
The Impacts of Field Management on Soil and Tea Root Microbiomes


Abstract
1. Introduction
2. Materials and Methods
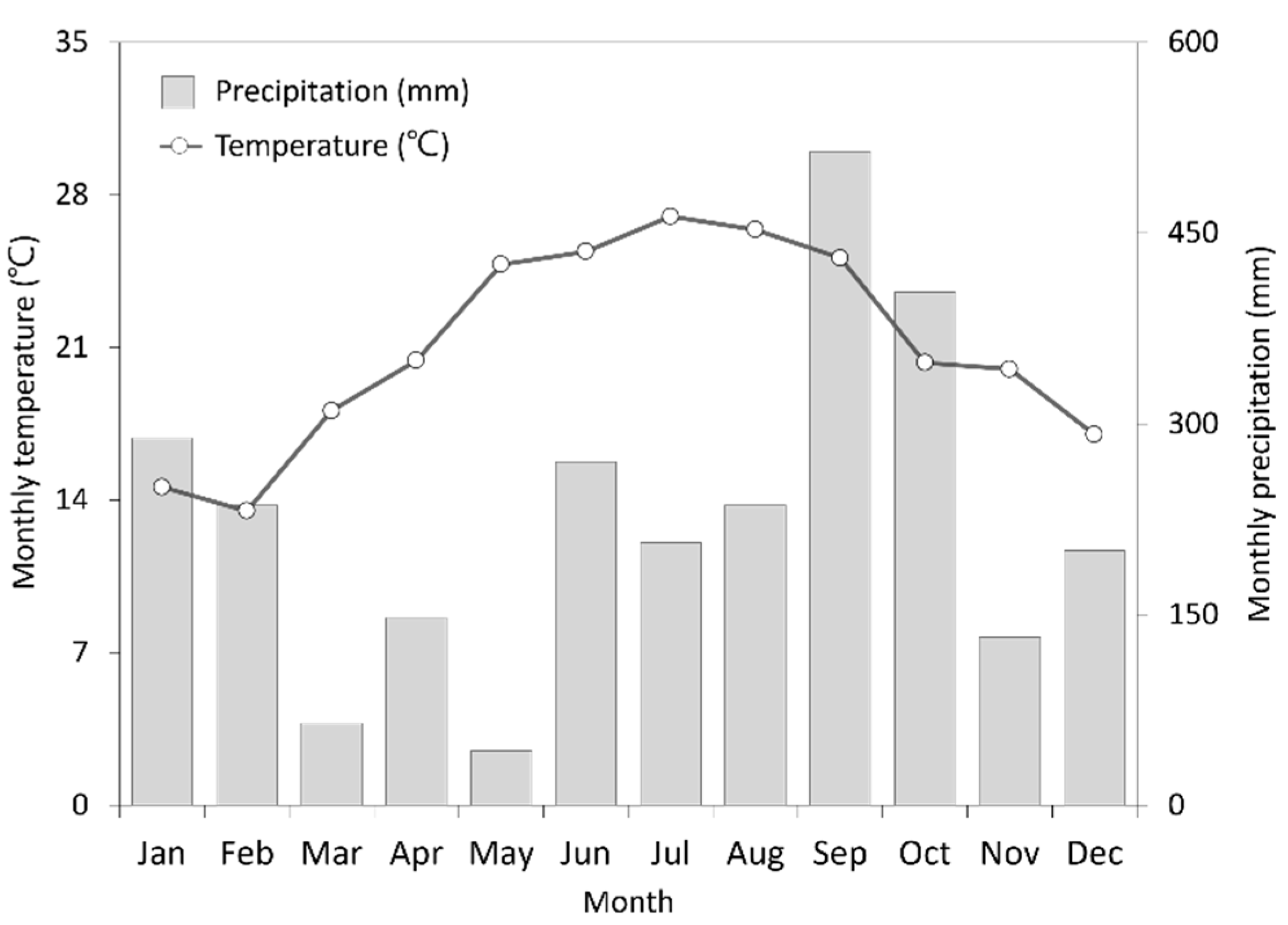
2.1. Experimental Sites
2.2. Sample Collection and Microbial DNA Isolation
2.3. 16S rRNA Gene Library Preparation and Sequencing
2.4. Data Analysis
2.5. Statistical Analysis
3. Results
3.1. Summary of 16S rRNA Amplicon Sequencing
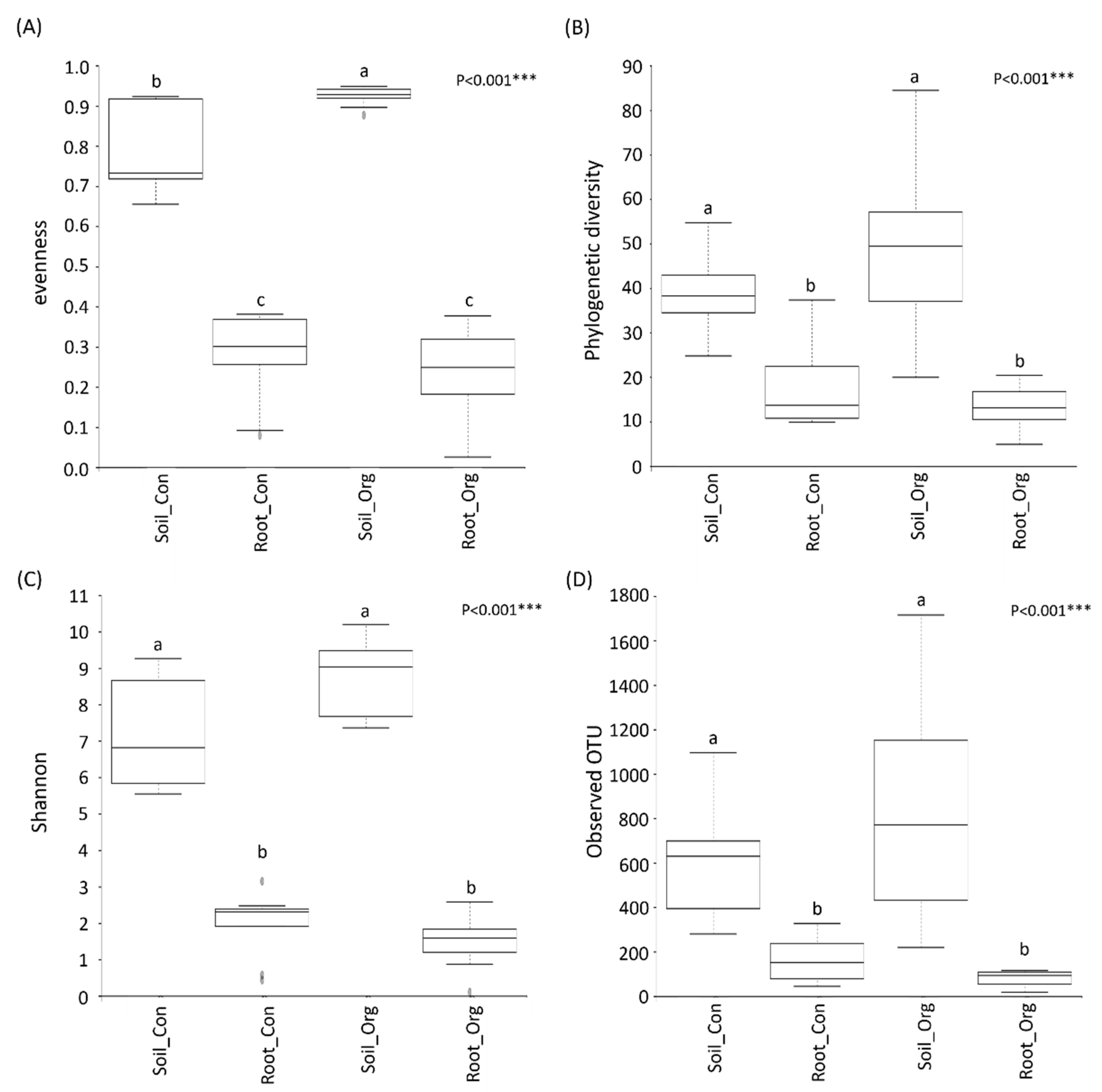
3.2. The Effects of Cropping System on Soil and Root Bacterial Diversity
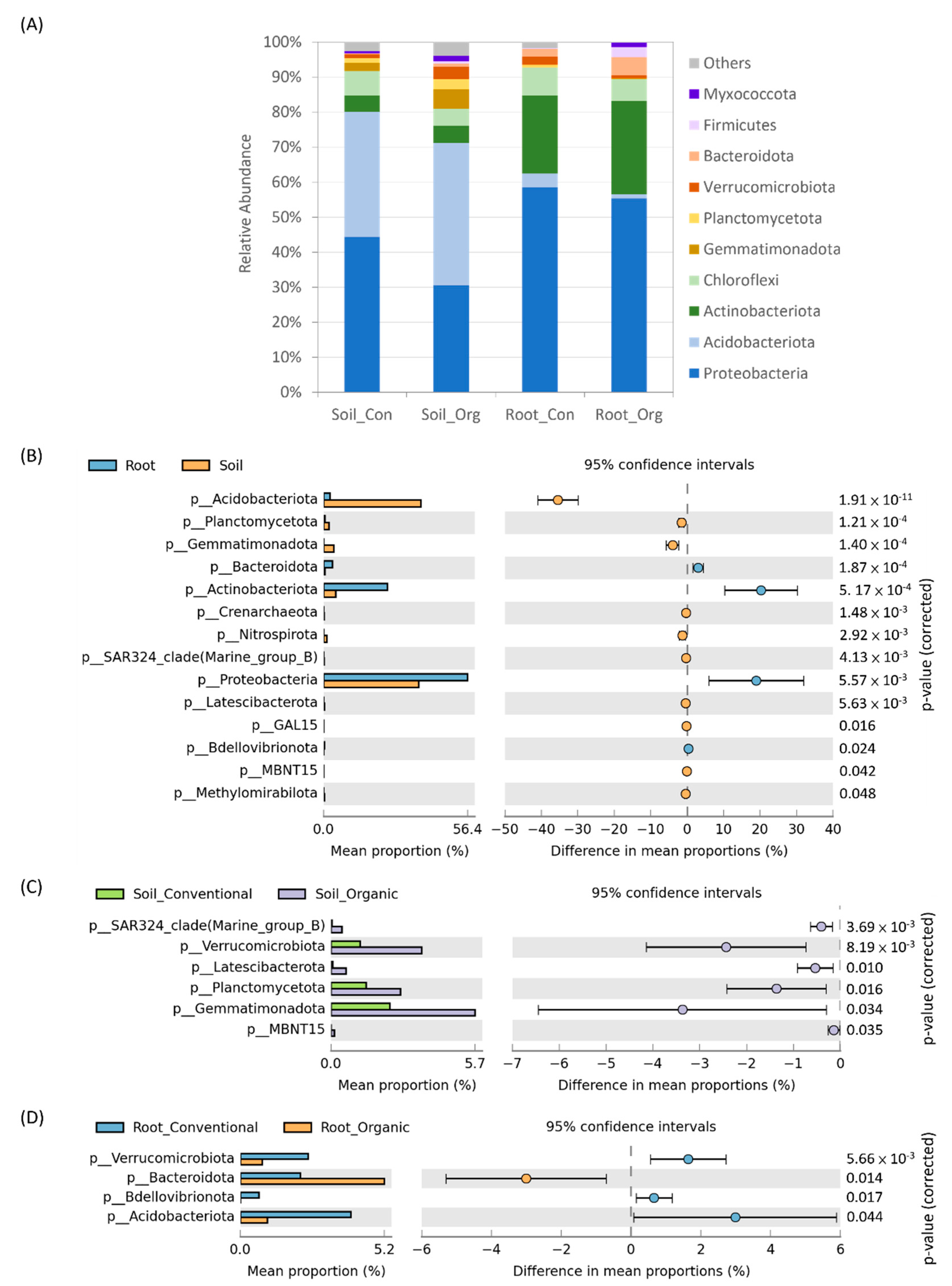
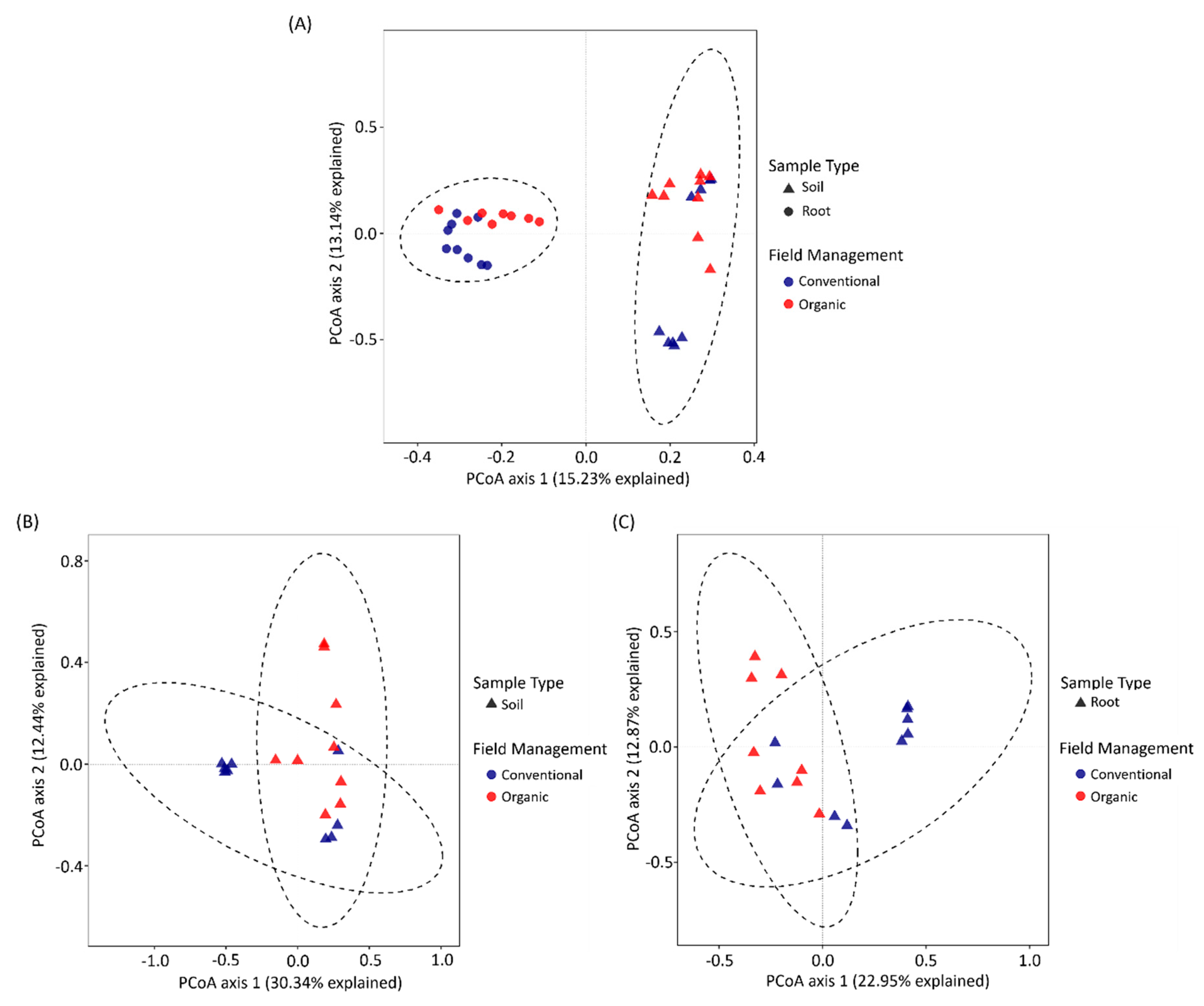
3.3. The Difference between Soil and Tea Root Bacterial Community Structure
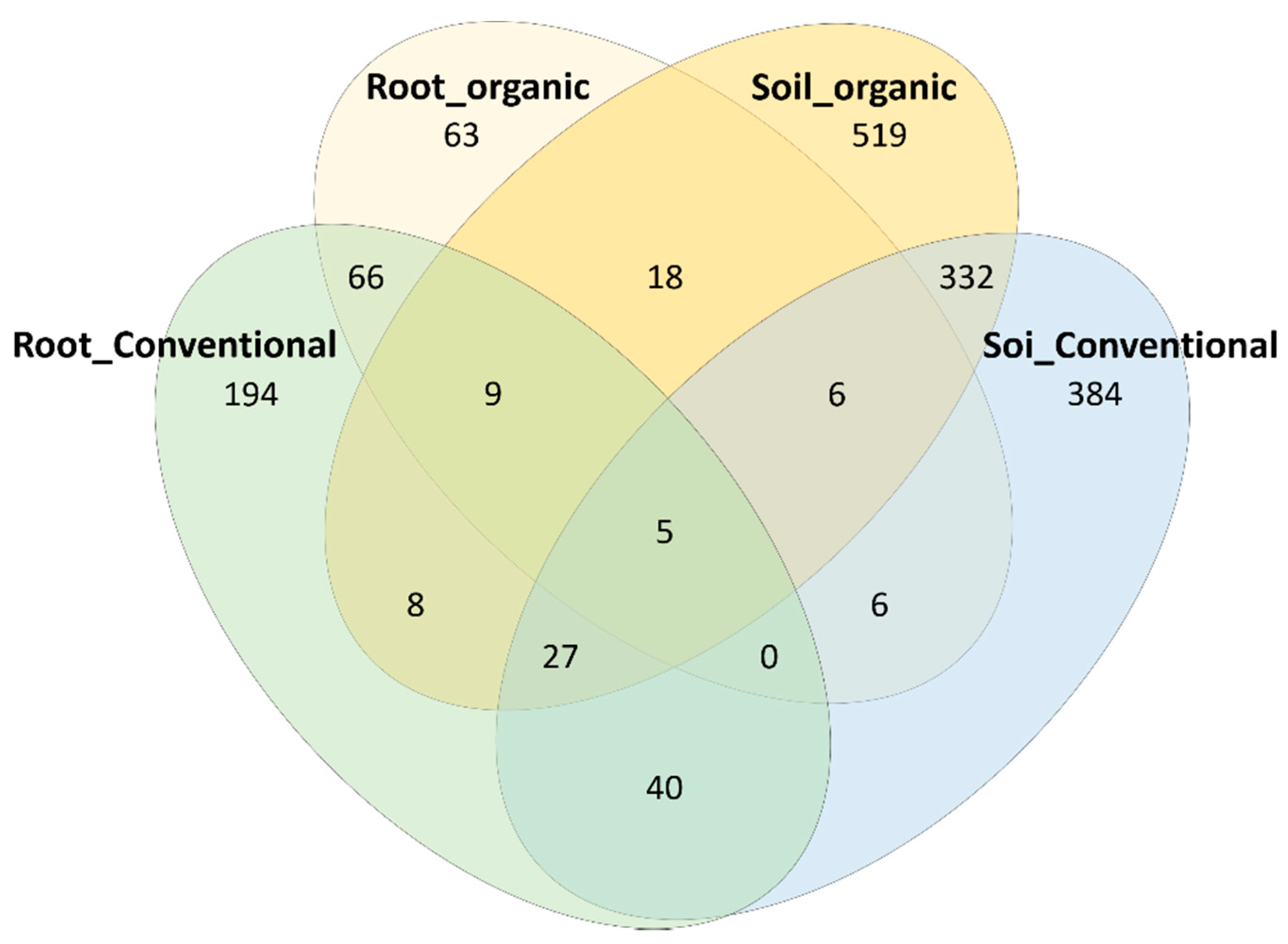
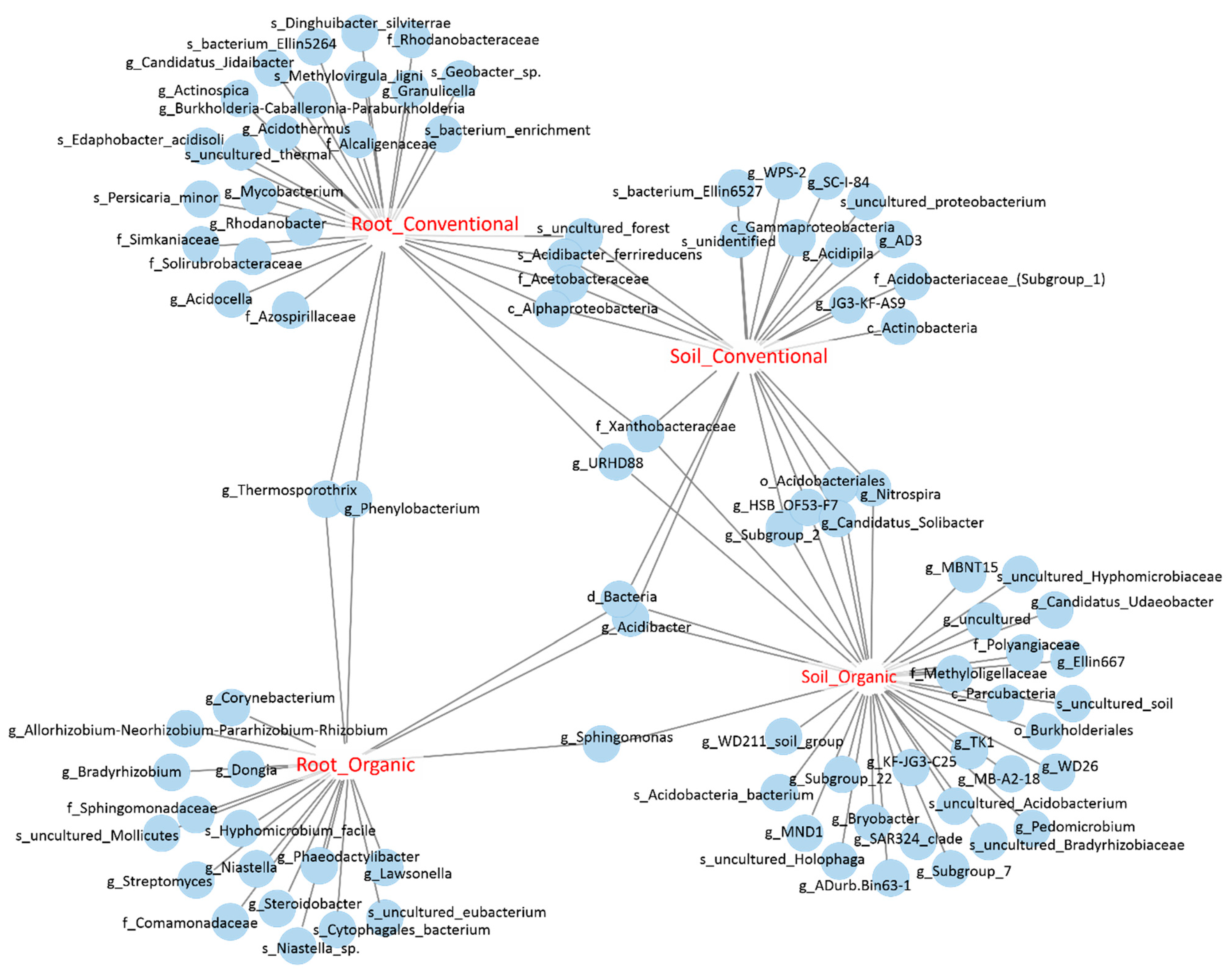
3.4. Identification of OTUs That Are Sensitive to Field Management and Growth Environments
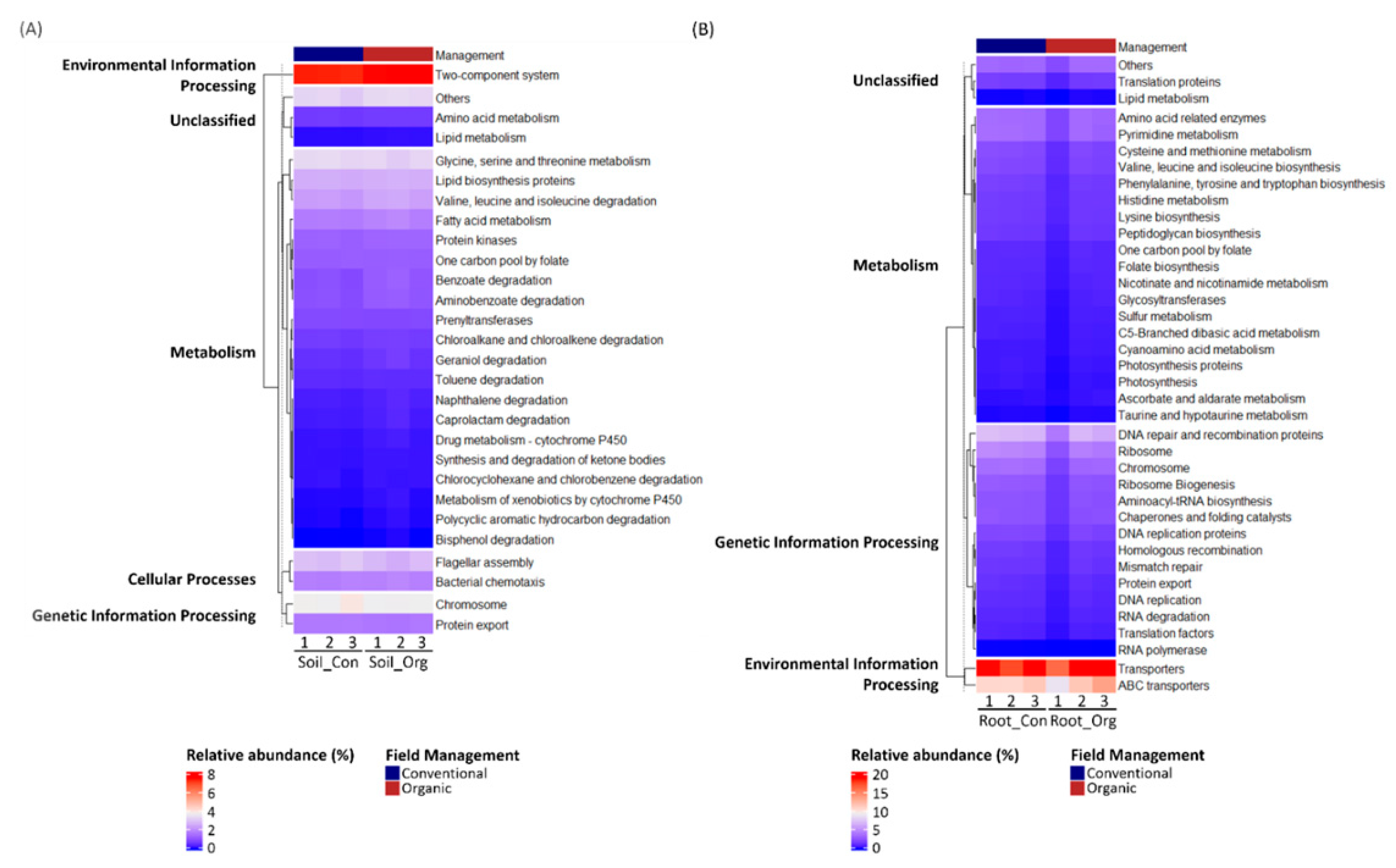
3.5. Functional Prediction of Bacterial Communities
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tilman, D. Global environmental impacts of agricultural expansion: The need for sustainable and efficient practices. Proc. Natl. Acad. Sci. USA 1999, 96, 5995–6000. [Google Scholar] [CrossRef]
- Pimentel, D.; Harvey, C.; Resosudarmo, P.; Sinclair, K.; Kurz, D.; McNair, M.; Crist, S.; Shpritz, L.; Fitton, L.; Saffouri, R.; et al. Environmental and Economic Costs of Soil Erosion and Conservation Benefits. Science 1995, 267, 1117–1123. [Google Scholar] [CrossRef]
- Pimentel, D.; Hepperly, P.; Hanson, J.; Douds, D.; Seidel, R. Environmental, Energetic, and Economic Comparisons of Organic and Conventional Farming Systems. Bioscience 2005, 55, 573–582. [Google Scholar] [CrossRef]
