2. Methodology and Theory
2.3. Mathematical Overview of Weighted Trajectory Analysis
Weighted trajectory analysis plots the health status of treatment arms as a function of time. Time values must be discrete but can correspond to days, weeks, months, or any chosen interval. For each time value on the x-axis, there is a corresponding score on the y-axis: a weighted health status. The higher the weighted health status, the healthier the group is. This score is scaled by the initial size of the treatment arm to facilitate simple comparison of groups with unequal size.
Consider a group of n patients with toxicity grades ranging from grade zero (asymptomatic/mild toxicity) to grade five (death related to an adverse event). The weighted health status at time point j is denoted by , where j = 0, 1, …, z. For each treatment arm, has a maximum value of 1 and a minimum value of 0. Suppose we begin a trial with all patients having no disease burden at grade zero: = = 1. A trial with the highest possible morbidity requires all patients to experience grade five toxicity (death): at this point, will drop to 0.
We let represent the severity score for the ith patient at time j, i = 1, …, n. The severity score is identical to their ordinal score for the variable of interest. If the range of the ordinal variable of interest does not have 0 as one extreme end, all values must be shifted to set 0 as the starting score (the polarity may also be reversed so that 0 represents peak health status). All patients begin the trial at grade zero, which reflects = 0. If a patient labeled with index 50 has a grade-three injury at the seventh time point, their severity score = 3.
Scaling for the WTA curve is performed through normalizing to a minimum of 0 and a maximum of 1 by using the initial weight of the treatment arm. This weight,
, is the product of the starting patient count
and the range of the ordinal variable of interest
r:
Suppose the initial size of the group,
, is 100 patients. The range
r for the ordinal variable (toxicity grade) is 5. Then,
is 500. The value of the weight changes over time due to patient censoring reflected by a drop in
. The general equation for
is provided in
Section 2.5 and is used in the weighted logrank test. However, for scaling and plotting
U, only the initial weight of a given treatment arm,
, is required.
The initial value
is a perfect score of 1.
Subsequent values of
U deviate based on observed event occurrences
. We define event occurrence as a change in the variable of interest for a given patient
i at time
j:
Therefore, the observed event score for a group of
n patients is defined as
with patients censored following time
j not contributing to the sum. Events and resulting changes in treatment arm trajectory are always scaled by
. Using this event definition,
can be calculated iteratively from
:
Alternatively,
for any given time point can be computed as follows:
Values for
at a given time point can be negative, and these represent cases in which the treatment arm improved in overall health status. From Equations (
6) and (
7), it follows that a negative value of
produces an increase in the weighted health status
.
2.5. The Weighted Logrank Test—Analytical Method
We define an event as a change in the severity score of a given condition. Let
be the severity score for the
ith individual in group A at time
, where
and
. Define
as the change in the severity score from time
to
.
Without loss of generality, we consider a severity score ranging from stage zero to stage four. As a result, has a total of nine possible values () if the observation of this person is uncensored at
Let L be the total number of possible values taken by the change variable . When a severity score takes values from 0 to 4, ;
Let W be the ordered non-decreasing list of the L possible change values. When a severity score takes values from 0 to 4, ;
Let be the lth element of ;
Let
be the number of subjects in group A at
whose change values equal
:
where
when
and 0 otherwise;
Let be the number of subjects in group B at whose change values equal ;
is the total number of patients whose change values equal at .
The information at
can be summarized in a 2 × 10 table:
| Observed values of () | −4 | −3 | −2 | −1 | 0 | 1 | 2 | 3 | 4 | | At risk at |
| Group A | | | | | | | | | | | |
| Group B | | | | | | | | | | | |
| | | | | | | | | | | | |
Under the null hypothesis , follows a multivariate hypergeometric distribution conditional on the margins .
We can show that the mean and variance of
, where
, are
For distinct
, we can derive the covariance of
and
These moment results are derived from the definition of multivariate hypergeometric distribution. To account for the direction and the magnitude of the change variable, we define the
observed weighted changes as
When a severity score is defined as a range from 0 to 4, the weight
takes the values of
for
. The expected value of
can be written as
When the event is coded as a binary outcome, this weighted change
is reduced to the
defined above. Using the results in Equations (
17) and (
18), we can write the variance of the weighted score
as
where
is defined in Equation (
18) when
and in Equation (
17) when
.
Similarly, we can aggregate the observed/expected weighted changes across all
K time points and define a
Z test statistic. The weighted logrank test statistic is defined as
which follows the standard normal distribution
under the null hypothesis
Equivalently,
i.e., the square of the
Z test statistic follows a chi-square distribution with one degree of freedom.
The asymptotic result in Equation (
22) is based on the assumption that the total number of distinct failure times recorded in the pooled samples (i.e.,
K) is sufficiently large. For smaller trials with shorter follow-up periods, this analytical method can provide conservative conclusions and result in type II errors below the designated significance level, as demonstrated in
Section 3.3. To complement the analytical method, we also propose a bootstrap-based approach for calculating
p-values, which, despite requiring greater computational effort, remains accurate and sensitive independent of trial sizes.
3. Simulation Study One—Toxicity
In our first clinical trial simulation study, we demonstrate the functionality of WTA and present its advantages over KM analysis. We establish the strength of our novel method through a rigorous power comparison between KM estimation, the GEE, and both analytical and simulated approaches to WTA.
The design was a phase III comparison of toxicity outcomes from chemotherapy between two treatment arms (control and treatment, 1:1 allocation). The variable of interest was CTCAE toxicity: grades range from one (mild/no toxicity) to five (death from toxicity) [
6]. For example, the grades of oral mucositis are: (1) asymptomatic/mild, (2) moderate pain or ulcer that does not interfere with oral intake, (3) severe pain interfering with oral intake, (4) life threatening consequences indicating urgent intervention, and (5) death. For the purposes of WTA, the ordinal range of 1–5 was shifted to 0–4, with censoring thus taking place at grade four.
The simulation study was generated using Python 3.7 [
8]. Study simulations are a stochastic process in which randomly generated numbers are programmed to mirror fluctuating toxicities experienced by groups of patients undergoing chemotherapy cycles with daily measurements of treatment toxicity. Each instance of the simulation requires a specified hazard ratio and sample size prior to the stochastic generation of toxicity.
Table 2 provides a snapshot of the results for a single simulated clinical trial.
Each patient (represented by an ID number) has a risk of developing treatment toxicity over time. This risk is determined by their treatment group and the numbers of days they have spent in the study. The values within
Table 2 were assigned as follows:
Treatment group: randomly assigned as zero or one with the constraint of having an equal number of patients allocated to each group;
Duration: the number of days a patient remains within the trial was programmed as a random value within a uniform distribution of 0 to 50 days;
Toxicity grade: computed for each patient on a daily basis for the extent of their assigned duration. To model the trajectory of toxicity grade over time, we made the following simplifying assumptions:
- (a)
On any given day, patients can rise or fall by a single toxicity grade;
- (b)
Transitions in toxicity grade are random, but a larger hazard ratio suggests a greater chance of exacerbation and lower chance of recovery;
- (c)
A patient is censored once their pre-assigned duration within the trial has elapsed or they reach maximum toxicity, in this case representing death, whichever occurs first.
A hazard ratio for control:treatment was modeled for the control group to have a higher toxicity burden through time compared to the treatment group (the value was programmed as 1.0 or higher). For the control group, the probability of exacerbation was a base probability of 0.10 multiplied by the hazard ratio. If exacerbation did not occur and the current stage was above the minimum, the probability of recovery would be a base probability of 0.05 divided by the hazard ratio. Patients in the treatment group fluctuated based on base probabilities alone. Once a patient reached the maximum toxicity or their maximum assigned duration, they were censored.
6. Discussion
WTA was created to (a) evaluate phase III clinical trials that assess outcomes defined by various ordinal grades (or stages) of severity; (b) permit continued analysis of participants following changes in the variable of interest; and (c) demonstrate the ability of an intervention to both prevent the exacerbation of outcomes and improve recovery and the time course of these effects. Its development was inspired by a pressure injury study—a disease process characterized by several stages of severity—for which Kaplan–Meier estimates would fail to capture the complete trajectory. Despite its limitations, KM estimation provides crucial advantages, such as patient censoring, rapid interpretation of survival plots, and a simple hypothesis test. To this end, we sought to create a statistical method that built on the foundations of Kaplan–Meier analysis but would overcome the inherent limitations of the technique.
We built the WTA toolkit based on expansion and extension of the Kaplan–Meier methodology. We adapted KM estimation to support analysis of ordinal variables by redefining events as changes in disease scores rather than assigning “1” and omitting the patient from further analysis. We adapted KM estimation to permit fluctuating outcomes (worsening and improvement of the ordinal outcome) by plotting a novel weighted health status as opposed to probability. We retained the ability to censor patients at the time of non-informative status. These changes warranted a novel significance test, for which we developed a modification of Peto et al.’s logrank test [
3] This analytical approach is rather conservative in its type I error rates for smaller trials, but the rate approaches 0.05 within the limit of massive trials with many distinct failure times. Thus, we developed a computational approach that is more resource-intensive but remains precise and accurate independent of trial size.
In order to explore and demonstrate the utility of WTA, we applied WTA to two randomized clinical trial simulation studies. The first clinical setting was chemotherapy toxicity, a trial in which the variable of interest ranged from one to five (shifted to zero to four), stage transitions were singular and started at zero, and up to 50 discrete time points were measured for each patient. The second setting was schizophrenia disease course, a more complex trial in which the variable of interested ranged from zero to six, stage transitions were often multiple and started at two, and up to 84 discrete time points were measured for each patient. We performed sensitivity and power comparisons across both sample size and hazard ratio. Through 1000-fold validation, WTA showed greater sensitivity and power, often requiring fewer than half the patients for comparable power to KM estimation. WTA also showed increased power compared to the GEE, likely secondary to its more robust nonparametric methodology compared to the semi-parametric GEE, at the cost of the GEE’s ability to model covariate effects. This demonstrates that designing a phase III clinical trial using our novel method as the primary endpoint can substantially lower cost, duration, and the risk of type II errors.
We also applied WTA to real-world clinical trial data. The first application was the assessment of time-dependent toxicity grades in melanoma patients receiving one of two immunotherapy treatment regimens. Although toxicities are generally reported in oncology trials as the worst grade experienced by each individual patient, this fails to capture those toxicities that resolve with treatment modification or targeted intervention. As such, the published literature suggests the prohibitive toxicity of the most effective therapy, while practitioners’ experience is that high-grade toxicities are often transient and treatable. The WTA we conducted confirmed that treatment-related toxicities of combination therapy resolved to rates close to that seen with less effective monotherapy regimens. The second application was the re-evaluation of a published phase III registration trial of an anti-angiogenic drug for the treatment of metastatic breast cancer. Although this study failed to demonstrate statistically significant improvement in the pre-defined primary endpoint, a number of secondary endpoints suggested the possibility of meaningful clinical benefit from the antiangiogenic therapy. By using an ordinal scale to describe the spectrum of clinical outcomes after therapy, spanning complete disease response, partial response, disease stability, disease progression, and death, WTA demonstrated that, although patients derived a modest benefit from antiangiogenic therapy when compared to control therapy, the difference was neither clinically nor statistically significant. The resulting graph captured the full clinical course of patients in a single figure. This result underscores that WTA did not inappropriately provide an overly sensitive analytic tool and justifies the regulatory stance that the intervention did not warrant approval for the market. Overall, the novel method affords greater specificity and reduces the likelihood of type I errors.
