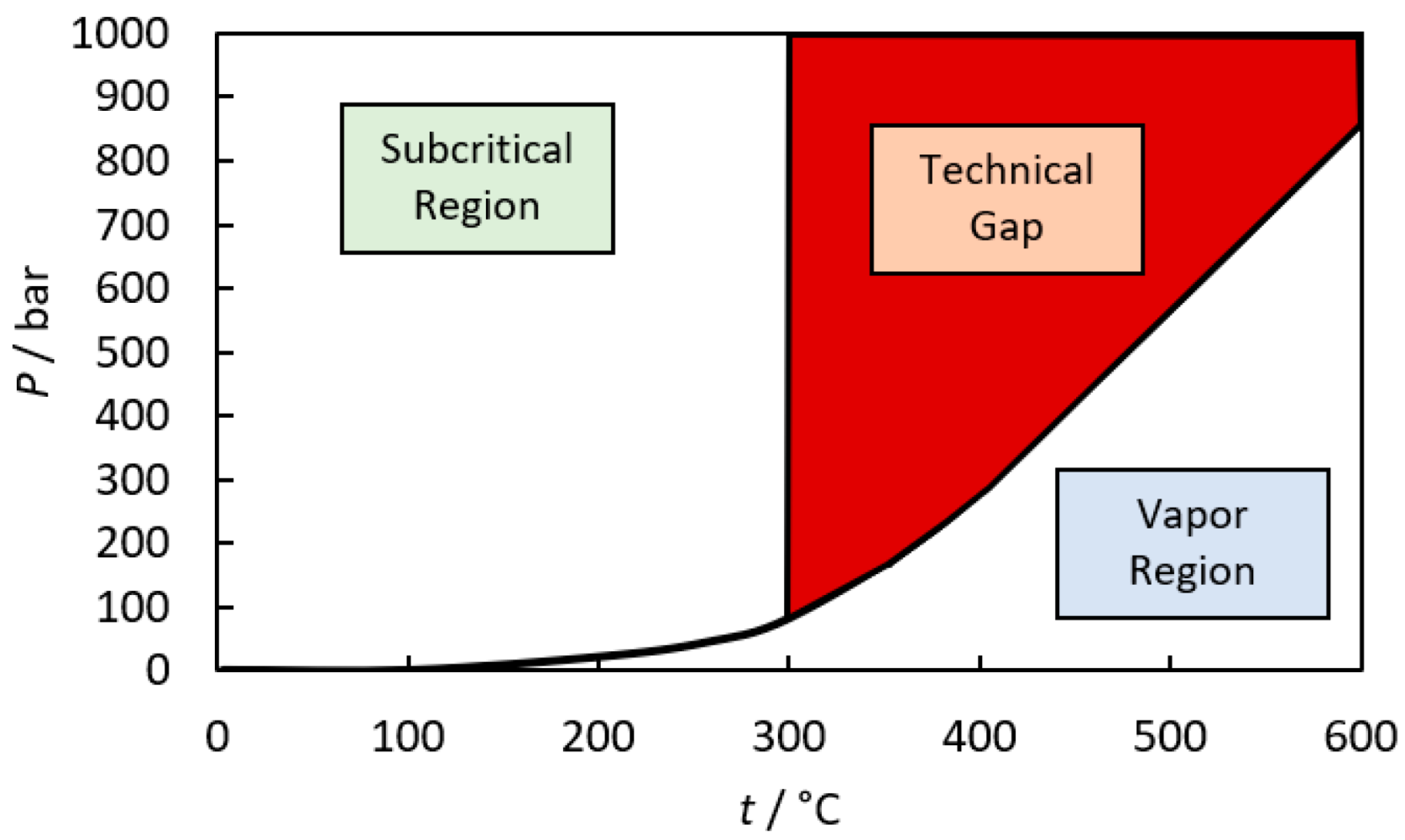
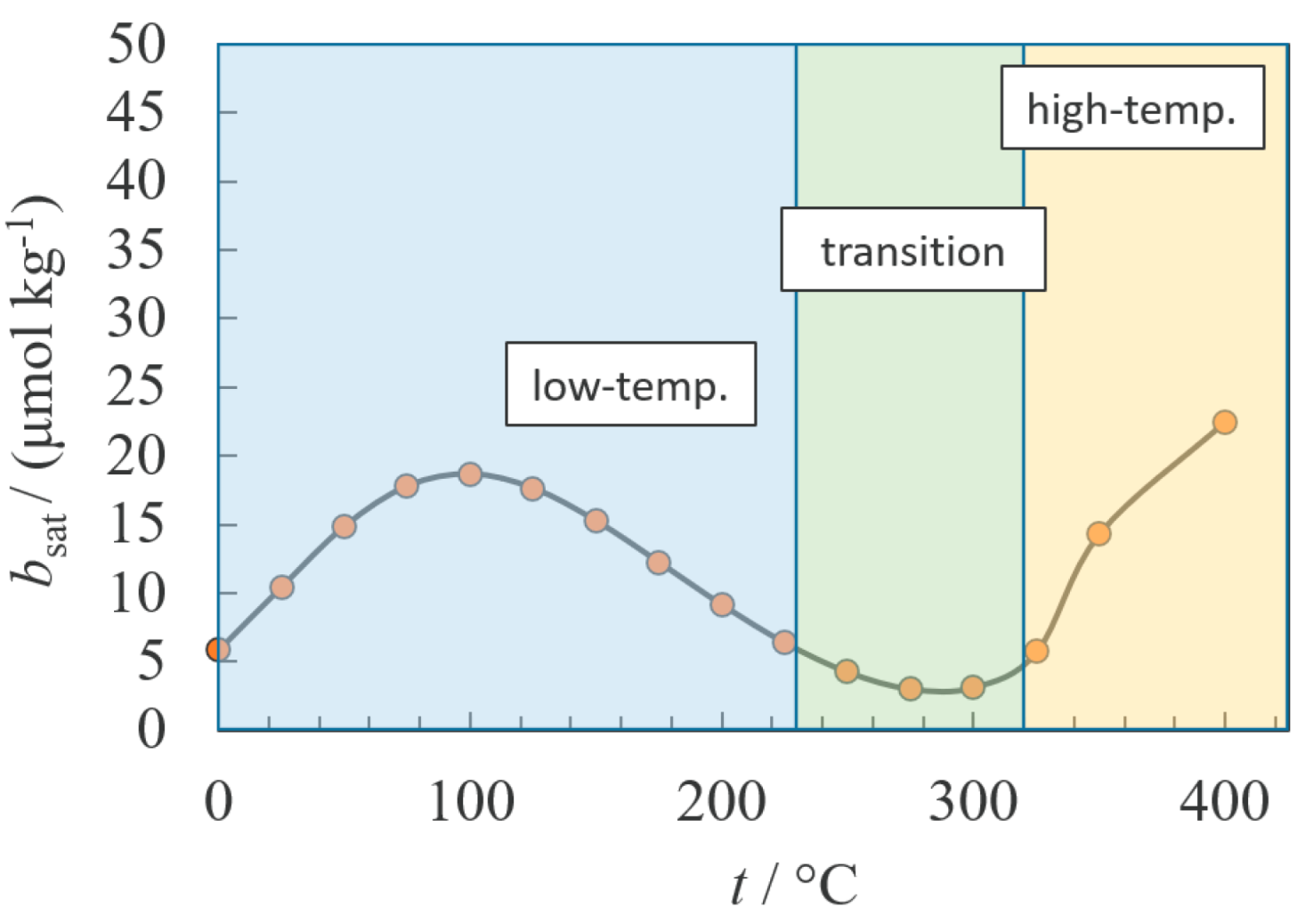
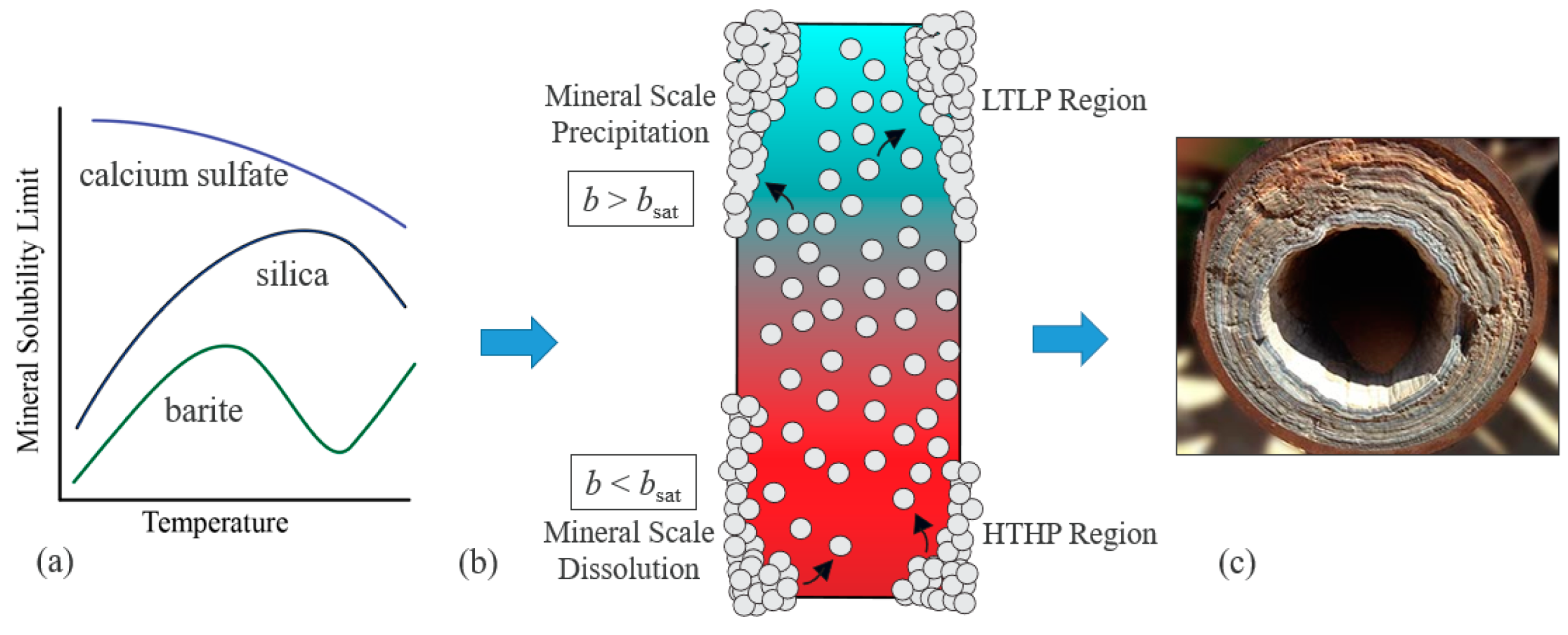
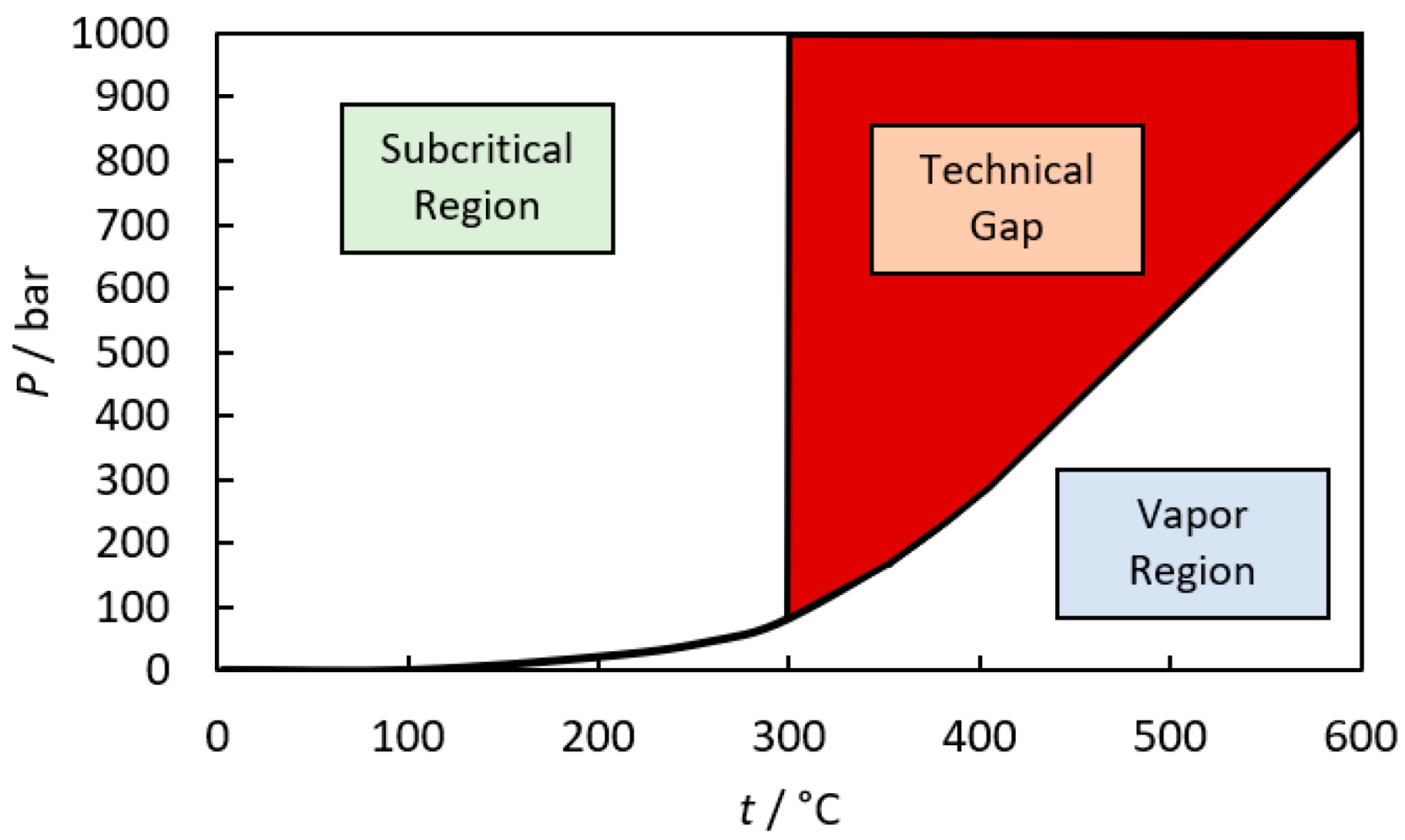
:1. Introduction
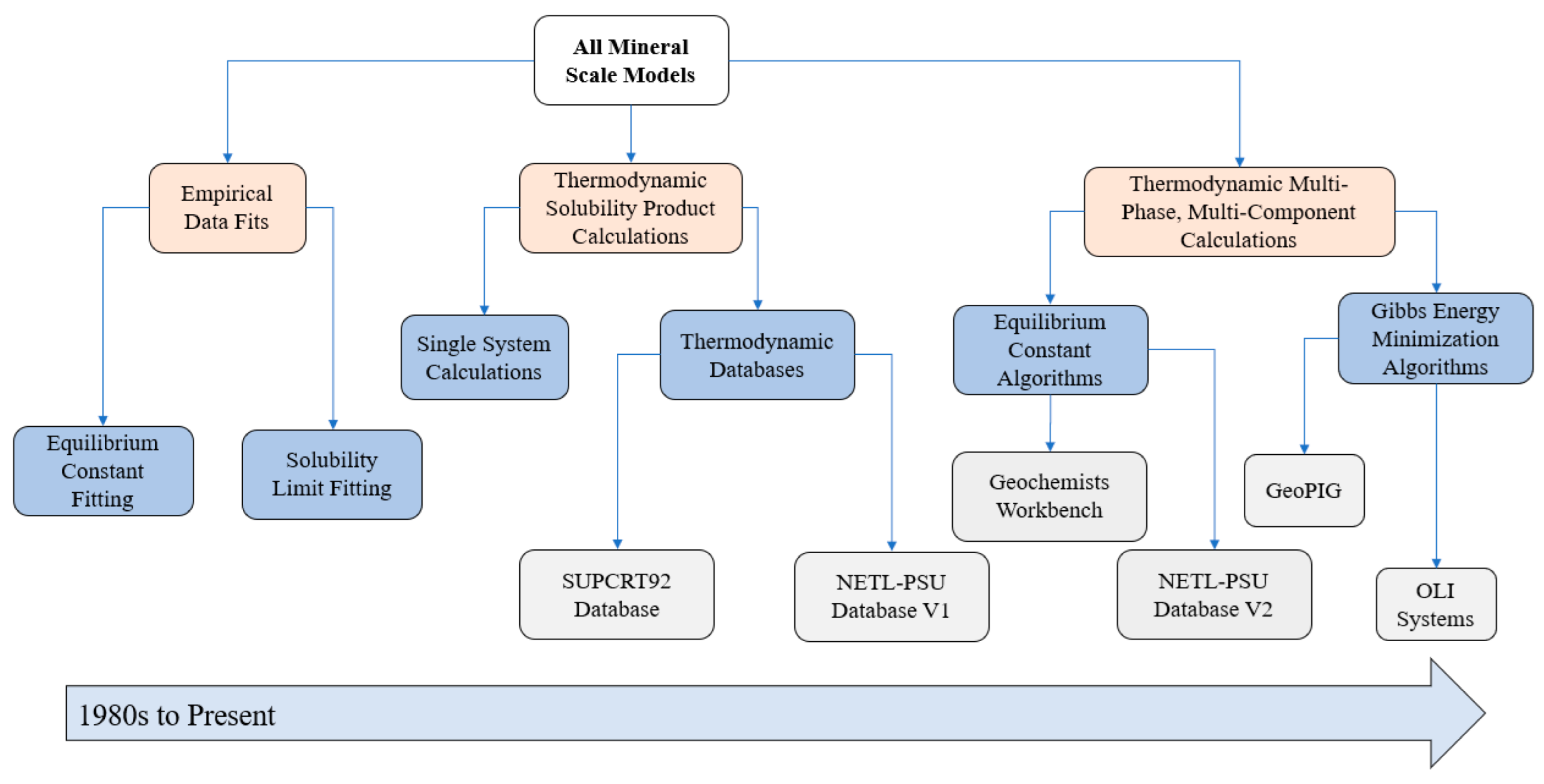
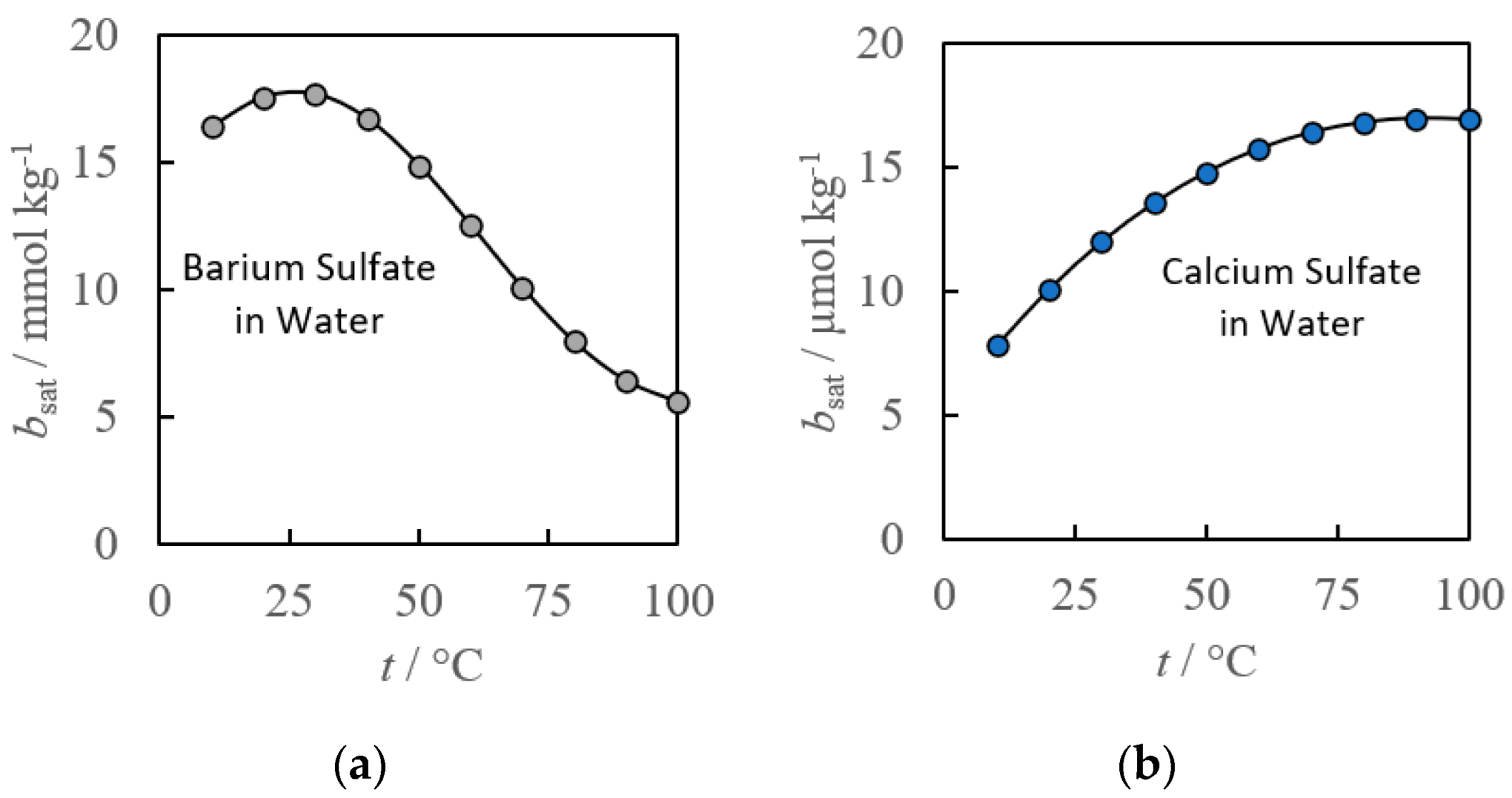
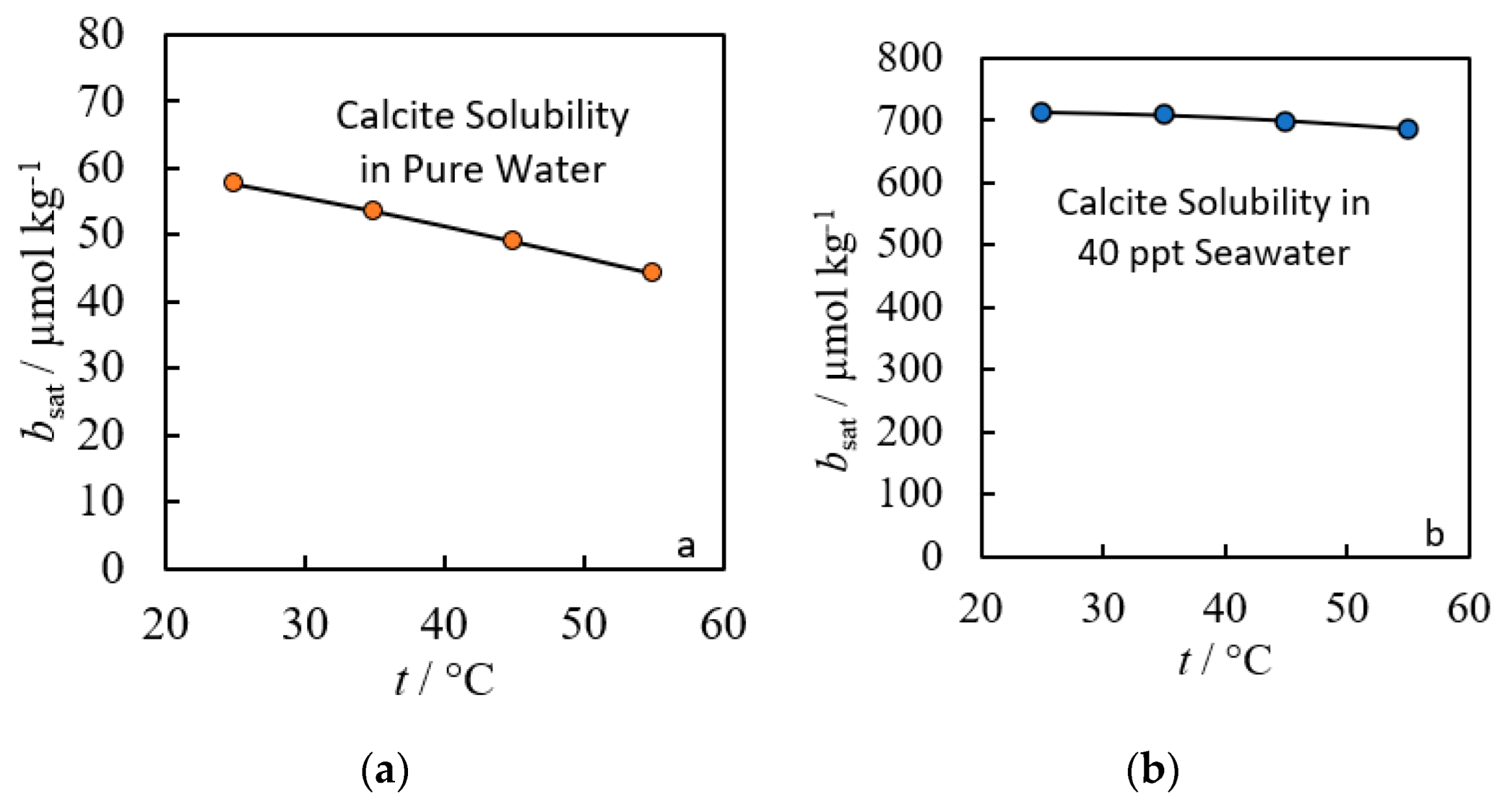
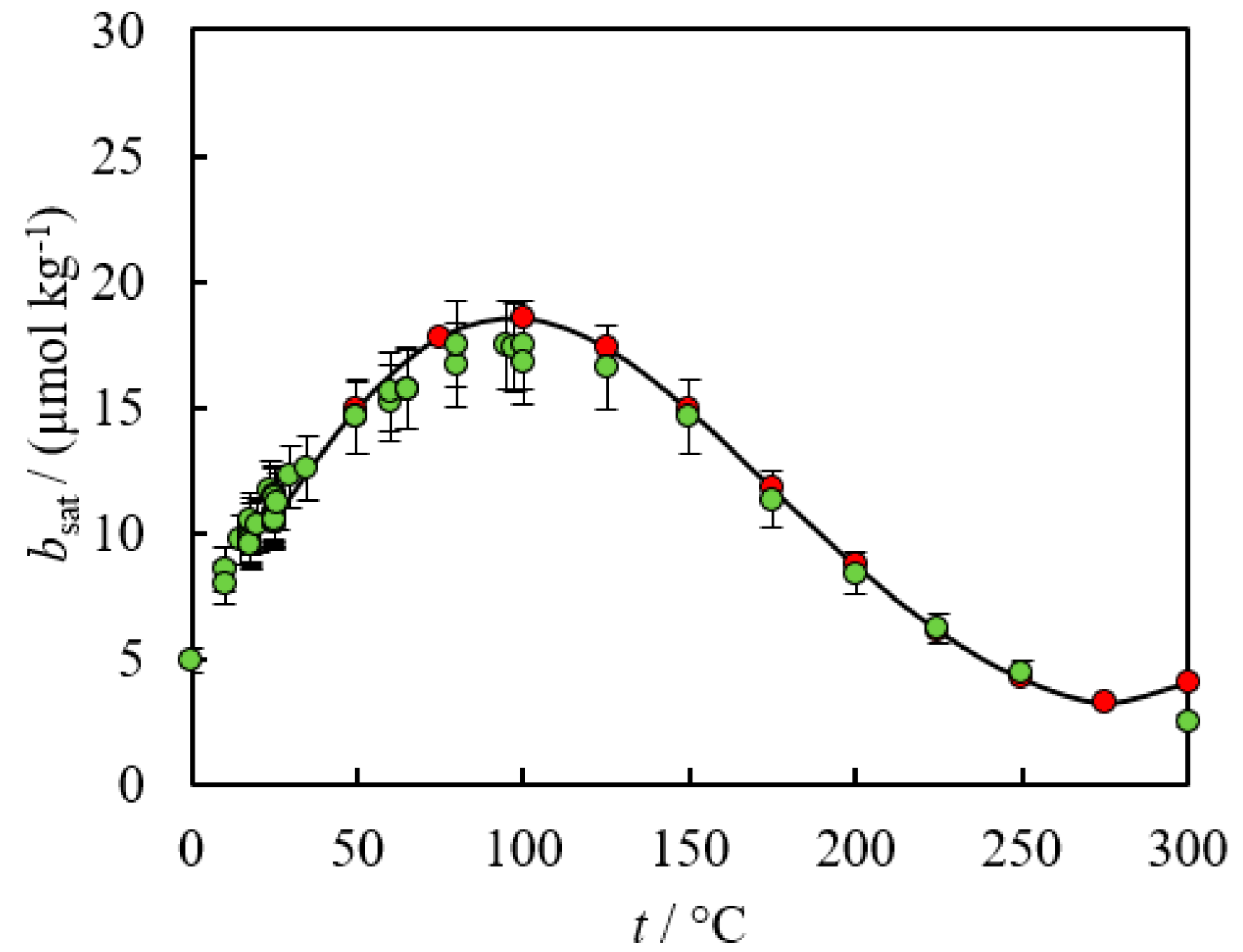
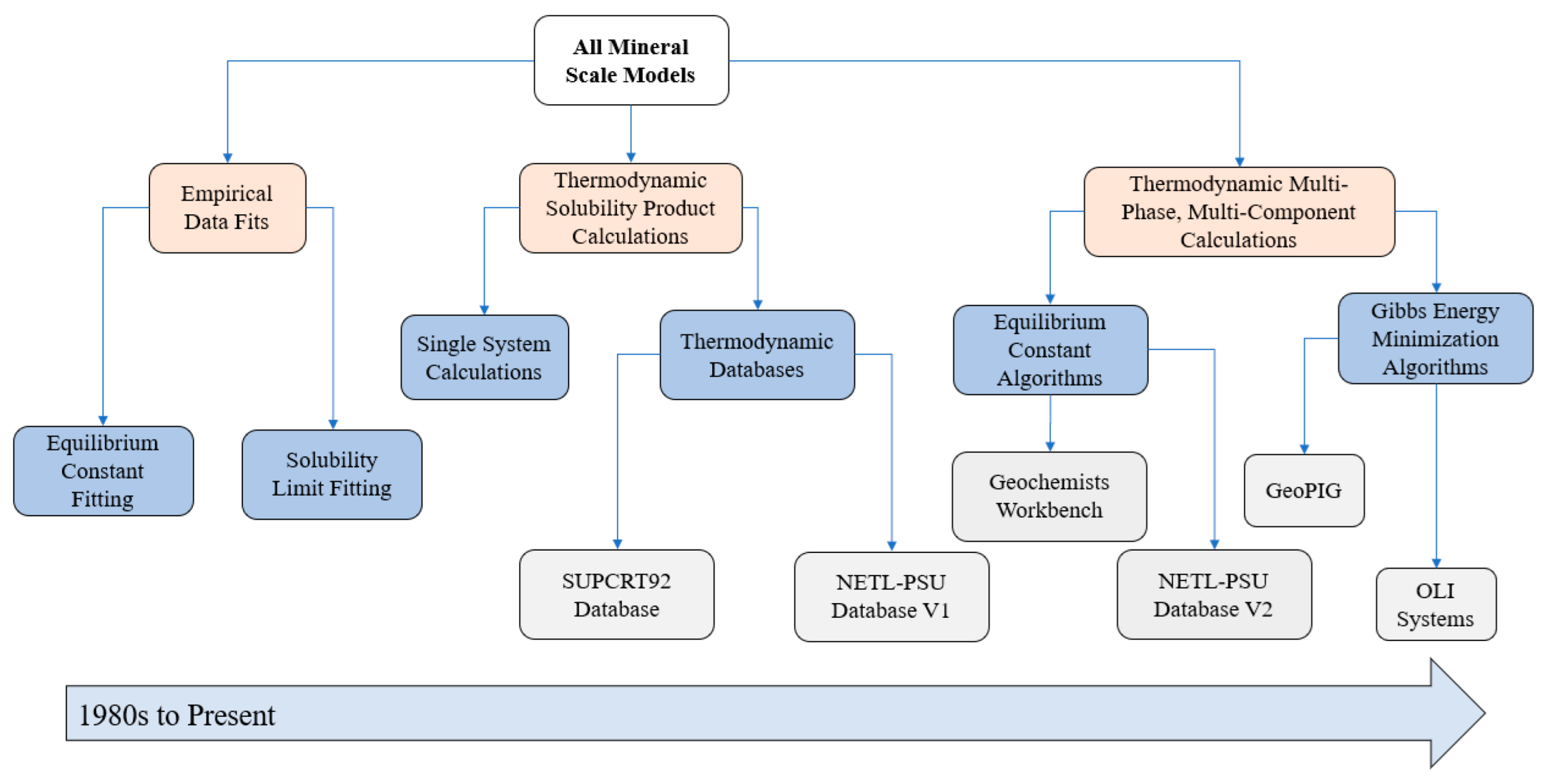
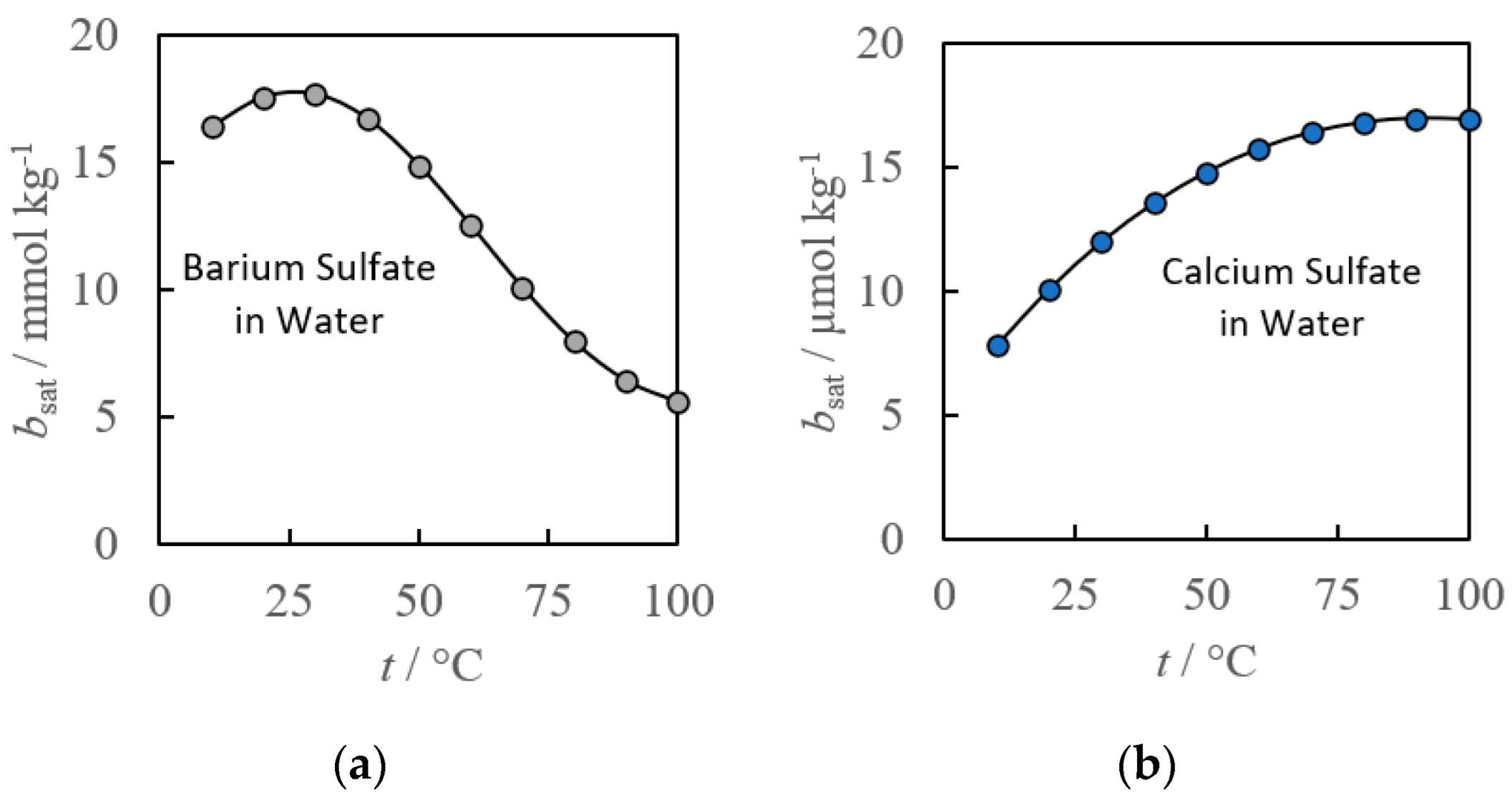
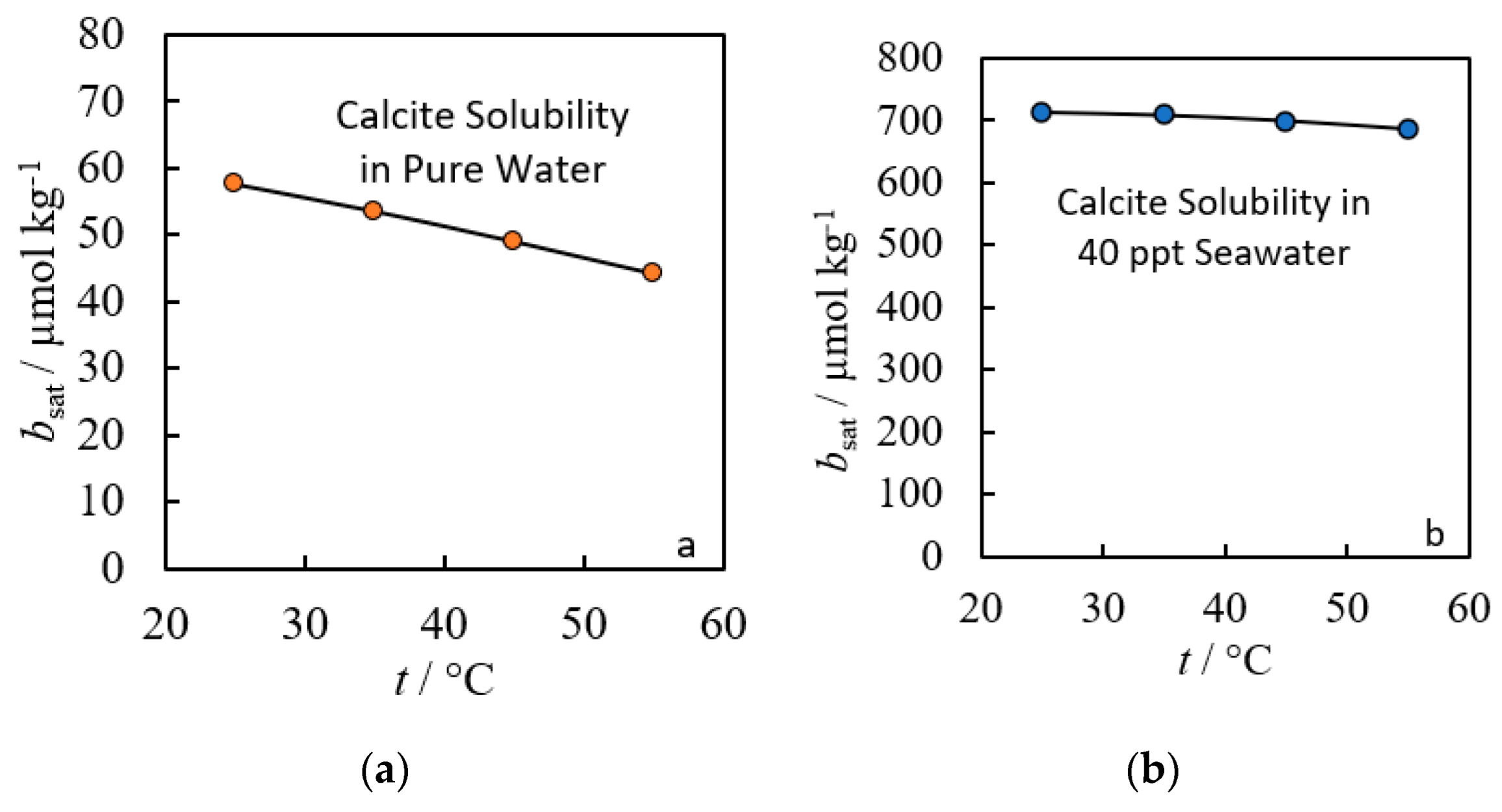
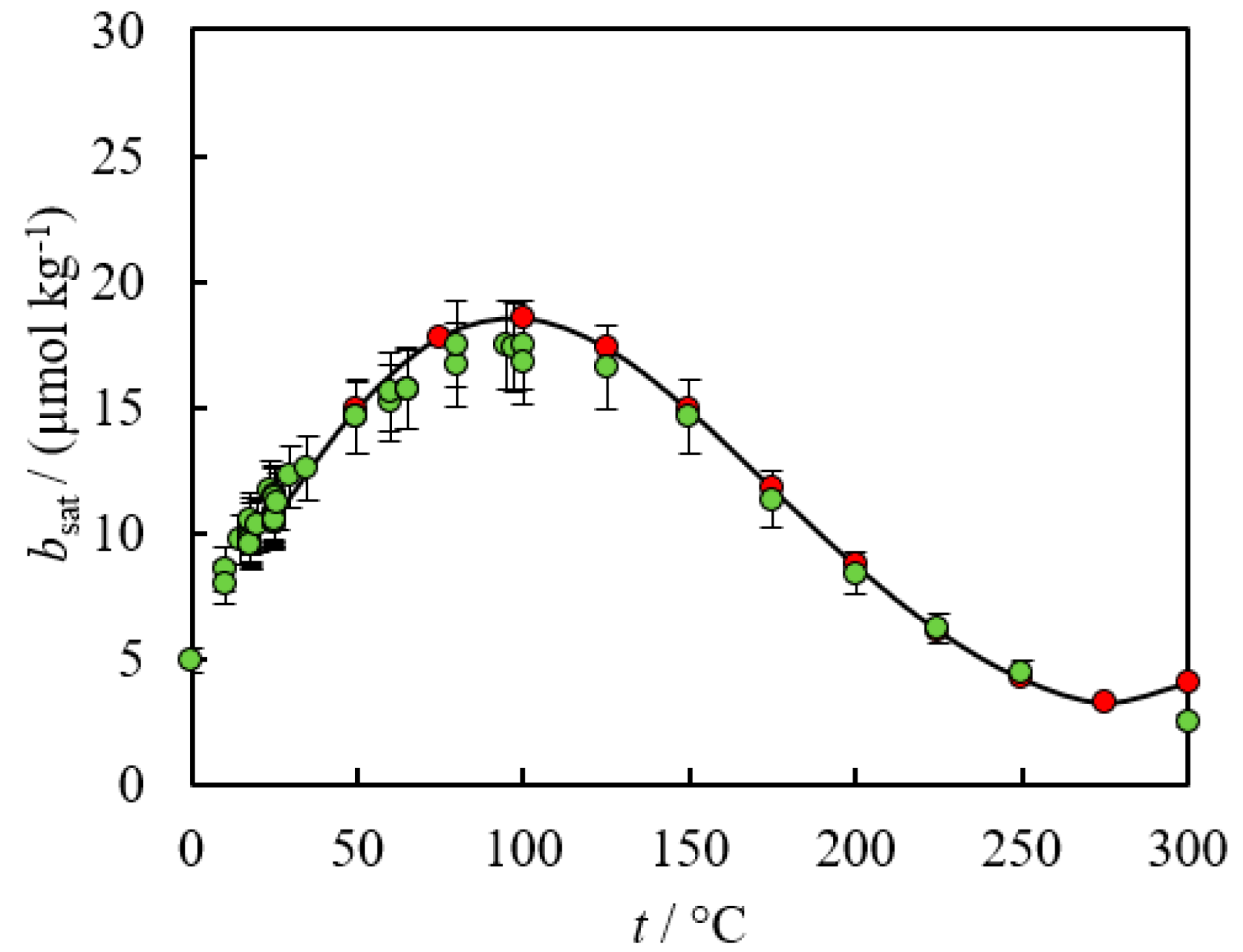
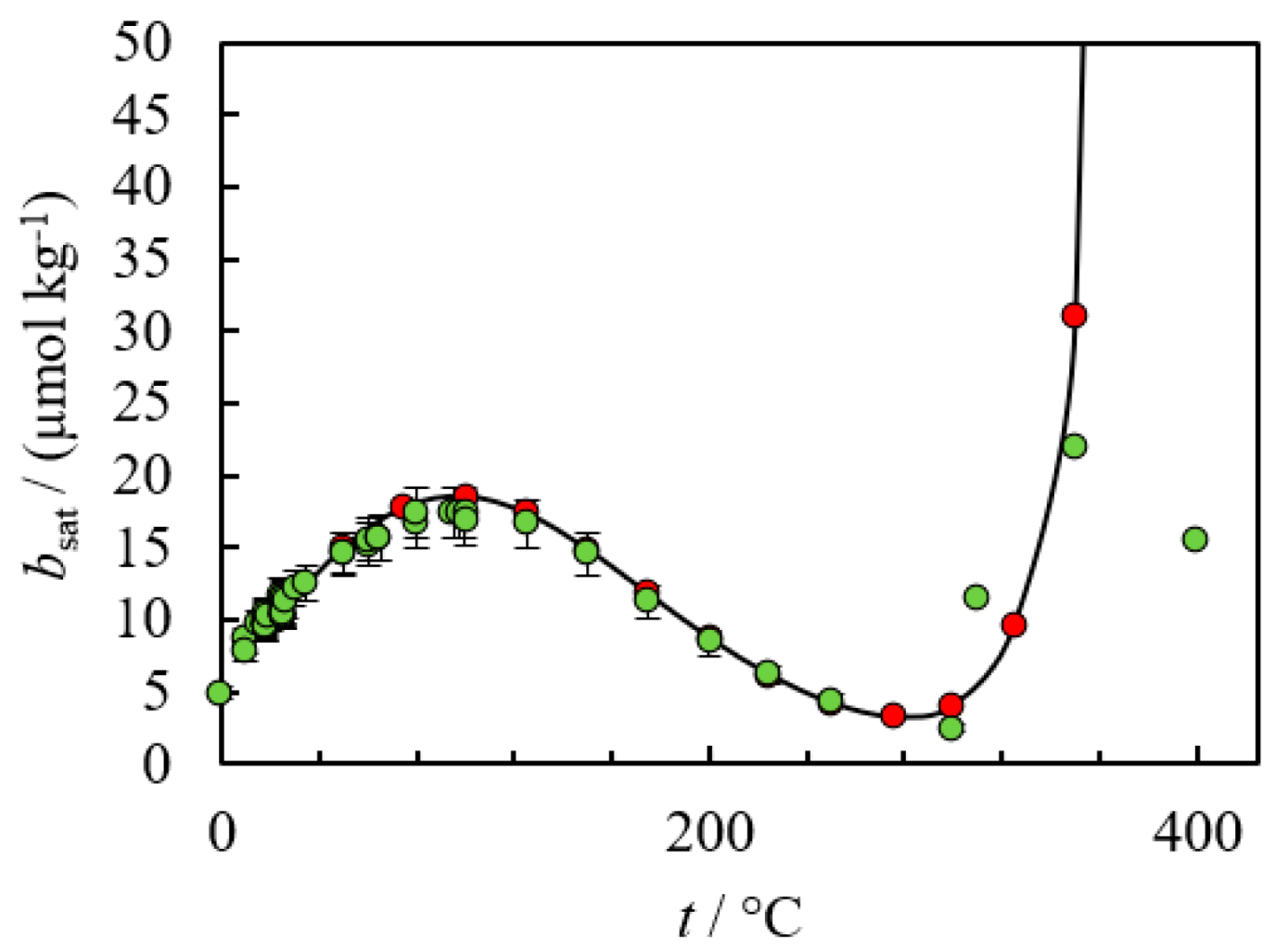
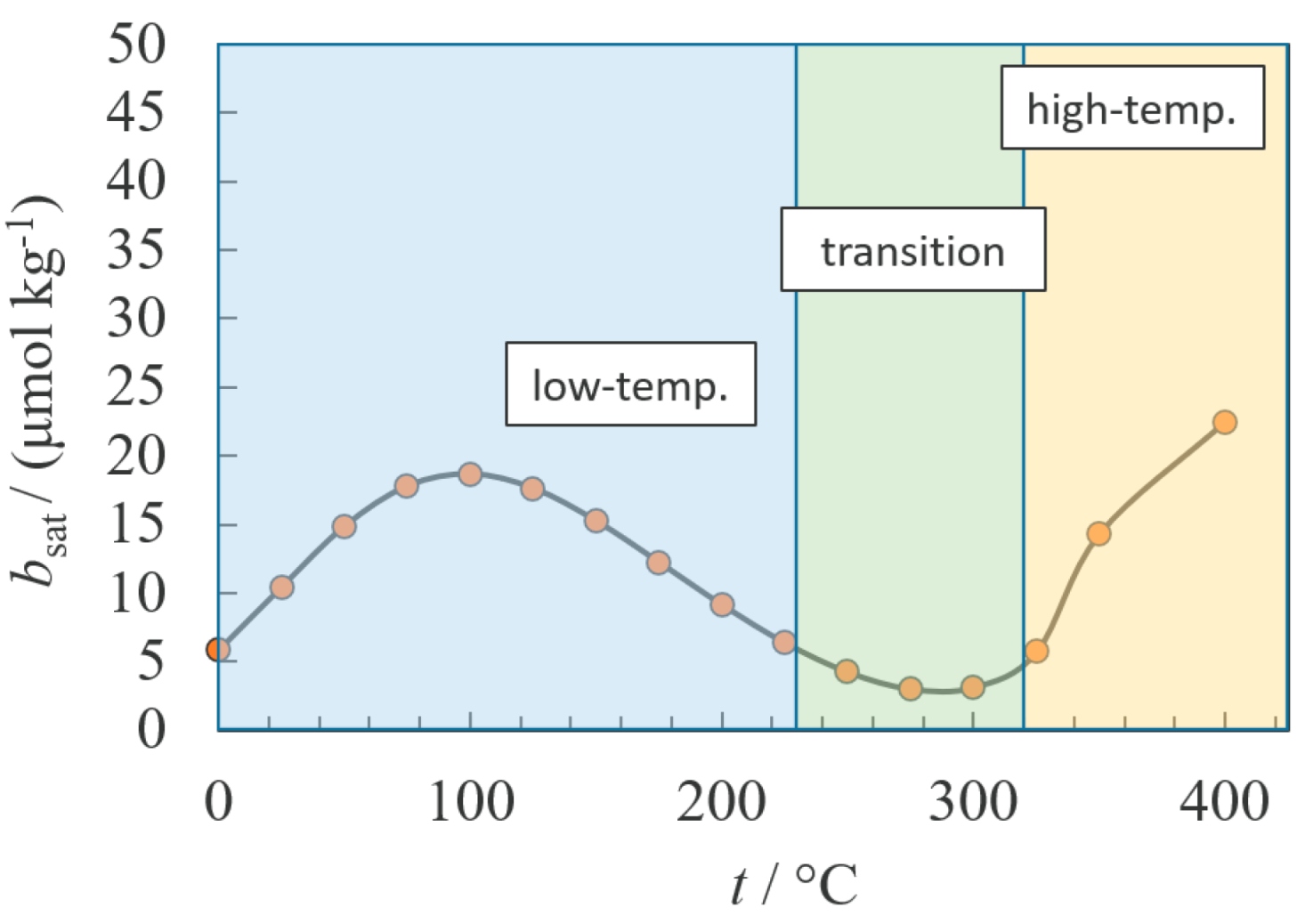
2. Empirical Fitting Methods
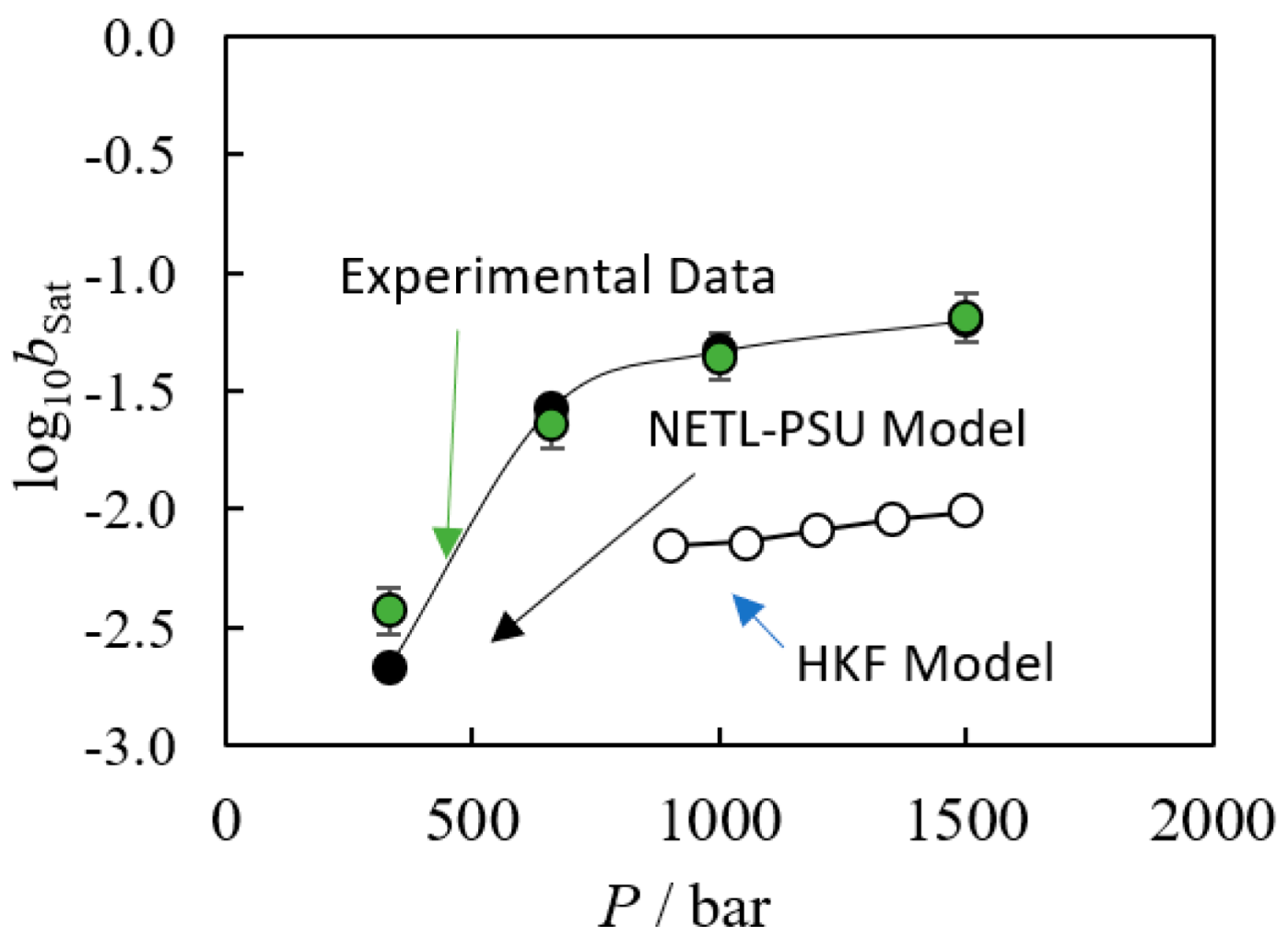
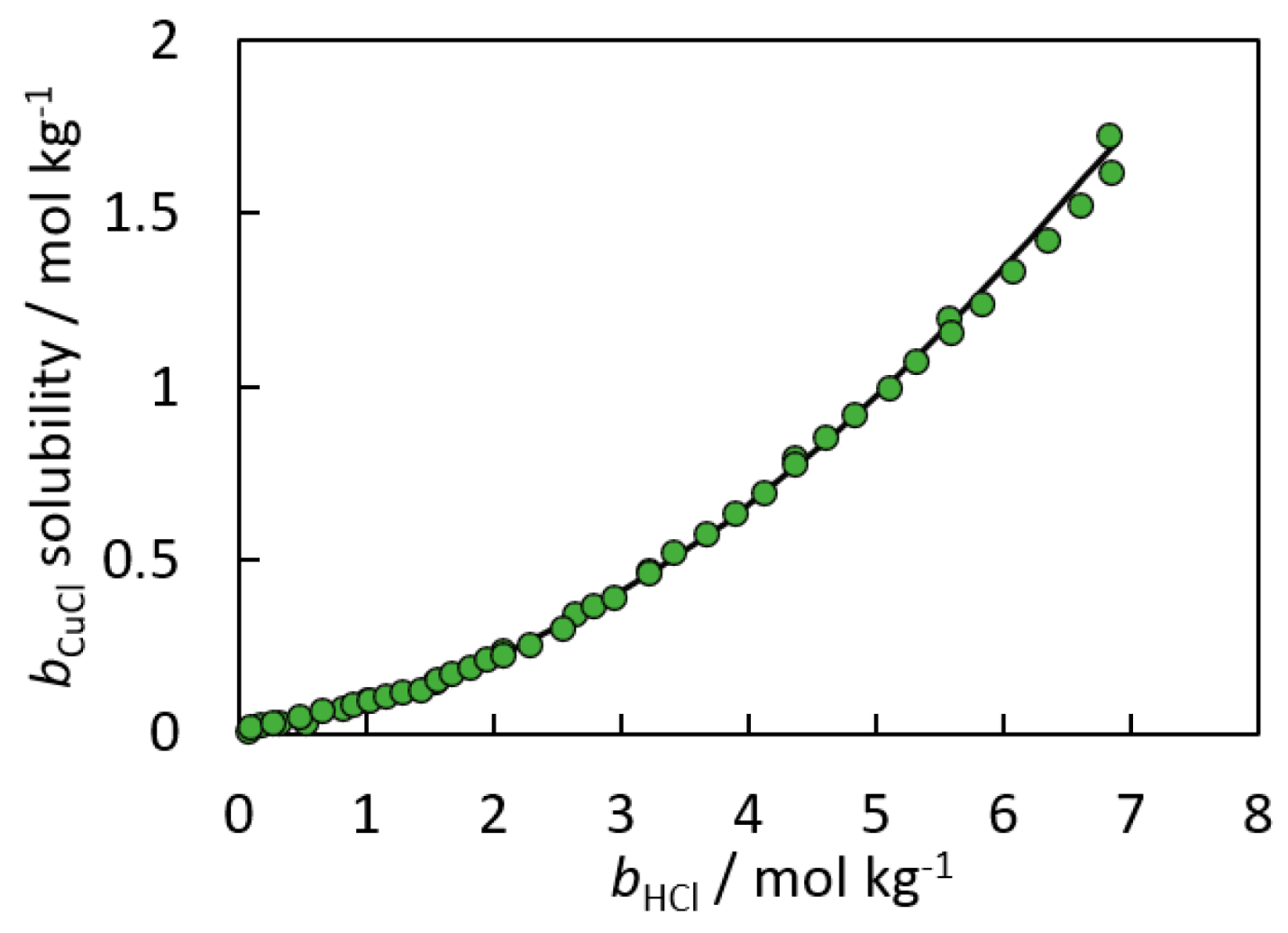
3. Solubility Product Methods
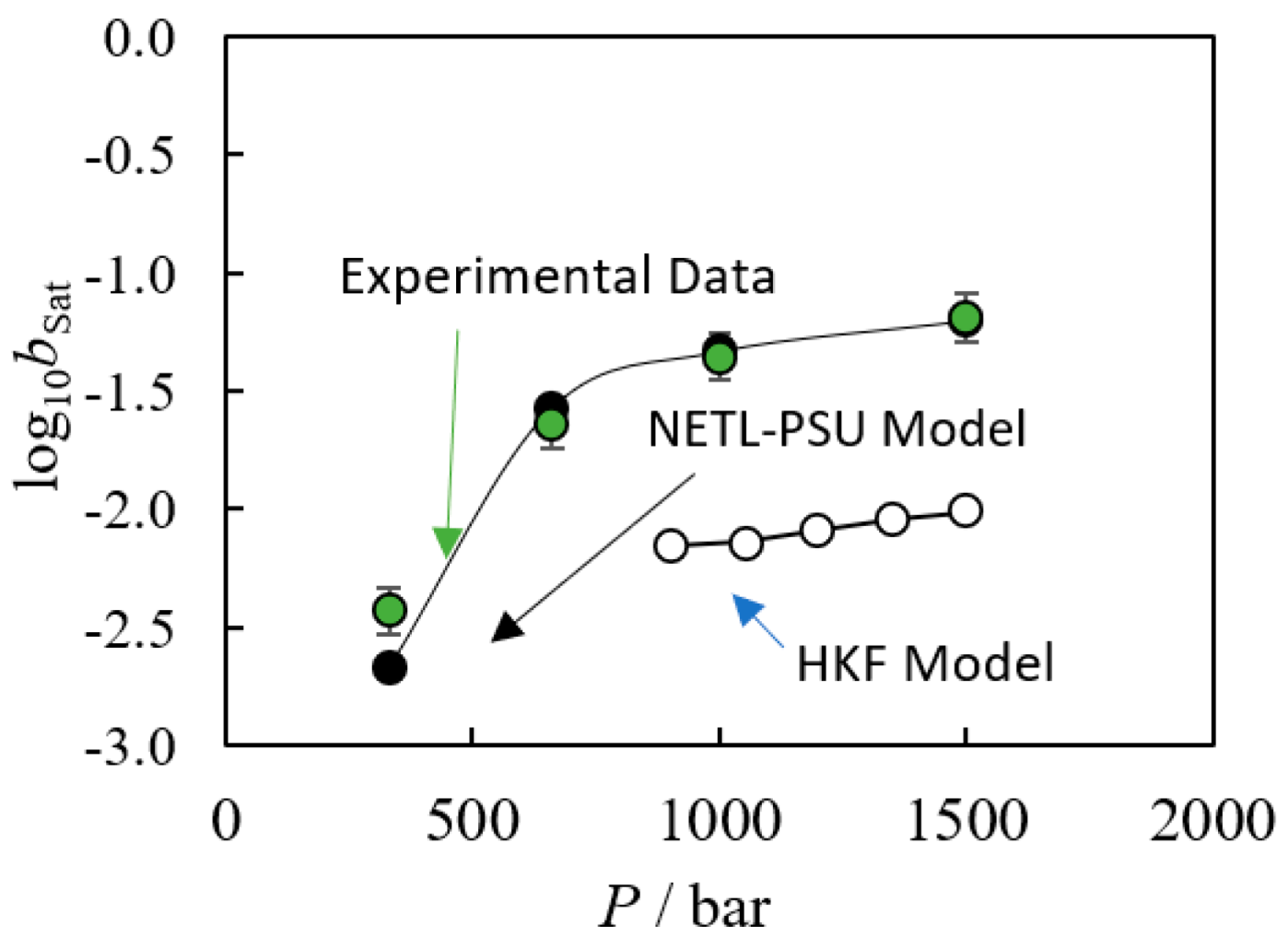
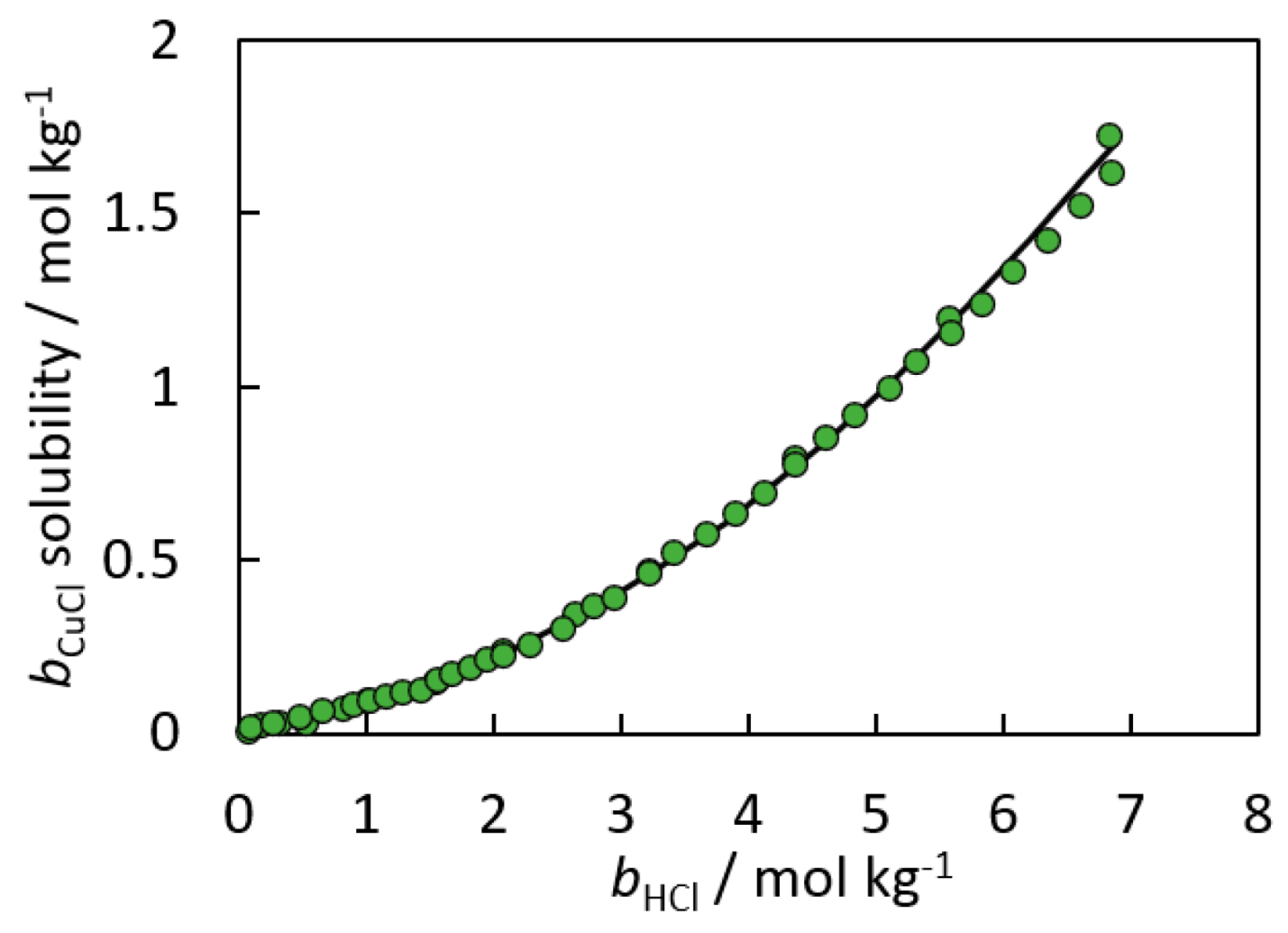
4. Speciation Model Methods
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Brown, M. Full Scale Attack. Review 1998, 30, 30–32. [Google Scholar]
- Kamal, M.S.; Hussein, I.; Mahmoud, M.; Sultan, A.S.; Saad, M.A.S. Oilfield Scale Formation and Chemical Removal: A Review. J. Pet. Sci. Eng. 2018, 171, 127–139. [Google Scholar] [CrossRef]
- Olajire, A.A. A Review of Oilfield Scale Management Technology for Oil and Gas Production. J. Pet. Sci. Eng. 2015, 135, 723–737. [Google Scholar] [CrossRef]
- Krumgalz, B.S. Temperature Dependence of Mineral Solubility in Water. Part 3. Alkaline and Alkaline Earth Sulfates. J. Phys. Chem. Ref. Data 2018, 47, 023101. [Google Scholar] [CrossRef]
- Mucci, A. The Solubility of Calcite and Aragonite in Seawater at Various Salinities, Temperatures, and One Atmosphere Total Pressure. Am. J. Sci. 1983, 283, 780–799. [Google Scholar] [CrossRef]
- Lvov, S.N.; Hall, D.M.; Bandura, A.V.; Gamwo, I.K. A Semi-Empirical Molecular Statistical Thermodynamic Model for Calculating Standard Molar Gibbs Energies of Aqueous Species above and below the Critical Point of Water. J. Mol. Liq. 2018, 270, 62–73. [Google Scholar] [CrossRef]
- Helgeson, H.C. Thermodynamics of Hydrothermal Systems at Elevated Temperatures and Pressures. Am. J. Sci. 1969, 267, 729–804. [Google Scholar] [CrossRef]
