
The Use of the Random Number Generator and Artificial Intelligence Analysis for Dimensionality Reduction of Follicular Lymphoma Transcriptomic Data

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
3. Results
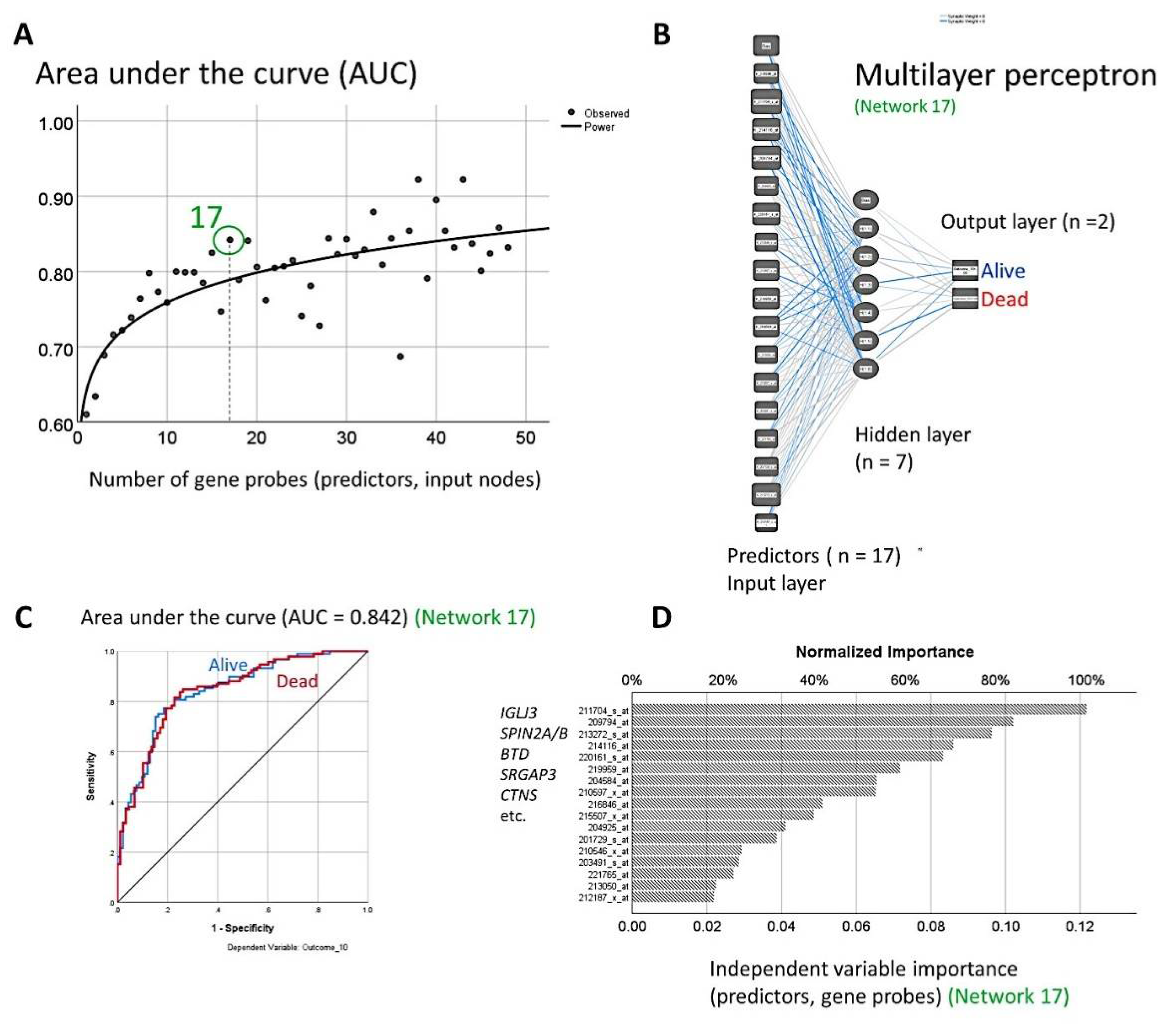
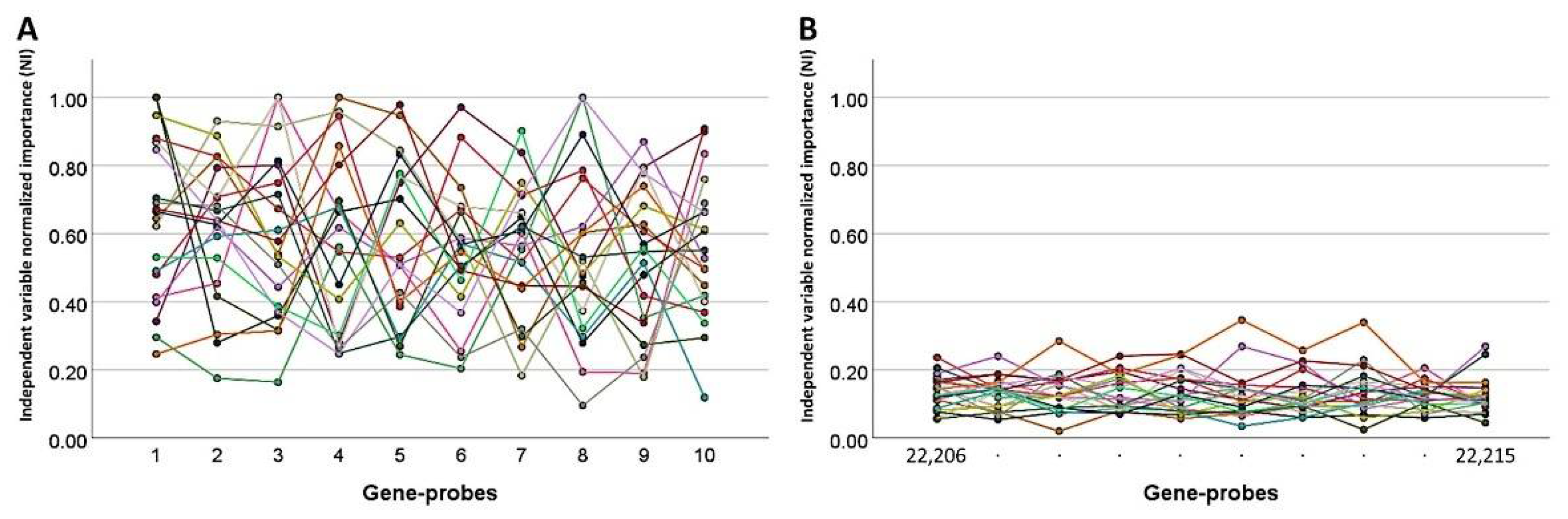
3.1. Generation of 120 Different Predictive Models for Overall Survival Using MLP Neural Networks
3.2. Dimensionality Reduction for Predicting the Overall Survival
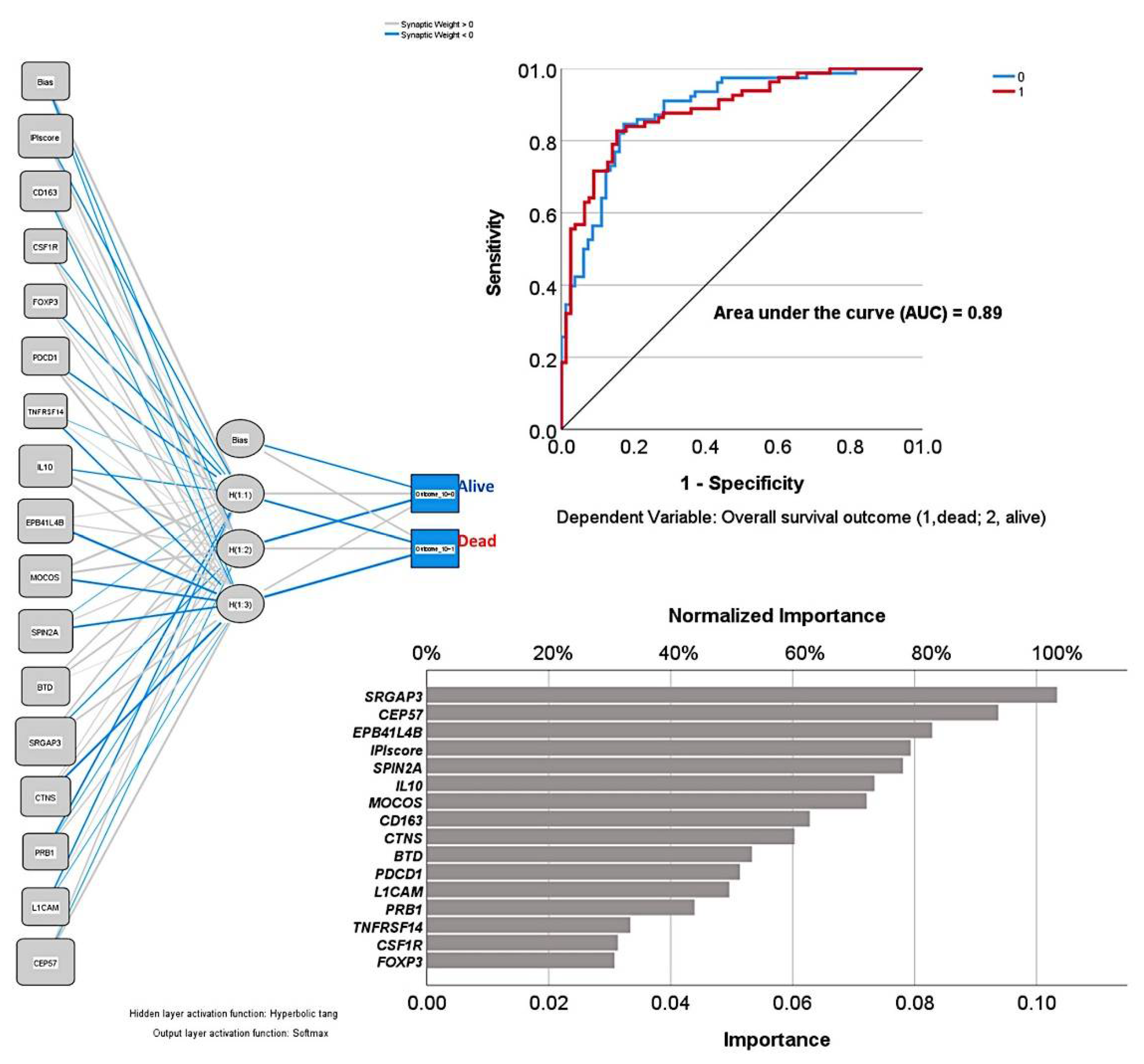
3.3. Correlation with the International Prognostic Index (IPI) and the Immune Response (IR)
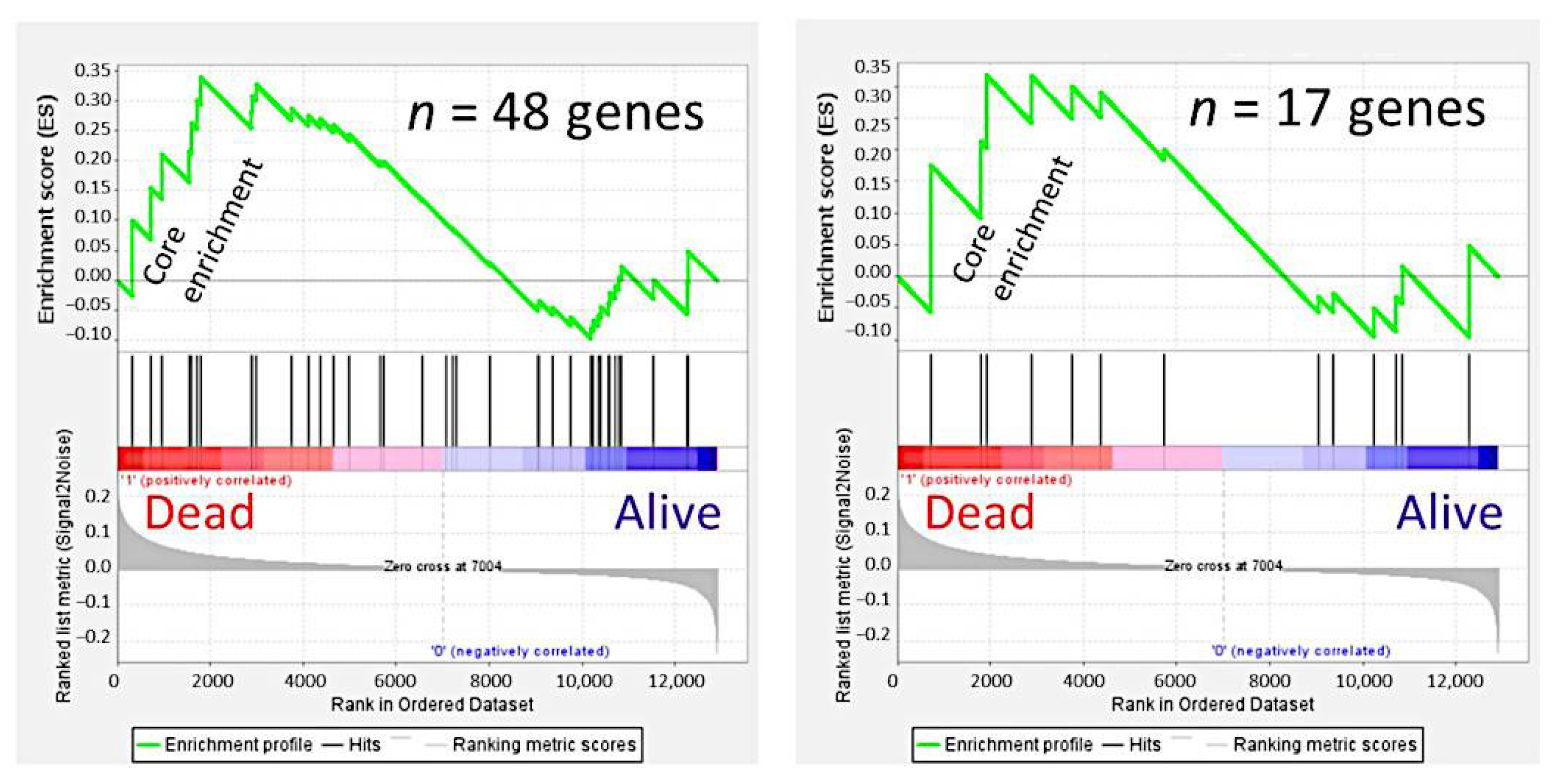
3.4. Gene Set Enrichment Analysis (GSEA)
3.5. Final Integrative Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 211748_x_at | 212187_x_at | 219971_at | 203788_s_at | 203892_at | 214461_at | 202540_s_at | 205272_s_at | 207436_x_at | 208791_at | |
|---|---|---|---|---|---|---|---|---|---|---|
| 211748_x_at | 0.3106 | |||||||||
| 212187_x_at | 0.2817 | 0.2752 | ||||||||
| 219971_at | 0.1735 | 0.1674 | 0.4121 | |||||||
