
Applied Metagenomic Profiling of Domestic Cat Feces from Cali, Colombia: An Exploratory Approach

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Site, Sample Collection, and Processing
2.2. Bacterial Composition
2.3. Statistical Distribution of Read Counts
2.4. Shotgun Metagenomic Analysis
2.5. Pathogenic Bacteria Genome Reconstruction
3. Results
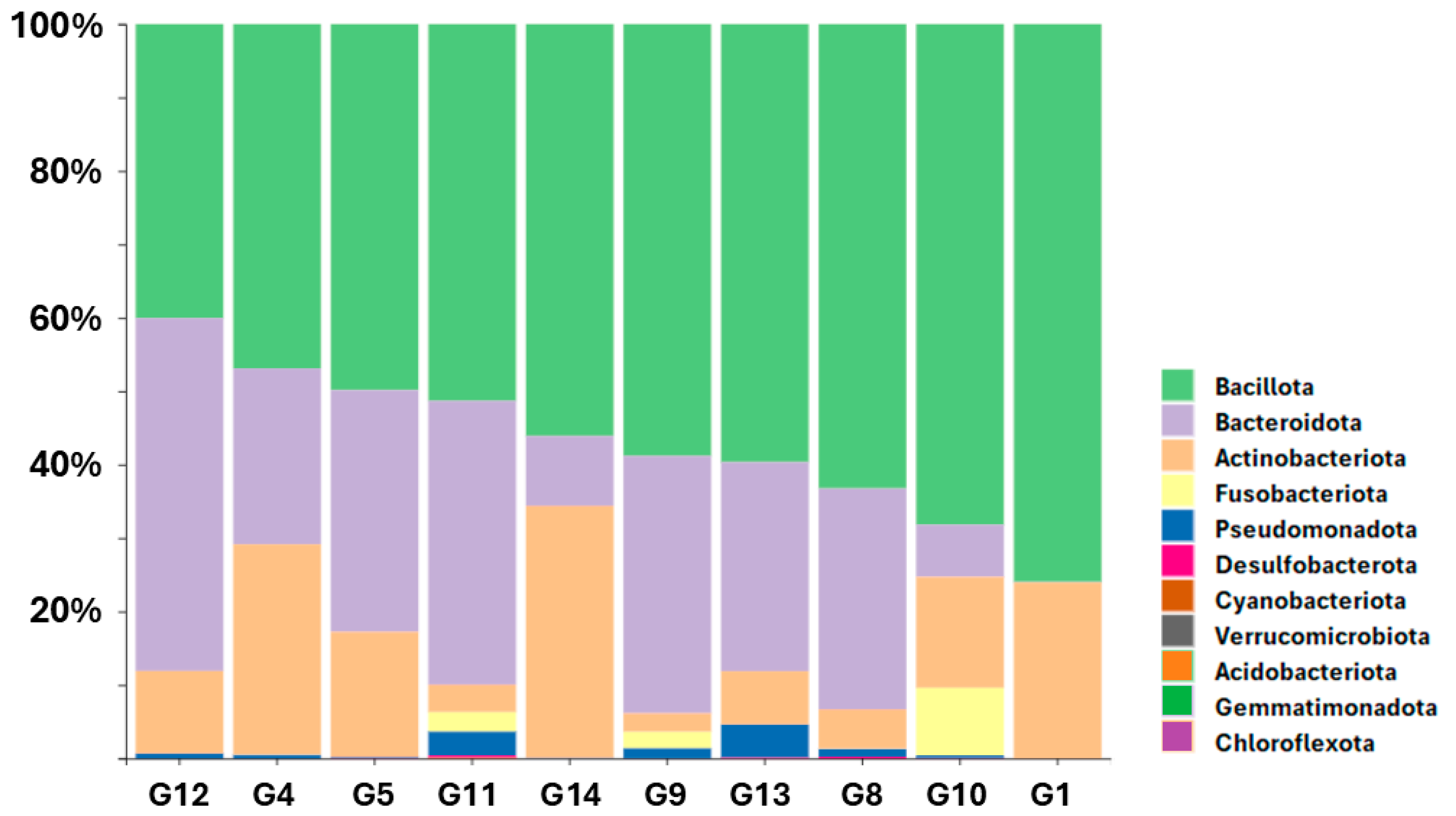
3.1. Relative Abundance of Bacterial Phyla Across Samples
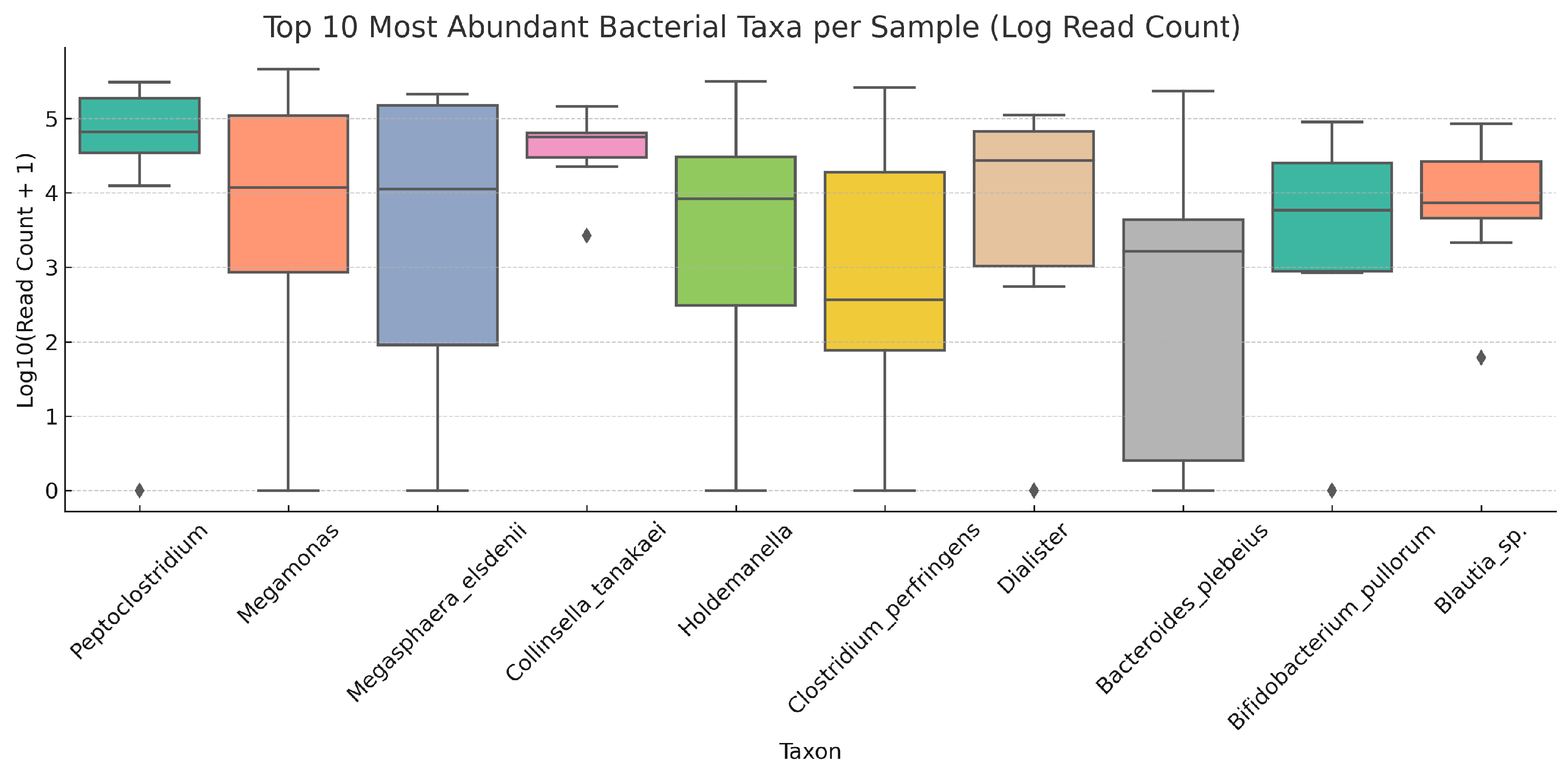
3.1.1. Distribution and Variability of the Most Abundant Taxa Across Samples
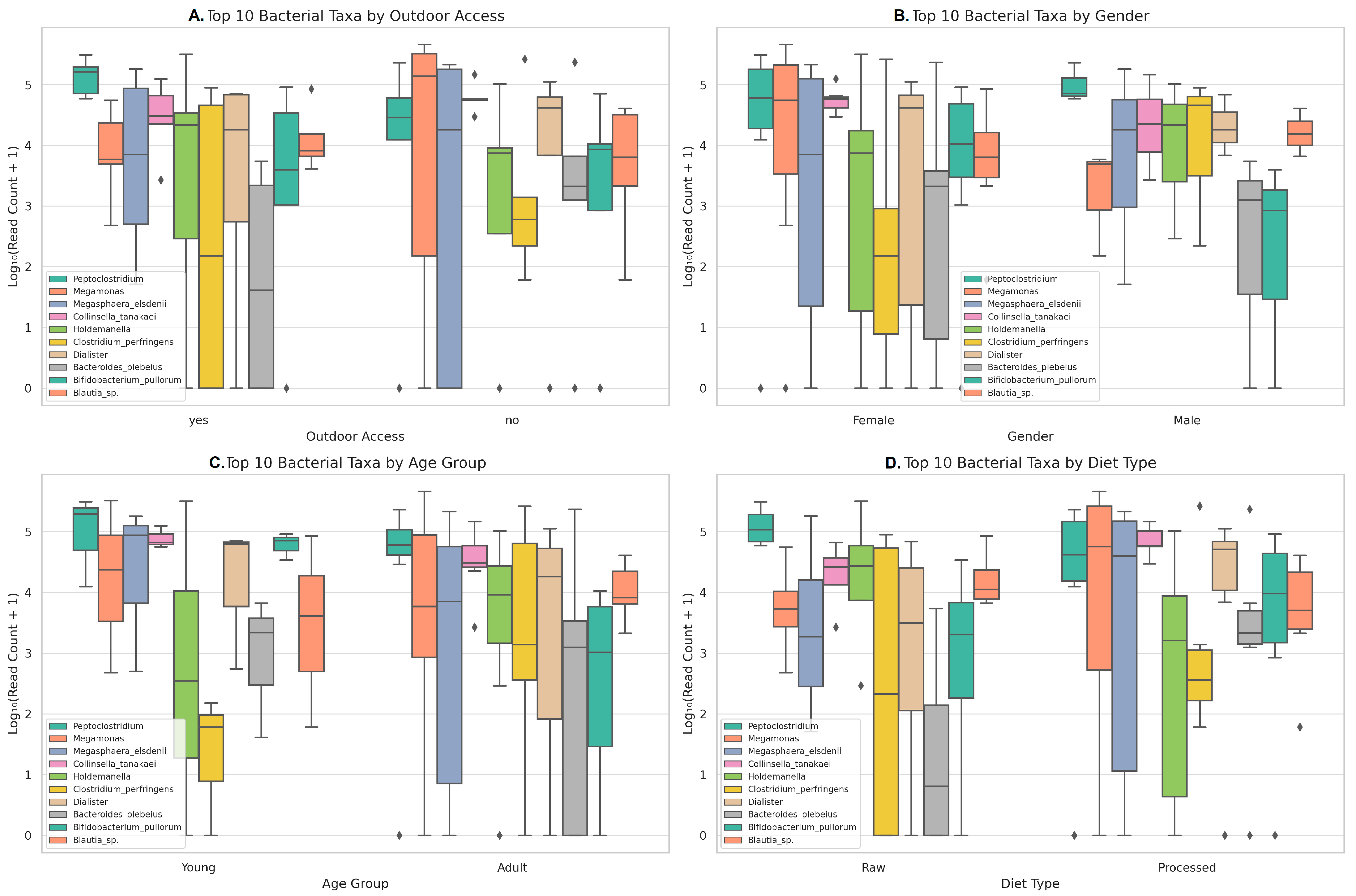
3.1.2. Distribution and Variability of the Most Abundant Taxa According to Metadata Variables
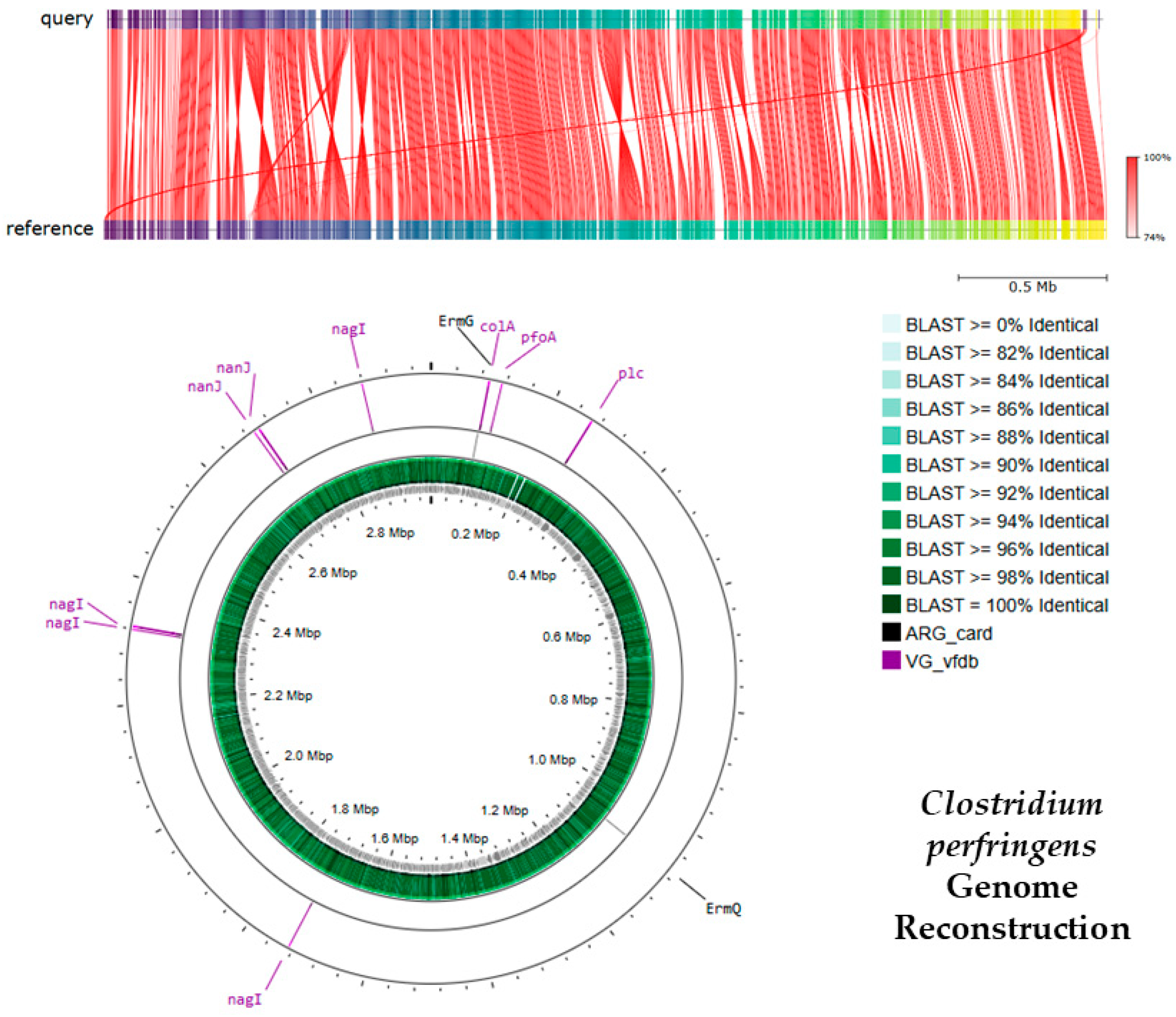
3.1.3. Antibiotic Resistance and Virulence Genes Identified in C. perfringens
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SCFA | Short-Chain Fatty Acid |
| ARG | Antibiotic Resistance Gene |
| VF | Virulence Factor |
| rRNA | Ribosomal Ribonucleic Acid |
| MLSb | Macrolide–Lincosamide–Streptogramin B |
| BSH | Bile Salt Hydrolase |
| DNA | Deoxyribonucleic Acid |
| PCR | Polymerase Chain Reaction |
| NCBI | National Center for Biotechnology Information |
| AMR | Antimicrobial Resistance |
| NGS | Next-Generation Sequencing |
| OTU | Operational Taxonomic Unit |
| LCA | Lowest Common Ancestor |
| QC | Quality Control |
| IQR | Interquartile Range |
References
- Suchodolski, J.S. Analysis of the gut microbiome in dogs and cats. Vet. Clin. Pathol. 2022, 50 (Suppl. S1), 6–17. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, S.S. Value of Probiotics in Canine and Feline Gastroenterology. Vet. Clin. Small Anim. Pract. 2021, 51, 171–217. [Google Scholar] [CrossRef] [PubMed]