- Johnson, J.W.; Oelkers, E.H.; Helgeson, H.C. SUPCRT92: A Software Package for Calculating the Standard Molal Thermodynamic Properties of Minerals, Gases, Aqueous Species, and Reactions from 1 to 5000 Bars and 0 to 1000 °C. Comput. Geosci. 1992, 18, 899–947. [Google Scholar] [CrossRef]
- Kan, A.T.; Dai, J.Z.; Deng, G.; Ruan, G.; Li, W.; Harouaka, K.; Lu, Y.T.; Wang, X.; Zhao, Y.; Tomson, M.B. Recent Advances in Scale Prediction, Approach, and Limitations. SPE J. 2019, 24, 2209–2220. [Google Scholar] [CrossRef]
- Shvarov, Y.V. HCh: New Potentialities for the Thermodynamic Simulation of Geochemical Systems Offered by Windows. Geochem. Int. 2008, 46, 834–839. [Google Scholar] [CrossRef]
- OLI Systems Inc. Selecting AQ, MSE or MSE-SRK for Your Scaling Calculation; OLI Systems Inc.: Parsippany, NJ, USA, 2017. [Google Scholar]
- Wang, P.; Springer, R.D.; Anderko, A.; Young, R.D. Modeling Phase Equilibria and Speciation in Mixed-Solvent Electrolyte Systems. Fluid Phase Equilib. 2004, 222–223, 11–17. [Google Scholar] [CrossRef]
- FQE Chemicals. Pipe contaminated with Barium Sulfate and NORM scale. Available online: https://fqechemicals.com/contaminants/barium-sulfate-scale/ (accessed on 10 August 2022).
- Greg Anderson Rock-Water Systems. In Thermodynamics of Natural Systems; Cambridge University Press: Cambridge, UK, 2005; pp. 473–497.
- Zhang, P.; Kan, A.T.; Tomson, M.B. Oil Field Mineral Scale Control. In Mineral Scales and Deposits; Elsevier: Amsterdam, The Netherlands, 2015; pp. 603–617. ISBN 9780444627520. [Google Scholar]
- Kan, A.T.; Tomson, M.B. Scale Prediction for Oil and Gas Production. SPE J. 2012, 17, 362–378. [Google Scholar] [CrossRef]
- Fan, C.; Kan, A.T.; Zhang, P.; Lu, H.; Work, S.; Yu, J.; Tomson, M.B. Scale Prediction and Inhibition for Oil and Gas Production at High Temperature/High Pressure. SPE J. 2012, 17, 379–392. [Google Scholar] [CrossRef]
- Kan, A.T.; Dai, Z.; Tomson, M.B. The State of the Art in Scale Inhibitor Squeeze Treatment. Pet. Sci. 2020, 17, 1579–1601. [Google Scholar] [CrossRef]
