


Novel Epigallocatechin Gallate (EGCG) Analogs with Improved Biochemical Properties for Targeting Extracellular and Intracellular Staphylococcus aureus


Abstract
1. Introduction
2. Materials and Methods
2.1. Compounds and Antibiotics
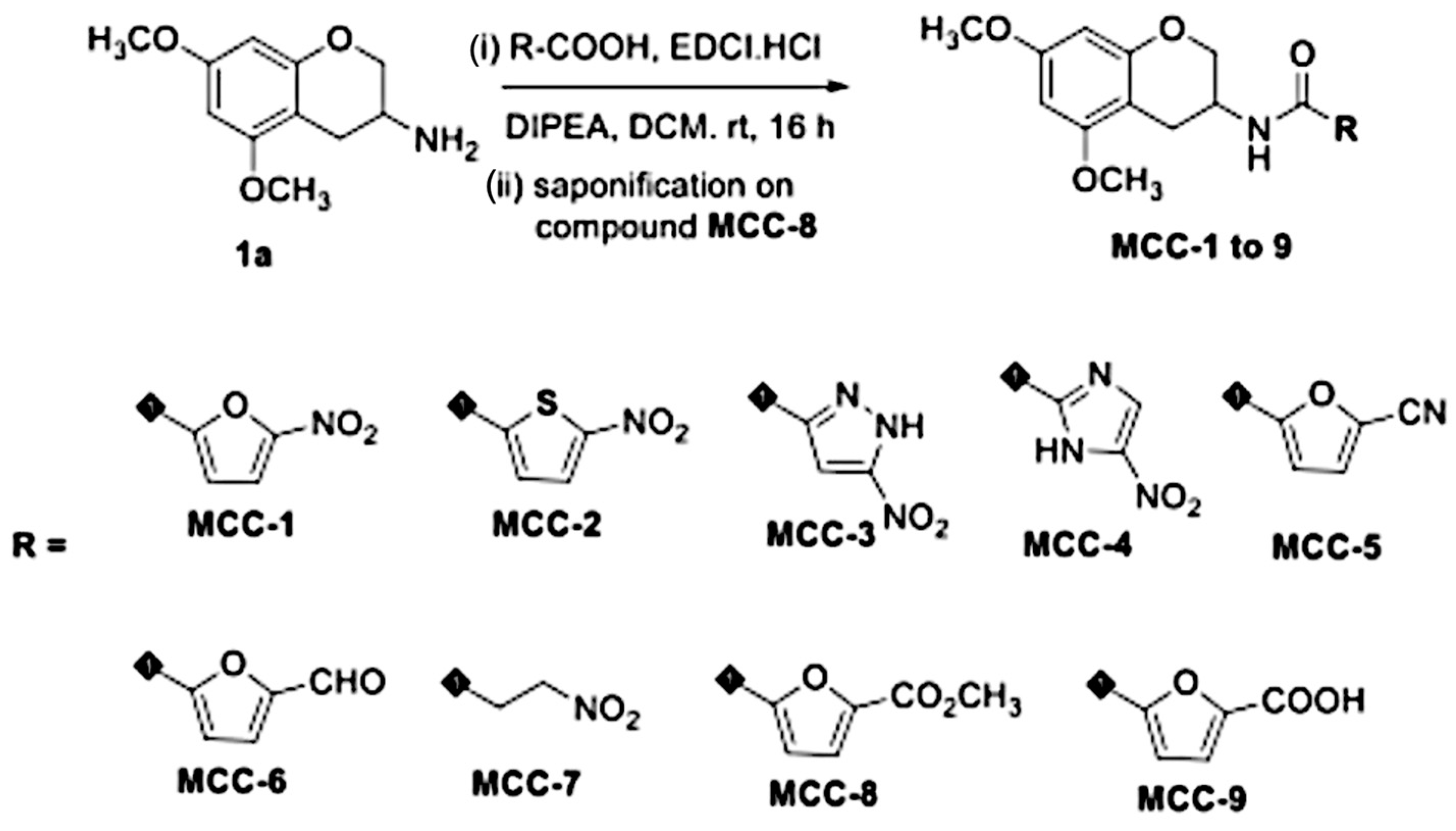
2.2. Synthesis and Evaluation of the Chemical Properties of EGCG Analogs
2.3. Bacterial Strains
2.4. Antimicrobial Susceptibility Testing
2.5. Intracellular Killing Assay
3. Results
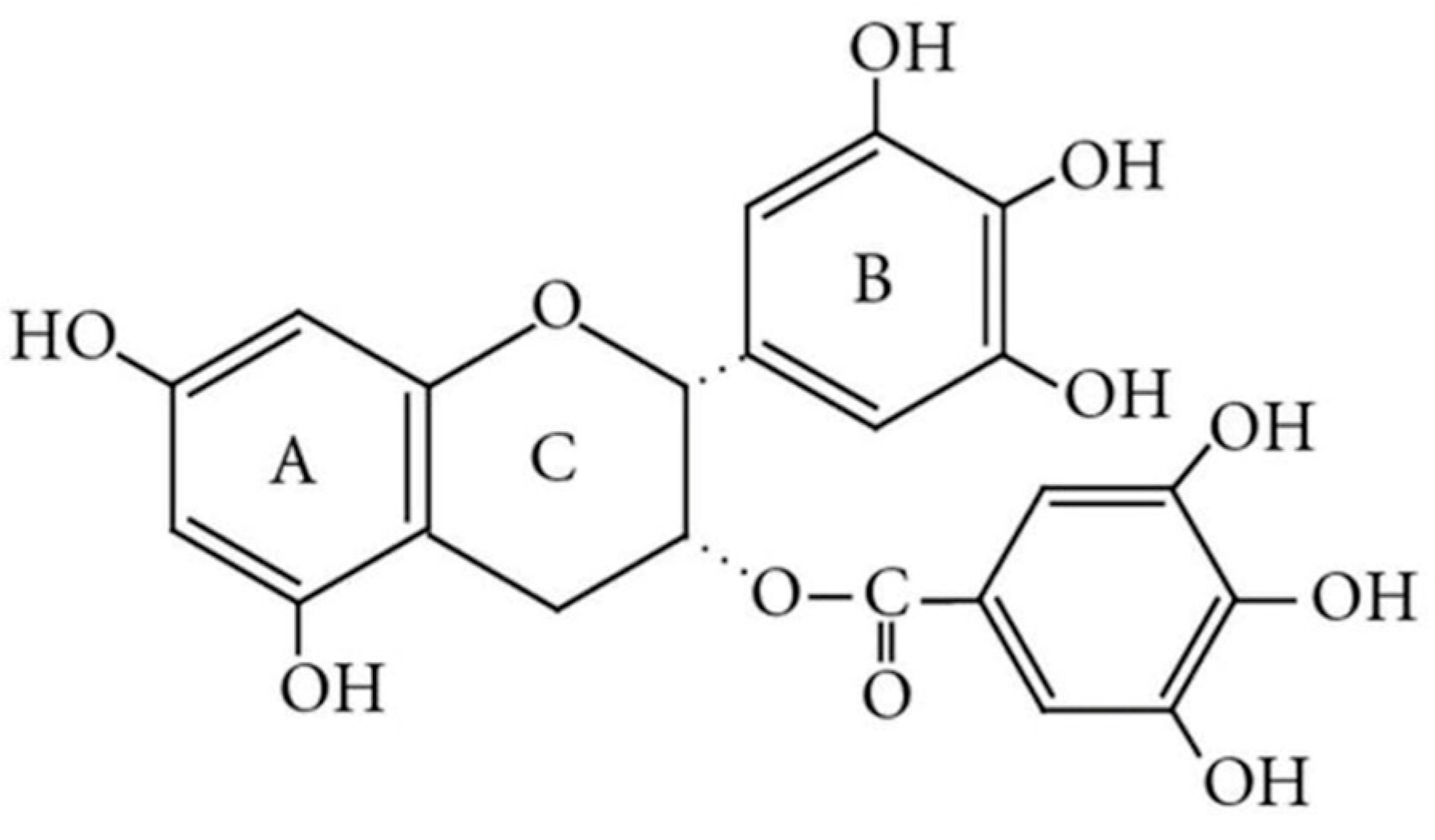
3.1. Preliminary Screening and Selection of EGCG Analogs
3.2. EGCG-Lead Analog MCC-1 Exhibited More Favorable ADME (Absorption, Distribution, Metabolism, and Excretion) Properties than That of EGCG In Vitro
3.3. EGCG Analogs Demonstrate Activity Alone Against Both Clinical and Reference Strains of MRSA and MSSA
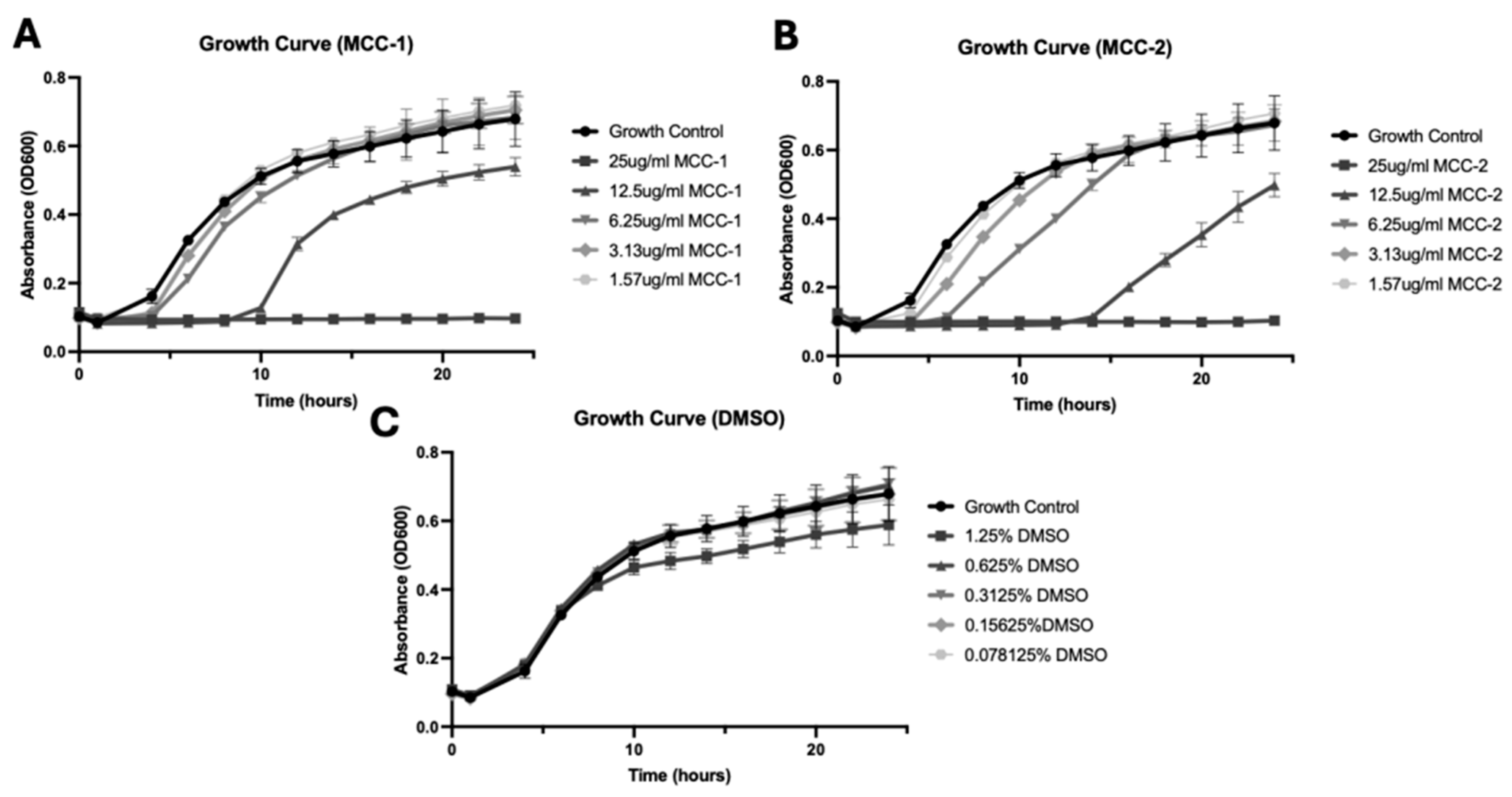
3.4. MCC-1 and MCC-2 Exhibit Dose-Dependent Growth Inhibition of S. aureus
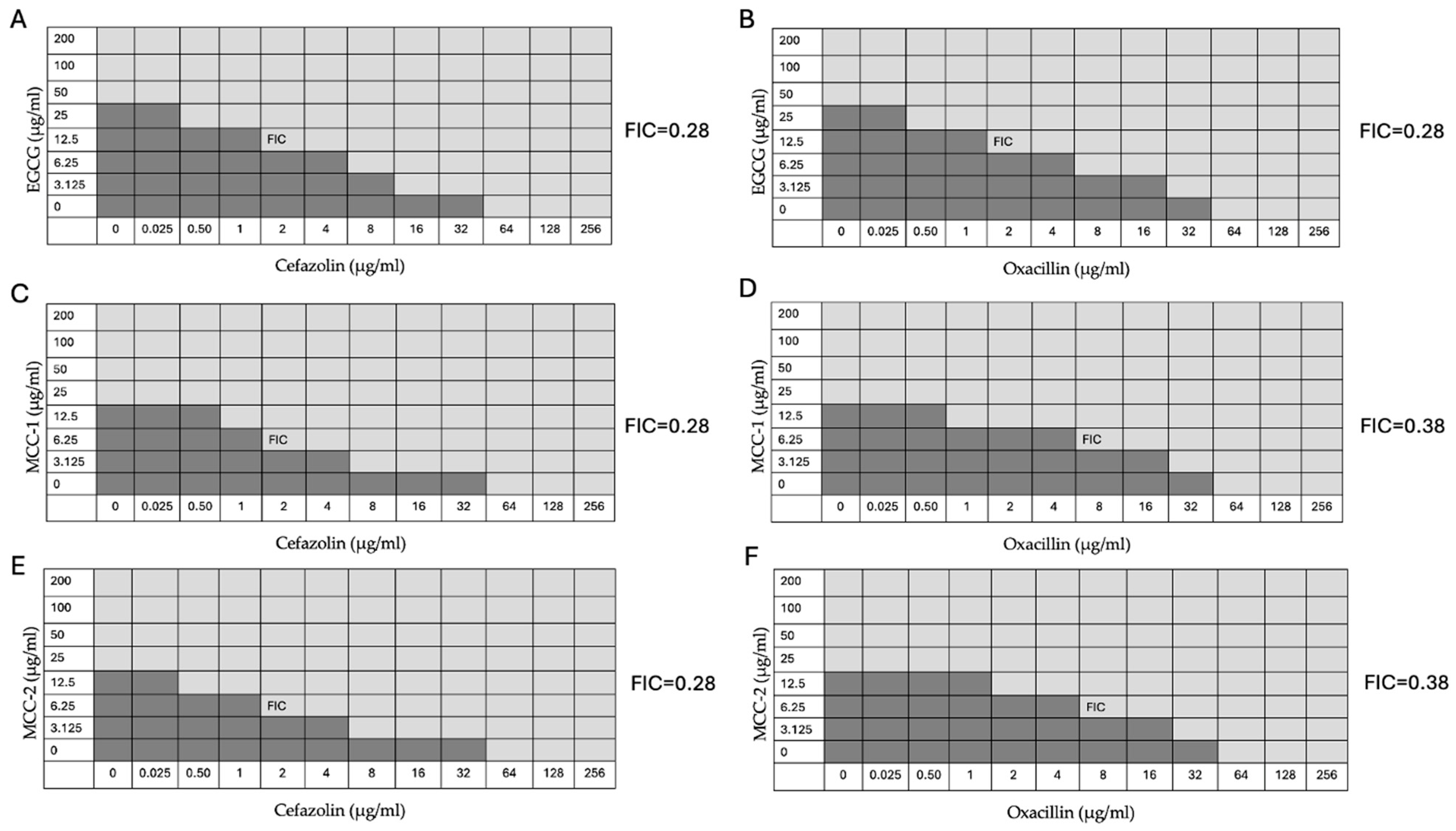
3.5. EGCG, MCC-1, and MCC-2 Restore Susceptibility of MRSA to β-lactam Antibiotics
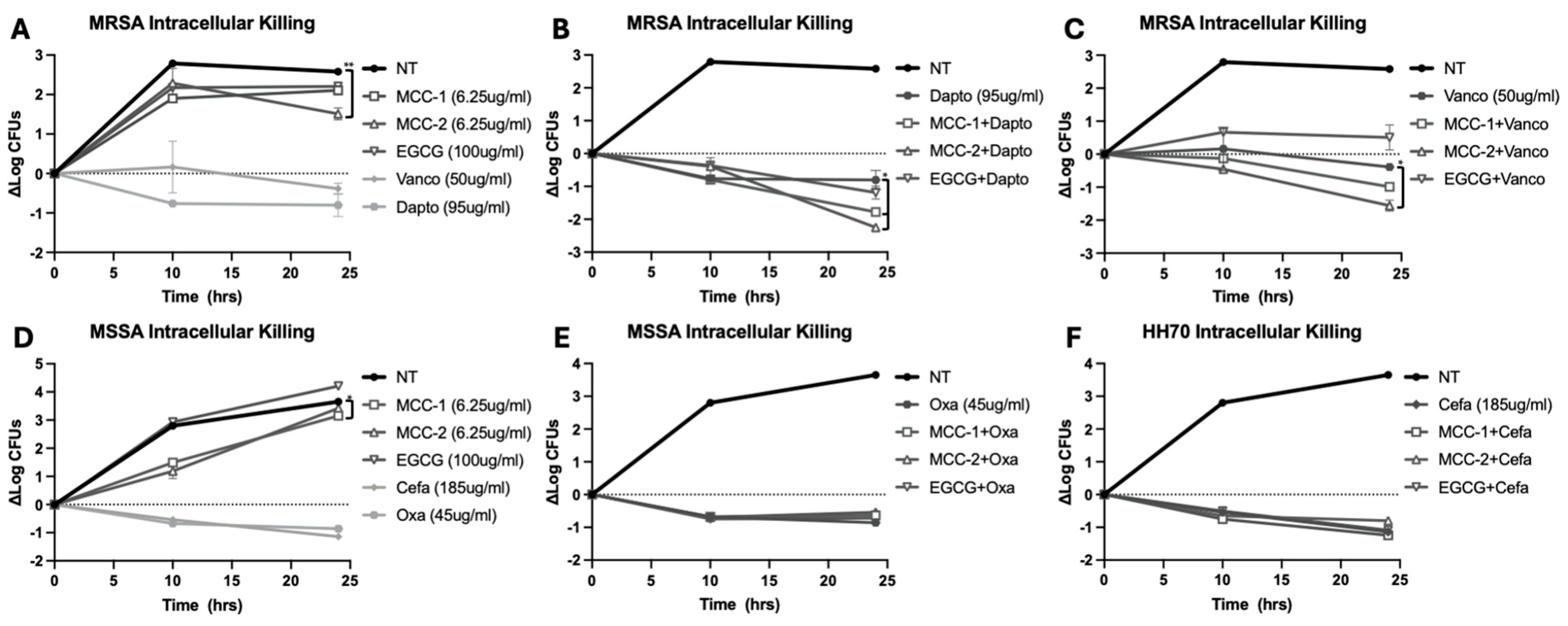
3.6. MCC-1 and MCC-2 at Subinhibitory Concentrations Decrease Intracellular S. aureus Alone and in Combination with Antibiotics
4. Discussion
5. Patents
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Naber, C.K. Staphylococcus aureus Bacteremia: Epidemiology, Pathophysiology, and Management Strategies. Clin. Infect. Dis. 2009, 48, S231–S237. [Google Scholar] [CrossRef] [PubMed]
- Ikuta, K.S.; Swetschinski, L.R.; Robles Aguilar, G.; Sharara, F.; Mestrovic, T.; Gray, A.P.; Davis Weaver, N.; Wool, E.E.; Han, C.; Gershberg Hayoon, A.; et al. Global Mortality Associated with 33 Bacterial Pathogens in 2019: A Systematic Analysis for the Global Burden of Disease Study 2019. Lancet 2022, 400, 2221–2248. [Google Scholar] [CrossRef] [PubMed]