In aggregate, we feel the strengths of the weighted trajectory analysis statistic are its ability to capture detailed trajectory outcomes in a simple summary plot, its greater power, and its ability to map exacerbation and improvement. These strengths are built upon key advantages that make KM estimation a favored tool for clinical trial evaluation: namely, the ability to censor patients and compare treatment arms using a simple hypothesis test. WTA-dependent trial design can substantially reduce sample size requirements, increasing the practicality and lowering the cost of phase III clinical trials. However, we acknowledge several limitations of this method. WTA does not facilitate Cox regression analysis or generate the equivalent of a hazard ratio. WTA is a new technique and does not yet have a clinical or regulatory track record. WTA relies on the assumption of non-informative censoring, and investigation into alternative approaches to censoring, such as inverse-probability-of-censoring weighting (IPCW), remains important future work [
16]. Lastly, WTA requires an assumption that the change between adjacent ordinal severities is equally important independent of the levels transitioned by applying a direct numerical weight. This conversion is not always medically appropriate: taking the example of pressure injuries, a transition from stage zero to one may necessitate a topical ointment, whereas a transition from stage three to four may warrant surgical repair. Thus, the method relies on a simplifying assumption and future research will be conducted to evaluate nonlinear scoring systems. For multi-stage systems, this method remains more precise than collapsing scores to binary systems in order to use KM estimation. Alternative statistical methods, such as multi-state modeling, are recommended to elicit the transition intensities of each unique level as necessary. To encourage the evaluation and improvement of WTA, software is in development to permit biostatisticians to further test and apply WTA and potentially expand its utility.
In summary, we report the development and validation of a flexible new analytic tool for analysis of clinical datasets that permits high-sensitivity assessment of ordinal time-dependent outcomes. We see multiple clinical applications and have successfully applied the new tool in the analysis of both simulated and real-world studies with complex illness trajectories. Future directions with weighted trajectory analysis include the addition of confidence intervals to group trajectories, the addition of nonlinear weights to mirror disease burden, exploration of alternative censoring assumptions, and a regression method analogous to the Cox model.
Author Contributions
Conceptualization, U.C. and J.R.M.; methodology, U.C. and K.Z.; software, U.C.; validation, U.C. and K.Z.; formal analysis, U.C.; investigation, U.C.; resources, J.W. and J.R.M.; data curation, U.C.; writing—original draft preparation, U.C.; writing—review and editing, K.Z., J.W. and J.R.M.; visualization, U.C.; supervision, J.R.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The data that support the findings of this research are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Acknowledgments
The authors thank Britsol Myers Squibb for access to their melanoma clinical trial dataset and the TRIO-012/ROSE study team, along with the TRIO Science Committee, for access to their database.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kaplan, E.L.; Meier, P. Nonparametric Estimation from Incomplete Observations. J. Am. Stat. Assoc. 1958, 5, 457–481. [Google Scholar] [CrossRef]
- Peto, R.; Pike, M.; Armitage, P.; Breslow, N.E.; Cox, D.R.; Howard, S.V.; Mantel, N.; McPherson, K.; Peto, J.; Smith, P.G. Design and analysis of randomized clinical trials requiring prolonged observation of each patient. I. Introduction and design. Br. J. Cancer 1976, 34, 585–612. [Google Scholar] [CrossRef] [PubMed]
- Peto, R.; Pike, M.; Armitage, P.; Breslow, N.E.; Cox, D.R.; Howard, S.V.; Mantel, N.; McPherson, K.; Peto, J.; Smith, P.G. Design and analysis of randomized clinical trials requiring prolonged observation of each patient. II. Analysis and examples. Br. J. Cancer 1977, 35, 1–39. [Google Scholar] [CrossRef] [PubMed]
- Oken, M.M.; Creech, R.H.; Tormey, D.C.; Horton, J.; Davis, T.E.; McFadden, E.T.; Carbone, P.P. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am. J. Clin. Oncol. 1982, 5, 649–655. [Google Scholar] [CrossRef] [PubMed]
- American Heart Association. Classes of Heart Failure. Published 2 June 2022. Available online: https://www.heart.org/en/health-topics/heart-failure/what-is-heart-failure/classes-of-heart-failure (accessed on 29 September 2022).
- U.S. Department of Health and Human Services. Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0. Published 27 November 2017. Available online: https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/ctcae_v5_quick_reference_5x7.pdf (accessed on 23 March 2020).
- Liang, K.; Zeger, S.L. Longitudinal data analysis using generalized linear models. Biometrika 1986, 73, 13–22. [Google Scholar] [CrossRef]
- Python Software Foundation. Python Language Reference, Version 3.7. Available online: http://www.python.org (accessed on 16 March 2020).
- Davidson-Pilon, C. Lifelines: Survival analysis in Python. J. Open Source Softw. 2019, 4, 1317. [Google Scholar] [CrossRef]
- IBM Corp. IBM SPSS Statistics for Windows; Version 26.0; IBM Corp.: Armonk, NY, USA, 2017. [Google Scholar]
- Wang, D.Y.; Salem, J.E.; Cohen, J.V.; Chandra, S.; Menzer, C.; Ye, F.; Zhao, S.; Das, S.; Beckermann, K.E.; Ha, L.; et al. Fatal toxic effects associated with immune checkpoint inhibitors: A systematic review and meta-analysis. JAMA Oncol. 2018, 4, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Mackey, J.R.; Ramos-Vazquez, M.; Lipatov, O.; McCarthy, N.; Krasnozhon, D.; Semiglazov, V.; Manikhas, A.; Gelmon, K.; Konecny, G.; Webster, M.; et al. Primary results of ROSE/TRIO-12, a randomized placebo-controlled phase III trial evaluating the addition of ramucirumab to first-line docetaxel chemotherapy in metastatic breast cancer. J. Clin. Oncol. 2015, 33, 141–148. [Google Scholar] [CrossRef]
- Schwartz, L.H.; Litière, S.; De Vries, E.; Ford, R.; Gwyther, S.; Mandrekar, S.; Shankar, L.; Bogaerts, J.; Chen, A.; Dancey, J.; et al. RECIST 1.1-Update and clarification: From the RECIST committee. Eur. J. Cancer 2016, 62, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Robins, J.M.; Rotnitzky, A.; Zhao, L.P. Analysis of Semiparametric Regression Models for Repeated Outcomes in the Presence of Missing Data. J. Am. Stat. Assoc. 1995, 90, 106–121. [Google Scholar] [CrossRef]
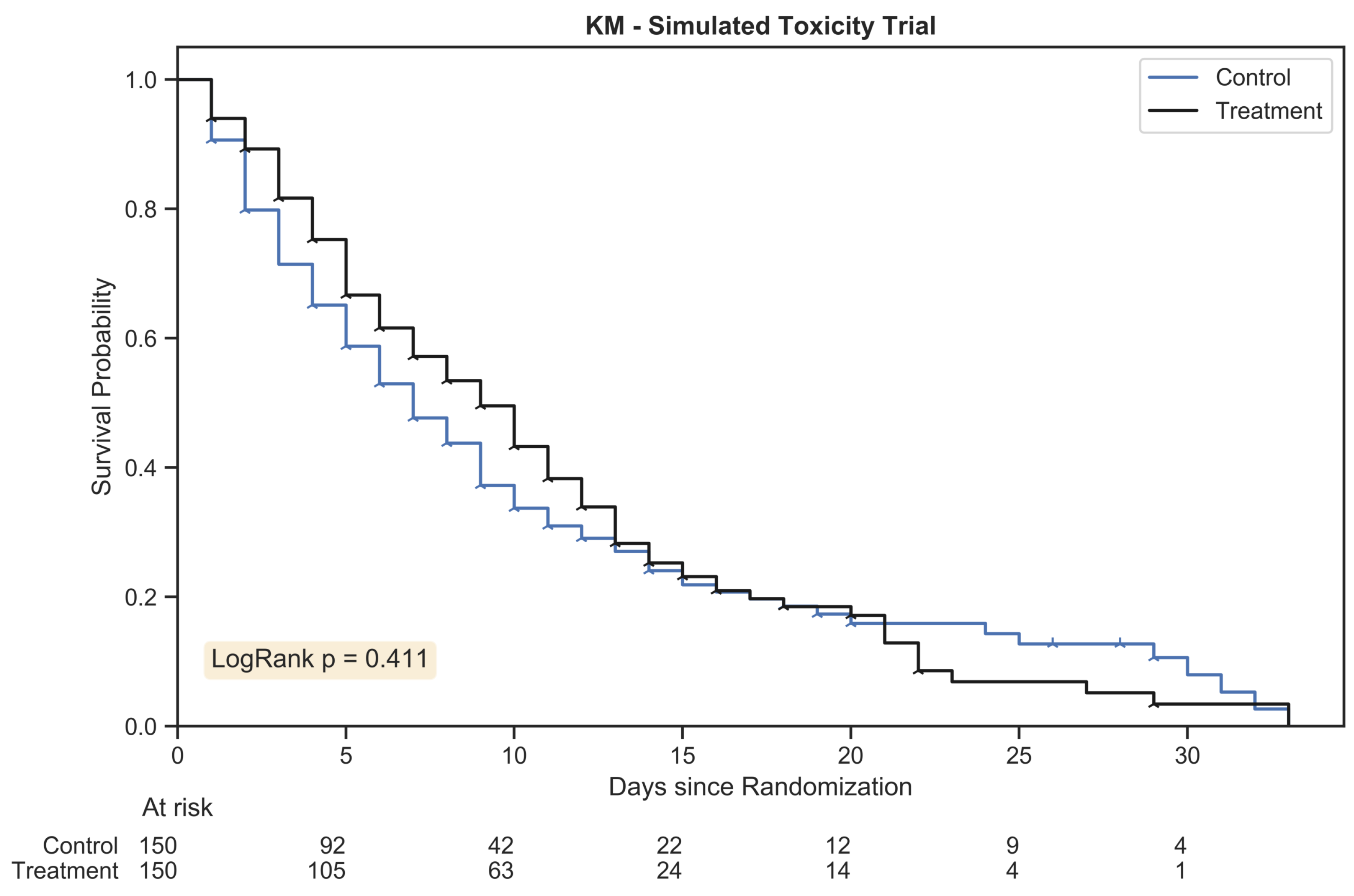
Figure 1.