- Ishaq, S.; Johnson, S.P.; Miller, Z.J.; Lehnhoff, E.A.; Olivo, S.; Yeoman, C.J.; Menalled, F.D. Impact of Cropping Systems, Soil Inoculum, and Plant Species Identity on Soil Bacterial Community Structure. Microb. Ecol. 2016, 73, 417–434. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; Xu, F.; Song, T.; Du, H.; Gui, Y.; Xu, M.; Cao, Y.; Dang, X.; Rensing, C.; et al. Combining Irrigation Scheme and Phosphorous Application Levels for Grain Yield and Their Impacts on Rhizosphere Microbial Communities of Two Rice Varieties in a Field Trial. J. Agric. Food Chem. 2019, 67, 10577–10586. [Google Scholar] [CrossRef]
- Novara, A.; Catania, V.; Tolone, M.; Gristina, L.; Laudicina, V.A.; Quatrini, P. Cover Crop Impact on Soil Organic Carbon, Nitrogen Dynamics and Microbial Diversity in a Mediterranean Semiarid Vineyard. Sustainability 2020, 12, 3256. [Google Scholar] [CrossRef]
- Benitez, M.-S.; Osborne, S.L.; Lehman, R.M. Previous Crop and Rotation History Effects On Maize Seedling Health and Associated Rhizosphere Microbiome. Sci. Rep. 2017, 7, 15709. [Google Scholar] [CrossRef]
- Morrison-Whittle, P.; Lee, S.A.; Goddard, M.R. Fungal Communities Are Differentially Affected by Conventional and Biodynamic Agricultural Management Approaches in Vineyard Ecosystems. Agric. Ecosyst. Environ. 2017, 246, 306–313. [Google Scholar] [CrossRef]
- Wemheuer, F.; Kaiser, K.; Karlovsky, P.; Daniel, R.; Vidal, S.; Wemheuer, B. Bacterial Endophyte Communities of Three Agricultural Important Grass Species Differ in Their Response Towards Management Regimes. Sci. Rep. 2017, 7, 40914. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.-Y.; Lin, C.-Y.; Chang, S.-J.; Lin, W.-Y. The Dynamics of Endophytic Bacterial Community Structure in Rice Roots under Different Field Management Systems. Agronomy 2020, 10, 1623. [Google Scholar] [CrossRef]