- Holland, T.L.; Bayer, A.S.; Fowler, V.G., Jr. Persistent Methicillin-Resistant Staphylococcus aureus Bacteremia: Resetting the Clock for Optimal Management. Clin. Infect. Dis. 2022, 75, 1668–1674. [Google Scholar] [CrossRef]
- van Hal, S.J.; Jensen, S.O.; Vaska, V.L.; Espedido, B.A.; Paterson, D.L.; Gosbell, I.B. Predictors of Mortality in Staphylococcus aureus Bacteremia. Clin. Microbiol. Rev. 2012, 25, 362–386. [Google Scholar] [CrossRef]
- Minejima, E.; Mai, N.; Bui, N.; Mert, M.; Mack, W.J.; She, R.C.; Nieberg, P.; Spellberg, B.; Wong-Beringer, A. Defining the Breakpoint Duration of Staphylococcus aureus Bacteremia Predictive of Poor Outcomes. Clin. Infect. Dis. 2020, 70, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Minejima, E.; Bensman, J.; She, R.C.; Mack, W.J.; Tuan Tran, M.; Ny, P.; Lou, M.; Yamaki, J.; Nieberg, P.; Ho, J.; et al. A Dysregulated Balance of Proinflammatory and Anti-Inflammatory Host Cytokine Response Early During Therapy Predicts Persistence and Mortality in Staphylococcus aureus Bacteremia. Crit. Care Med. 2016, 44, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Surewaard, B.G.J.; Deniset, J.F.; Zemp, F.J.; Amrein, M.; Otto, M.; Conly, J.; Omri, A.; Yates, R.M.; Kubes, P. Identification and Treatment of the Staphylococcus aureus Reservoir in Vivo. J. Exp. Med. 2016, 213, 1141–1151. [Google Scholar] [CrossRef]
- Pidwill, G.R.; Gibson, J.F.; Cole, J.; Renshaw, S.A.; Foster, S.J. The Role of Macrophages in Staphylococcus aureus Infection. Front. Immunol. 2021, 11, 620339. [Google Scholar] [CrossRef]