- Rojas, C.A.; Gardy, J.; Eisen, J.A.; Ganz, H.H.; Rasko, D. Recovery of 52 bacterial genomes from the fecal microbiome of the domestic cat (Felis catus) using Hi-C proximity ligation and shotgun metagenomics. Microbiol. Resour. Announc. 2023, 12, e0060123. [Google Scholar] [CrossRef] [PubMed]
- Ing, N.H.; Steiner, J.M. The Use of Diets in the Diagnosis and Treatment of Common Gastrointestinal Diseases in Dogs and Cats. Adv. Exp. Med. Biol. 2024, 1446, 39–53. [Google Scholar] [CrossRef]
- Ganz, H.H.; Jospin, G.; Rojas, C.A.; Martin, A.L.; Dahlhausen, K.; Kingsbury, D.D.; Osborne, C.X.; Entrolezo, Z.; Redner, S.; Ramirez, B.; et al. The Kitty Microbiome Project: Defining the Healthy Fecal “Core Microbiome” in Pet Domestic Cats. Vet. Sci. 2022, 9, 635. [Google Scholar] [CrossRef]
- Butowski, C.F.; Thomas, D.G.; Cave, N.J.; Bermingham, E.N.; Rosendale, D.I.; Hea, S.-Y.; Stoklosinski, H.M.; Young, W. In Vitro Assessment of Hydrolysed Collagen Fermentation Using Domestic Cat (Felis catus) Faecal Inocula. Animals 2022, 12, 498. [Google Scholar] [CrossRef]
- Summers, S.; Quimby, J.; Gagné, J.; Lappin, M. The Effect of Dietary Protein Concentration on the Fecal Microbiome and Serum Concentrations of Gut-Derived Uremic Toxins in Healthy Adult Cats. Vet. Sci. 2023, 10, 497. [Google Scholar] [CrossRef]
- Rojas, C.A.; Entrolezo, Z.; Jarett, J.K.; Jospin, G.; Kingsbury, D.D.; Martin, A.; Eisen, J.A.; Ganz, H.H. Microbiome Responses to Fecal Microbiota Transplantation in Cats with Chronic Digestive Issues. Vet. Sci. 2023, 10, 561. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 1091. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Schwengers, O.; Jelonek, L.; Dieckmann, M.A.; Beyvers, S.; Blom, J.; Goesmann, A. Bakta: Rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Microb. Genom. 2021, 7, 000685. [Google Scholar] [CrossRef]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Chen, L.; Zheng, D.; Liu, B.; Yang, J.; Jin, Q. VFDB 2016: Hierarchical and refined dataset for big data analysis—10 years on. Nucleic Acids Res. 2016, 44, D694–D697. [Google Scholar] [CrossRef]
