
Drosophila Eye Gene Regulatory Network Inference Using BioGRNsemble: An Ensemble-of-Ensembles Machine Learning Approach



Abstract
1. Introduction
2. Materials and Methods
2.1. Dataset Information
2.1.1. RNA-Seq Dataset
2.1.2. Dataset Exploration & Preprocessing
2.2. Machine Learning Algorithms
2.2.1. GENIE3
2.2.2. GRNBoost2
2.2.3. The BioGRNSemble
3. Results
3.1. Tools and Libraries
3.2. Experimental Setup
- The Potier et al. [5] RNA-seq dataset was used after data cleaning, noise-filtering, and bias reduction.
- A list of 652 known transcription factors of the Drosophila was fed into GENIE3 and GRNBoost2 separately with similar hyperparameter settings.
- The resulting intersection of common links was then filtered further based on a minimum importance score threshold of 0.25.
- TFLink [16], an online database containing regulatory interactions between over 18,000 target genes and 527 transcription factor genes, was used to validate the GRN prediction accuracy. TFLink consists of genes present in organisms commonly used for research, among them being mice, E. coli, Homo sapiens, and Drosophila melanogaster.
3.3. Key Metrics
- Number of estimators: Also known as the number of trees to be generated for predictions, this hyperparameter can quickly become computationally intensive as it is increased. Through trial and error, we set the number of estimators for both GRNBoost2 and GENIE3 to be 25 to balance time efficiency and prediction performance.
- Correlation/Importance score: The probability or significance of a regulatory link’s existence between the transcription factor and target gene, represented by a float value between 0.0 and 1.0. For output by both GENIE3 and GRNBoost2, a higher correlation score indicates a ‘likelier’ regulatory link.
- Average correlation/importance score: The value produced by averaging the correlation scores generated by GENIE3 and GRNBoost2. The BioGRNSemble pipeline utilizes this methodology to yield a pruned, smaller regulatory network for further analysis.
- Minimum Correlation/Importance threshold: The minimum correlation score, a value between 0.0 and 1.0, was used filter out links deemed ‘weak’ by the machine learning algorithms. For example, a minimum correlation score of 0.25 considers links with correlation scores only above that value.
3.4. GRN Prediction Performance
3.5. Top Paired Genes
3.5.1. GENIE3
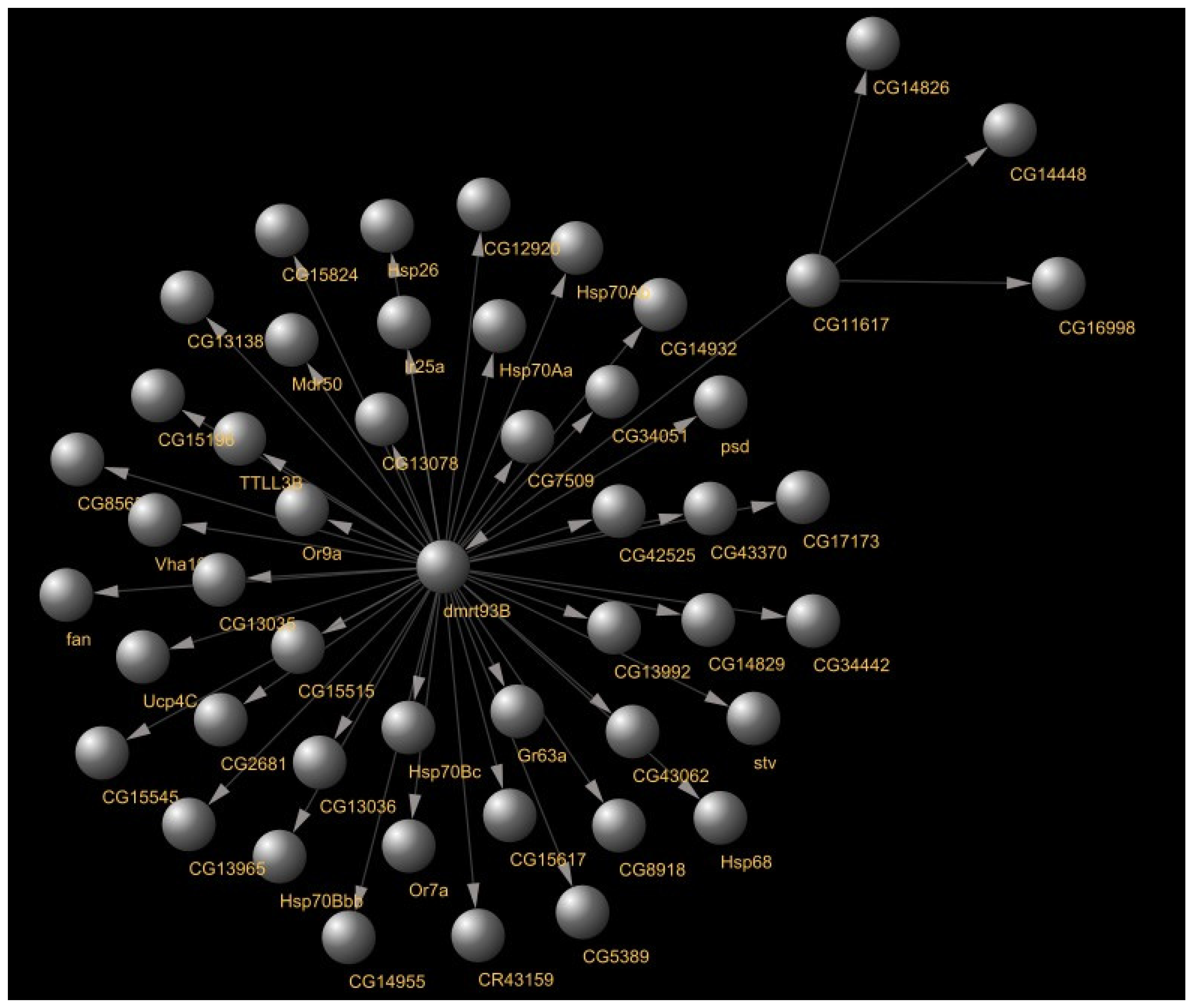
- Dmrt93B: A transcription factor gene responsible for sex configuration of the Drosophila [17] expressed in the frontal ganglion region of the Drosophila brain. It was predicted to be regulated by CG11617, another transcription gene expressed in the larval muscle system, in wing cells, responsible for muscle development [18]. Figure 7 showcases dmrt93B regulatory gene links as predicted by GENIE3.