- Beadell, B.; Yamauchi, J.; Wong-Beringer, A. Comparative in Vitro Efficacy of Antibiotics against the Intracellular Reservoir of Staphylococcus aureus. J. Antimicrob. Chemother. 2024, 79, dkae241. [Google Scholar] [CrossRef]
- Chacko, S.M.; Thambi, P.T.; Kuttan, R.; Nishigaki, I. Beneficial Effects of Green Tea: A Literature Review. Chin. Med. 2010, 5, 13. [Google Scholar] [CrossRef]
- Zhao, W.-H.; Hu, Z.-Q.; Okubo, S.; Hara, Y.; Shimamura, T. Mechanism of Synergy between Epigallocatechin Gallate and β-Lactams against Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2001, 45, 1737–1742. [Google Scholar] [CrossRef]
- Kitichalermkiat, A.; Katsuki, M.; Sato, J.; Sonoda, T.; Masuda, Y.; Honjoh, K.-I.; Miyamoto, T. Effect of Epigallocatechin Gallate on Gene Expression of Staphylococcus aureus. J. Glob. Antimicrob. Resist. 2020, 22, 854–859. [Google Scholar] [CrossRef]
- Liu, C.; Hao, K.; Liu, Z.; Liu, Z.; Guo, N. Epigallocatechin Gallate (EGCG) Attenuates Staphylococcal Alpha-Hemolysin (Hla)-Induced NLRP3 Inflammasome Activation via ROS-MAPK Pathways and EGCG-Hla Interactions. Int. Immunopharmacol. 2021, 100, 108170. [Google Scholar] [CrossRef]
- Warden, B.A.; Smith, L.S.; Beecher, G.R.; Balentine, D.A.; Clevidence, B.A. Catechins Are Bioavailable in Men and Women Drinking Black Tea throughout the Day. J. Nutr. 2001, 131, 1731–1737. [Google Scholar] [CrossRef]
- Chow, H.H.; Cai, Y.; Alberts, D.S.; Hakim, I.; Dorr, R.; Shahi, F.; Crowell, J.A.; Yang, C.S.; Hara, Y. Phase I Pharmacokinetic Study of Tea Polyphenols Following Single-Dose Administration of Epigallocatechin Gallate and Polyphenon E. Cancer Epidemiol. Biomark. Prev. 2001, 10, 53–58. [Google Scholar]