- Lu, J.; Rincon, N.; Wood, D.E.; Breitwieser, F.P.; Pockrandt, C.; Langmead, B.; Salzberg, S.L.; Steinegger, M. Metagenome analysis using the Kraken software suite. Nat. Protoc. 2024, 17, 2815–2839. [Google Scholar] [CrossRef]
- Breitwieser, F.P.; Salzberg, S.L.; Schwartz, R. Pavian: Interactive analysis of metagenomics data for microbiome studies and pathogen identification. Bioinformatics 2020, 36, 1303–1304. [Google Scholar] [CrossRef] [PubMed]
- Tun, H.M.; Brar, M.S.; Khin, N.; Jun, L.; Hui, R.K.-H.; Dowd, S.E.; Leung, F.C.-C. Gene-centric metagenomics analysis of feline intestinal microbiome using 454 junior pyrosequencing. J. Microbiol. Methods 2012, 88, 369–376. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Hooda, S.; Minamoto, Y.; Suchodolski, J.S.; Swanson, K.S. Current state of knowledge: The canine gastrointestinal microbiome. Anim. Health Res. Rev. 2012, 13, 78–88. [Google Scholar] [CrossRef]
- Thomas, F.; Hehemann, J.-H.; Rebuffet, E.; Czjzek, M.; Michel, G. Environmental and gut bacteroidetes: The food connection. Front. Microbiol. 2011, 2, 93. [Google Scholar] [CrossRef] [PubMed]
- Bermingham, E.N.; Maclean, P.; Thomas, D.G.; Cave, N.J.; Young, W. Key bacterial families (Clostridiaceae, Erysipelotrichaceae and Bacteroidaceae) are related to the digestion of protein and energy in dogs. PeerJ 2017, 5, e3019. [Google Scholar] [CrossRef]
- Milani, C.; Duranti, S.; Bottacini, F.; Casey, E.; Turroni, F.; Mahony, J.; Belzer, C.; Delgado Palacio, S.; Arboleya Montes, S.; Mancabelli, L.; et al. The First Microbial Colonizers of the Human Gut: Composition, Activities, and Health Implications of the Infant Gut Microbiota. Microbiol. Mol. Biol. Rev. 2017, 81, e00036-17. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef]