- Hartman, K.; Van Der Heijden, M.G.A.; Wittwer, R.A.; Banerjee, S.; Walser, J.-C.; Schlaeppi, K. Cropping Practices Manipulate Abundance Patterns of Root and Soil Microbiome Members Paving The Way to Smart Farming. Microbiome 2018, 6, 14. [Google Scholar] [CrossRef]
- Chong, K.P.; Ho, T.Y.; Jalloh, M.B. Soil Nitrogen Phosphorus and Tea Leaf Growth in Organic and Conventional Farming of Selected Fields at Sabah Tea Plantation Slope. J. Sustain. Dev. 2009, 1, 117–122. [Google Scholar] [CrossRef][Green Version]
- Han, W.-Y.; Xu, J.-M.; Wei, K.; Shi, R.-Z.; Ma, L.-F. Soil Carbon Sequestration, Plant Nutrients and Biological Activities Affected by Organic Farming System in Tea (Camellia sinensis (L.) O. Kuntze) fields. Soil Sci. Plant Nutr. 2013, 59, 727–739. [Google Scholar] [CrossRef]
- Das, S.; Borua, P.K.; Bhagat, R.M. Soil Nitrogen and Tea Leaf Properties in Organic and Conventional Farming Systems Under Humid Sub-Tropical Conditions. Org. Agric. 2016, 6, 119–132. [Google Scholar] [CrossRef]
- Ghosh, B.C.; Palit, S.; Gupta, S.D.; Swain, D.K. Studies on Tea Quality Grown Through Conventional and Organic Management Practices: Its Impact on Antioxidant and Antidiarrhoeal Activity. Trans. ASABE 2008, 51, 2227–2238. [Google Scholar] [CrossRef]
- Bagchi, A.; Ch, B.; Ghosh, R.; Swain, D.K.; Bera, N. Organic Farming Practice for Quality Improvement of Tea and Its Anti Parkinsonism Effect on Health Defense. J. Phys. Chem. Biophys. 2015, 5, 178. [Google Scholar] [CrossRef]
- Qiu, S.-L.; Wang, L.-M.; Huang, D.-F.; Lin, X.-J. Effects of Fertilization Regimes on Tea Yields, Soil Fertility, and Soil Microbial Diversity. Chil. J. Agric. Res. 2014, 74, 333–339. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME Allows Analysis of High-Throughput Community Sequencing Data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Hung, Y.-M.; Lu, T.