- Chow, H.-H.S.; Cai, Y.; Hakim, I.A.; Crowell, J.A.; Shahi, F.; Brooks, C.A.; Dorr, R.T.; Hara, Y.; Alberts, D.S. Pharmacokinetics and Safety of Green Tea Polyphenols after Multiple-Dose Administration of Epigallocatechin Gallate and Polyphenon E in Healthy Individuals. Clin. Cancer Res. 2003, 9, 3312–3319. [Google Scholar]
- Ullmann, U.; Haller, J.; Decourt, J.; Girault, N.; Girault, J.; Richard-Caudron, A.; Pineau, B.; Weber, P. A Single Ascending Dose Study of Epigallocatechin Gallate in Healthy Volunteers. J. Int. Med. Res. 2003, 31, 88–101. [Google Scholar] [CrossRef]
- Shinde, S.; Lee, L.H.; Chu, T. Inhibition of Biofilm Formation by the Synergistic Action of EGCG-S and Antibiotics. Antibiotics 2021, 10, 102. [Google Scholar] [CrossRef]
- Zhao, J.; Qian, D.; Zhang, L.; Wang, X.; Zhang, J. Improved Antioxidative and Antibacterial Activity of Epigallocatechin Gallate Derivative Complexed by Zinc Cations and Chitosan. RSC Adv. 2024, 14, 10410–10415. [Google Scholar] [CrossRef]
- Wang, C.; Xiao, R.; Yang, Q.; Pan, J.; Cui, P.; Zhou, S.; Qiu, L.; Zhang, Y.; Wang, J. Green Synthesis of Epigallocatechin Gallate-Ferric Complex Nanoparticles for Photothermal Enhanced Antibacterial and Wound Healing. Biomed. Pharmacother. 2024, 171, 116175. [Google Scholar] [CrossRef] [PubMed]
- Tyzack, J.D.; Hunt, P.A.; Segall, M.D. Predicting Regioselectivity and Lability of Cytochrome P450 Metabolism Using Quantum Mechanical Simulations. J. Chem. Inf. Model. 2016, 56, 2180–2193. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, A.; Hall, J.; Blagg, B.S.J. Synthesis and Structure Activity Relationships of EGCG Analogues, A Recently Identified Hsp90 Inhibitor. J. Org. Chem. 2013, 78, 7859–7884. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhang, S.-Y.; He, G.; Ai, Z.; Nack, W.A.; Chen, G. Copper-Catalyzed Carboxamide-Directed Ortho Amination of Anilines with Alkylamines at Room Temperature. Org. Lett. 2014, 16, 1764–1767. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.K.; Mondal, N.B.; Banerjee, S.; Mazumder, U.K. Determination of Drug-like Properties of a Novel Antileishmanial Compound: In Vitro Absorption, Distribution, Metabolism, and Excretion Studies. Indian. J. Pharmacol. 2009, 41, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Pasha, M.K.; Jayaraman, R.; Reddy, V.P.; Yeo, P.; Goh, E.; Williams, A.; Goh, K.C.; Kantharaj, E. Preclinical Metabolism and Pharmacokinetics of SB1317 (TG02), a Potent CDK/JAK2/FLT3 Inhibitor. Drug Metab. Lett. 2012, 6, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Hubatsch, I.; Ragnarsson, E.G.E.; Artursson, P. Determination of Drug Permeability and Prediction of Drug Absorption in Caco-2 Monolayers. Nat. Protoc. 2007, 2, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- Banker, M.J.; Clark, T.H.; Williams, J.A. Development and Validation of a 96-Well Equilibrium Dialysis Apparatus for Measuring Plasma Protein Binding. J. Pharm. Sci. 2003, 92, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Davies, B.; Morris, T. Physiological Parameters in Laboratory Animals and Humans. Pharm. Res 1993, 10, 1093–1095. [Google Scholar] [CrossRef] [PubMed]