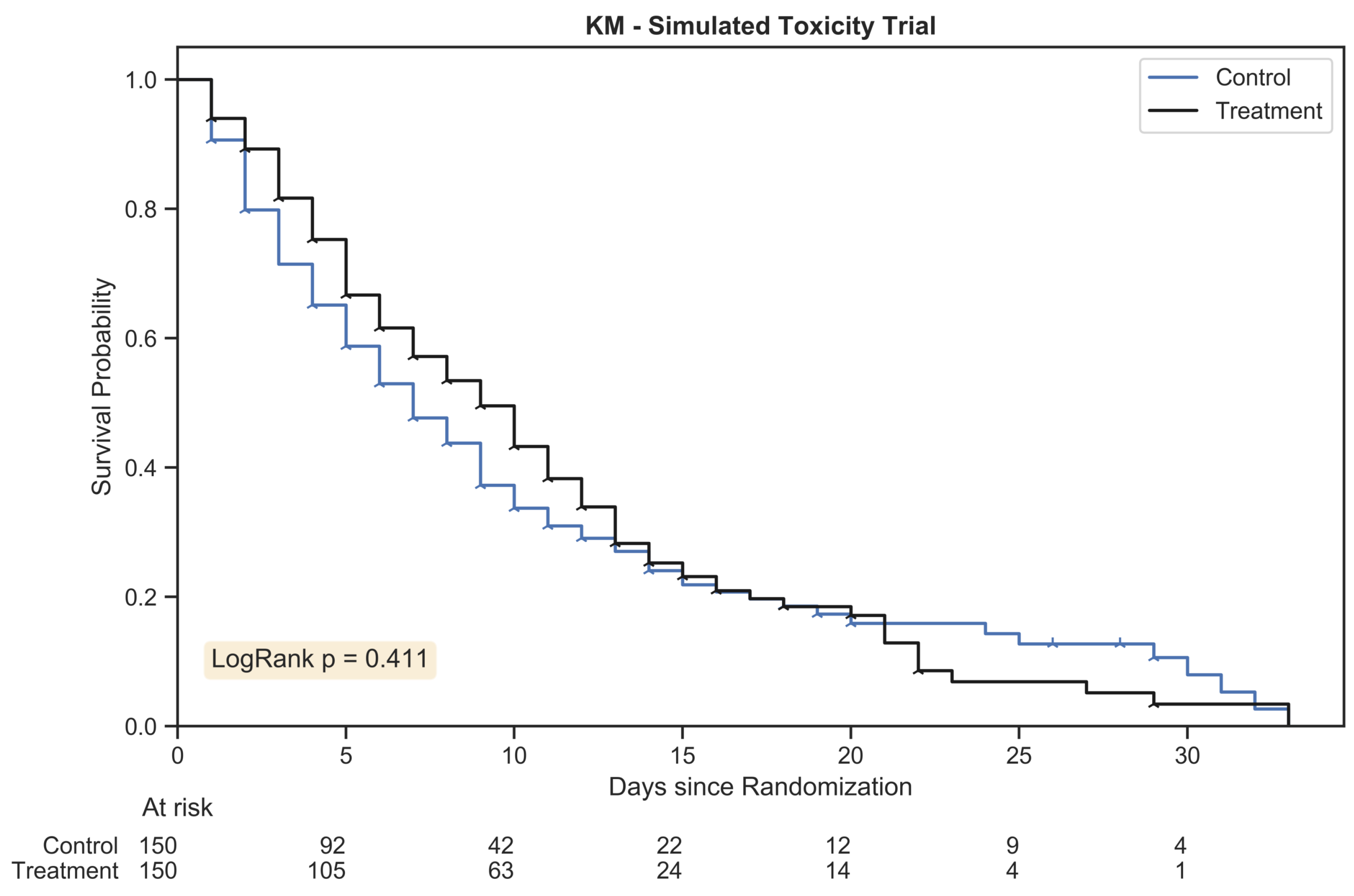
The Kaplan Meier estimator plot for a randomly generated chemotherapy toxicity trial of 300 patients with 1:1 allocation. An event was considered the onset of chemotherapy toxicity (beyond stage zero) and patients were censored once their assigned duration had been reached. The hazard ratio between treatment arms was 1.25:1.
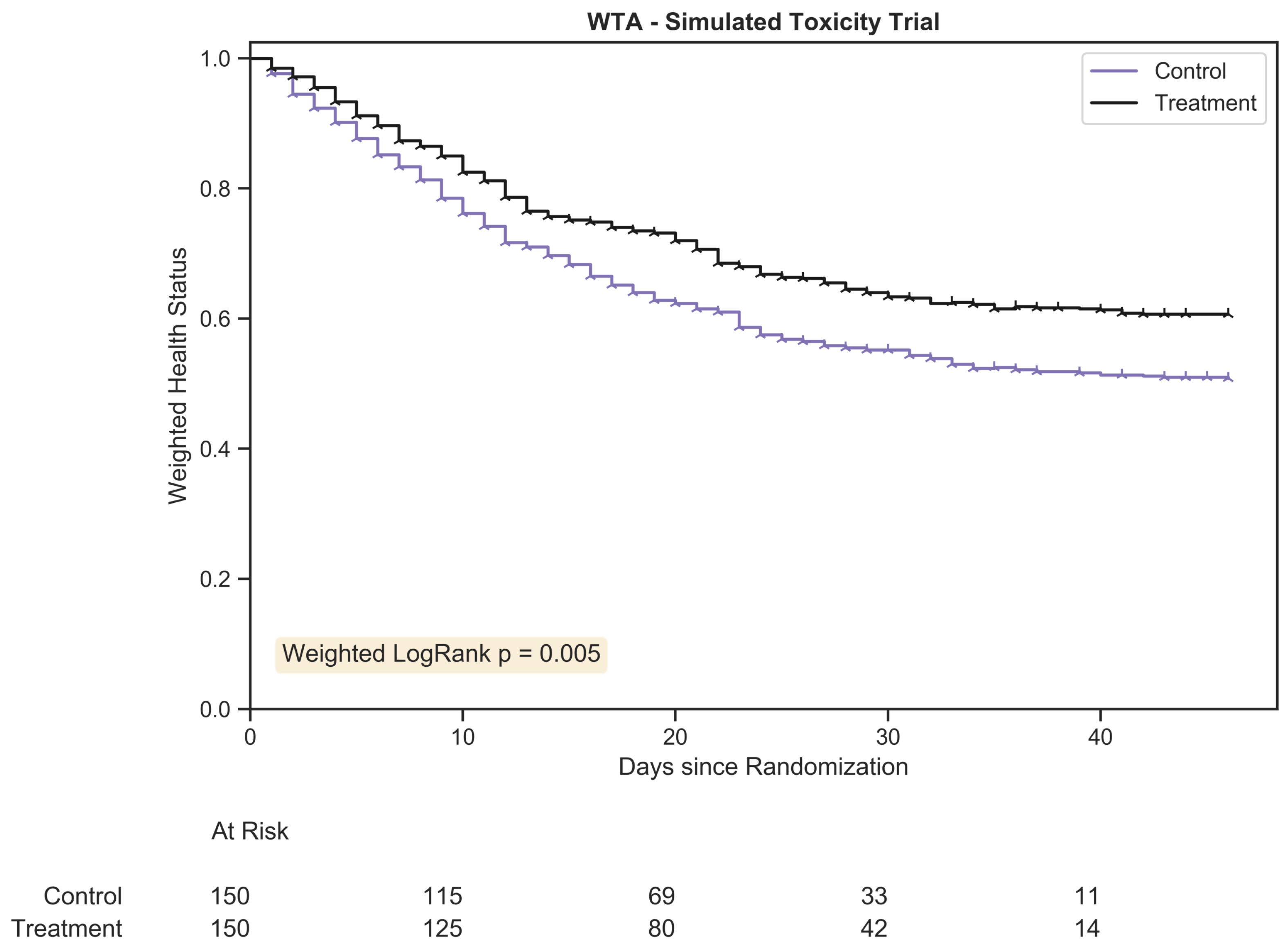
Figure 2.
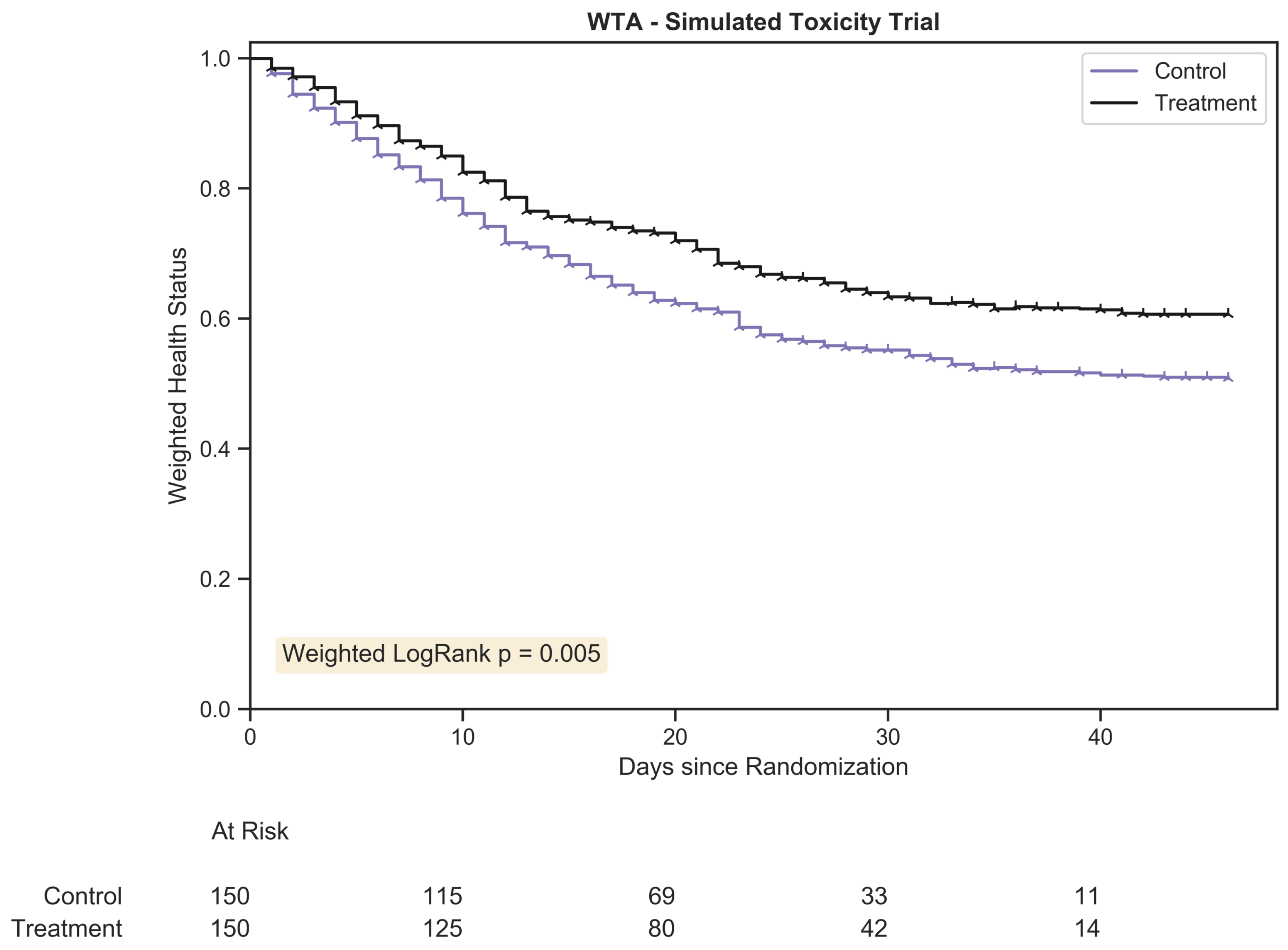
The weighted trajectory analysis plot for a randomly generated chemotherapy toxicity trial of 300 patients with 1:1 allocation. The weighted health status of both groups dropped due to increasing morbidity from chemotherapy toxicity following randomization. The hazard ratio between treatment arms was 1.25:1.
