

Predictions of Programmed Cell Death Ligand 1 Blockade Therapy Success in Patients with Non-Small-Cell Lung Cancer

Abstract
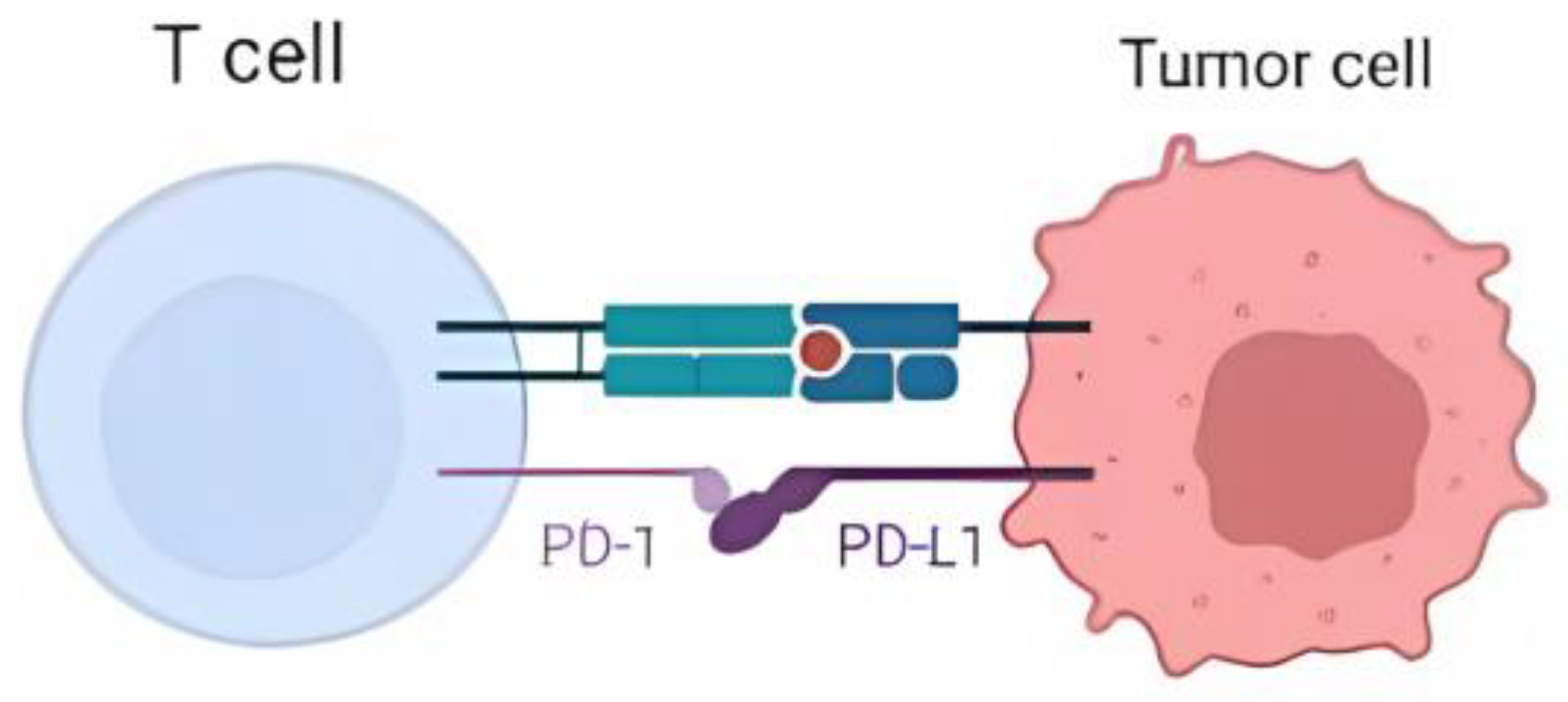
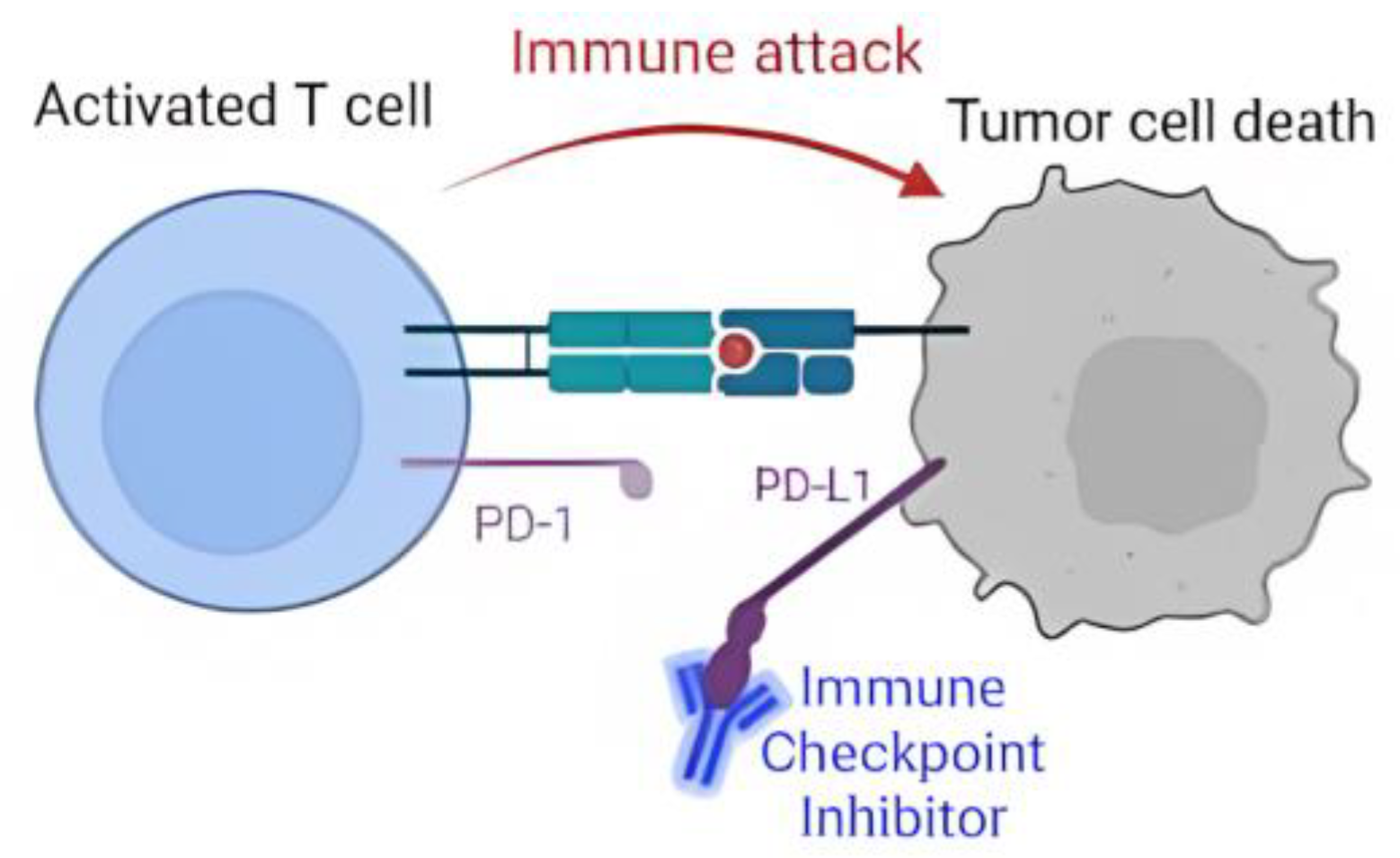
:1. Introduction
Spread of Non-Small-Cell Lung Cancer
2. Materials and Methods
2.1. Materials
2.2. Methodology
3. Results and Discussions
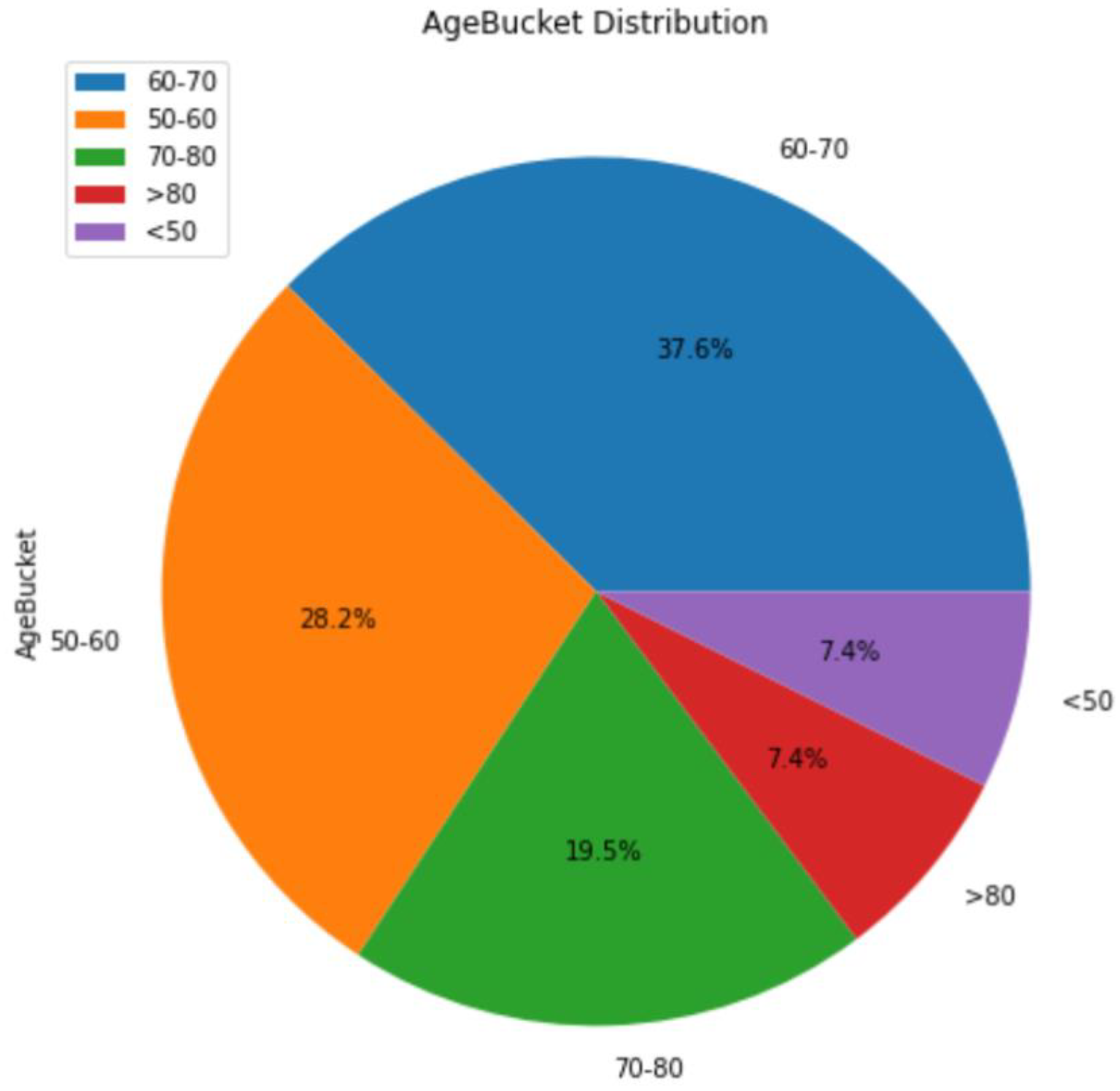
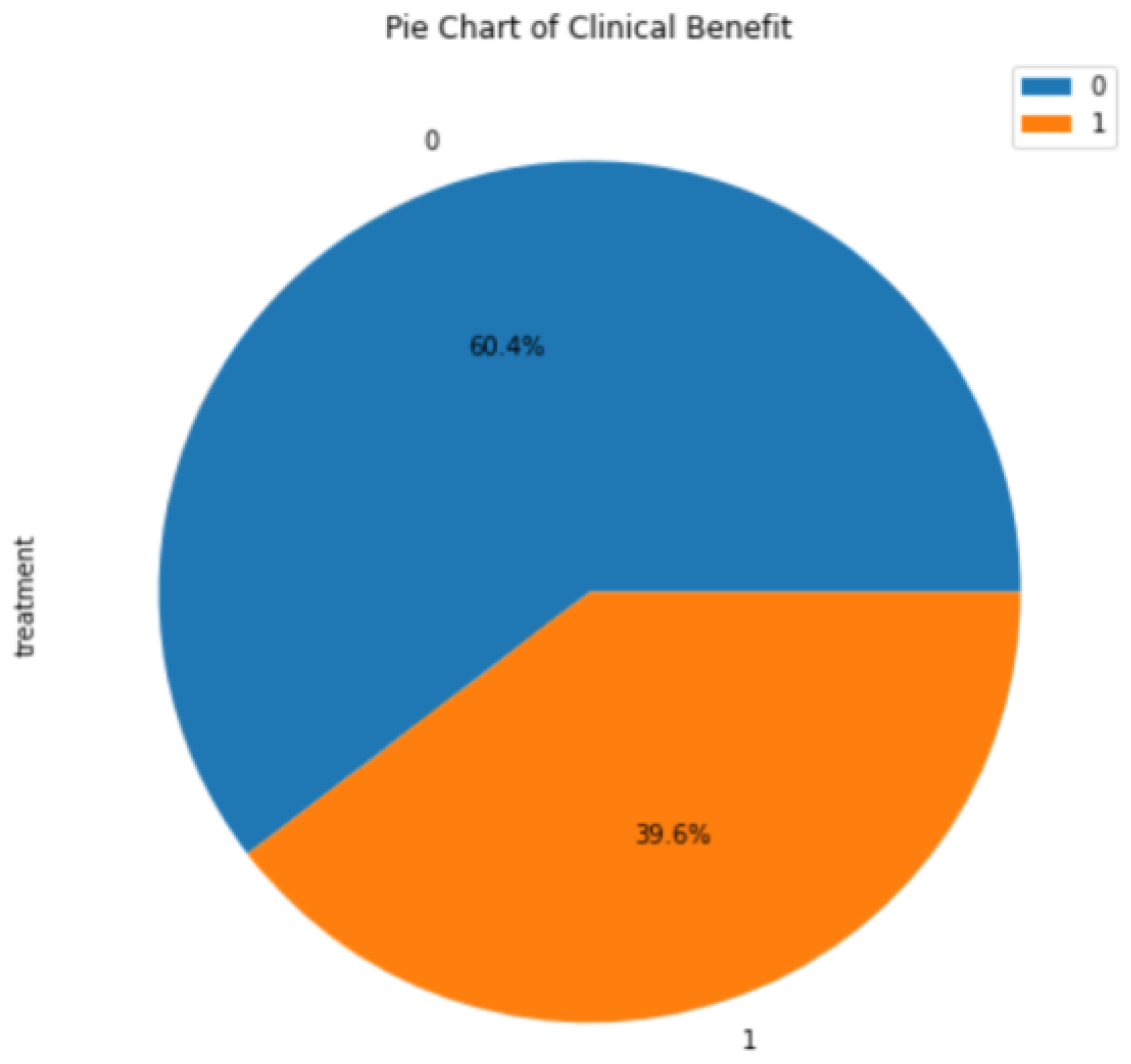
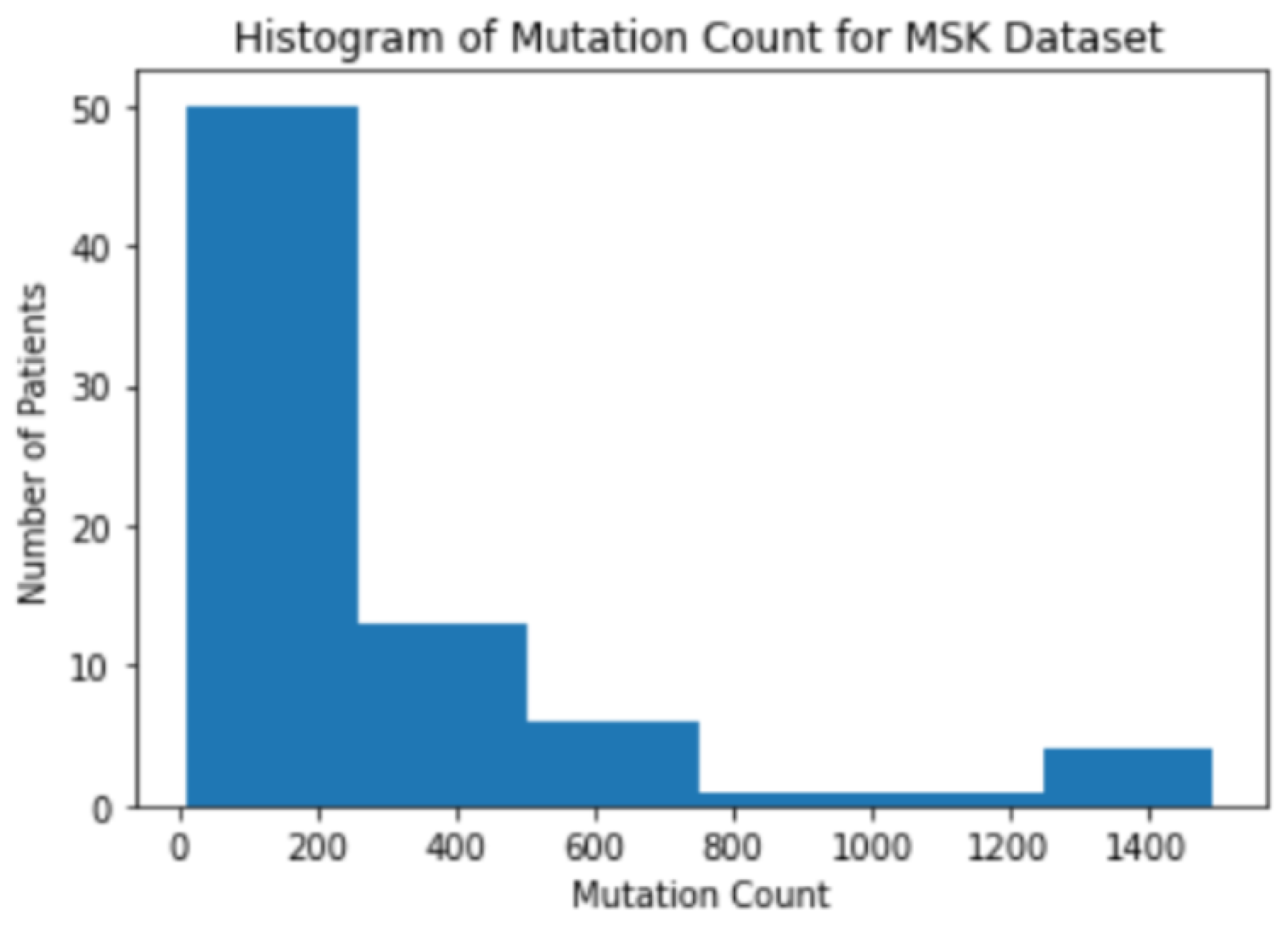