- Mpelwa, M.; Tang, S.F. State of the Art of Synthetic Threshold Scale Inhibitors for Mineral Scaling in the Petroleum Industry: A Review. Pet. Sci. 2019, 16, 830–849. [Google Scholar] [CrossRef]
- Helgeson, H.C.; Kirkham, D.H.; Flowers, G.C. Theoretical Prediction of the Thermodynamic Behavior of Aqueous Electrolytes at High Pressures and Temperatures: Calculation of Activity Coefficients, Osmotic Coefficients, and Apparent Molal and Standard and Relative Partial Molal Properties to 600 °C And. Am. J. Sci. 1981, 281, 1249–1516. [Google Scholar] [CrossRef]
- Plummer, N.; Busenberg, E. The Solubilities of Calcite, Aragonite and Vaterite in CO2-H2O Solutions between 0 and 90 C, and an Evaluation of the Aqueous Model for the System CaCO3-CO2-H2O. Geochim. Cosmochim. Acta 1981, 46, 1011–1040. [Google Scholar] [CrossRef]
- Krumgalz, B.S. Temperature Dependence of Mineral Solubility in Water. Part I. Alkaline and Alkaline Earth Chlorides. J. Phys. Chem. Ref. Data 2017, 46, 043101. [Google Scholar] [CrossRef]
- Krumgalz, B.S. Temperature Dependence of Mineral Solubility in Water. Part 2. Alkaline and Alkaline Earth Bromides. J. Phys. Chem. Ref. Data 2018, 47, 013101. [Google Scholar] [CrossRef]
- Marion, G.M.; Millero, F.J.; Feistel, R. Precipitation of Solid Phase Calcium Carbonates and Their Effect on Application of Seawater SA-T-P Models. Ocean Sci. 2009, 5, 285–291. [Google Scholar] [CrossRef]
- Djamali, E.; Chapman, W.G.; Cox, K.R. A Systematic Investigation of the Thermodynamic Properties of Aqueous Barium Sulfate up to High Temperatures and High Pressures. J. Chem. Eng. Data 2016, 61, 3585–3594. [Google Scholar] [CrossRef]
- Monnin, C. A Thermodynamic Model for the Solubility of Barite and Celestite in Electrolyte Solutions and Seawater to 200 °C and to 1 Kbar. Chem. Geol. 1999, 153, 187–209. [Google Scholar] [CrossRef]
- Robie, R.; Hemingway, B.; Fisher, J. Thermodynamic Properties of Minerals and Related Substances at 298.15 K and 1 bar Pressure and at Higher Temperatures. US Geol. Surv. Bull. 1984, 1452. [Google Scholar] [CrossRef]