Figure 3.
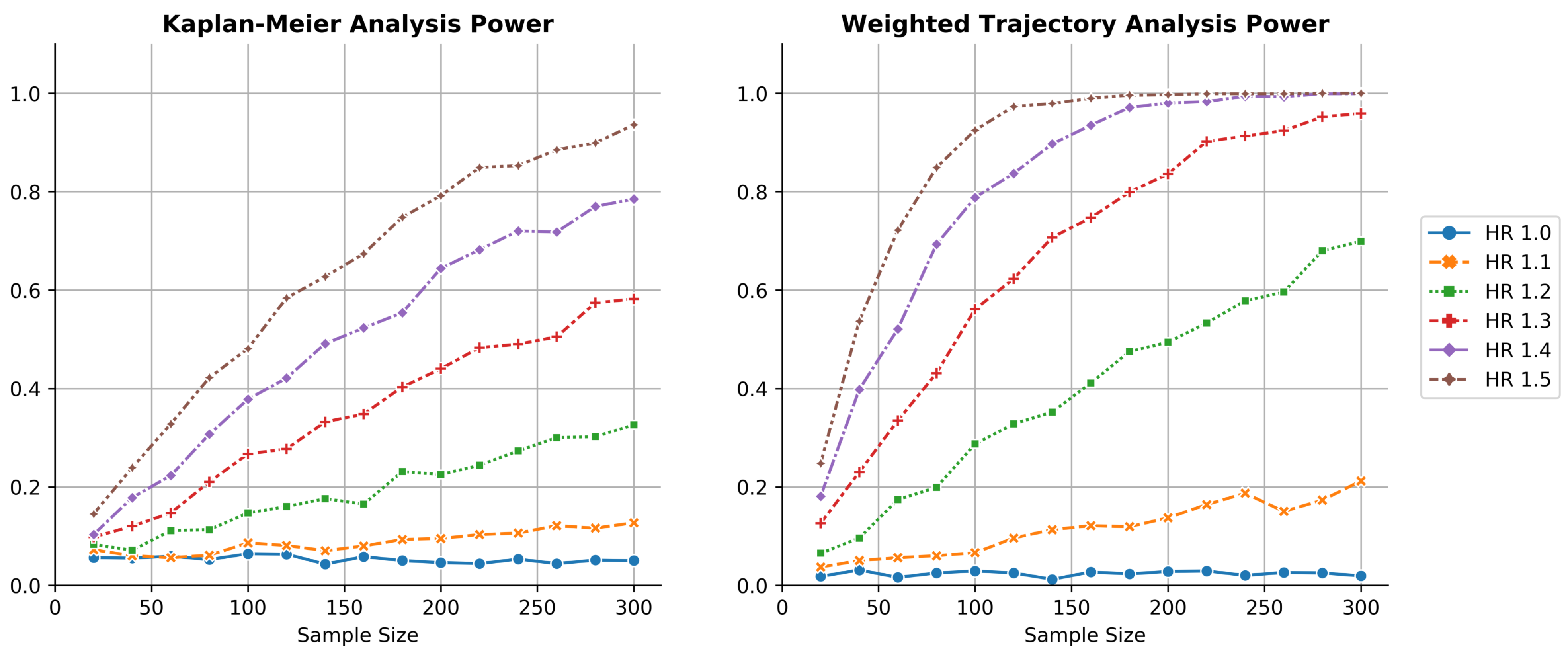
Thousandfold simulations of power as a function of sample size for both KM estimation and WTA across several hazard ratios. WTA demonstrated consistently higher power, reflecting a smaller sample size requirement during trial design. The type I error rate of WTA was approximately 0.025, indicating the method was conservative. The type I error approached 0.05 within the limit of larger trials with more distinct failure times.
Figure 4.
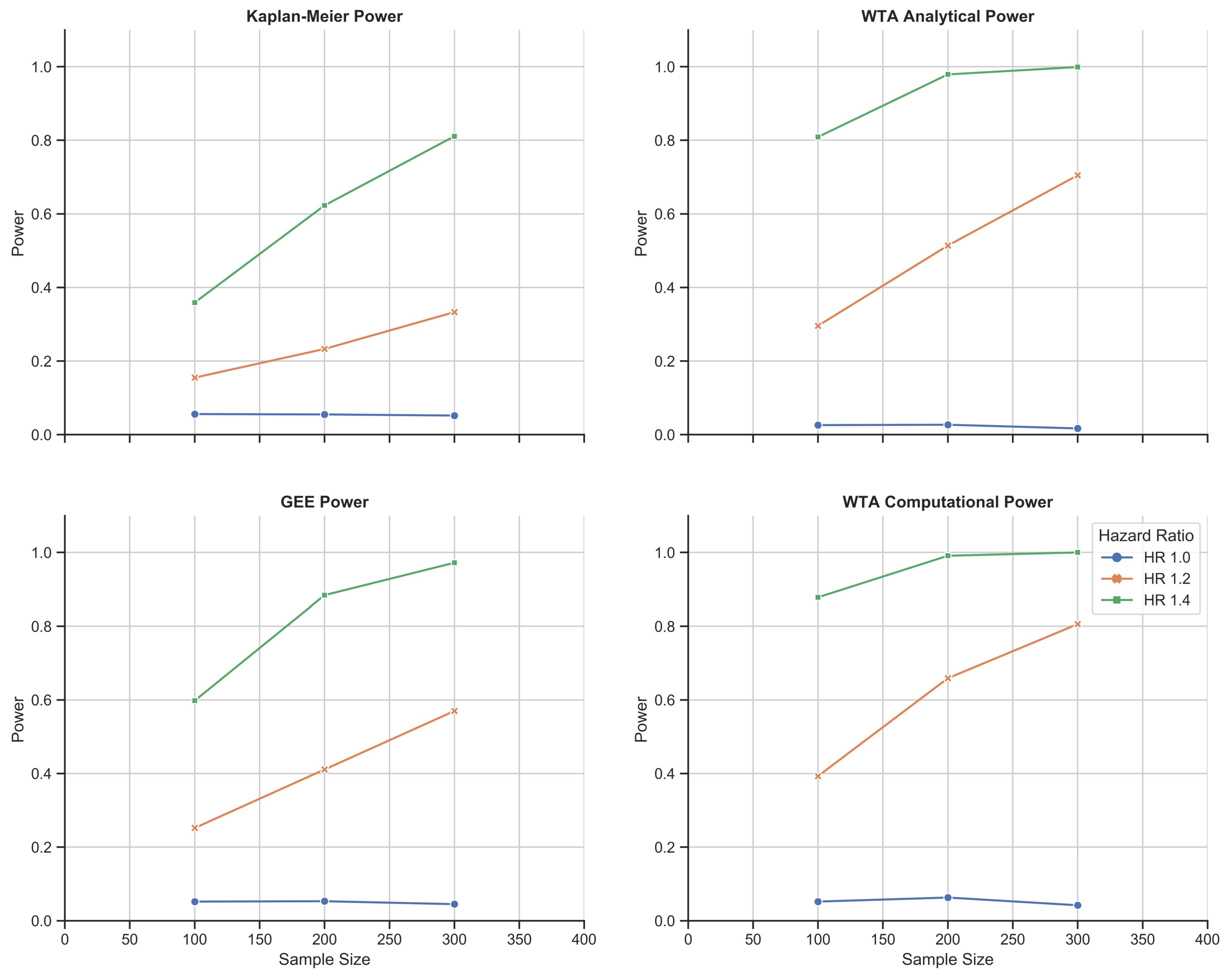
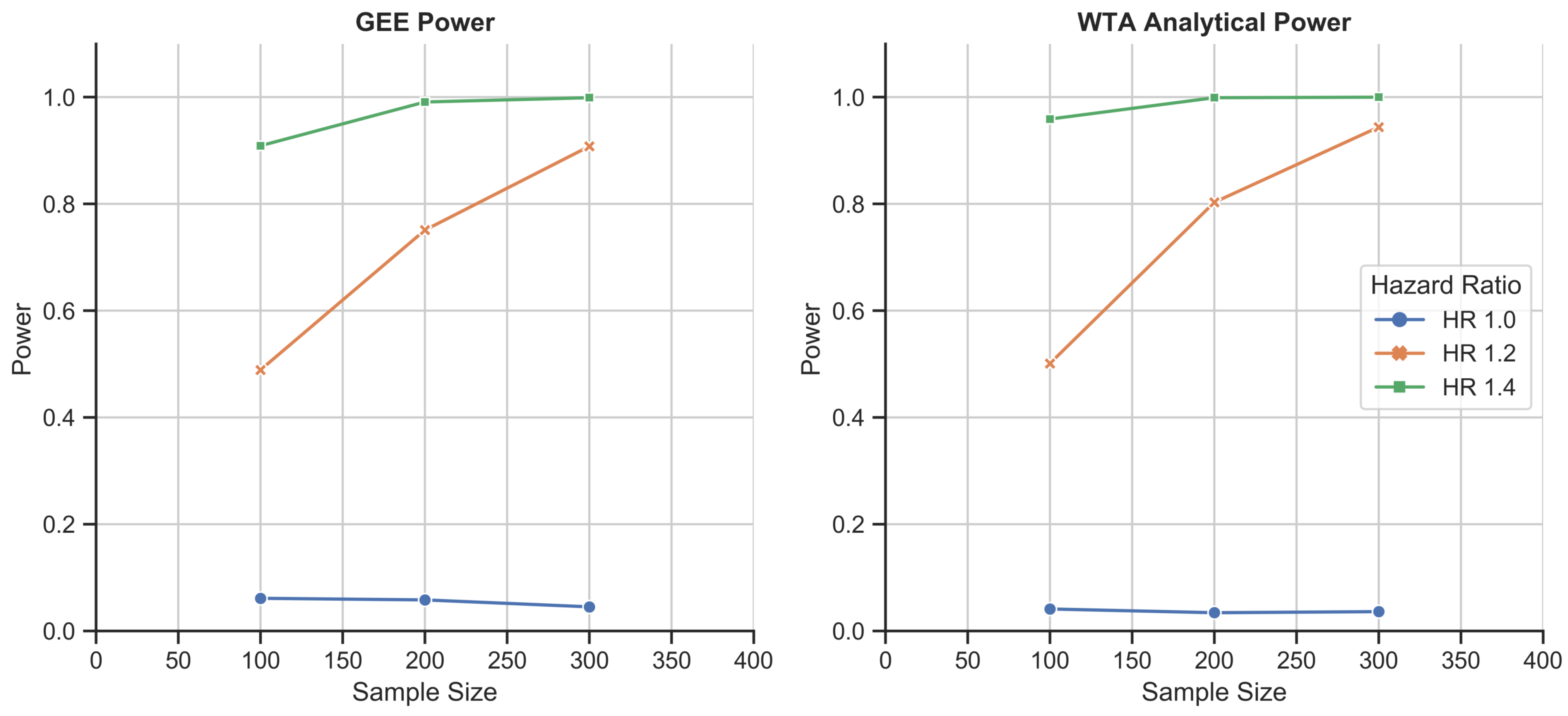
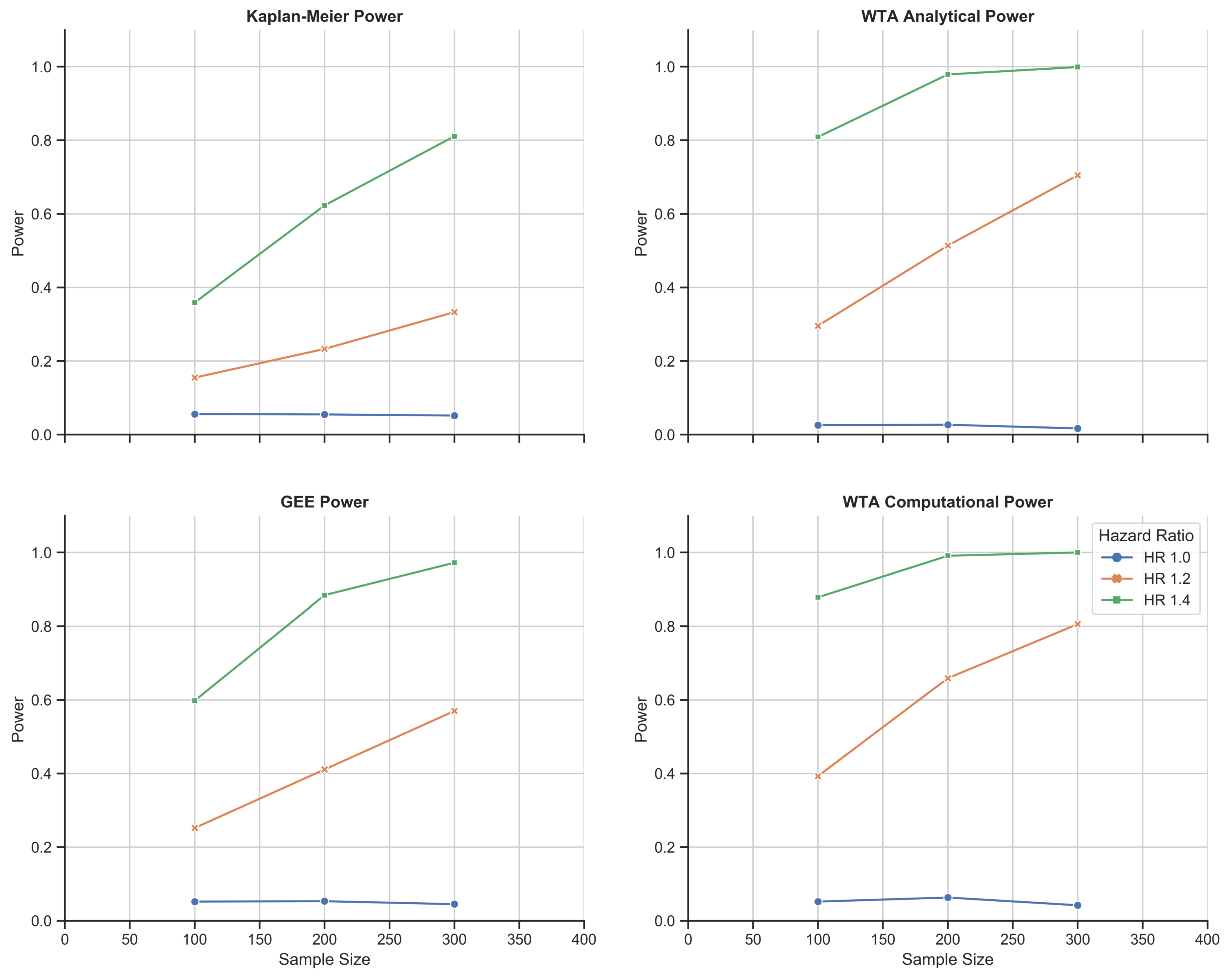
Chemotherapy toxicity simulation study: 1000-fold simulations of power as a function of sample size for KM estimation, the GEE, and WTA in both its analytical