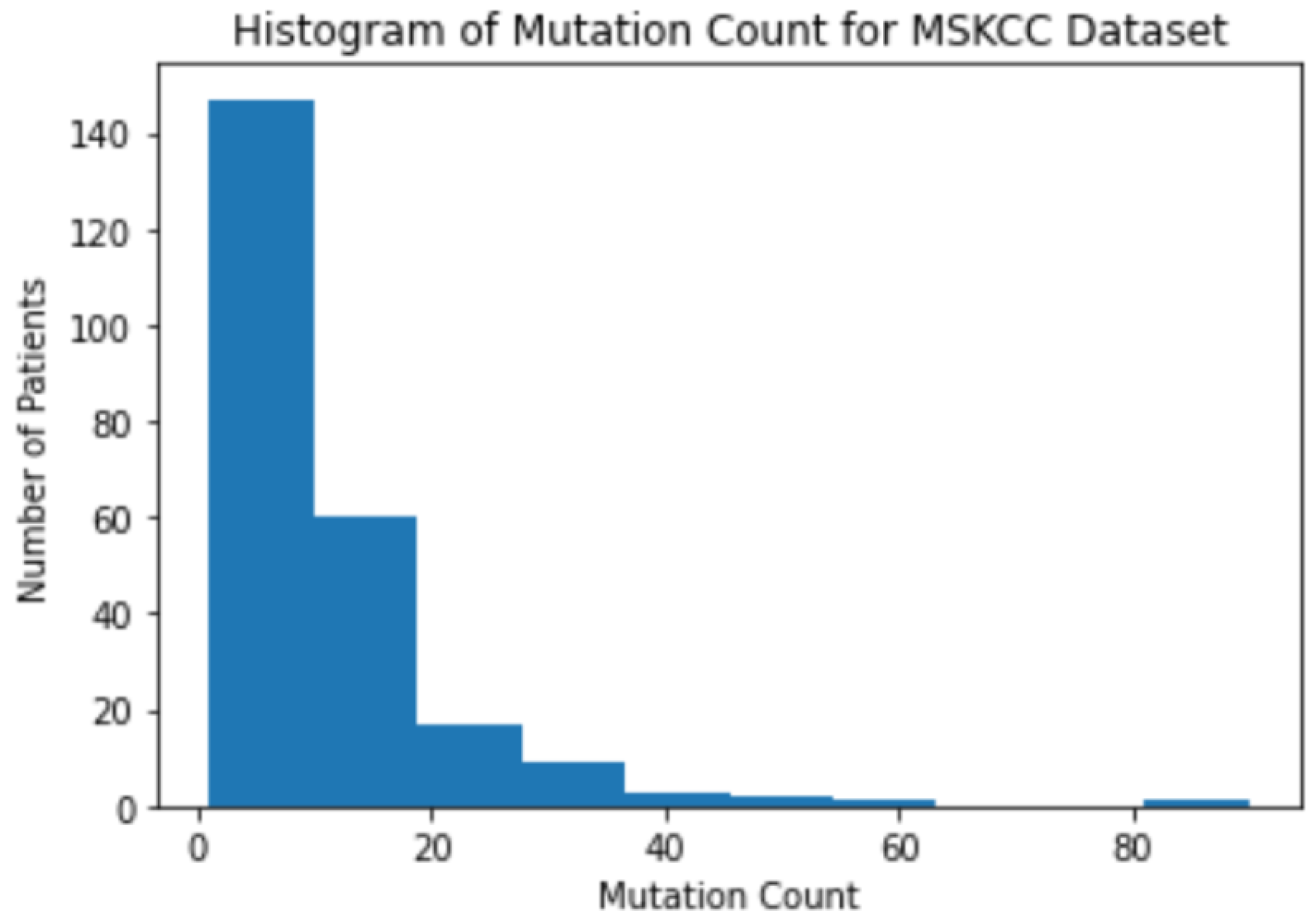
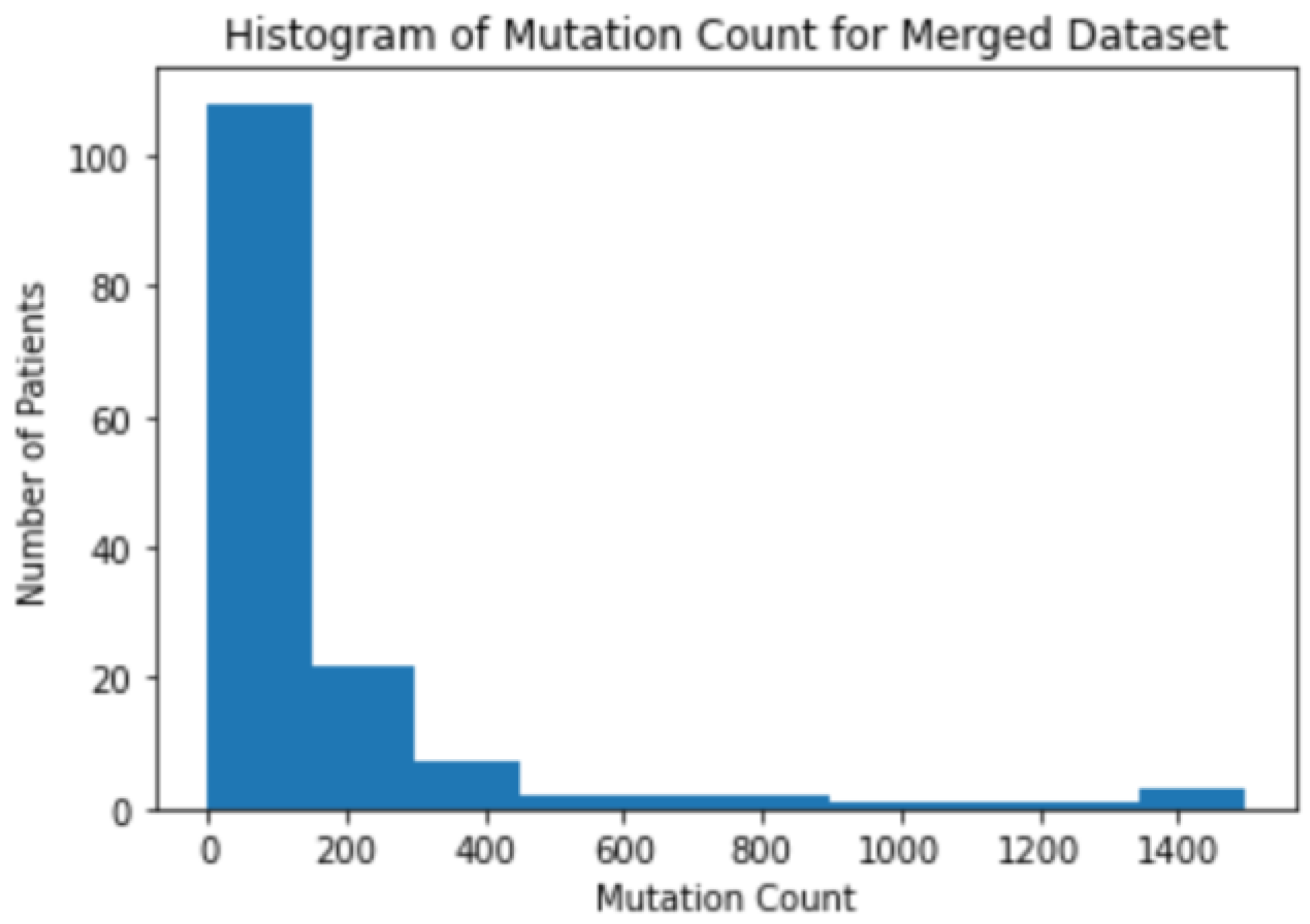
3.1. Background Information for Datasets
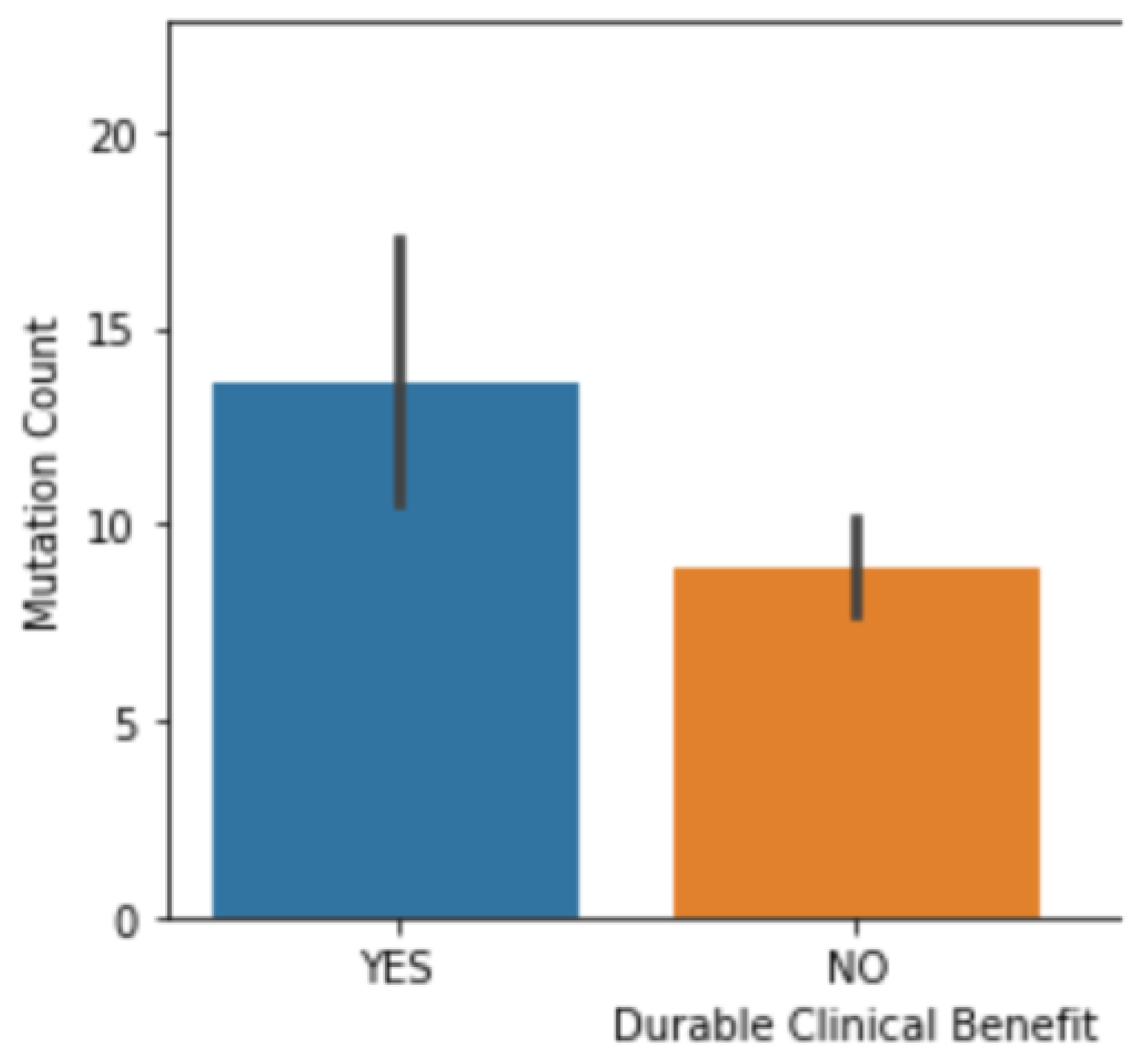
3.2. The Correlation between Mutation Counts and Clinical Benefits
3.3. Correlation Coefficient Test
3.4. Leave-One-Out Cross-Validation
3.5. Machine Learning Models
4. Conclusions
4.1. Conclusions
4.2. Future Investigations
4.3. Applications
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Siegel, R.L.; Jemal, A. Lung cancer statistics. Adv. Exp. Med. Biol. 2016, 893, 1–19. [Google Scholar]