-P.; Tsai, M.-H.; Lai, L.-C.; Chuang, E.Y. EasyMAP: A User-Friendly Online Platform for Analyzing 16S Ribosomal DNA Sequencing Data. New Biotechnol. 2021, 63, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Anderson, M.J. A New Method for Non-Parametric Multivariate Analysis of Variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar] [CrossRef]
- Lahti, L.; Shetty, S.A. Tools for Microbiome Analysis in R. Available online: https://microbiome.github.io/tutorials/ (accessed on 15 December 2020).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive Functional Profiling of Microbial Communities Using 16S rRNA Marker Gene Sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for Integration and Interpretation of Large-Scale Molecular Data Sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef] [PubMed]
- De Cáceres, M.; Legendre, P.; Moretti, M. Improving Indicator Species Analysis by Combining Groups of Sites. Oikos 2010, 119, 1674–1684. [Google Scholar] [CrossRef]
- Csardi, G.; Nepusz, T. The igraph Software Package for Complex Network Research. Int. J. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical Analysis of Taxonomic and Functional Profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- Rodríguez-Blanco, A.; Sicardi, M.; Frioni, L. Plant Genotype and Nitrogen Fertilization Effects on Abundance and Diversity of Diazotrophic Bacteria Associated with Maize (Zea mays L.). Biol. Fertil. Soils 2015, 51, 391–402. [Google Scholar] [CrossRef]
- Chávez-Romero, Y.; Navarro-Noya, Y.E.; Reynoso-Martínez, S.C.; Sarria-Guzmán, Y.; Govaerts, B.; Verhulst, N.; Dendooven, L.; Luna-Guido, M. 16S Metagenomics Reveals Changes in The Soil Bacterial Community Driven by Soil Organic C, N-Fertilizer and Tillage-Crop Residue Management. Soil Tillage Res. 2016, 159, 1–8. [Google Scholar] [CrossRef]