and computational forms. WTA outperformed KM estimation and the GEE with consistently higher power and, thus, a smaller sample size requirement. In addition, the computational approach with WTA outperformed the analytical approach in return for a more time- and resource-intensive methodology. The computational approach also met a standard type I error rate of 0.05 that was robust to changes in trial size.
Figure 5.
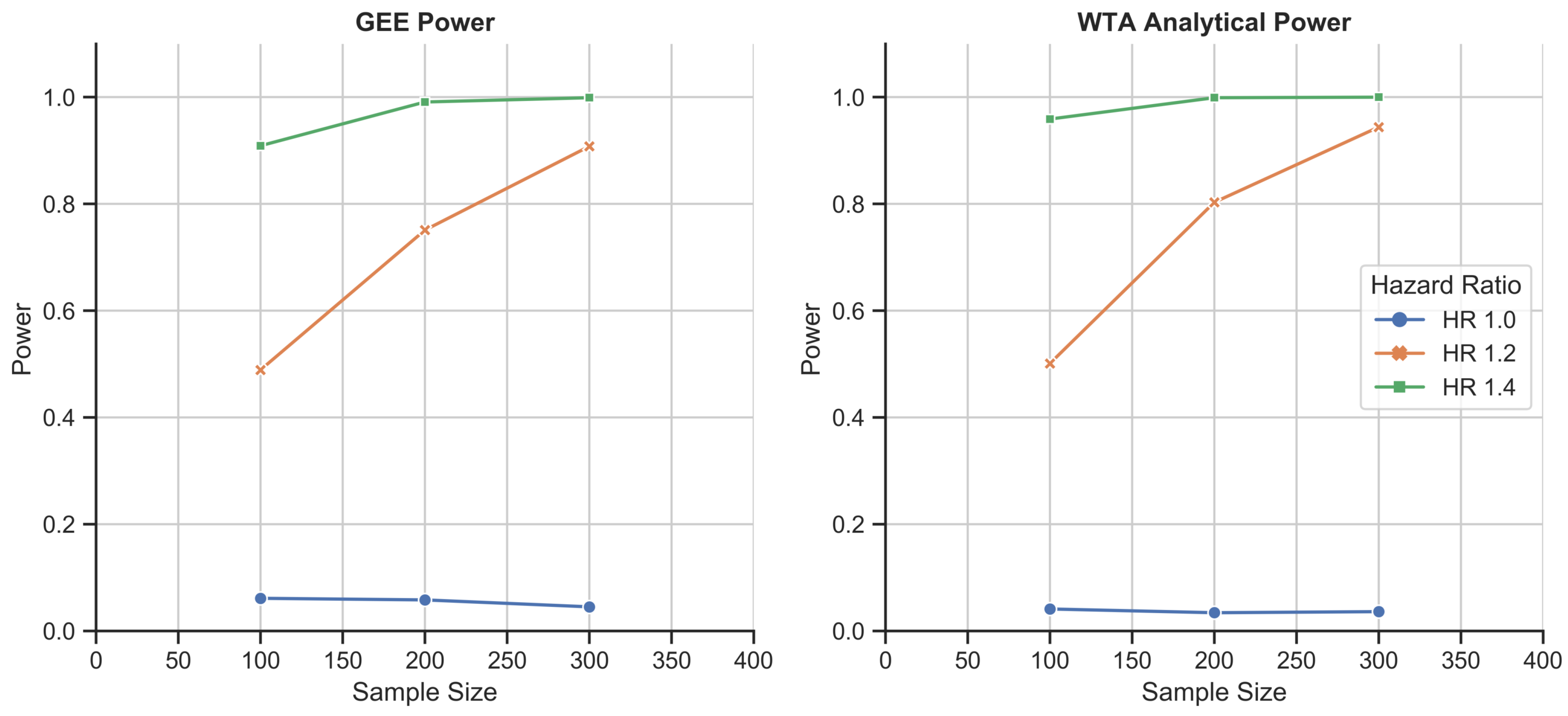
Schizophrenia disease course simulation study: 1000-fold simulations of power as a function of sample size for the GEE and WTA in its analytical form. WTA again outperformed the GEE and demonstrated a type I error rate of 0.037, closer to the 0.05 standard due to the larger size of each trial.
Figure 6.
