
RNA-Seq Unveils Cross-Talk among Stress Response Mechanisms during Leaf Color Transformation in ALS Resistant Sorghums

Abstract
1. Introduction
2. Materials and Methods
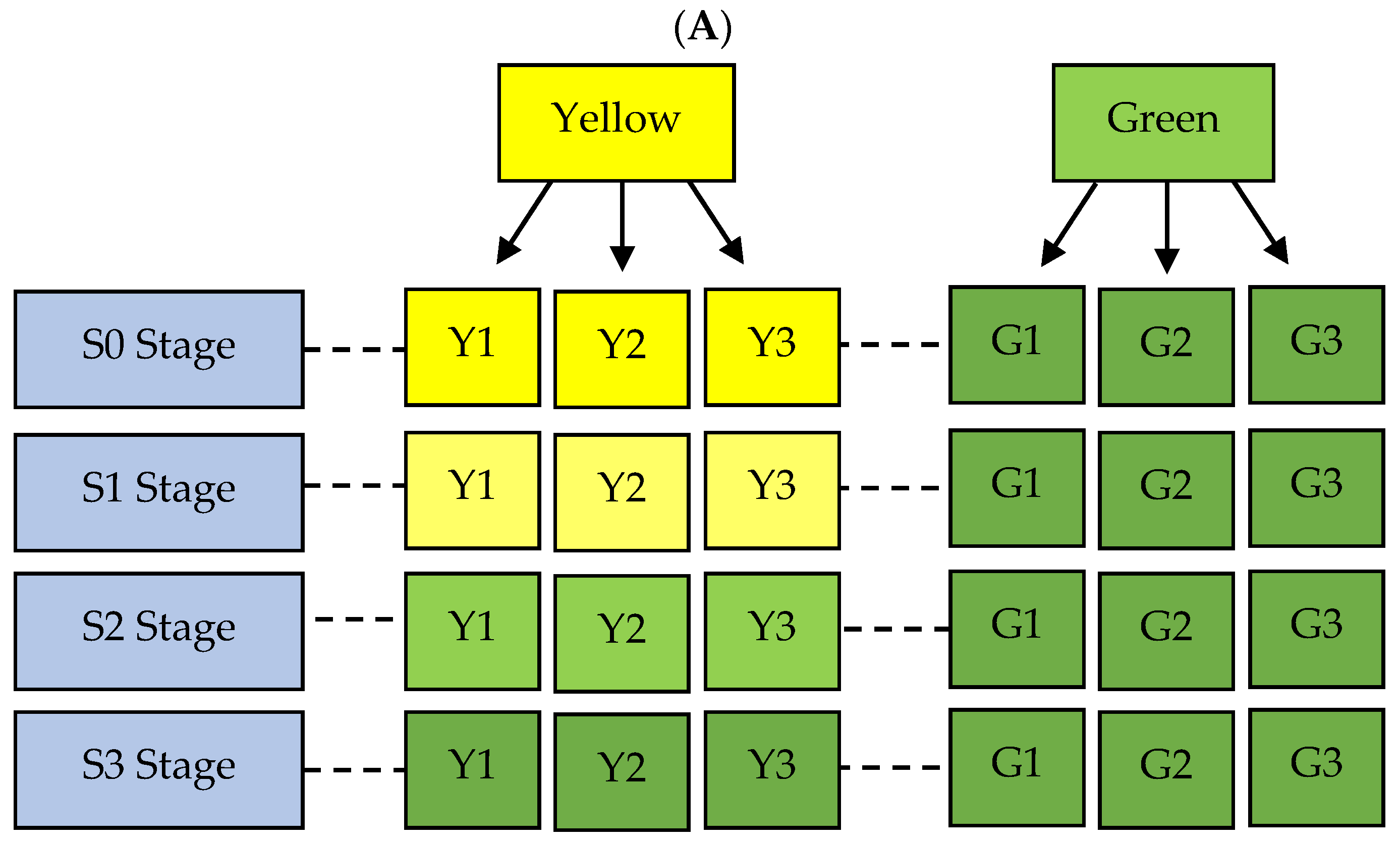
2.1. Genetic Materials
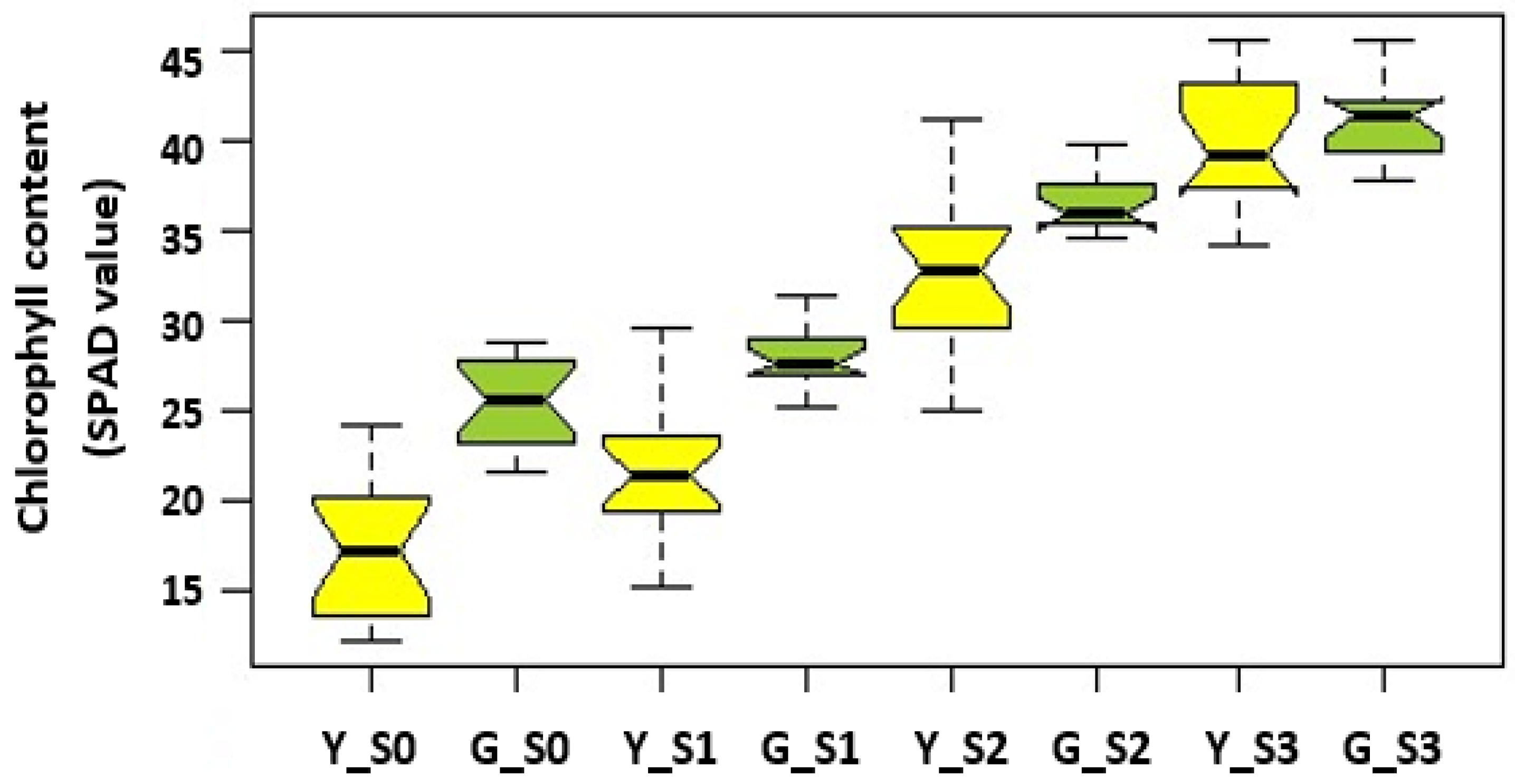
2.2. Experimental Design, Tissue Collection, and RNA Extraction
2.3. cDNA Library Construction and Sequencing
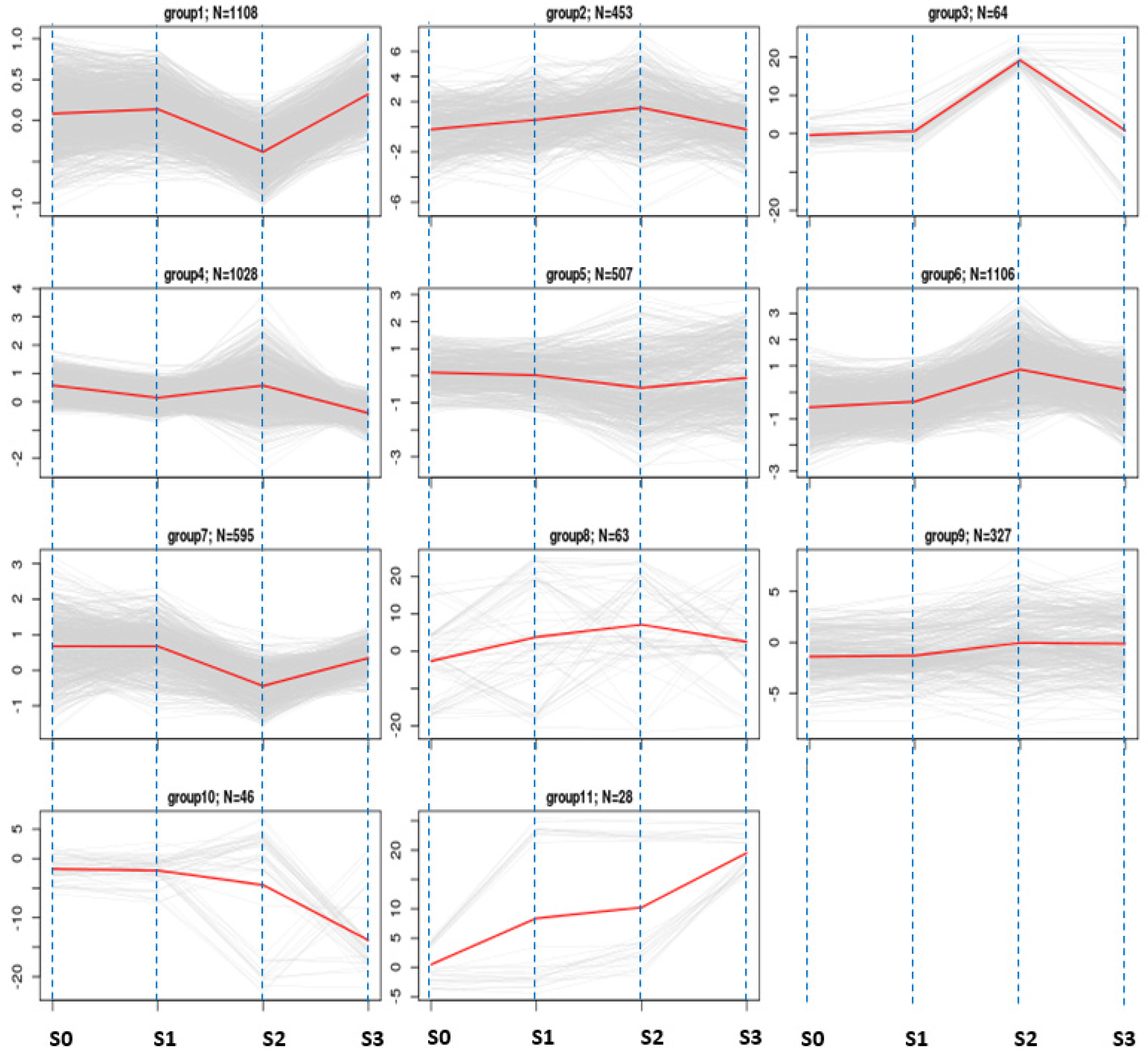
2.4. Differential Gene Expression Analysis and Gene Clustering
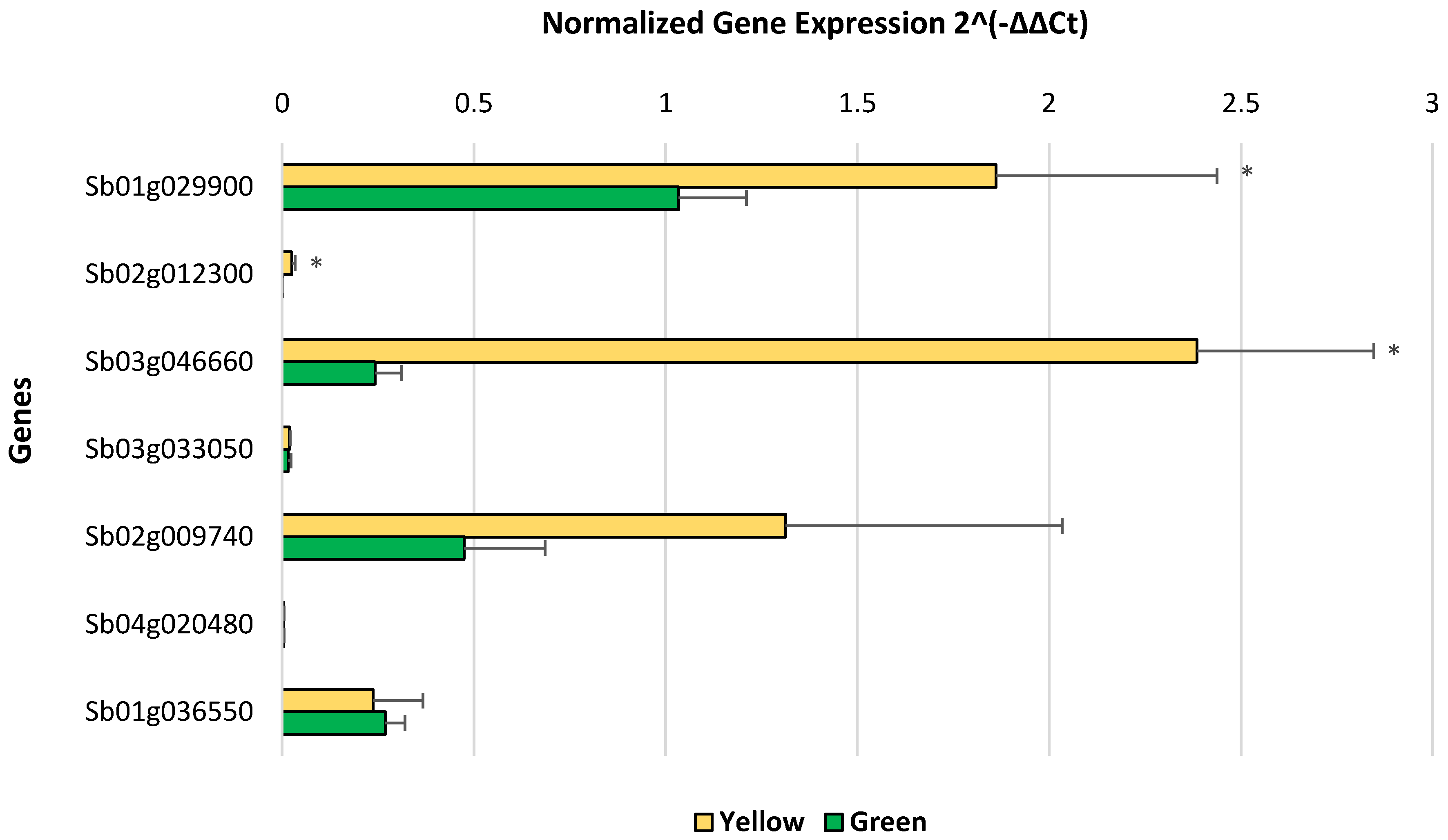
2.5. qRT-PCR Validation of RNA-Seq Results
2.6. GO Enrichment
2.7. SorghumCyc Pathway Analysis Followed by Visualization via Mapman
3. Results and Discussion
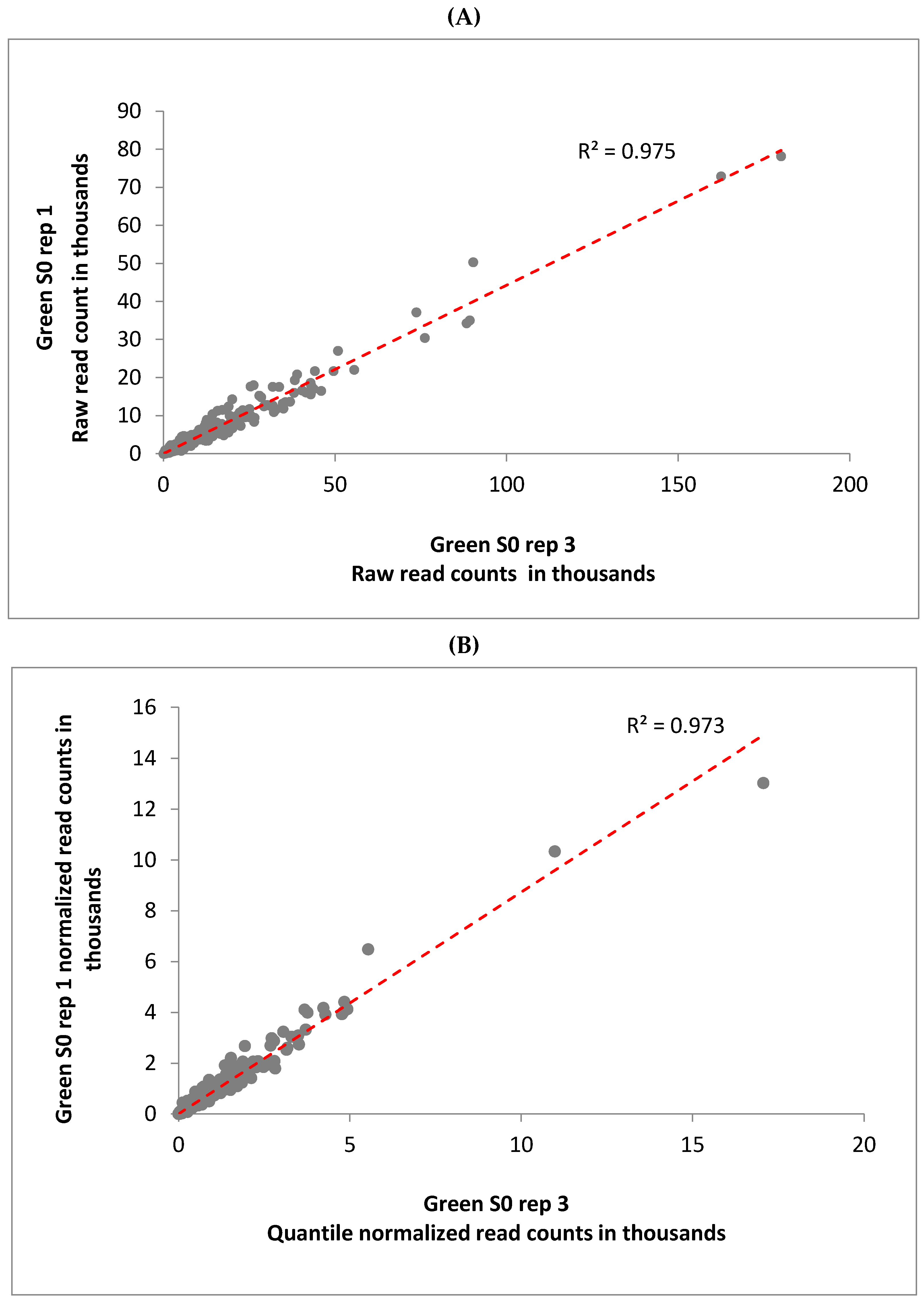
3.1. Mapping of Transcriptome to the Sorghum Genome
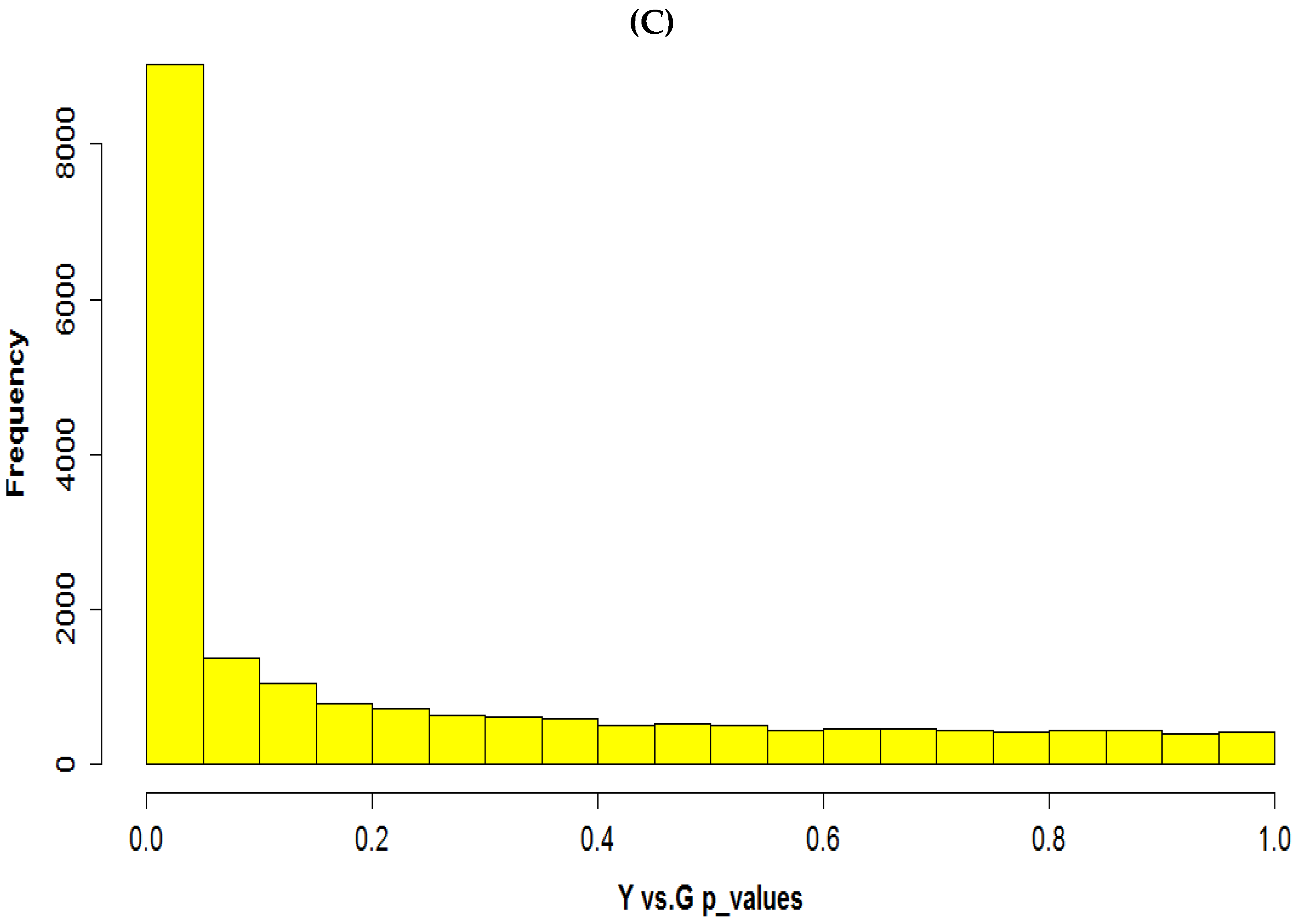
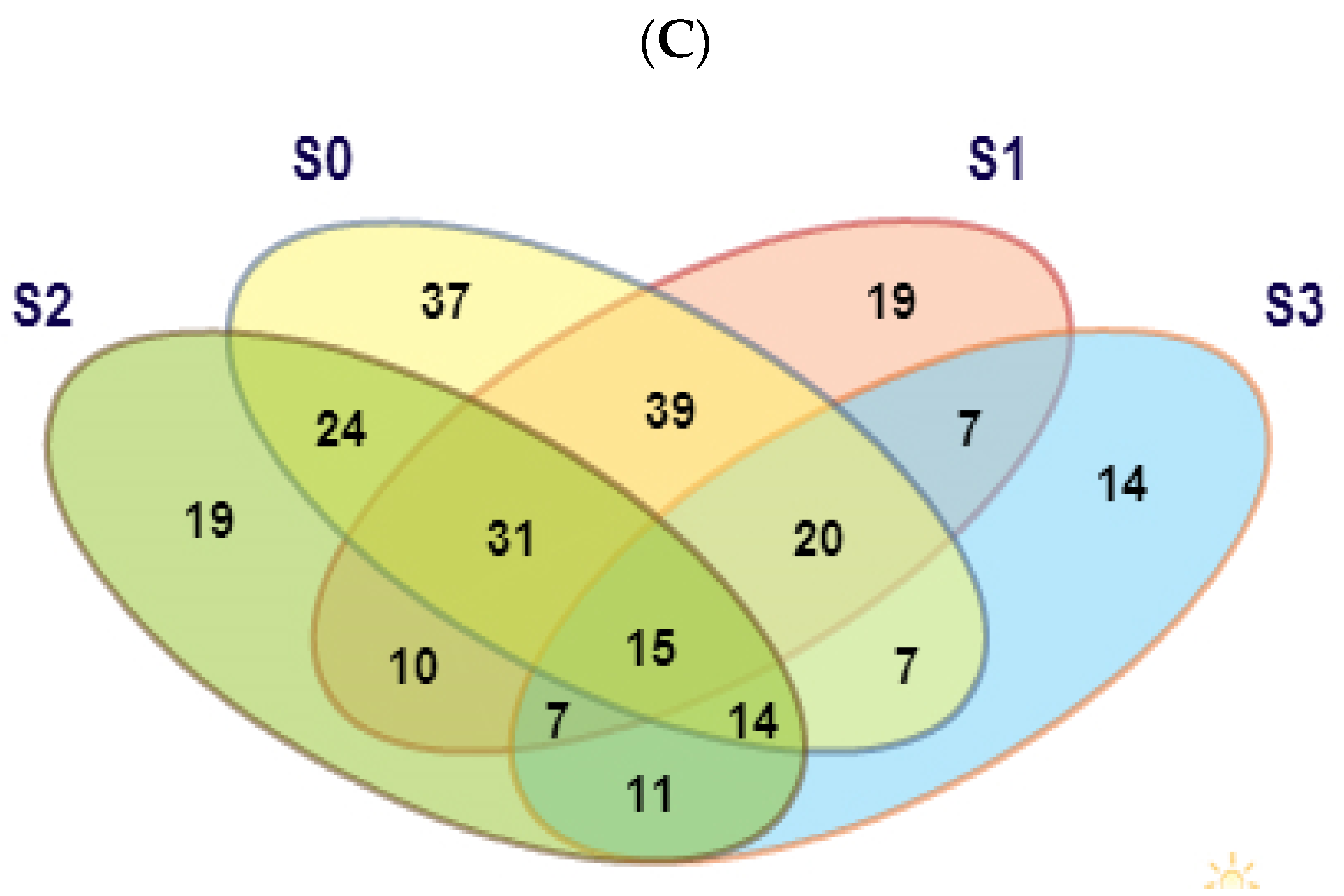