| 203788_s_at | −0.0123 | −0.0129 | −0.0106 | 0.2564 | ||||||
| 203892_at | 0.0289 | 0.0205 | 0.0366 | 0.0398 | 0.2449 | |||||
| 214461_at | 0.0358 | 0.0197 | −0.0193 | 0.0213 | 0.0014 | 0.3596 | ||||
| 202540_s_at | −0.0227 | −0.0267 | −0.0402 | −0.0227 | −0.0487 | 0.0061 | 0.2111 | |||
| 205272_s_at | 0.0171 | 0.0166 | 0.0245 | −0.0139 | 0.3841 | −0.0534 | −0.1129 | 2.0909 | ||
| 207436_x_at | −0.0199 | −0.0204 | −0.0959 | 0.0190 | −0.0261 | 0.0134 | 0.1088 | −0.0626 | 0.1878 | |
| 208791_at | 0.3040 | 0.2744 | 0.1746 | −0.0392 | 0.0364 | 0.0512 | −0.0148 | 0.0290 | −0.0163 | 0.6622 |
| Num. | Gene Probe | Gene Symbol | B | p Value | Hazard Risk (HR) | 95.0% CI for HR | MLP | |
|---|---|---|---|---|---|---|---|---|
| Lower | Upper | NI | ||||||
| 1 | 216846_at | IGLJ3 | 2.0 | 2.35 × 10−7 | 7.5 | 3.5 | 16.1 | 1.000 |
| 2 | 211704_s_at | SPIN2A/B | 0.6 | 0.083002 | 1.9 | 0.9 | 3.8 | 0.881 |
| 3 | 214116_at | BTD | −1.6 | 7.6 × 10−8 | 0.2 | 0.1 | 0.4 | 0.876 |
| 4 | 209794_at | SRGAP3 | −1.1 | 0.033109 | 0.3 | 0.1 | 0.9 | 0.839 |
| 5 | 204925_at | CTNS | 1.7 | 0.000263 | 5.3 | 2.2 | 13.1 | 0.838 |