- Barter, Z.E.; Bayliss, M.K.; Beaune, P.H.; Boobis, A.R.; Carlile, D.J.; Edwards, R.J.; Houston, J.B.; Lake, B.G.; Lipscomb, J.C.; Pelkonen, O.R.; et al. Scaling Factors for the Extrapolation of in Vivo Metabolic Drug Clearance from in Vitro Data: Reaching a Consensus on Values of Human Microsomal Protein and Hepatocellularity per Gram of Liver. Curr. Drug Metab. 2007, 8, 33–45. [Google Scholar] [CrossRef]
- EM100 Connect—CLSI M100 ED34:2024. Available online: https://em100.edaptivedocs.net/GetDoc.aspx?doc=CLSI%20M100%20ED34:2024&scope=user (accessed on 10 September 2024).
- Bellio, P.; Fagnani, L.; Nazzicone, L.; Celenza, G. New and Simplified Method for Drug Combination Studies by Checkerboard Assay. MethodsX 2021, 8, 101543. [Google Scholar] [CrossRef]
- Meletiadis, J.; Pournaras, S.; Roilides, E.; Walsh, T.J. Defining Fractional Inhibitory Concentration Index Cutoffs for Additive Interactions Based on Self-Drug Additive Combinations, Monte Carlo Simulation Analysis, and In Vitro-In Vivo Correlation Data for Antifungal Drug Combinations against Aspergillus fumigatus. Antimicrob. Agents Chemother. 2010, 54, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Peyrusson, F.; Nguyen, T.K.; Buyck, J.M.; Lemaire, S.; Wang, G.; Seral, C.; Tulkens, P.M.; Van Bambeke, F. In Vitro Models for the Study of the Intracellular Activity of Antibiotics. Methods Mol. Biol. 2021, 2357, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Backert, S.; Hofreuter, D. Molecular Methods to Investigate Adhesion, Transmigration, Invasion and Intracellular Survival of the Foodborne Pathogen Campylobacter Jejuni. J. Microbiol. Methods 2013, 95, 8–23. [Google Scholar] [CrossRef]