3.2. Determination of Differentially Expressed Genes and Validation via qRT-PCR
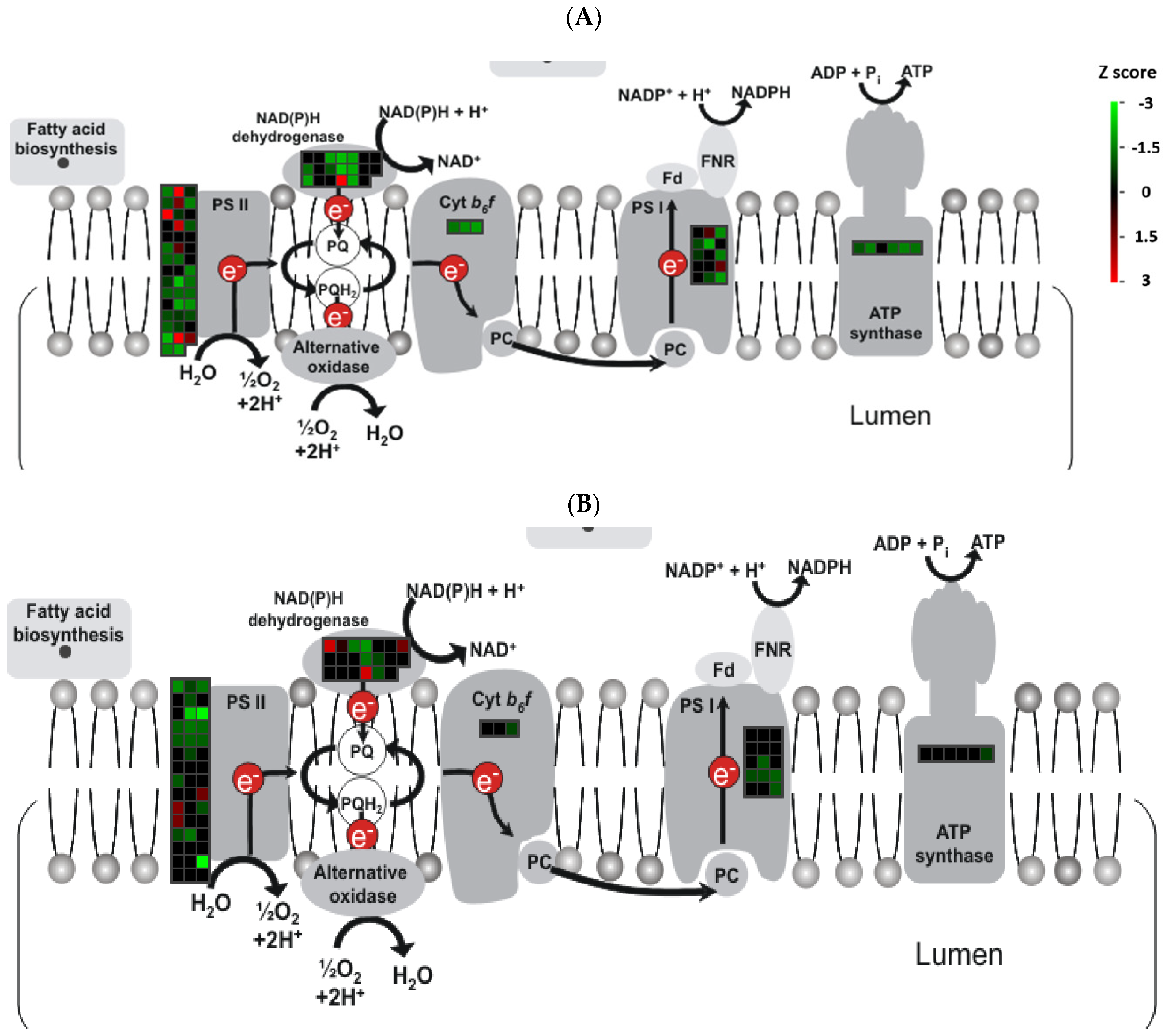
3.3. Transcript Analysis in Response to Herbicide Effect
3.4. Activated Hormonal Networks in Yellow Plants
3.5. Defense Responses Closely Mimic Gene Regulation under Drought and Salinity Stresses
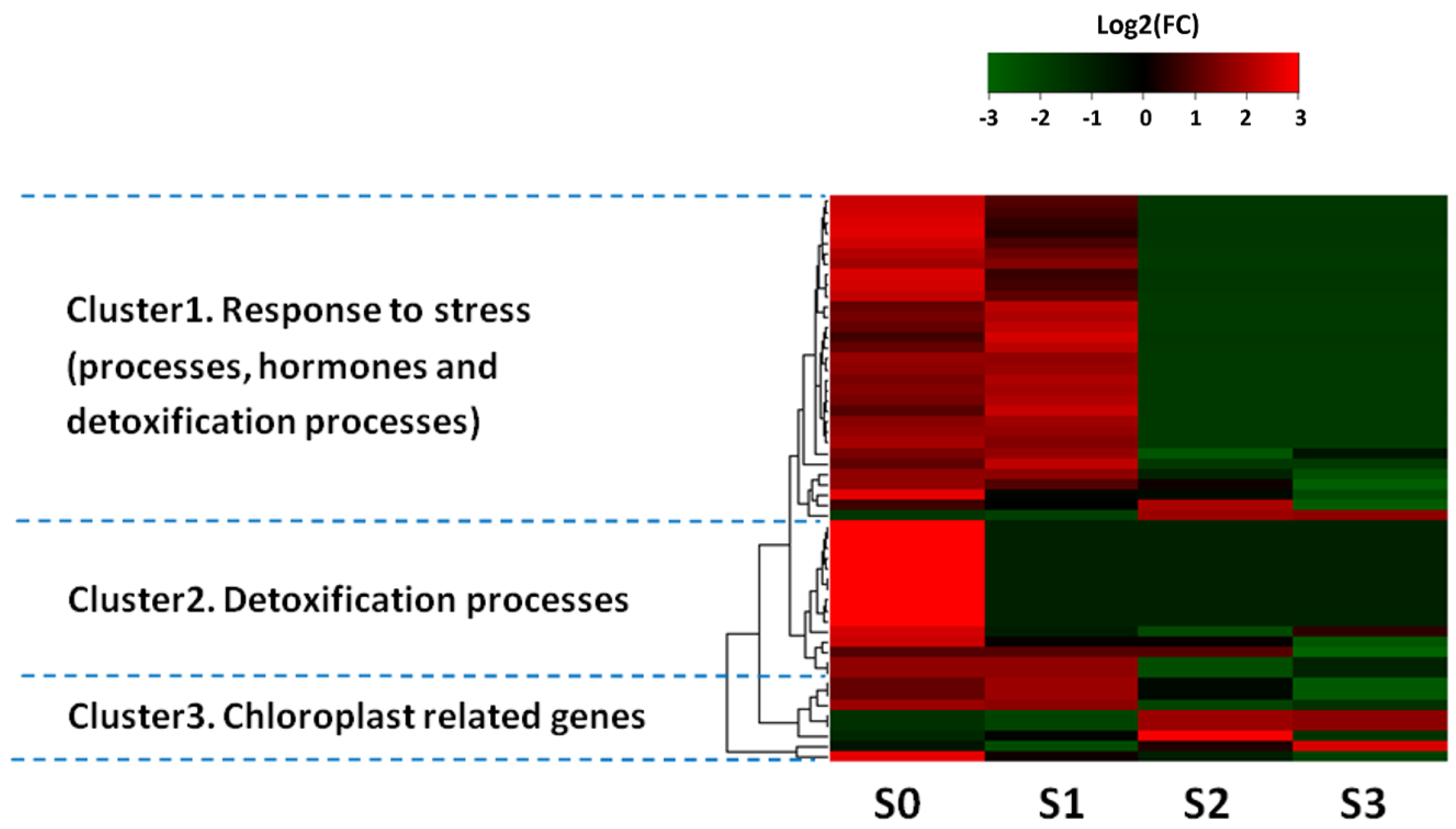
3.6. Clustering Pattern of Differentially Expressed Genes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hamman, L.; Dhuyvetter, K.; Boland, M.A. Economic Issues with Grain Sorghum; MF-2512; K-State Research and Extension Publication: Manhattan, KS, USA, 2001. [Google Scholar]
- Ariola, P.E.; Ellstrand, N. Crop-to-weed gene flow in the genus Sorghum (Poaceae): Spontaneous interspecific hybridization between Johnsongrass, Sorghum halepense, and crop sorghum, S. Bicolor. Am. J. Bot. 1996, 83, 1153–1160. [Google Scholar] [CrossRef]