- Shin, N.-R.; Whon, T.W.; Bae, J.-W. Pseudomonadota: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Tetz, G.; Tetz, V. Bacteriophage infections of microbiota can lead to leaky gut in an experimental rodent model. Gut Pathog. 2016, 8, 33. [Google Scholar] [CrossRef]
- Shade, A.; Peter, H.; Allison, S.D.; Baho, D.L.; Berga, M.; Bürgmann, H.; Huber, D.H.; Langenheder, S.; Lennon, J.T.; Martiny, J.B.H.; et al. Fundamentals of microbial community resistance and resilience. Front. Microbiol. 2012, 3, 417. [Google Scholar] [CrossRef]
- Paredes-Sabja, D.; Shen, A.; Sorg, J.A. Clostridioides difficile spore biology: Sporulation, germination, and spore structural proteins. Trends Microbiol. 2014, 22, 406–416. [Google Scholar] [CrossRef]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef]
- Counotte, G.H.M.; Prins, R.A.; Janssen, R.H.A.M.; Debie, M.J.A. Role of Megasphaera elsdenii in the Fermentation of dl-[2-C]lactate in the Rumen of Dairy Cattle. Appl. Environ. Microbiol. 1981, 42, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Krautkramer, K.A.; Kreznar, J.H.; Romano, K.A.; Vivas, E.I.; Barrett-Wilt, G.A.; Rabaglia, M.E.; Keller, M.P.; Attie, A.D.; Rey, F.E.; Denu, J.M. Diet-Microbiota Interactions Mediate Global Epigenetic Programming in Multiple Host Tissues. Mol. Cell 2016, 64, 982–992. [Google Scholar] [CrossRef]
- Cook, A.K. Feline Infectious Diarrhea. Top. Companion Anim. Med. 2008, 23, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Mao, B.; Gu, J.; Wu, J.; Cui, S.; Wang, G.; Zhao, J.; Zhang, H.; Chen, W. Blautia—A new functional genus with potential probiotic properties? Gut Microbes 2021, 13, 1875796. [Google Scholar] [CrossRef] [PubMed]