- Hallmann, J.; Quadt-Hallmann, A.; Mahaffee, W.F.; Kloepper, J.W. Bacterial Endophytes in Agricultural Crops. Can. J. Microbiol. 1997, 43, 895–914. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Schlaeppi, K.; Spaepen, S.; Van Themaat, E.V.L.; Schulze-Lefert, P. Structure and Functions of the Bacterial Microbiota of Plants. Annu. Rev. Plant Biol. 2013, 64, 807–838. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, C.R.; Copeland, J.; Wang, P.W.; Guttman, D.S.; Kotanen, P.M.; Johnson, M.T.J. Assembly and Ecological Function of The Root Microbiome Across Angiosperm Plant Species. Proc. Natl. Acad. Sci. USA 2018, 115, E1157–E1165. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Von-Wobeser, E.; Rocha-Estrada, J.; Shapiro, L.R.; De La Torre, M. Enrichment of Verrucomicrobia, Actinobacteria and Burkholderiales drives selection of bacterial community from soil by maize roots in a traditional milpa agroecosystem. PLoS ONE 2018, 13, e0208852. [Google Scholar] [CrossRef] [PubMed]
- Bünger, W.; Jiang, X.; Müller, J.; Hurek, T.; Reinhold-Hurek, B. Novel Cultivated Endophytic Verrucomicrobia Reveal Deep-Rooting Traits of Bacteria to Associate with Plants. Sci. Rep. 2020, 10, 8692. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N.; Ladau, J.; Clemente, J.C.; Leff, J.W.; Owens, S.M.; Pollard, K.S.; Knight, R.; Gilbert, J.A.; McCulley, R.L. Reconstructing the Microbial Diversity and Function of Pre-Agricultural Tallgrass Prairie Soils in the United States. Science 2013, 342, 621–624. [Google Scholar] [CrossRef] [PubMed]