- Tesso, T.T.; Kershner, K.; Ochanda, N.; Al-Khatib, K.; Tuinstra, M.R. Registration of 34 Sorghum Germplasm Lines Resistant to Acetolactate Synthase-Inhibitor Herbicides. J. Plant Regist. 2011, 5, 215–219. [Google Scholar] [CrossRef]
- Kershner, K.S. Herbicide Resistance in Grain Sorghum. Ph.D. Thesis, Kansas State University, Manhattan, KS, USA, 2010. [Google Scholar]
- Tuinstra, M.R.; Al-Khatib, K. Acetolactate Synthase Herbicide Resistant Sorghum. US Patent EP 2511373 A1, 17 October 2012. [Google Scholar]
- Weerasooriya, D.K.; Tesso, T.; Tuinstra, M.; Al-Khatib, K. Reduced Seedling Vigor and Interveinal Chlorosis Associated with ALS Mutation in Sorghum Are Affected by Background Genotype. In Proceedings of the Agronomy abstracts, International Annual Meetings of ASA, CSSA, SSSA, Cincinnati, OH, USA, 21–24 October 2012. [Google Scholar]
- Paterson, A.H.; Bowers, J.E.; Bruggmann, R.; Dubchak, I.; Grimwood, J.; Gundlach, H.; Haberer, G.; Hellsten, U.; Mitros, T.; Poliakov, A.; et al. The Sorghum bicolor genome and the diversification of grasses. Nature 2009, 457, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Fraley, C.; Raftery, A.E.; Murphy, T.B.; Scrucca, L. MCLUST Version 4 for R: Normal Mixture Modeling for Model-Based Clustering, Classification, and Density Estimation; University of Washington: Washington, DC, USA, 2012. [Google Scholar]
- Pavli, O.I.; Ghikas, D.V.; Katsiotis, A.; Skaracis, G.N. Differential expression of heat shock protein genes in sorghum (Sorghum bicolor L.) genotypes under heat stress. Aust. J. Crop Sci. 2011, 5, 511–515. [Google Scholar]
- Shih, C.-H.; Chu, I.K.; Yip, W.K.; Lo, C. Differential Expression of Two Flavonoid 3′-Hydroxylase cDNAs Involved in Biosynthesis of Anthocyanin Pigments and 3-Deoxyanthocyanidin Phytoalexins in Sorghum. Plant Cell Physiol. 2006, 47, 1412–1419. [Google Scholar] [CrossRef]