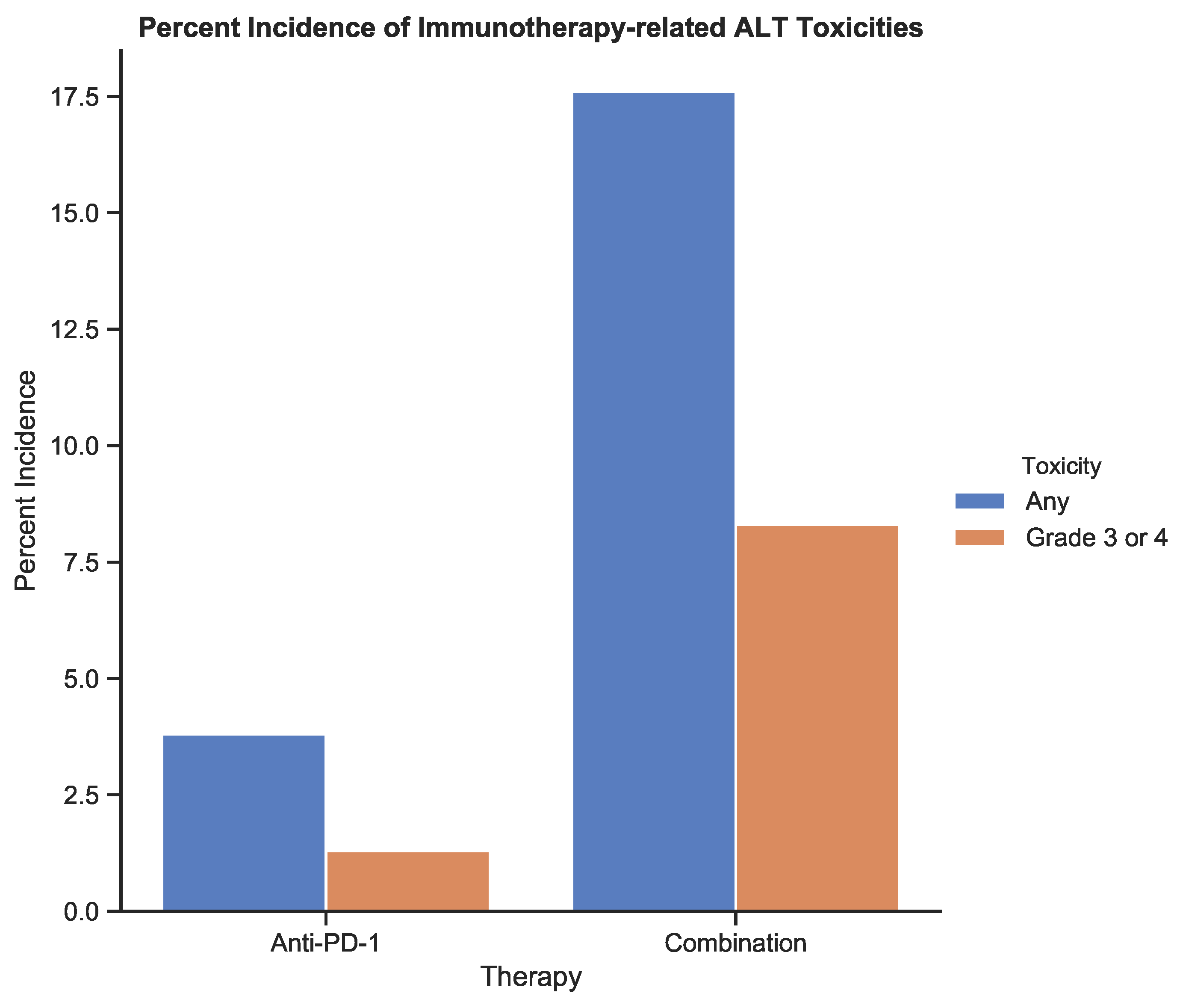
The incidence of treatment-related toxicities associated with an increase in alanine aminotransferase (ALT) for patients receiving anti-PD-1 therapy and combination therapy. Toxicities were graded using CTCAE v5.0 [
6]. Data from
Table 3 from the study by Larkin et al. (2015) [
12].
Figure 7.
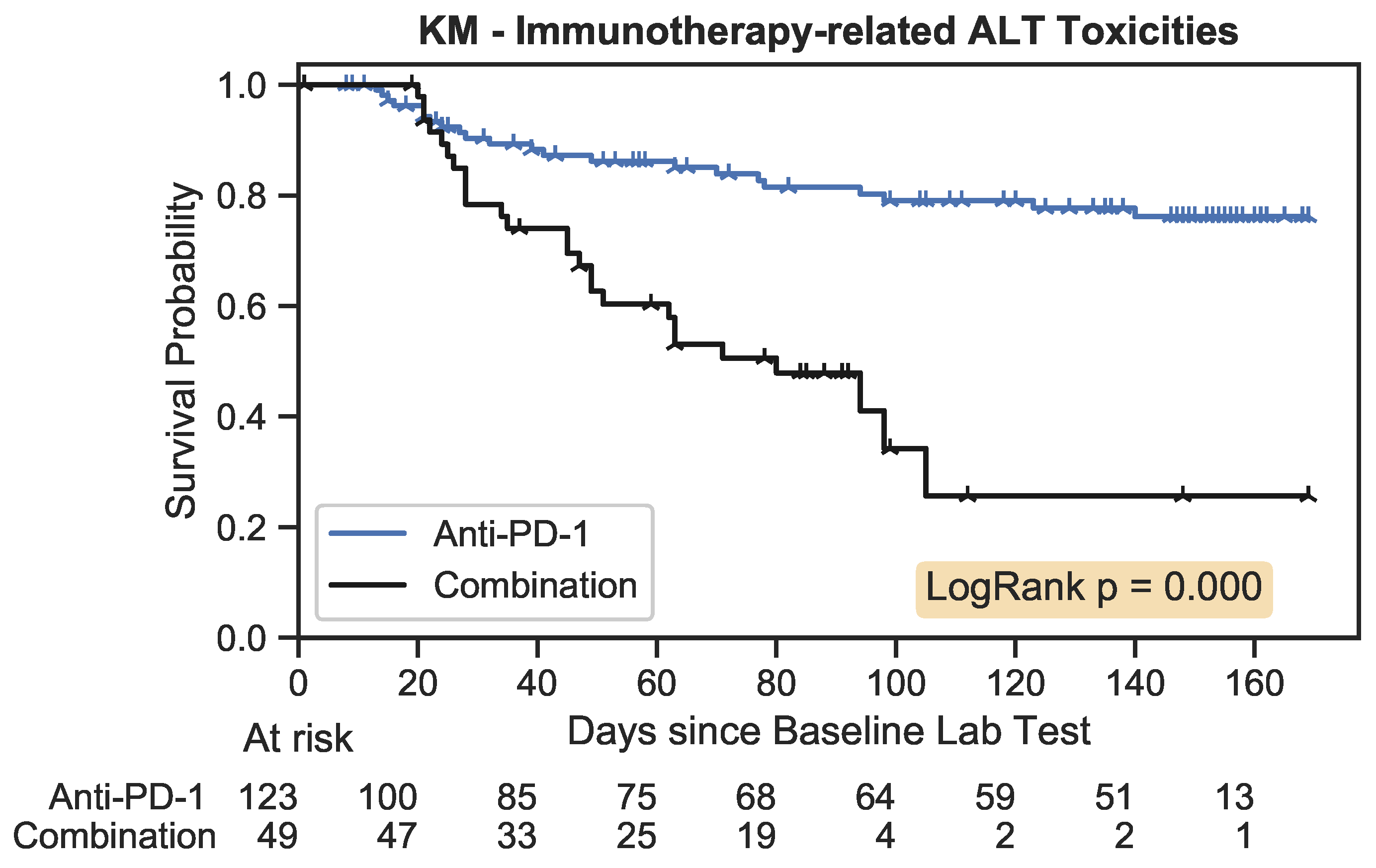
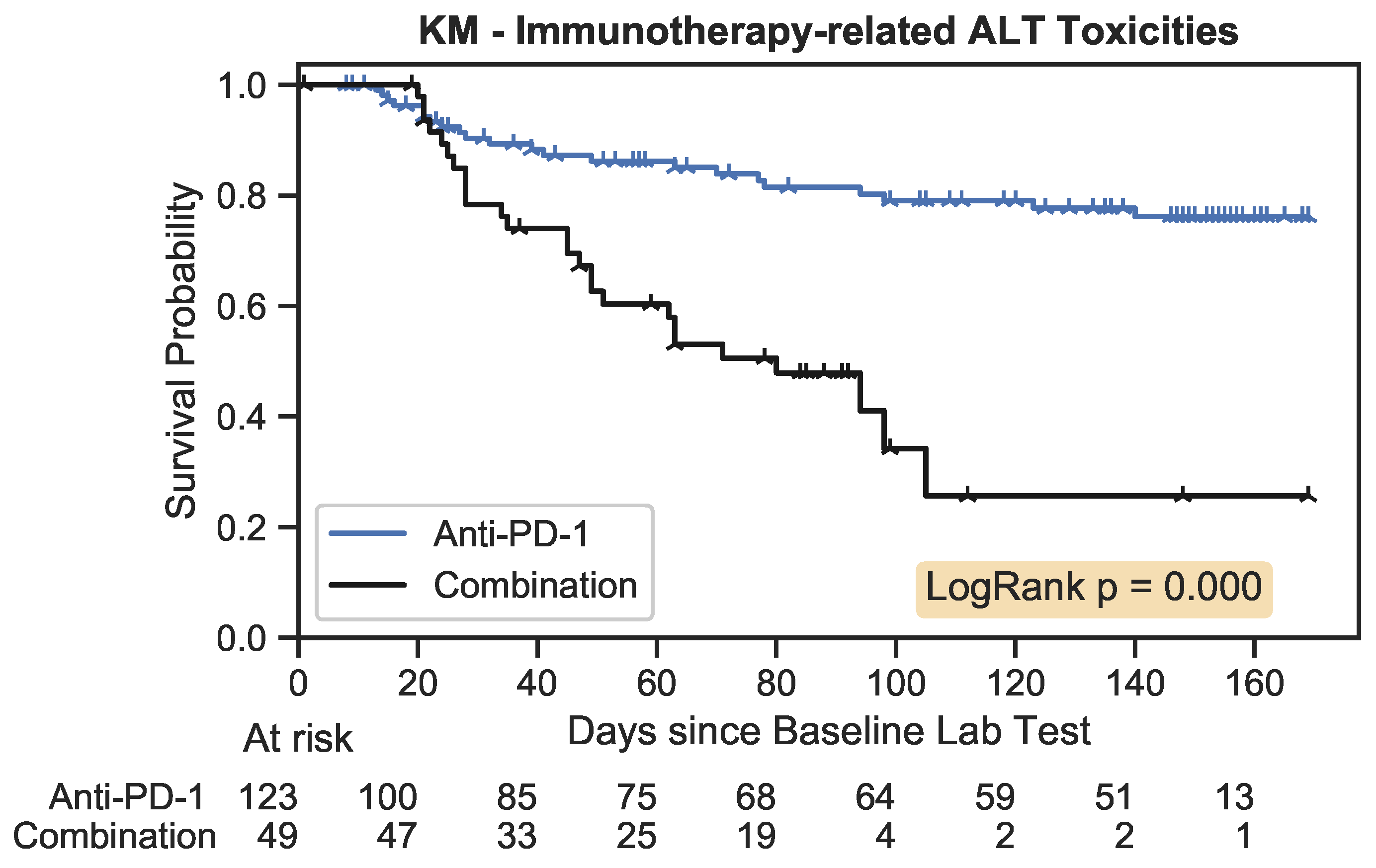
The Kaplan–Meier estimator plot for immunotherapy-related toxicities associated with an increase in ALT. An event was considered the onset of a nonzero toxicity grade.
Figure 8.