- GATAe: A transcription factor gene involved in stem cell maintenance and is expressed in the endoderm region [19].
- Shn: A transcription factor involved in gut development and wing imaginal disc structuring [21].
- CG13510: A gene that regulates zinc ion binding. Usually present in the head region of adult Drosophila [22].
3.5.2. GRNBoost2
- TFAM: A transcription factor involved in mitochondrial regulation. The variations within this gene are often implicated in diseases like Alzheimer’s and Parkinson’s disease [23].
- CG2116: A transcription factor playing a role in dendrite formation [24].
- CG10979: Usually expressed in gut and germline cells, plays a role in zinc production [25].
3.5.3. BioGRNSemble
- Srp [26]: Known as the “Serpent” gene, this transcription factor plays a role in nervous system maintenance and muscle formation.
- Pho [27]: This gene is involved in maintaining embryonic genes necessary for organ growth.
- Ato [28]: Commonly expressed in reproductive gamete cells, this transcription factor is essential for maintaining egg cell expression and growth.
4. Discussion
4.1. Regulatory Network Analysis
4.2. TFLink Validation Analysis
4.3. Limitations
- The GRNBoost2 algorithm is sensitive to hyperparameter settings and may need to be further tuned to provide consistent results and denser regulatory networks comparable to those of GENIE3.
- Due to the drastic differences between GENIE3 and GRNBoost2 correlation scores, averaging may not be sufficient to prevent the skewing of scores towards either model’s output, as seen in the BioGRNsemble results being very similar to those of GENIE3. An alternative method can involve extracting the top n gene links with the highest scores from each model, merging them, and then performing an averaging for the rest of the common links to produce the final, intersected output.
- Without a gold standard ground truth network, it can be labor-intensive to verify the accuracy of each predicted connection. Unfortunately, there is yet to be such a network for the Drosophila eye, and the closest we have is the network constructed by Potier et al. [5], which does not seem to be available publicly. While TFLink is extremely helpful for verification, additional literature may need to be studied to discover more transcription gene–target gene links that may or may not have been established.
5. Conclusions and Future Extension
- Exploring alternative scoring systems, fine-tuning hyperparameters, and employing more advanced data preprocessing techniques.
- Integrating convolutional neural network-based deep learning models alongside GENIE3 and GRNBoost2.
- Testing BioGRNsemble on additional species, such as mice, zebrafish, and E. coli.