- Sharma, A.; Puhar, A. Gentamicin Protection Assay to Determine the Number of Intracellular Bacteria during Infection of Human TC7 Intestinal Epithelial Cells by Shigella Flexneri. Bio Protoc. 2019, 9, e3292. [Google Scholar] [CrossRef]
- Yoda, Y.; Hu, Z.-Q.; Shimamura, T.; Zhao, W.-H. Different Susceptibilities of Staphylococcus and Gram-Negative Rods to Epigallocatechin Gallate. J. Infect. Chemother. 2004, 10, 55–58. [Google Scholar] [CrossRef]
- Brown, S.; Xia, G.; Luhachack, L.G.; Campbell, J.; Meredith, T.C.; Chen, C.; Winstel, V.; Gekeler, C.; Irazoqui, J.E.; Peschel, A.; et al. Methicillin Resistance in Staphylococcus aureus Requires Glycosylated Wall Teichoic Acids. Proc. Natl. Acad. Sci. USA 2012, 109, 18909–18914. [Google Scholar] [CrossRef]
- Yang, Y.; Han, X.; Chen, Y.; Wu, J.; Li, M.; Yang, H.; Xu, W.; Wei, L. EGCG Induces Pro-Inflammatory Response in Macrophages to Prevent Bacterial Infection through the 67LR/P38/JNK Signaling Pathway. J. Agric. Food Chem. 2021, 69, 5638–5651. [Google Scholar] [CrossRef]
- Trübe, P.; Hertlein, T.; Mrochen, D.M.; Schulz, D.; Jorde, I.; Krause, B.; Zeun, J.; Fischer, S.; Wolf, S.A.; Walther, B.; et al. Bringing Together What Belongs Together: Optimizing Murine Infection Models by Using Mouse-Adapted Staphylococcus aureus Strains. Int. J. Med. Microbiol. 2019, 309, 26–38. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EGCG | 0.65 |
| MCC-1 | 2.93 |
| MCC-2 | 3.18 |
| MCC-3 | 2.56 |
| MCC-4 | 2.57 |
| MCC-5 | 2.57 |
| MCC-6 | 2.50 |
| MCC-7 | 1.94 |
| MCC-8 | 2.69 |
| MCC-9 | 1.95 |
| MCC-10 | 3.58 |
| In Vitro ADME Assay | Target Values | EGCG | MCC-1 | MCC-2 |
|---|---|---|---|---|
| LogD | 2–4 | >4 | 2.67 | 3.64 |
| PPB/Bound | <95% | 92.4 ± 3.1 | 86.90 | ND |