- Phi, T.; Elgaddafi, R.; Al Ramadan, M.; Fahd, K.; Ahmed, R.; Teodoriu, C. Well Integrity Issues: Extreme High-Pressure High-Temperature Wells and Geothermal Wells a Review. In SPE Thermal Well Integrity and Design Symposium; OnePetro: Richardson, TX, USA, 2019. [Google Scholar] [CrossRef]
- Friðleifsson, G.; Elders, W.A.; Zierenberg, R.A.; Fowler, A.P.G.; Weisenberger, T.B.; Mesfin, K.G.; Sigurðsson, Ó.; Níelsson, S.; Einarsson, G.; Óskarsson, F.; et al. The Iceland Deep Drilling Project at Reykjanes: Drilling into the Root Zone of a Black Smoker Analog. J. Volcanol. Geotherm. Res. 2020, 391, 106435. [Google Scholar] [CrossRef]
- Su, H.; Yan, M.; Wang, S. Recent Advances in Supercritical Water Gasification of Biowaste Catalyzed by Transition Metal-Based Catalysts for Hydrogen Production. Renew. Sustain. Energy Rev. 2022, 154, 111831. [Google Scholar] [CrossRef]
- Shock, E.L.; Helgeson, H.C. Calculation of the Thermodynamic and Transport Properties of Aqueous Species at High Pressures and Temperatures: Correlation Algorithms for Ionic Species and Equation of State Predictions to 5 Kb and 1000 °C. Geochim. Cosmochim. Acta 1988, 52, 2009–2036. [Google Scholar] [CrossRef]
- Haas, J.R.; Shock, E.L.; Sassani, D.C. Rare-Earth Elements in Hydrothermal Systems—Estimates of Standard Partial Molal Thermodynamic Properties of Aqueous Complexes of the Rare-Earth Elements at High-Pressures and Temperatures. Geochim. Cosmochim. Acta 1995, 59, 4329–4350. [Google Scholar] [CrossRef]
- Sverjensky, D.A.; Shock, E.L.; Helgeson, H.C. Prediction of the Thermodynamic Properties of Aqueous Metal Complexes to 1000 Degrees C and 5 Kb. Geochim. Cosmochim. Acta 1997, 61, 1359–1412. [Google Scholar] [CrossRef]
- Shock, E.L.L.; Sassani, D.C.; Willis, M.; Sverjensky, D.A. Inorganic Species in Geologic Fluids: Correlations among Standard Molal Thermodynamic Properties of Aqueous Ions and Hydroxide Complexes. Geochim. Cosmochim. Acta 1997, 61, 907–950. [Google Scholar] [CrossRef]