- Turroni, F.; Milani, C.; Duranti, S.; Ferrario, C.; Lugli, G.A.; Mancabelli, L.; van Sinderen, D.; Ventura, M. Bifidobacteria and the infant gut: An example of co-evolution and natural selection. Cell. Mol. Life Sci. 2018, 75, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Wernimont, S.M.; Radosevich, J.; Jackson, M.I.; Ephraim, E.; Badri, D.V.; MacLeay, J.M.; Jewell, D.E.; Suchodolski, J.S. The Effects of Nutrition on the Gastrointestinal Microbiome of Cats and Dogs: Impact on Health and Disease. Front. Microbiol. 2020, 11, 1266. [Google Scholar] [CrossRef]
- Song, S.J.; Lauber, C.; Costello, E.K.; Lozupone, C.A.; Humphrey, G.; Berg-Lyons, D.; Caporaso, J.G.; Knights, D.; Clemente, J.C.; Nakielny, S.; et al. Cohabiting family members share microbiota with one another and with their dogs. eLife 2013, 2, e00458. [Google Scholar] [CrossRef]
- Rood, J.I.; Adams, V.; Lacey, J.; Lyras, D.; McClane, B.A.; Melville, S.B.; Moore, R.J.; Popoff, M.R.; Sarker, M.R.; Songer, J.G.; et al. Expansion of the Clostridium perfringens toxin-based typing scheme. Anaerobe 2018, 53, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Uzal, F.A.; Vidal, J.E.; McClane, B.A.; Gurjar, A.A.C. perfringens Toxins Involved in Mammalian Veterinary Diseases. Open Toxinology J. 2010, 2, 24–42. [Google Scholar] [CrossRef]
- Markle, J.G.M.; Frank, D.N.; Mortin-Toth, S.; Robertson, C.E.; Feazel, L.M.; Rolle-Kampczyk, U.; von Bergen, M.; McCoy, K.D.; Macpherson, A.J.; Danska, J.S. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 2013, 339, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Roswall, J.; Olsson, L.M.; Kovatcheva-Datchary, P.; Nilsson, S.; Tremaroli, V.; Simon, M.-C.; Kiilerich, P.; Akrami, R.; Krämer, M.; Uhlén, M.; et al. Developmental trajectory of the healthy human gut microbiota during the first 5 years of life. Cell Host Microbe 2021, 29, 765–776.e3. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes 2012, 3, 289–306. [Google Scholar] [CrossRef]