- Dugas, D.V.; Monaco, M.K.; Olsen, A.; Klein, R.R.; Kumari, S.; Ware, D.; Klein, P.E. Functional Annotation of the Transcriptome of Sorghum bicolor in Response to Osmotic Stress and Abscisic Acid. BMC Genom. 2011, 12, 514. [Google Scholar] [CrossRef]
- Youens-Clark, K.; Buckler, E.; Casstevens, T.; Chen, C.; Declerck, G.; Derwent, P.; Dharmawardhana, P.; Jaiswal, P.; Kersey, P.; Karthikeyan, A.S.; et al. Gramene database in 2010: Updates and extensions. Nucleic Acids Res. 2010, 39, D1085–D1094. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C. New Inzen-Z Grain Sorghum Hybrids; Kansas State Research and Extension: Manhattan, KS, USA, 2014. [Google Scholar]
- Vanderlip, R.; Roozeboom, K.; Fjell, D.L.; Shroyer, J.; Kok, H.; Regehr, D.; Whitney, D.A.; Regehr, D.H.; Alam, M.; Jardine, D.J.; et al. Grain Sorghum Handbook; Kansas State University Agricultural Experiment Station and Cooperative Extension Service: Manhattan, KS, USA, 1998. [Google Scholar]
- Brooks, H.L.; Jardine, D.J.; Regehr, D.L.; Fjell, D.L.; Whitney, D.A.; Webb, K.; Vanderlip, R. Diagnosing Sorghum Production Problems, Research and Extension Service; Kansas State University: Manhattan, KS, USA, 1999. [Google Scholar]
- Isakeit, T. Downy Mildew Fungus and Grain Sorghum. The Texas A&M University System. 2012. Available online: http://texassorghum.org (accessed on 9 December 2014).
- Krausz, J.P.; Collins, S.D.; Frederiksen, R.A.; Duncan, R.R.; Kaufman, H.W.N. Odvody Disease Response of Grain Sorghum Hybrids. The Texas A&M University System. Available online: http://texassorghum.org (accessed on 9 December 2014).
- Vincelli, P.; Hershman, D.E. Disease of Grain Sorghum. University of Kentucky, Issue 10–85. Available online: http://www.ca.uky.edu (accessed on 9 November 2014).