- Navarrete, A.; Soares, T.; Rossetto, R.; Van Veen, J.A.; Tsai, S.M.; Kuramae, E.E. Verrucomicrobial community structure and abundance as indicators for changes in chemical factors linked to soil fertility. Antonie Leeuwenhoek 2015, 108, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Bonanomi, G.; De Filippis, F.; Cesarano, G.; La Storia, A.; Ercolini, D.; Scala, F. Organic farming induces changes in soil microbiota that affect agro-ecosystem functions. Soil Biol. Biochem. 2016, 103, 327–336. [Google Scholar] [CrossRef]
- Lupatini, M.; Korthals, G.W.; De Hollander, M.; Janssens, T.K.; Kuramae, E.E. Soil Microbiome Is More Heterogeneous in Organic Than in Conventional Farming System. Front. Microbiol. 2017, 7, 2064. [Google Scholar] [CrossRef]
- Green, S.; Prakash, O.; Jasrotia, P.; Overholt, W.A.; Cardenas, E.; Hubbard, D.; Tiedje, J.M.; Watson, D.B.; Schadt, C.W.; Brooks, S.; et al. Denitrifying Bacteria from the Genus Rhodanobacter Dominate Bacterial Communities in the Highly Contaminated Subsurface of a Nuclear Legacy Waste Site. Appl. Environ. Microbiol. 2012, 78, 1039–1047. [Google Scholar] [CrossRef]
- Citak, S.; Sonmez, S. Effects of chemical fertilizer and different organic manures application on soil pH, EC and organic matter content. J. Food Agri. Environ. 2011, 9, 739–741. [Google Scholar]
- Reid, T.E.; Kavamura, V.N.; Abadie, M.; Torres-Ballesteros, A.; Pawlett, M.; Clark, I.M.; Harris, J.; Mauchline, T.H. Inorganic Chemical Fertilizer Application to Wheat Reduces the Abundance of Putative Plant Growth-Promoting Rhizobacteria. Front. Microbiol. 2021, 12, 642587. [Google Scholar] [CrossRef]
- Wang, L.; Li, J.; Yang, F.; E, Y.; Raza, W.; Huang, Q.; Shen, Q. Application of Bioorganic Fertilizer Significantly Increased Apple Yields and Shaped Bacterial Community Structure in Orchard Soil. Microb. Ecol. 2016, 73, 404–416. [Google Scholar] [CrossRef]