| 6 | 220161_s_at | EPB41L4B | 1.6 | 4.7 × 10−9 | 5.0 | 2.9 | 8.5 | 0.767 |
| 7 | 210546_x_at | CTAG1A | 1.0 | 3.23 × 10−6 | 2.7 | 1.8 | 4.0 | 0.757 |
| 8 | 210597_x_at | PRB1 | 1.0 | 1.46 × 10−5 | 2.7 | 1.7 | 4.3 | 0.701 |
| 9 | 219959_at | MOCOS | 0.7 | 0.001377 | 2.0 | 1.3 | 3.0 | 0.689 |
| 10 | 204584_at | L1CAM | −0.9 | 0.000622 | 0.4 | 0.2 | 0.7 | 0.661 |
| 11 | 213050_at | COBL | −1.1 | 0.001523 | 0.3 | 0.2 | 0.7 | 0.646 |
| 12 | 215507_x_at | - | −0.5 | 0.082379 | 0.6 | 0.4 | 1.1 | 0.645 |
| 13 | 203491_s_at | CEP57 | −0.5 | 0.016744 | 0.6 | 0.4 | 0.9 | 0.629 |
| 14 | 221765_at | UGCG | −0.4 | 0.069669 | 0.7 | 0.5 | 1.0 | 0.599 |
| 15 | 201729_s_at | KIAA0100 | 3.1 | 6.79 × 10−6 | 22.4 | 5.8 | 86.5 | 0.547 |
| 16 | 213272_s_at | TMEM159 | 1.3 | 0.003146 | 3.8 | 1.6 | 9.4 | 0.525 |
| 17 | 212187_x_at | PTGDS | 1.9 | 0.000211 | 6.7 | 2.4 | 18.2 | 0.521 |
| 18 | 220600_at | ELP6 | 0.9 | 0.002653 | 2.4 | 1.4 | 4.3 | 0.519 |
| 19 | 205839_s_at | BZRAP1 | −0.9 | 0.015594 | 0.4 | 0.2 | 0.8 | 0.503 |
| 20 | 204738_s_at | KRIT1 | −1.3 | 0.000619 | 0.3 | 0.1 | 0.6 | 0.493 |