- Gene Mutations in Non-Small-Cell Lung Cancer. Available online: https://www.webmd.com/lung-cancer/story/nsclc-gene-mutations (accessed on 29 July 2022).
- Cheng, P.-C.; Cheng, Y.-C. Correlation between familial cancer history and epidermal growth factor receptor mutations in Taiwanese never smokers with non-small cell lung cancer: A case-control study. J. Thorac. Dis. 2015, 7, 281–287. [Google Scholar] [PubMed]
- Ettinger, D.S.; Akerley, W.; Bepler, G.; Blum, M.G.; Chang, A.; Cheney, R.T.; Yang, S.C. Non-small-cell lung cancer. Nat. Rev. Dis. Primers 2015, 1, 15009. [Google Scholar]
- NCI Dictionary of Cancer Terms. National Cancer Institute, 2 February 2011. Available online: https://www.cancer.gov/publications/dictionaries/cancer-terms/def/pd-l1 (accessed on 29 July 2022).
- PDL1 (Immunotherapy) Tests. Available online: https://medlineplus.gov/lab-tests/pdl1-immunotherapy-tests/ (accessed on 29 July 2022).
- Gavrielatou, N.; Shafi, S.; Gaule, P.; Rimm, D.L. PD-L1 expression scoring: Non-interchangeable, non-interpretable, neither, or both. J. Natl. Cancer Inst. 2021, 113, 1613–1614. [Google Scholar] [CrossRef]
- Immune Checkpoint Inhibitors. National Cancer Institute, 24 September 2019. Available online: https://www.cancer.gov/about-cancer/treatment/types/immunotherapy/checkpoint-inhibitors (accessed on 29 July 2022).
- Doroshow, D.B.; Bhalla, S.; Beasley, M.B.; Sholl, L.M.; Kerr, K.M.; Gnjatic, S.; Hirsch, F.R. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2021, 18, 345–362. [Google Scholar] [CrossRef] [PubMed]
- Akinleye, A.; Rasool, Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J. Hematol. Oncol. 2019, 12, 92. [Google Scholar] [CrossRef] [PubMed]
- Verma, V.; Sprave, T.; Haque, W.; Simone, C.B.; Chang, J.Y.; Welsh, J.W.; Thomas, C.R. A systematic review of the cost and cost-effectiveness studies of immune checkpoint inhibitors. J. Immunother. Cancer 2018, 6, 128. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Nathanson, T.; Rizvi, H.; Creelan, B.C.; Sanchez-Vega, F.; Ahuja, A.; Wolchok, J.D. Genomic features of response to combination immunotherapy in patients with advanced non-small-cell lung cancer. Cancer Cell 2018, 33, 843–852.e4. [Google Scholar] [CrossRef] [PubMed]
- CBioPortal for Cancer Genomics. 2018. Available online: https://www.cbioportal.org/study/clinicalData?id=nsclc_pd1_msk_2018 (accessed on 22 October 2023).