- Ma, B.; Lv, X.; Cai, Y.; Chang, S.X.; Dyck, M. Liming does not counteract the influence of long-term fertilization on soil bacterial community structure and its co-occurrence pattern. Soil Biol. Biochem. 2018, 123, 45–53. [Google Scholar] [CrossRef]
- Enebe, M.C.; Babalola, O.O. Effects of inorganic and organic treatments on the microbial community of maize rhizosphere by a shotgun metagenomics approach. Ann. Microbiol. 2020, 70, 49. [Google Scholar] [CrossRef]
- Pan, F.; Meng, Q.; Wang, Q.; Luo, S.; Chen, B.; Khan, K.Y.; Yang, X.; Feng, Y. Endophytic bacterium Sphingomonas SaMR12 promotes cadmium accumulation by increasing glutathione biosynthesis in Sedum alfredii Hance. Chemosphere 2016, 154, 358–366. [Google Scholar] [CrossRef]
- Backer, R.; Rokem, J.S.; Ilangumaran, G.; Lamont, J.; Praslickova, D.; Ricci, E.; Subramanian, S.; Smith, D.L. Plant Growth-Promoting Rhizobacteria: Context, Mechanisms of Action, and Roadmap to Commercialization of Biostimulants for Sustainable Agriculture. Front. Plant Sci. 2018, 9, 1473. [Google Scholar] [CrossRef]
- Luo, Y.; Wang, F.; Huang, Y.; Zhou, M.; Gao, J.; Yan, T.; Sheng, H.; An, L. Sphingomonas sp. Cra20 Increases Plant Growth Rate and Alters Rhizosphere Microbial Community Structure of Arabidopsis thaliana Under Drought Stress. Front. Microbiol. 2019, 10, 1221. [Google Scholar] [CrossRef]
- Amaresan, N.; Kumar, K.; Naik, J.H.; Bapatla, K.G.; Mishra, R.K. Streptomyces in plant growth promotion: Mechanisms and role. In New and Future Developments in Microbial Biotechnology and Bioengineering; Singh, B.P., Gupta, V.K., Passari, A.K., Eds.; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Mishra, S.; Lin, Z.; Pang, S.; Zhang, W.; Bhatt, P.; Chen, S. Recent Advanced Technologies for the Characterization of Xenobiotic-Degrading Microorganisms and Microbial Communities. Front. Bioeng. Biotechnol. 2021, 9, 632059. [Google Scholar] [CrossRef]