- Validating unknown gene regulatory links through established literature or wet lab experiments.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reiter, L.T.; Potocki, L.; Chien, S.; Gribskov, M.; Bier, E. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 2001, 11, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Kacsoh, B.; Greene, C.S.; Bosco, G. Machine Learning Analysis Identifies Drosophila Grunge/Atrophin as an Important Learning and Memory Gene Required for Memory Retention and Social Learning. G3 2017, 7, 3705–3718. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cho, D.-Y.; Lee, H.; Fear, J.; Oliver, B.; Przytycka, T.M. Reprogramming of regulatory network using expression uncovers sex-specific gene regulation in Drosophila. Nat. Commun. 2019, 9, 4061. [Google Scholar] [CrossRef] [PubMed]
- Razaghi-Moghadam, Z.; Nikoloski, Z. Supervised learning of gene-regulatory networks based on graph distance profiles of transcriptomics data. NPJ 2020, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Potier, D.; Davie, K.; Hulselmans, G.; Sanchez, M.N.; Haagen, L.; Huynh-Thu, V.A.; Koldere, D.; Celik, A.; Geurts, P.; Christiaens, V.; et al. Mapping Gene Regulatory Networks in Drosophila Eye Development by Large-Scale Transcriptome Perturbations and Motif Inference. Cell Rep. 2014, 9, 2290–2303. [Google Scholar] [CrossRef] [PubMed]
- Schena, M.; Shalon, D.; Davis, R.W.; Brown, P.O. Quantitative Monitoring of Gene Expression Patterns with a Complementary DNA Microarray. Science 1995, 270, 5235. [Google Scholar] [CrossRef] [PubMed]
- Huynh-Thu, V.A.; Irrthum, A.; Wehenkel, L.; Geurts, P. Inferring Regulatory Networks from Expression Data Using Tree-Based Methods. PLoS ONE 2010, 5, e12776. [Google Scholar] [CrossRef] [PubMed]
- Moerman, T.; Santos, S.A.; Gonzalez-Blas, C.B.; Simm, J.; Moreau, Y.; Aerts, J.; Aerts, S. GRNBoost2 and Arboreto: Efficient and scalable inference of gene regulatory networks. Bioinformatics 2019, 35, 2159–2161. [Google Scholar] [CrossRef] [PubMed]
- Cytoscape. Available online: https://cytoscape.org (accessed on 10 February 2024).
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- McKinney, W. Data structures for statistical computing in Python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010; pp. 51–56. [Google Scholar] [CrossRef]
- Harris, C.R.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; Kern, R.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Waskom, M.L. Seaborn: Statistical data visualization. J. Open Source Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]
- Aibar, S.; Gonzalez-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.-C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef] [PubMed]
- TFLink. Available online: https://tflink.net (accessed on 20 April 2024).
- Casado-Navarro, R.; Serrano-Saiz, E. DMRT Transcription Factors in the Control of Nervous System Sexual Differentiation. Front. Neuroanat. 2022, 16, 937596. [Google Scholar] [CrossRef] [PubMed]
- NIH. CG11617. Available online: https://www.ncbi.nlm.nih.gov/gene/33183 (accessed on 21 April 2024).
- NIH. GATAe [Drosophila Melanogaster]. Available online: https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=DetailsSearch&Term=41945 (accessed on 21 April 2024).
- NIH. Fkh Fork Head [Drosophila Melanogaster (Fruit Fly)]. Available online: https://www.ncbi.nlm.nih.gov/gene/43383 (accessed on 21 April 2024).
- FlyBase. Dmel\shn. Available online: https://flybase.org/reports/FBgn0003396.htm (accessed on 22 April 2024).
- Alliance of Genome Resources. CG13510 Gene. Available online: https://www.alliancegenome.org/gene/FB:FBgn0034758 (accessed on 22 April 2024).
- NCBI. TFAM. Available online: https://www.ncbi.nlm.nih.gov/gene/7019 (accessed on 22 April 2024).
- NCBI. CG2116. Available online: https://www.ncbi.nlm.nih.gov/gene/31735 (accessed on 23 April 2024).
- NCBI. CG10979. Available online: https://www.ncbi.nlm.nih.gov/gene/40720 (accessed on 23 April 2024).
- SDB. Serpent. Available online: https://www.sdbonline.org/sites/fly/gene/serpent.htm (accessed on 23 April 2024).
- UniProt. Pho_Drome. Available online: https://www.uniprot.org/uniprotkb/Q8ST83/entry (accessed on 23 April 2024).