- CBioPortal for Cancer Genomics. 2018. Available online: https://www.cbioportal.org/study/clinicalData?id=nsclc_mskcc_2018 (accessed on 22 October 2023).
- 1.9. Naive Bayes. Scikit. (n.d.-b). Available online: https://scikit-learn.org/stable/modules/naive_bayes.html (accessed on 22 October 2023).
- 1.10. Decision Trees. Scikit. (n.d.-c). Available online: https://scikit-learn.org/stable/modules/tree.html (accessed on 22 October 2023).
- 1.1. Linear Models. Scikit. (n.d.-a). Available online: https://scikit-learn.org/stable/modules/linear_model.html#logistic-regression (accessed on 22 October 2023).
- CDC. Current Cigarette Smoking among Adults in the United States, Centers for Disease Control and Prevention. 16 March 2022. Available online: https://www.cdc.gov/tobacco/data_statistics/fact_sheets/adult_data/cig_smoking/index.htm (accessed on 29 July 2022).
- Bailey, C.; MIIA Health Trust Manager. Gene Therapies Offer Breakthrough Results but Extraordinary Costs. Massachusetts Municipal Association (MMA), 18 March 2020. Available online: https://www.mma.org/gene-therapies-offer-breakthrough-results-but-extraordinary-costs/ (accessed on 29 July 2022).
- Kim, H.; Liew, D.; Goodall, S. Cost-effectiveness and financial risks associated with immune checkpoint inhibitor therapy. Br. J. Clin. Pharmacol. 2020, 86, 1703–1710. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Xin, W.; Tong, Y.; Sun, J.; Xu, G.; Ye, Z.; Rao, Y. Cost effectiveness of immune checkpoint inhibitors for treatment of non-small cell lung cancer: A systematic review. PLoS ONE 2020, 15, e0238536. [Google Scholar] [CrossRef] [PubMed]
- Iivanainen, S.; Koivunen, J.P. Possibilities of improving the clinical value of immune checkpoint inhibitor therapies in cancer care by optimizing patient selection. Int. J. Mol. Sci. 2020, 21, 556. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, K.K.; Voabil, P.; Schumacher, T.N.; Voest, E.E. Genomics- and transcriptomics-based patient selection for cancer treatment with immune checkpoint inhibitors: A review. JAMA Oncol. 2016, 2, 1490–1495. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Xin, W.; Tong, Y.; Sun, J.; Xu, G.; Ye, Z.; Rao, Y. Adverse effects of immune-checkpoint inhibitors: Epidemiology, management and surveillance. Nat. Rev. Clin. Oncol. 2019, 16, 563–580. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Correlation Coefficient |
|---|---|
| Nonsynonymous Mutation Burden | 0.3730 |
| Predicted Neoantigen Burder | 0.3392 |
| Mutation Count | 0.3261 |
| Tumor Mutation Burden | 0.2655 |
| PD-L1 | 0.2362 |
| Smoking History | 0.1445 |
| Models/Datasets | MSK | MSKCC | Merged |
|---|---|---|---|
| GNB | 0.4107 | 0.2500 | 0.3025 |
| Decision Tree | 0.5000 | 0.3529 | 0.4958 |
| Logistic Regression | 0.3928 | 0.2352 | 0.3109 |
| Models/Datasets | MSK | MSKCC | Merged |
|---|---|---|---|
| GNB | 71.43% | 77.78% | 73.33% |
| Decision Tree | 85.71% | 55.56% | 73.33% |
| Logistic Regression | 78.57% | 77.78% | 70.00% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, T.; Qawasmeh, T.; McCalla, S. Predictions of Programmed Cell Death Ligand 1 Blockade Therapy Success in Patients with Non-Small-Cell Lung Cancer. BioMedInformatics 2023, 3, 1060-1070. https://doi.org/10.3390/biomedinformatics3040063
Gupta T, Qawasmeh T, McCalla S. Predictions of Programmed Cell Death Ligand 1 Blockade Therapy Success in Patients with Non-Small-Cell Lung Cancer. BioMedInformatics. 2023; 3(4):1060-1070. https://doi.org/10.3390/biomedinformatics3040063
Chicago/Turabian StyleGupta, Taksh, Tamara Qawasmeh, and Serena McCalla. 2023. "Predictions of Programmed Cell Death Ligand 1 Blockade Therapy Success in Patients with Non-Small-Cell Lung Cancer" BioMedInformatics 3, no. 4: 1060-1070. https://doi.org/10.3390/biomedinformatics3040063
APA StyleGupta, T., Qawasmeh, T., & McCalla, S. (2023). Predictions of Programmed Cell Death Ligand 1 Blockade Therapy Success in Patients with Non-Small-Cell Lung Cancer. BioMedInformatics, 3(4), 1060-1070. https://doi.org/10.3390/biomedinformatics3040063