- Bermingham, E.N.; Young, W.; Kittelmann, S.; Kerr, K.R.; Swanson, K.S.; Roy, N.C.; Thomas, D.G. Dietary format alters fecal bacterial populations in the domestic cat (Felis catus). Microbiologyopen 2013, 2, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef]
- Leclercq, R. Mechanisms of resistance to macrolides and lincosamides: Nature of the resistance elements and their clinical implications. Clin. Infect. Dis. 2002, 34, 482–492. [Google Scholar] [CrossRef]
- Brouwer, M.S.; Roberts, A.P.; Hussain, H.; Williams, R.J.; Allan, E.; Mullany, P. Horizontal gene transfer converts non-toxigenic Clostridioides difficile strains into toxin producers. Nat. Commun. 2013, 4, 2601. [Google Scholar] [CrossRef]
- Rood, J.I. Virulence genes of C. perfringens. Annu. Rev. Microbiol. 1998, 52, 333–360. [Google Scholar] [CrossRef]
- Verherstraeten, S.; Goossens, E.; Valgaeren, B.; Pardon, B.; Timbermont, L.; Haesebrouck, F.; Ducatelle, R.; Deprez, P.; Wade, K.R.; Tweten, R.; et al. Perfringolysin O: The Underrated Clostridium perfringens Toxin? Toxins 2015, 7, 1702–1721. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, J.; Nagahama, M.; Oda, M.C. Clostridium perfringens alpha-toxin: Characterization and mode of action. J. Biochem. 2004, 136, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Uzal, F.A.; McClane, B.A. Clostridium perfringens Sialidases: Potential Contributors to Intestinal Pathogenesis and Therapeutic Targets. Toxins 2016, 8, 341. [Google Scholar] [CrossRef]
- Camargo, A.; Ramírez, J.D.; Kiu, R.; Hall, L.J.; Muñoz, M. Unveiling the pathogenic mechanisms of Clostridium perfringens toxins and virulence factors. Emerg. Microbes Infect. 2024, 13, 2341968. [Google Scholar] [CrossRef] [PubMed]