| 21 | 221196_x_at | BRCC3 | 1.2 | 0.001986 | 3.3 | 1.6 | 7.2 | 0.470 |
| 22 | 215788_at | CFAP74 | −0.7 | 0.02512 | 0.5 | 0.3 | 0.9 | 0.465 |
| 23 | 203892_at | WFDC2 | 1.3 | 0.001288 | 3.6 | 1.6 | 7.7 | 0.459 |
| 24 | 219349_s_at | EXOC2 | −0.8 | 0.045831 | 0.4 | 0.2 | 1.0 | 0.452 |
| 25 | 208791_at | CLU | 1.2 | 0.000159 | 3.4 | 1.8 | 6.5 | 0.450 |
| 26 | 215287_at | STRN | 0.9 | 0.002545 | 2.4 | 1.4 | 4.2 | 0.443 |
| 27 | 219361_s_at | AEN | 1.6 | 0.000405 | 4.8 | 2.0 | 11.4 | 0.439 |
| 28 | 207436_x_at | SORBS1 | −0.8 | 0.07232 | 0.4 | 0.2 | 1.1 | 0.436 |
| 29 | 219815_at | GAL3ST4 | −1.9 | 2.15 × 10−5 | 0.1 | 0.1 | 0.4 | 0.418 |
| 30 | 215183_at | - | −1.3 | 0.001 | 0.3 | 0.1 | 0.6 | 0.416 |
| 31 | 207356_at | DEFB4A | −1.4 | 0.000927 | 0.2 | 0.1 | 0.6 | 0.405 |
| 32 | 218268_at | TBC1D15 | 2.2 | 0.000131 | 8.9 | 2.9 | 27.5 | 0.388 |
| 33 | 215867_x_at | CA12 | −1.8 | 4.6 × 10−5 | 0.2 | 0.1 | 0.4 | 0.342 |
| 34 | 214876_s_at | TUBGCP5 | −0.9 | 0.000816 | 0.4 | 0.2 | 0.7 | 0.332 |
| 35 | 213539_at | CD3D | −2.8 | 5.16 × 10−8 | 0.1 | 0.0 | 0.2 | 0.332 |