- Shock, E.L.; Helgeson, H.C. Calculation of the Thermodynamic and Transport Properties of Aqueous Species at High Pressures and Temperatures: Standard Partial Molal Properties of Organic Species. Geochim. Cosmochim. Acta 1990, 54, 915–945. [Google Scholar] [CrossRef]
- Pokrovskii, V.A.; Helgeson, H.C. Calculation of the Standard Partial Molal Thermodynamic and Activity Coefficients of Aqueous KC1 at Temperatures and Pressures. Geochim. Cosmochim. Acta 1997, 61, 2175–2183. [Google Scholar] [CrossRef]
- Hall, D.M.; Lvov, S.N.; Gamwo, I.K. Prediction of Barium Sulfate Deposition in Petroleum and Hydrothermal Systems. In Solid–Liquid Separation Technologies: Applications for Produced Water; CRC Press Taylor & Francis: Boca Raton, FL, USA, 2022. [Google Scholar]
- Morey, G.W.; Hesselgesser, J.M. The Solubility of Some Minerals in Superheated Steam at High Pressures. Econ. Geol. 1951, 46, 821–835. [Google Scholar] [CrossRef]
- Mansoori, G.A.; Carnahan, N.F.; Starling, K.E.; Leland, T.W., Jr. Equilibrium Thermodynamic Properties of the Mixture of Hard Spheres. J. Chem. Phys. 1971, 54, 1523–1525. [Google Scholar] [CrossRef]
- Triolo, R.; Grigera, J.; Blum, L. Simple Electrolytes in the Mean Spherical Approximation. J. Phys. Chem. 1976, 80, 1858–1861. [Google Scholar] [CrossRef]
- Blum, L. Solution of a Model for the Solvent-Electrolyte Interactions in the Mean Spherical Approximation. J. Chem. Phys. 1974, 61, 2129. [Google Scholar] [CrossRef]
- Bandura, A.V.; Holovko, M.F.; Lvov, S.N. The Chemical Potential of a Dipole in Dipolar Solvent at Infinite Dilution: Mean Spherical Approximation and Monte Carlo Simulation. J. Mol. Liq. 2018, 270, 52–61. [Google Scholar] [CrossRef]
- Lvov, S.N.; Wood, R.H. Equation of State of Aqueous NaCl Solutions over a Wide Range of Temperatures, Pressures and Concentrations. Fluid Phase Equilib. 1990, 60, 273–287. [Google Scholar] [CrossRef]
- Heberling, F.; Trainor, T.P.; Lützenkirchen, J.; Eng, P.; Denecke, M.A.; Bosbach, D. Structure and Reactivity of the Calcite-Water Interface. J. Colloid Interface Sci. 2011, 354, 843–857. [Google Scholar] [CrossRef]