| Papp (Caco2) 10−6 cm/s | ≤1 | 0.13 | 0.91 | 0.89 |
| Sol. (µM) | >10 µM | >300 | 16.89 | 2.88 |
| HLM t½min | >30 min | ND | 17.43 | 14.54 |
| MLM t½min | >30 min | ND | <4.51 | <4.51 |
| USA300 (MRSA) | HH35 (MRSA) | HH37 (MRSA) | HH70 (MSSA) | LAC164 (MSSA) | |
|---|---|---|---|---|---|
| EGCG | 50 | 100 | 50 | 50 | 50 |
| MCC-1 | 25 | 50 | 25 | 25 | 25 |
| MCC-2 | 25 | 50 | 25 | 25 | 25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grosso, R.; Nguyen, V.; Ahmed, S.K.; Wong-Beringer, A. Novel Epigallocatechin Gallate (EGCG) Analogs with Improved Biochemical Properties for Targeting Extracellular and Intracellular Staphylococcus aureus. Appl. Microbiol. 2024, 4, 1568-1581. https://doi.org/10.3390/applmicrobiol4040107
Grosso R, Nguyen V, Ahmed SK, Wong-Beringer A. Novel Epigallocatechin Gallate (EGCG) Analogs with Improved Biochemical Properties for Targeting Extracellular and Intracellular Staphylococcus aureus. Applied Microbiology. 2024; 4(4):1568-1581. https://doi.org/10.3390/applmicrobiol4040107
Chicago/Turabian StyleGrosso, Riley, Vy Nguyen, Syed Kaleem Ahmed, and Annie Wong-Beringer. 2024. "Novel Epigallocatechin Gallate (EGCG) Analogs with Improved Biochemical Properties for Targeting Extracellular and Intracellular Staphylococcus aureus" Applied Microbiology 4, no. 4: 1568-1581. https://doi.org/10.3390/applmicrobiol4040107
APA StyleGrosso, R., Nguyen, V., Ahmed, S. K., & Wong-Beringer, A. (2024). Novel Epigallocatechin Gallate (EGCG) Analogs with Improved Biochemical Properties for Targeting Extracellular and Intracellular Staphylococcus aureus. Applied Microbiology, 4(4), 1568-1581. https://doi.org/10.3390/applmicrobiol4040107