| 36 | 207752_x_at | PRB1 | −0.7 | 0.002323 | 0.5 | 0.3 | 0.8 | 0.292 |
| 37 | 210312_s_at | IFT20 | −1.8 | 0.002342 | 0.2 | 0.1 | 0.5 | 0.273 |
| 38 | 41397_at | ZNF821 | −1.2 | 0.000852 | 0.3 | 0.1 | 0.6 | 0.260 |
| 39 | 204547_at | RAB40B | 0.8 | 0.019527 | 2.2 | 1.1 | 4.4 | 0.252 |
| 40 | 214465_at | ORM1 | 1.3 | 1.14 × 10−5 | 3.7 | 2.1 | 6.6 | 0.242 |
| 41 | 201131_s_at | CDH1 | −0.7 | 0.008031 | 0.5 | 0.3 | 0.8 | 0.231 |
| 42 | 210039_s_at | PRKCQ | 1.0 | 0.089034 | 2.7 | 0.9 | 8.2 | 0.224 |
| 43 | 207377_at | PPP1R2P9 | −0.8 | 0.037392 | 0.4 | 0.2 | 1.0 | 0.210 |
| 44 | 206994_at | CST4 | 0.7 | 0.006194 | 1.9 | 1.2 | 3.1 | 0.200 |
| 45 | 214408_s_at | RFPL1S | 0.7 | 0.014436 | 2.0 | 1.1 | 3.5 | 0.200 |
| 46 | 216699_s_at | KLK1 | −0.7 | 0.045702 | 0.5 | 0.3 | 1.0 | 0.119 |
| 47 | 221004_s_at | ITM2C | −0.5 | 0.077654 | 0.6 | 0.4 | 1.1 | 0.080 |
| 48 | 211262_at | PCSK6 | −1.0 | 0.02704 | 0.4 | 0.1 | 0.9 | 0.028 |
References