- Zavaleta-Mancera, H.A.; Thomas, B.J.; Thomas, H.; Scott, I.M. Regreening of senescent Nicotiana leaves II. Redifferentiation Plast. J. Exp. Bot. 1999, 50, 1683–1689. [Google Scholar]
- Prebeg, T.; Wrischer, M.; Fulgosi, H.; Ljubešić, N. Ultrastructural Characterization of the Reversible Differentiation of Chloroplasts in Cucumber Fruit. J. Plant Biol. 2008, 51, 122–131. [Google Scholar] [CrossRef]
- Huff, A. Nutritional control of regreening and degreening in citrus peel segments. Plant Physiol. 1983, 73, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, C.D.; Lim, S.; Salzman, R.A.; Kagiampakis, I.; Morishige, D.T.; Weers, B.D.; Klein, R.R.; Pratt, L.H.; Cordonnier-Pratt, M.M.; Klein, P.E.; et al. Sorghum bicolor’s transcriptome response to dehydration, high salinity and ABA. Plant Mol. Biol. 2005, 58, 699–720. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Lu, G.; Fan, D.; Zhu, C.; Li, W.; Zhao, Q.; Feng, Q.; Zhao, Y.; Guo, Y.; Li, W.; et al. Function annotation of the rice transcriptome at single-nucleotide resolution by RNA-Seq. Genome Res. 2010, 20, 1238–1249. [Google Scholar] [CrossRef]
- Guo, Y.; Sheng, Q.; Li, J.; Ye, F.; Samuels, D.C.; Shyr, Y. Large Scale Comparison of Gene Expression Levels by Microarrays and RNAseq Using TCGA Data. PLoS ONE 2013, 8, e071462. [Google Scholar] [CrossRef]
- Marrs, K. The functions and regulation of glutathione S transferases in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1996, 47, 127–158. [Google Scholar] [CrossRef]
- Alscher, R.G. Biosynthesis and antioxidant function of GSH in plants. Physiol. Plantarum. 1989, 77, 457–464. [Google Scholar] [CrossRef]
- Grant, C.M.; MacIver, F.H.; Dawes, I.W. Glutathione is an essential metabolite required for resistance to oxidative stress in the yeast Saccharomyces cerevisiae. Curr. Genet. 1996, 29, 511–515. [Google Scholar] [CrossRef]
- Nordman, T.; Xia, L.; Bjorkhem-Bergman, L.; Damdimopoulos, A.; Nalvarte, I.; Arner, E.S.J.; Spyrou, G.; Eriksson, L.C.; Bjornstedt, M.; Olsson, J.M. Regeneration of the antioxidant ubiquinol by lipoamide dehydrogenase, thioredoxin reductase and glutathione reductase. Biofactors 2003, 18, 45–5010. [Google Scholar] [CrossRef] [PubMed]
- Wingate, V.P.M.; Lawton, M.A.; Lamb, C.J. Glutathione causes a massive and selective induction of plant defence genes. Plant Physiol. 1988, 31, 205–211. [Google Scholar]
- Baier, M.; Dietz, K.J. The plant 2-cys peroxiredoxin BAS1 is a nuclear encoded chloroplast protein: Its expression regulation, phylogenetic origin and implications for its specific physiological function in plants. Plant J. 1997, 12, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Kilian, J.; Whitehead, D.; Horak, J.; Wanke, D.; Weinl, S.; Batistic, O.; D’Angelo, C.; Bornberg-Bauer, E.; Kudla, J.; Harter, K. The AtGenExpress global stress expression data set: Protocols, evaluation and model data analysis of UV-B light, drought and cold stress responses. Plant J. 2007, 50, 347–363. [Google Scholar] [CrossRef] [PubMed]
- Goda, H.; Sasaki, E.; Akiyama, K.; Maruyama-Nakashita, A.; Nakabayashi, K.; Li, W.; Ogawa, M.; Yamauchi, Y.; Preston, J.; Aoki, K.; et al. The AtGenExpress hormone and chemical treatment dataset: Experimental design, data evaluation, model data analysis and data access. Plant J. 2008, 55, 526–542. [Google Scholar] [CrossRef] [PubMed]
- Zeller, G.; Henz, S.R.; Widmer, C.K.; Sachsenberg, T.; Rätsch, G.; Weigel, D.; Laubinger, S. Stress-induced changes in the Arabidopsis thaliana transcriptome analyzed using whole-genome tiling arrays. Plant J. 2009, 58, 1068–1082. [Google Scholar] [CrossRef] [PubMed]
- Nemhauser, J.L.; Hong, F.; Chory, J. Different plant hormones regulate similar processes through largely nonoverlapping transcriptional responses. Cell 2006, 126, 467–475. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.A.; Benková, E. Cytokinin cross-talking during biotic and abiotic stress responses. Front. Plant Sci. 2013, 4, 451. [Google Scholar] [CrossRef]
- Tran, L.-S.P.; Urao, T.; Qin, F.; Maruyama, K.; Kakimoto, T.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Functional analysis of AHK1/ATHK1 and cytokinin receptor histidine kinases in response to abscisic acid, drought, and salt stress in Arabidopsis. Proc. Natl. Acad. Sci. USA 2007, 104, 20623–20628. [Google Scholar] [CrossRef]
- Kang, N.Y.; Cho, C.; Kim, N.Y.; Kim, J. Cytokinin receptor-dependent and receptor-independent pathways in the dehydration response of Arabidopsis thaliana. J. Plant Physiol. 2012, 169, 1382–1391. [Google Scholar] [CrossRef]
- Kamada, I.; Yamauchi, S.; Youssefian, S.; Sano, H. Transgenic tobacco plants expressing grpg1,a gene encoding a ras-related GTP-binding protein from rice, show distinct morphological characteristics. Plant J. 1992, 2, 799–807. [Google Scholar] [CrossRef]
- Sano, H.; Seo, S.; Orudgev, E.; Youssefian, S.; Ishizuka, K.; Ohashi, Y. Expression of the gene for a small GTP binding protein in transgenic tobacco elevates endogenous cytokinin levels, abnormally induces salicylic acid in response to wounding, and increases resistance to tobacco mosaic virus infection. Proc. Natl. Acad. Sci. USA 1994, 91, 10556–10560. [Google Scholar] [CrossRef]
- Masuta, C.; Tanaka, H.; Uehara, K.; Kuwata, S.; Koiwai, A.; Noma, M. Broad resistance to plant viruses in transgenic plants conferred by anti-sense inhibition of a host gene essential in S-adenosylmethionine-dependent transmethylation reactions. Proc. Natl. Acad. Sci. USA 1995, 92, 6117–6121. [Google Scholar] [CrossRef]
- Yuan, S.; Lin, H.H. Role of salicylic acid in plant abiotic stress. J. Biosci. 2008, 63, 313–320. [Google Scholar] [CrossRef]
- Nawrath, C.; Heck, S.; Parinthawong, N.; Metraux, J.P. EDS5, an essential component of salicylic acid-dependent signaling for disease resistance in Arabidopsis, is a member of the MATE transporter family. Plant Cell. 2002, 14, 275–286. [Google Scholar] [CrossRef]