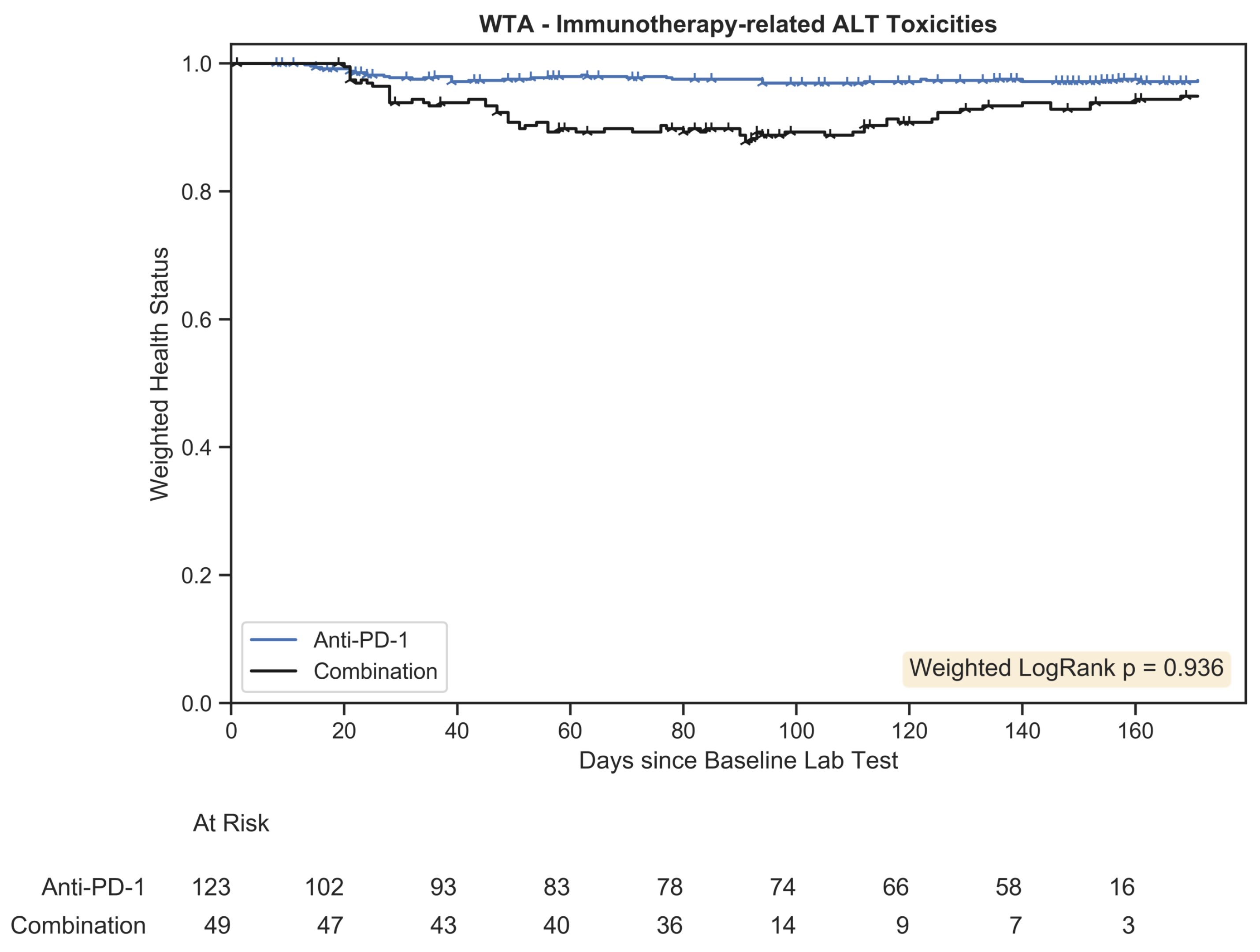
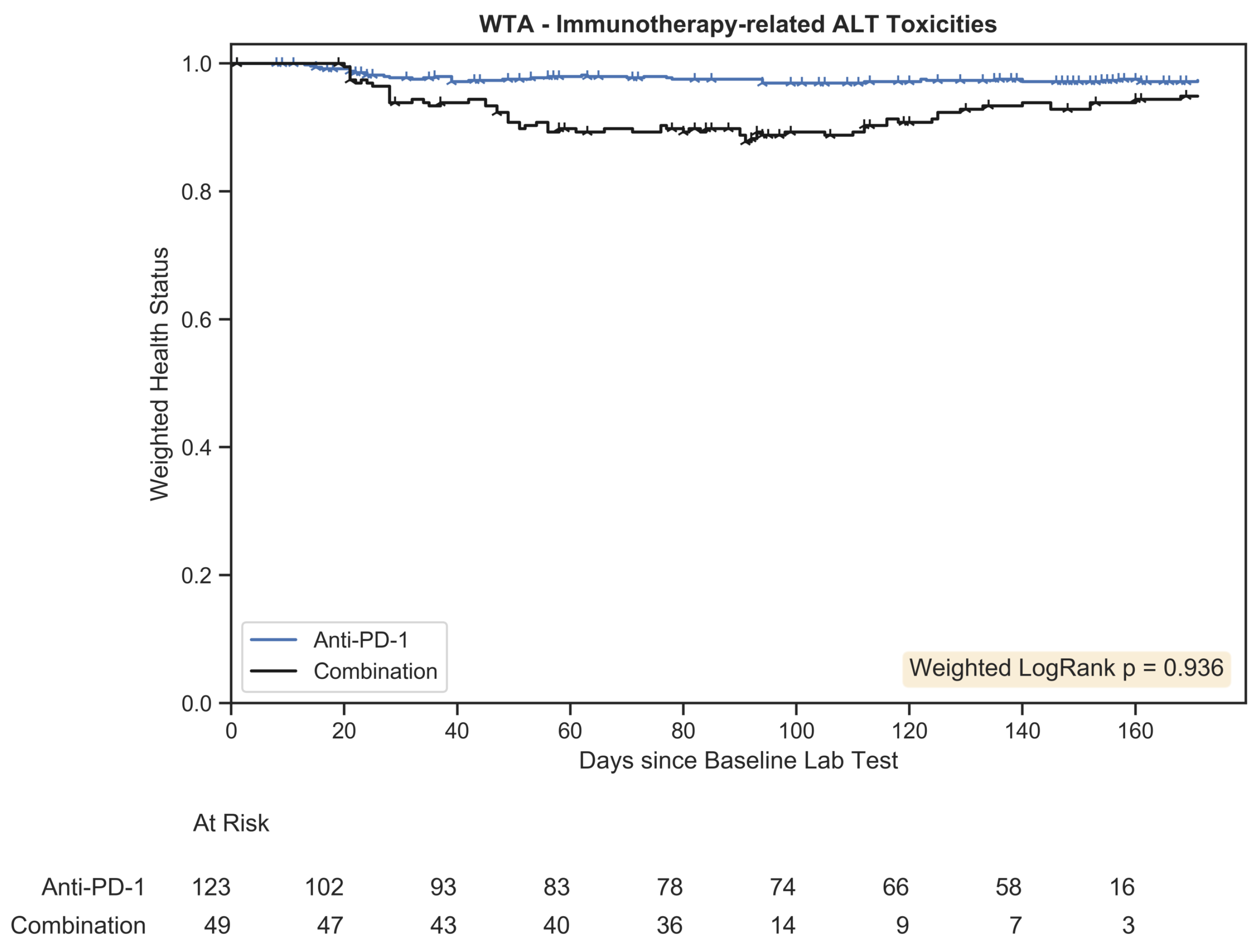
Weighted trajectory analysis plot for immunotherapy-related toxicities associated with an increase in ALT. The weighted health status of the combination group initially diverged from the anti-PD-1 group but subsequent recovery led to similar longitudinal outcomes.
Figure 9.
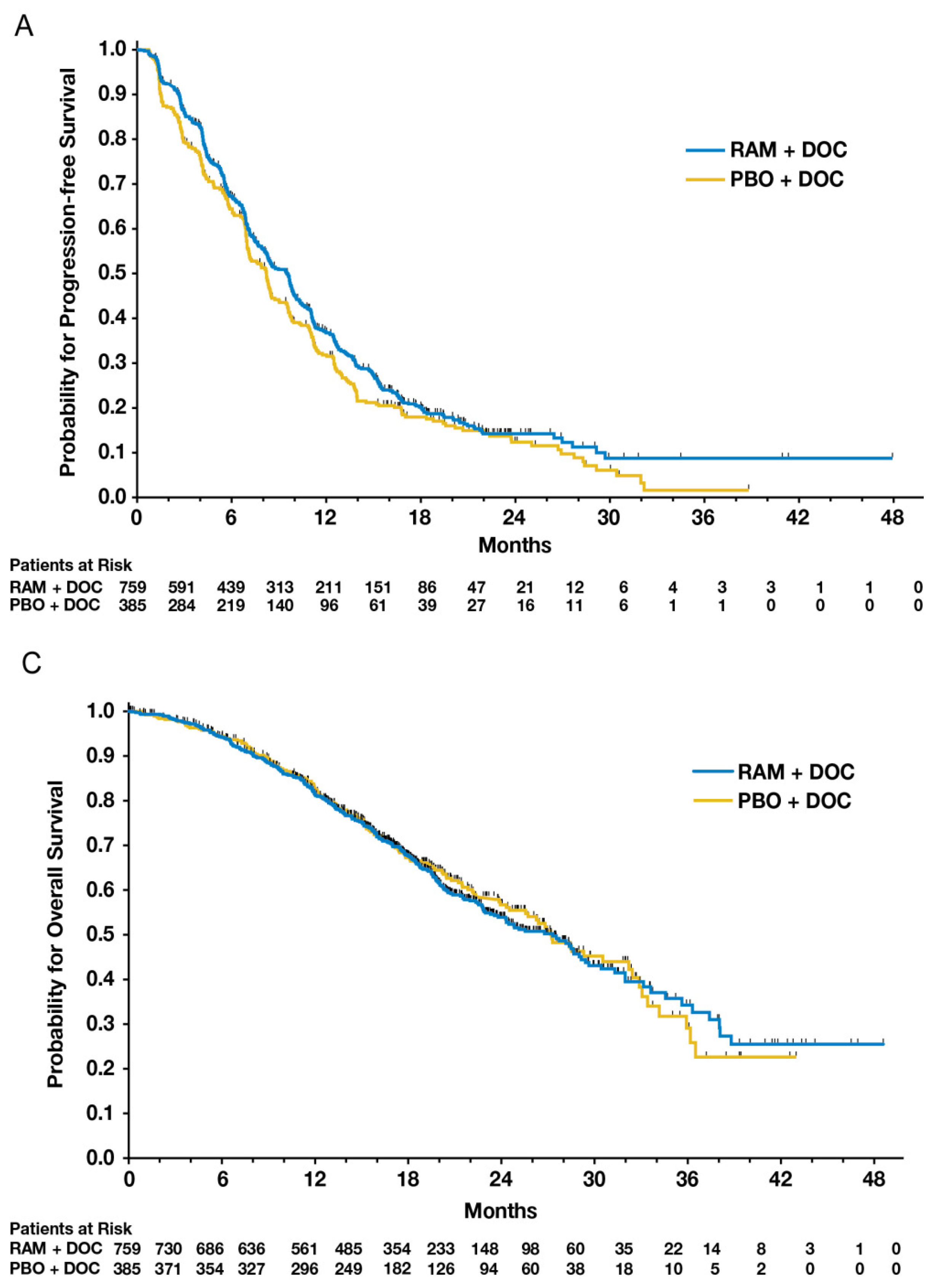
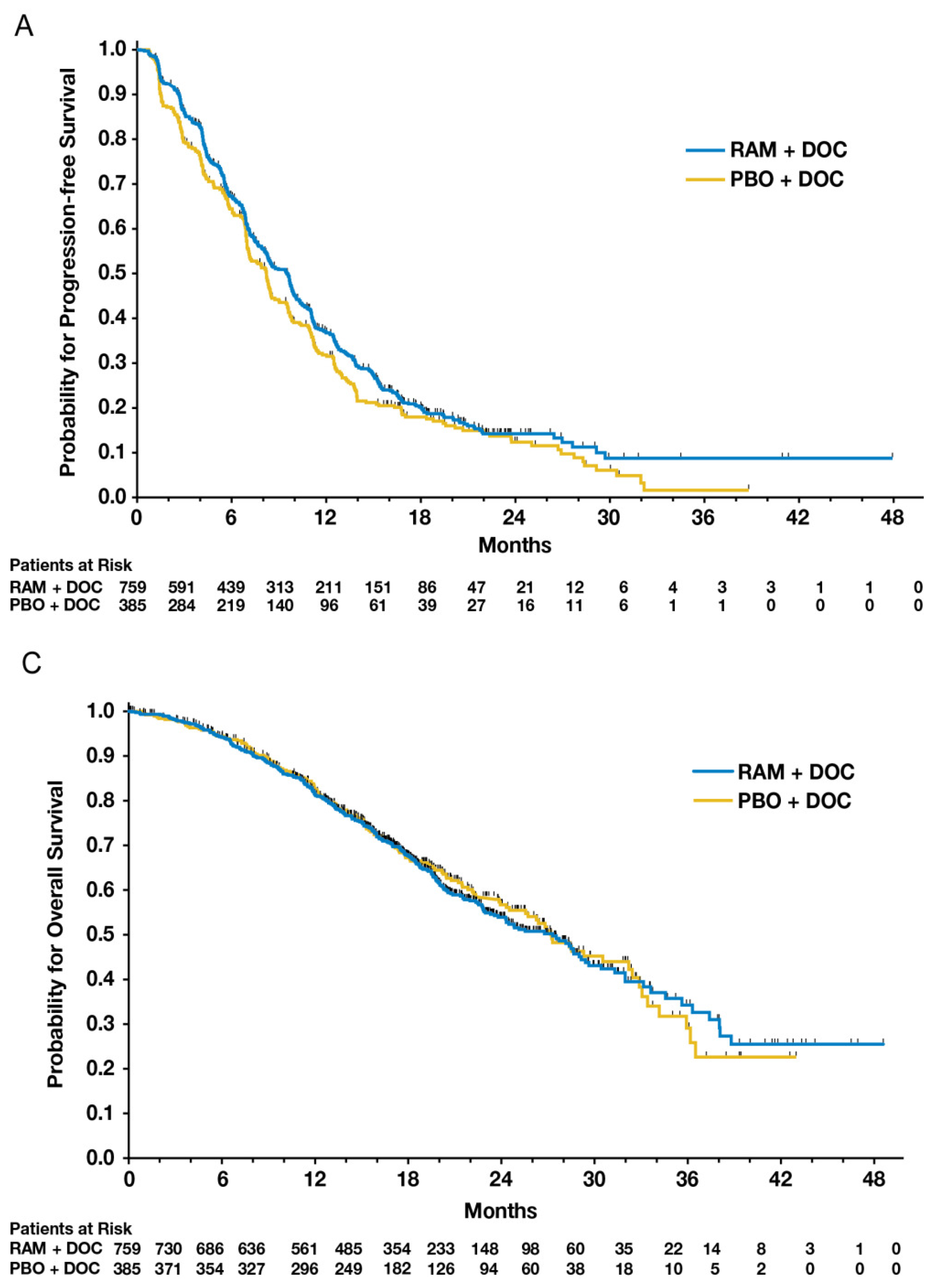
Figure 2A,C from Mackey et al.’s 2014 paper comparing ramucirumab to a placebo added to standard docetaxel chemotherapy [
14]. The figures provide patient outcomes using KM estimates of progression-free survival (PFS) and overall survival (OS), respectively.
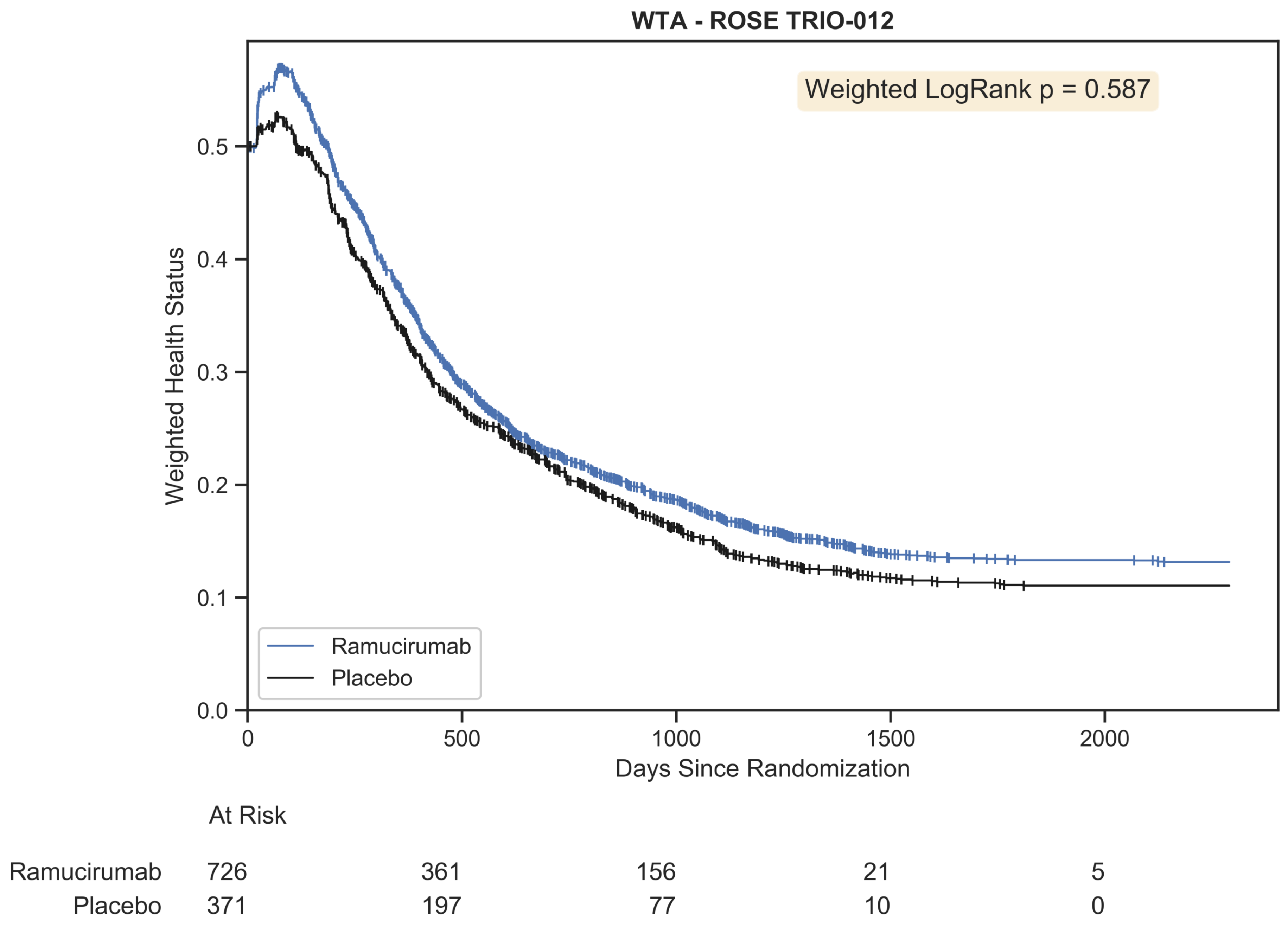
Figure 10.
Weighted trajectory analysis of the original ROSE/TRIO-012 dataset using an ordinal scale that merges RECIST criteria with mortality. The trajectory of patient outcomes demonstrates that partial and complete response initially outweighed progressive disease and mortality for the first few chemotherapy cycles. Following this peak, patient prognosis was generally poor, as both treatment arms experienced growing disease burden and death.
Table 1.
Feature comparison between the Kaplan–Meier estimator and weighted trajectory analysis.
| Feature | Kaplan–Meier Estimator | Weighted Trajectory Analysis |
|---|
| Event | Outcome with binary coding. A patient must begin at “0” and is removed from analysis following an event (“1”) | An event is a change in clinical severity and does not remove a patient from further analysis. Must be discrete with a finite range that depends on the variable of interest |
| Variable of interest | Death, metastases, local recurrence, stroke, and more. Can include variables outside of medicine, such as postgraduate employment | Graded/staged outcomes: ECOG performance, toxicities, NYHA heart failure class, questionnaire scores, and more; also includes variables outside of medicine |