- Campo, E.; Swerdlow, S.H.; Harris, N.L.; Pileri, S.; Stein, H.; Jaffe, E.S. The 2008 WHO classification of lymphoid neoplasms and beyond: Evolving concepts and practical applications. Blood 2011, 117, 5019–5032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreras, J.; Lopez-Guillermo, A.; Fox, B.C.; Colomo, L.; Martinez, A.; Roncador, G.; Montserrat, E.; Campo, E.; Banham, A.H. High numbers of tumor-infiltrating FOXP3-positive regulatory T cells are associated with improved overall survival in follicular lymphoma. Blood 2006, 108, 2957–2964. [Google Scholar] [CrossRef] [PubMed]
- Carreras, J.; Lopez-Guillermo, A.; Roncador, G.; Villamor, N.; Colomo, L.; Martinez, A.; Hamoudi, R.; Howat, W.J.; Montserrat, E.; Campo, E. High numbers of tumor-infiltrating programmed cell death 1-positive regulatory lymphocytes are associated with improved overall survival in follicular lymphoma. J. Clin. Oncol. 2009, 27, 1470–1476. [Google Scholar] [CrossRef]
- Freedman, A.S.; Aster, J.C. Clinical Manifestations, Pathologic Features, Diagnosis, and Prognosis of Follicular Lymphoma. Available online: https://www.uptodate.com/contents/search (accessed on 23 February 2022).