- García, A.V.; Thomsen, K.; Stenby, E.H. Prediction of Mineral Scale Formation in Geothermal and Oilfield Operations Using the Extended UNIQUAC Model. Part II. Carbonate-Scaling Minerals. Geothermics 2006, 35, 239–284. [Google Scholar] [CrossRef]
- Li, J.; Duan, Z. A Thermodynamic Model for the Prediction of Phase Equilibria and Speciation in the H2O-CO2-NaCl-CaCO3-CaSO4 System from 0 to 250 °C, 1 to 1000 Bar with NaCl Concentrations up to Halite Saturation. Geochim. Cosmochim. Acta 2011, 75, 4351–4376. [Google Scholar] [CrossRef]
- Amiri, M.; Moghadasi, J. The Effect of Temperature on Calcium Carbonate Scale Formation in Iranian Oil Reservoirs Using OLI ScaleChem Software. Pet. Sci. Technol. 2012, 30, 453–466. [Google Scholar] [CrossRef]
- Hajirezaie, S.; Wu, X.; Soltanian, M.R.; Sakha, S. Numerical Simulation of Mineral Precipitation in Hydrocarbon Reservoirs and Wellbores. Fuel 2019, 238, 462–472. [Google Scholar] [CrossRef]
- Ho, P.C.; Palmer, D.A. Ion Association of Dilute Aqueous Potassium Chloride and Potassium Hydroxide Solutions to 600 °C and 300 MPa Determined by Electrical Conductance Measurements. Geochim. Cosmochim. Acta 1997, 61, 3027–3040. [Google Scholar] [CrossRef]
- Ho, P.C.; Bianchi, H.; Palmer, D.A.; Wood, R.H. Conductivity of Dilute Aqueous Electrolyte Solutions at High Temperatures and Pressures Using a Flow Cell. J. Solution Chem. 2000, 29, 217–235. [Google Scholar] [CrossRef]
- Ho, P.; Palmer, D. Ion Association of Dilute Aqueous Sodium Hydroxide Solutions to 600 C and 300 MPa by Conductance Measurements. J. Solution Chem. 1996, 25, 711–729. [Google Scholar] [CrossRef]
- Fritz, J.J. Chloride Complexes of CuCl in Aqueous Solution. J. Phys. Chem. 1980, 84, 2241–2246. [Google Scholar] [CrossRef]
- Balashov, V.N.; Schatz, R.S.; Chalkova, E.; Akinfiev, N.N.; Fedkin, M.V.; Lvov, S.N. CuCl Electrolysis for Hydrogen Production in the Cu–Cl Thermochemical Cycle. J. Electrochem. Soc. 2011, 158, B266–B275. [Google Scholar] [CrossRef]