| Trajectory | Survival function always decreases | Bidirectional: severity function can decrease or increase |
| Censoring | Removes patients from subsequent analysis (for withdrawal, discharge, loss to follow up, etc.) | |
| Test for significance | Logrank test | Weighted logrank test |
| Y-axis | Survival probability | Weighted health status |
| X-axis | Time (discrete: days, weeks, months, etc.) |
| Y-intercept | 1.0 | Between 0 and 1.0, inclusive |
Table 2.
A snapshot of the final results of a simulated chemotherapy toxicity-grade trial.
| Patient ID | Treatment Arm | Duration | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|
| 1 | 1 | 10 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | |
| 2 | 1 | 10 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | |
| 3 | 0 | 11 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 4 | 1 | 6 | 0 | 0 | 0 | 0 | 0 | 0 | | | | | |
| 5 | 0 | 13 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 |
| 6 | 1 | 9 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | | |
| 7 | 0 | 18 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 2 | 2 |
| 8 | 1 | 6 | 0 | 0 | 0 | 0 | 0 | 0 | | | | | |
| 9 | 1 | 29 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| 10 | 0 | 4 | 0 | 0 | 0 | 0 | | | | | | | |
Table 4.
RECIST 1.1 mapped to ordinal severity scores.
| Outcome | Score |
|---|
| Complete response (CR) | 0 |
| Partial response (PR) | 1 |
| Stable disease (SD) | 2 |
| Progressive disease (PD) | 3 |
| Death | 4 |
| Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).




 ). The number of patients remaining within the study is tabulated below the plot at evenly spaced time intervals for each treatment arm.
). The number of patients remaining within the study is tabulated below the plot at evenly spaced time intervals for each treatment arm.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}