- TAIR. AT5G06160. Available online: https://www.arabidopsis.org/servlets/TairObject?type=locus&name=At5g06160 (accessed on 23 April 2024).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Cell 1 | Cell 2 | Cell 3 | Cell 4 |
|---|---|---|---|---|
| Gene 1 | 1.0 | 5.0 | 7.0 | 2.0 |
| Gene 2 | 0.0 | 3.0 | 2.0 | 1.0 |
| Gene 3 | 10.0 | 0.0 | 2.0 | 12.0 |
| Gene 4 | 2.0 | 1.0 | 0.0 | 0.0 |
| Algorithm | Minimum Threshold (0.0 to 1.0) | No. of Links | No. of Transcription Factors |
|---|---|---|---|
| GENIE3 | 0.0 | 579,908 | 548 |
| GENIE3 | 0.25 | 2141 | 395 |
| GENIE3 | 0.40 | 494 | 147 |
| GENIE3 | 0.50 | 210 | 62 |
| GENIE3 | 0.65 | 57 | 21 |
| GENIE3 | 0.75 | 18 | 6 |
| GRNBoost2 | 0.0 | 4,879,738 | 548 |
| GRNBoost2 | 0.25 | 9818 | 465 |
| GRNBoost2 | 0.40 | 873 | 267 |
| GRNBoost2 | 0.50 | 138 | 103 |
| GRNBoost2 | 0.65 | 3 | 3 |
| GRNBoost2 | 0.75 | 1 | 1 |
| BioGRNsemble | 0.0 | 534,843 | 131 |
| BioGRNsemble | 0.50 | 30 | 16 |
| Transcription Factor Gene | Target Gene | Correlation Score |
|---|---|---|
| dmrt93B | CG14932 | 0.85471 |
| dmrt93B | Gr63a | 0.84326 |
| dmrt93B | CG14955 | 0.83637 |
| dmrt93B | Hsp70Aa | 0.83425 |
| dmrt93B | Hsp70Bc | 0.83026 |
| GATAe | PH4alphaPV | 0.82495 |
| fkh | MRE23 | 0.82274 |
| shn | CG14471 | 0.81887 |
| dmrt93B | Hsp70Bbb | 0.81283 |
| CG13510 | CG13511 | 0.79432 |
| Transcription Factor Gene | Target Gene | Correlation Score |
|---|---|---|
| TFAM | Exo70 | 0.833288 |
| Tsf2 | CG9634 | 0.671760 |
| Atf-2 | CG23815 | 0.663000 |
| her | CG813 | 0.647054 |
| Cnc | CG15099 | 0.612437 |
| ems | Hsp60 | 0.612328 |
| CG1024 | Vir | 0.611803 |
| CG12219 | Wdn | 0.610983 |
| MBD-like | CG9797 | 0.600319 |
| B-H2 | so | 0.598897 |
| Transcription Factor Gene | Target Gene | Correlation Score |
|---|---|---|
| CG13510 | CG13511 | 0.672 |
| Dmrt93B | CG13138 | 0.658 |
| Salm | Salr | 0.641 |
| Dmrt93B | Gr63a | 0.635 |
| Fkh | MRE23 | 0.616 |
| Shn | CG14471 | 0.615 |
| Dmrt93B | CG14932 | 0.607 |
| Jra | CG14207 | 0.604 |
| Srp | CG30046 | 0.593 |
| Dmrt93B | Hsp70Aa | 0.593 |
| Model/Approach | No. of Validated Links | Total Predicted Links | No. of Transcription Factors | % of Prediction Dataset |
|---|---|---|---|---|
| GENIE3 | 3846 | 579,908 | 132 | 0.66% |
| GRNBoost2 | 28,957 | 4,879,738 | 211 | 0.59% |
| BioGRNsemble | 3703 | 534,843 | 131 | 0.69% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammed, A.J.; Khalifa, A. Drosophila Eye Gene Regulatory Network Inference Using BioGRNsemble: An Ensemble-of-Ensembles Machine Learning Approach. BioMedInformatics 2024, 4, 2186-2200. https://doi.org/10.3390/biomedinformatics4040117
Mohammed AJ, Khalifa A. Drosophila Eye Gene Regulatory Network Inference Using BioGRNsemble: An Ensemble-of-Ensembles Machine Learning Approach. BioMedInformatics. 2024; 4(4):2186-2200. https://doi.org/10.3390/biomedinformatics4040117
Chicago/Turabian StyleMohammed, Abdul Jawad, and Amal Khalifa. 2024. "Drosophila Eye Gene Regulatory Network Inference Using BioGRNsemble: An Ensemble-of-Ensembles Machine Learning Approach" BioMedInformatics 4, no. 4: 2186-2200. https://doi.org/10.3390/biomedinformatics4040117
APA StyleMohammed, A. J., & Khalifa, A. (2024). Drosophila Eye Gene Regulatory Network Inference Using BioGRNsemble: An Ensemble-of-Ensembles Machine Learning Approach. BioMedInformatics, 4(4), 2186-2200. https://doi.org/10.3390/biomedinformatics4040117