- Lvov, S.N.; Akinfiev, N.N.; Bandura, A.V.; Sigon, F.; Perboni, G. Multisys: Computer Code for Calculating Multicomponent Equilibria in High-Temperature Subcritical and Supercritical Aqueous Systems. In Steam, Water, and Hydrothermal Systems: Physics and Chemistry Meeting the Needs of Industry; Tremaine, P.R., Hill, P.G., Irish, D.E., Palakrishnan, P.V., Eds.; NRC Press: Ottawa, ON, USA, 2000; pp. 866–873. [Google Scholar]
- Du, P.C.; Mansoori, G.A. Phase Equilibrium of Multicomponent Mixtures: Continuous Mixture Gibbs Free Energy Minimization and Phase Rule. Chem. Eng. Commun. 1987, 54, 139–148. [Google Scholar] [CrossRef]
- Shobu, K.; Tabaru, T. Development of New Equilibrium Calculation Software: CaTCalc. Mater. Trans. 2005, 46, 1175–1179. [Google Scholar] [CrossRef]
- Bethke, C.M. Geochemical Reaction Modelling: Concepts and Applications; Oxford University Press: New York, NY, USA, 1996. [Google Scholar]
- Bethke, C.M. The Geochemists Workbench Version 4.0: A User’s Guide; University of Illinois: Urbana, IL, USA, 2002. [Google Scholar]
- Cleverley, J.S.; Bastrakov, E.N. K2GWB: Utility for Generating Thermodynamic Data Files for The Geochemist’s Workbench® at 0–1000 °C and 1–5000 Bar from UT2K and the UNITHERM Database. Comput. Geosci. 2005, 31, 756–767. [Google Scholar] [CrossRef]
- Shvarov, Y. A Suite of Programs, OptimA, OptimB, OptimC, and OptimS Compatible with the Unitherm Database, for Deriving the Thermodynamic Properties of Aqueous Species from Solubility, Potentiometry and Spectroscopy Measurements. Appl. Geochem. 2015, 55, 17–27. [Google Scholar] [CrossRef]
- OLI Systems. OLI Studio Stream Analyzer User Guide. V 9.5; OLI Systems: Parsippany, NJ, USA, 2018. [Google Scholar]
- Lencka, M.M.; Springer, R.D.; Wang, P.; Anderko, A. Modeling Mineral Scaling in Oil and Gas Environments up to Ultra High Pressures and Temperatures: Paper No. C2018-10828. In CORROSION; OnePetro: Richardson, TX, USA, 2018; pp. 1–15. [Google Scholar]
- Shvarov, Y.V. A General Equilibrium Criterion for an Isobaric-Isothermal Model of a Chemical System. Geochem. Int. 1981, 18, 38–45. [Google Scholar]