- Clarke, S.M.; Mur, J.; Wood, J.E.; Scott, I.M. Salicylic acid dependent signaling promotes basal thermotolerance but is not essential for acquired thermotolerance in Arabidopsis thaliana. Plant J. 2004, 38, 432–447. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Wang, Y.; Zhou, Y.; Tao, Y.; Mao, W.; Shi, K.; Asami, T.; Chen, Z.; Yu, J. Reactive oxygen species are involved in brassinosteroid-induced stress tolerance in cucumber. Plant Physiol. 2009, 150, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Bajguz, A.; Hayat, S. Effects of brassinosteroids on the plant responses to environmental stresses. Plant Physiol. Biochem. 2009, 47, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Holá, D. Brassinosteroids and photosynthesis. In Brassinosteroids: A Class of Plant Hormone; Hayat, S., Ahmad, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Wybraniec, S.; Stalica, P.; Spórna, A.; Nemzer, B.; Pietrzkowski, Z.; Michałowski, T. Antioxidant activity of betanidin: Electrochemical study in aqueous media. Agric. Food Chem. 2011, 59, 12163–12170. [Google Scholar] [CrossRef]
- Triantaphylide’s, C.; Krischke, M.; Hoeberichts, F.A.; Ksas, B.; Gresser, G.; Havaux, M.; Breusegem, F.V.; Mueller, M.J. Singlet Oxygen Is the Major Reactive Oxygen Species Involved in Photooxidative Damage to Plants. Plant Physiol. 2008, 148, 960–968. [Google Scholar] [CrossRef]
- Saijo, Y.; Hata, S.; Kyozuka, J.; Shimamoto, K.; Izui, K. Over-expression of a single Ca2+-dependent protein kinase confers both cold and salt/drought tolerance on rice plants. Plant J. 2000, 23, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, K.K.; Tripathi, A.K.; Pareek, A.; Singla-Pareek, S.L. Taming drought stress in rice through genetic engineering of transcription factors and protein kinases. Plant Stress. 2013, 7 (Suppl. S1), 60–72. [Google Scholar]
- Quan, R.; Shang, M.; Zhang, H.; Zhao, Y.; Zhang, J. Engineering of enhanced glycine betaine synthesis improves drought tolerance in maize. Plant Biotechnol. J. 2004, 2, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Lv, S.; Yang, A.; Zhang, K.; Wang, L.; Zhang, J. Increase of glycinebetaine synthesis improves drought tolerance in cotton. Mol. Breed. 2007, 20, 233–248. [Google Scholar] [CrossRef]
- Zinselmeier, C.; Jeong, B.R.; Boyer, J.S. Starch and the control of kernel number in maize at low water potentials. Plant Physiol. 1999, 121, 25–36. [Google Scholar] [CrossRef]
- Jones, L.; McQueen-Mason, S. A role for expansins in dehydration and rehydration of the resurrection plant Craterostigma plantagineum. FEBS Lett. 2004, 559, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Hoch, W.A.; Zeldin, E.L.; McCown, B.H. Physiological significance of anthocyanins during autumnal leaf senescence. Tree Physiol. 2001, 21, 1–8. [Google Scholar] [CrossRef]
- Maurel, C.; Javot, H.; Lauvergeat, V.; Gerbeau, P.; Tournaire, C.; Santoni, V.; Heyes, J. Molecular physiology of aquaporins in plants. Int. Rev. Cytol. 2002, 215, 105–148. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Trimmed Reads | Mapped Reads | Mapped % | Confident Mapped Reads | Confident Mapped % |
|---|---|---|---|---|---|
| G-S0-rep1 | 17,100,000 | 16,717,481 | 97.8 | 15,489,374 | 90.6 |
| G-S0-rep2 | 11,386,966 | 10,314,346 | 90.6 | 9,478,054 | 83.2 |
| G-S0-rep3 | 8,138,406 | 7,919,657 | 97.3 | 7,302,557 | 89.7 |
| G-S1-rep1 | 12,544,645 | 11,972,499 | 95.4 | 11,094,164 | 88.4 |
| G-S1-rep2 | 11,304,313 | 11,009,385 | 97.4 | 10,188,836 | 90.1 |
| G-S1-rep3 | 12,367,994 | 12,098,903 | 97.8 | 11,261,965 | 91.1 |
| G-S2-rep1 | 12,715,191 | 12,151,177 | 95.6 | 11,236,344 | 88.4 |
| G-S2-rep2 | 11,345,117 | 10,851,960 | 95.7 | 10,078,756 | 88.8 |
| G-S2-rep3 | 15,031,984 | 14,694,783 | 97.8 | 13,570,020 | 90.3 |
| G-S3-rep1 | 12,185,823 | 11,838,370 | 97.1 | 10,975,077 | 90.1 |
| G-S3-rep2 | 10,549,889 | 10,096,828 | 95.7 | 9,352,468 | 88.6 |
| G-S3-rep3 | 10,053,833 | 9,774,332 | 97.2 | 9,048,876 | 90 |
| Y-S0-rep1 | 12,932,513 | 12,585,942 | 97.3 | 11,656,006 | 90.1 |
| Y-S0-rep2 | 14,913,289 | 14,551,466 | 97.6 | 13,512,155 | 90.6 |
| Y-S0-rep3 | 14,578,395 | 14,244,619 | 97.7 | 13,222,902 | 90.7 |
| Y-S1-rep1 | 8,850,648 | 8,598,795 | 97.2 | 7,995,057 | 90.3 |
| Y-S1-rep2 | 11,699,313 | 11,428,736 | 97.7 | 10,566,808 | 90.3 |
| Y-S1-rep3 | 11,271,245 | 10,928,415 | 97 | 10,133,689 | 89.9 |
| Y-S2-rep1 | 14,450,551 | 14,051,613 | 97.2 | 13,064,471 | 90.4 |
| Y-S2-rep2 | 13,050,379 | 12,460,776 | 95.5 | 11,593,536 | 88.8 |
| Y-S2-rep3 | 11,872,101 | 11,542,378 | 97.2 | 10,755,236 | 90.6 |
| Y-S3-rep1 | 11,241,136 | 10,989,004 | 97.8 | 10,182,955 | 90.6 |
| Y-S3-rep2 | 11,541,148 | 11,235,620 | 97.4 | 10,434,927 | 90.4 |
| Y-S3-rep3 | 12,012,777 | 11,625,966 | 96.8 | 10,759,674 | 89.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weerasooriya, D.K.; Bandara, A.Y.; Liu, S.; Tesso, T.T. RNA-Seq Unveils Cross-Talk among Stress Response Mechanisms during Leaf Color Transformation in ALS Resistant Sorghums. Crops 2024, 4, 348-365. https://doi.org/10.3390/crops4030025
Weerasooriya DK, Bandara AY, Liu S, Tesso TT. RNA-Seq Unveils Cross-Talk among Stress Response Mechanisms during Leaf Color Transformation in ALS Resistant Sorghums. Crops. 2024; 4(3):348-365. https://doi.org/10.3390/crops4030025
Chicago/Turabian StyleWeerasooriya, Dilooshi K., Ananda Y. Bandara, Sanzhen Liu, and Tesfaye T. Tesso. 2024. "RNA-Seq Unveils Cross-Talk among Stress Response Mechanisms during Leaf Color Transformation in ALS Resistant Sorghums" Crops 4, no. 3: 348-365. https://doi.org/10.3390/crops4030025
APA StyleWeerasooriya, D. K., Bandara, A. Y., Liu, S., & Tesso, T. T. (2024). RNA-Seq Unveils Cross-Talk among Stress Response Mechanisms during Leaf Color Transformation in ALS Resistant Sorghums. Crops, 4(3), 348-365. https://doi.org/10.3390/crops4030025