- Garai, P.; Chandra, K.; Chakravortty, D. Bacterial peptide transporters: Messengers of nutrition to virulence. Virulence 2016, 8, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Piepenbreier, H.; Fritz, G.; Gebhard, S. Transporters as information processors in bacterial signalling pathways. Mol. Microbiol. 2017, 104, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y. Illumina-based Analysis of Endophytic Bacterial Diversity of Four Allium Species. Sci. Rep. 2019, 9, 15271. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Df | Sum of Sqs $ | R2 | F | Pr (>F) $$ | |
|---|---|---|---|---|---|
| Field management | 1 | 0.9596 | 0.06474 | 2.7405 | 0.001 *** |
| Sample type | 1 | 2.1726 | 0.14657 | 6.2046 | 0.001 *** |
| Field management X sample type | 1 | 0.8361 | 0.05641 | 2.3879 | 0.004 ** |
| Residual | 31 | 10.855 | 0.73229 | ||
| Total | 34 | 14.8233 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, G.-Y.; Chen, B.-J.; Hu, C.-Y.; Lin, W.-Y. The Impacts of Field Management on Soil and Tea Root Microbiomes. Appl. Microbiol. 2021, 1, 361-376. https://doi.org/10.3390/applmicrobiol1020025
Lin G-Y, Chen B-J, Hu C-Y, Lin W-Y. The Impacts of Field Management on Soil and Tea Root Microbiomes. Applied Microbiology. 2021; 1(2):361-376. https://doi.org/10.3390/applmicrobiol1020025
Chicago/Turabian StyleLin, Guan-Ying, Bo-Jhen Chen, Chih-Yi Hu, and Wei-Yi Lin. 2021. "The Impacts of Field Management on Soil and Tea Root Microbiomes" Applied Microbiology 1, no. 2: 361-376. https://doi.org/10.3390/applmicrobiol1020025
APA StyleLin, G.-Y., Chen, B.-J., Hu, C.-Y., & Lin, W.-Y. (2021). The Impacts of Field Management on Soil and Tea Root Microbiomes. Applied Microbiology, 1(2), 361-376. https://doi.org/10.3390/applmicrobiol1020025