- Shvarov, Y.V. Algorithmization of the Numeric Equilibrium Modeling of Dynamic Geochemical Processes. Geochem. Int. 1999, 37, 571–576. [Google Scholar]
- Hall, D.M.; Akinfiev, N.N.; LaRow, E.G.; Schatz, R.S.; Lvov, S.N. Thermodynamics and Efficiency of a CuCl(Aq)/HCl(Aq) Electrolyzer. Electrochim. Acta 2014, 143, 70–82. [Google Scholar] [CrossRef]
- Koukkari, P.; Pajarre, R. Calculation of Constrained Equilibria by Gibbs Energy Minimization. Comput. Coupling Phase Diagr. Thermochem. 2006, 30, 18–26. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Empirical Fits | Solubility Product | Speciation Models |
|---|---|---|---|
| Benefits | Easy to implement | Easy to implement | Predictive capabilities |
| Predictive capabilities | Works with multiphase systems | ||
| Provides solution compositions | |||
| Limitations | No predictive capabilities | Requires a thermodynamic database | Requires a thermodynamic database |
| Requires system-specific solubility data | Limited to the temperature and pressure range of the database | Limited to the temperature and pressure range of the database | |
| Limited to the temperature and pressure range of the fit | Limited to simple systems |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hall, D.M.; Lvov, S.N.; Gamwo, I.K. Thermodynamic Modeling of Mineral Scaling in High-Temperature and High-Pressure Aqueous Environments. Liquids 2022, 2, 303-317. https://doi.org/10.3390/liquids2040018
Hall DM, Lvov SN, Gamwo IK. Thermodynamic Modeling of Mineral Scaling in High-Temperature and High-Pressure Aqueous Environments. Liquids. 2022; 2(4):303-317. https://doi.org/10.3390/liquids2040018
Chicago/Turabian StyleHall, Derek M., Serguei N. Lvov, and Isaac K. Gamwo. 2022. "Thermodynamic Modeling of Mineral Scaling in High-Temperature and High-Pressure Aqueous Environments" Liquids 2, no. 4: 303-317. https://doi.org/10.3390/liquids2040018
APA StyleHall, D. M., Lvov, S. N., & Gamwo, I. K. (2022). Thermodynamic Modeling of Mineral Scaling in High-Temperature and High-Pressure Aqueous Environments. Liquids, 2(4), 303-317. https://doi.org/10.3390/liquids2040018