- Kiu, R.; Hall, L.J. An update on the human and animal enteric pathogen Clostridium perfringens. Emerg. Microbes Infect. 2018, 7, 141. [Google Scholar] [CrossRef]
- Camargo, A.; Páez-Triana, L.; Camargo, D.; García-Corredor, D.; Pulido-Medellín, M.; Camargo, M.; Ramírez, J.D.; Muñoz, M. Carriage of Clostridium perfringens in domestic and farm animals across the central highlands of Colombia: Implications for gut health and zoonotic transmission. Vet. Res. Commun. 2024, 48, 2857–2862. [Google Scholar] [CrossRef]
- Djordjevic, S.P.; Jarocki, V.M.; Seemann, T.; Cummins, M.L.; Watt, A.E.; Drigo, B.; Wyrsch, E.R.; Reid, C.J.; Donner, E.; Howden, B.P. Genomic surveillance for antimicrobial resistance—A One Health perspective. Nat. Rev. Genet. 2024, 25, 142–157. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Index | Total Reads | Outdoor Access | Gender | Age | Diet Type |
|---|---|---|---|---|---|
| G1 | 1040130 | Yes | Female | Young | Raw |
| G10 | 953230 | No | Male | Adult | Processed |
| G11 | 644640 | Yes | Female | Adult | Raw |
| G12 | 764420 | No | Female | Young | Processed |
| G13 | 744580 | Yes | Male | Adult | Raw |
| G14 | 714650 | Yes | Female | Young | Processed |
| G4 | 751440 | No | Female | Adult | Processed |
| G5 | 607410 | No | Female | Adult | Processed |
| G8 | 993870 | No | Female | Adult | Processed |
| G9 | 389070 | Yes | Male | Adult | Raw |
| Gene | Coverage (%) | Identity (%) | Database | Function | Resistance |
|---|---|---|---|---|---|
| C. perfringens Antibiotic Resistance Genes | |||||
| ErmG | 100 | 99.05 | ARG card | rRNA methyltransferase protects the ribosome from antibiotic binding | Lincosamides, Macrolides, Streptogramins |
| ErmQ | 100 | 100 | ARG card | Confers MLSb phenotype via ribosomal methylation | Lincosamides, Macrolides, Streptogramins |
| C. perfringens Virulence Genes | |||||
| pfoA | 100 | 98.74 | VG vfdb | Perfringolysin O (theta-toxin); pore-forming cytolysin | |
| Plc | 100 | 99.33 | VG vfdb | Phospholipase C (alpha-toxin) disrupts host membranes | |
| nagI | 94.5 | 98.72 | VG vfdb | Hyaluronidase (mu-toxin) degrades connective tissue | |
| nanJ | 97.56 | 97.64 | VG vfdb | Exo-alpha-sialidase; cleaves sialic acids for colonization | |
| colA | 100 | 98.7 | VG vfdb | Collagenase (kappa-toxin) breaks down collagen |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pimienta, M.; Florez-Rios, H.; Patiño-Montoya, A.; Florez, A.; Mejia, L.; Sedano, R.; Castillo, A. Applied Metagenomic Profiling of Domestic Cat Feces from Cali, Colombia: An Exploratory Approach. Appl. Microbiol. 2025, 5, 67. https://doi.org/10.3390/applmicrobiol5030067
Pimienta M, Florez-Rios H, Patiño-Montoya A, Florez A, Mejia L, Sedano R, Castillo A. Applied Metagenomic Profiling of Domestic Cat Feces from Cali, Colombia: An Exploratory Approach. Applied Microbiology. 2025; 5(3):67. https://doi.org/10.3390/applmicrobiol5030067
Chicago/Turabian StylePimienta, Monica, Hernan Florez-Rios, Angie Patiño-Montoya, Anyelo Florez, Lizeth Mejia, Raul Sedano, and Andres Castillo. 2025. "Applied Metagenomic Profiling of Domestic Cat Feces from Cali, Colombia: An Exploratory Approach" Applied Microbiology 5, no. 3: 67. https://doi.org/10.3390/applmicrobiol5030067
APA StylePimienta, M., Florez-Rios, H., Patiño-Montoya, A., Florez, A., Mejia, L., Sedano, R., & Castillo, A. (2025). Applied Metagenomic Profiling of Domestic Cat Feces from Cali, Colombia: An Exploratory Approach. Applied Microbiology, 5(3), 67. https://doi.org/10.3390/applmicrobiol5030067