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef]
- Pastore, A.; Jurinovic, V.; Kridel, R.; Hoster, E.; Staiger, A.M.; Szczepanowski, M.; Pott, C.; Kopp, N.; Murakami, M.; Horn, H.; et al. Integration of gene mutations in risk prognostication for patients receiving first-line immunochemotherapy for follicular lymphoma: A retrospective analysis of a prospective clinical trial and validation in a population-based registry. Lancet Oncol. 2015, 16, 1111–1122. [Google Scholar] [CrossRef]
- Cossman, J.; Neckers, L.M.; Hsu, S.; Longo, D.; Jaffe, E.S. Low-grade lymphomas. Expression of developmentally regulated B-cell antigens. Am. J. Pathol. 1984, 115, 117–124. [Google Scholar]
- Morton, L.M.; Wang, S.S.; Devesa, S.S.; Hartge, P.; Weisenburger, D.D.; Linet, M.S. Lymphoma incidence patterns by WHO subtype in the United States, 1992–2001. Blood 2006, 107, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Horning, S.J.; Rosenberg, S.A. The natural history of initially untreated low-grade non-Hodgkin’s lymphomas. N. Engl. J. Med. 1984, 311, 1471–1475. [Google Scholar] [CrossRef]
- Casulo, C.; Byrtek, M.; Dawson, K.L.; Zhou, X.; Farber, C.M.; Flowers, C.R.; Hainsworth, J.D.; Maurer, M.J.; Cerhan, J.R.; Link, B.K.; et al. Early Relapse of Follicular Lymphoma After Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone Defines Patients at High Risk for Death: An Analysis From the National LymphoCare Study. J. Clin. Oncol. 2015, 33, 2516–2522. [Google Scholar] [CrossRef]
- Solal-Celigny, P.; Roy, P.; Colombat, P.; White, J.; Armitage, J.O.; Arranz-Saez, R.; Au, W.Y.; Bellei, M.; Brice, P.; Caballero, D.; et al. Follicular lymphoma international prognostic index. Blood 2004, 104, 1258–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alig, S.; Jurinovic, V.; Pastore, A.; Haebe, S.; Schmidt, C.; Zoellner, A.K.; Dreyling, M.; Unterhalt, M.; Hoster, E.; Hiddemann, W.; et al. Impact of age on clinical risk scores in follicular lymphoma. Blood Adv. 2019, 3, 1033–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- IBM Cloud Education, Neural Networks. 17 August 2020. Available online: https://www.ibm.com/cloud/learn/neural-networks (accessed on 23 February 2022).
- IBM Corporation. IBM SPSS Neural Networks 1989–2019; IBM Corporation, North Castle Drive: Armonk, NY, USA, 2019. [Google Scholar]
- Carreras, J.; Kikuti, Y.Y.; Miyaoka, M.; Hiraiwa, S.; Tomita, S.; Ikoma, H.; Kondo, Y.; Ito, A.; Nakamura, N.; Hamoudi, R. A Combination of Multilayer Perceptron, Radial Basis Function Artificial Neural Networks and Machine Learning Image Segmentation for the Dimension Reduction and the Prognosis Assessment of Diffuse Large B-Cell Lymphoma. AI 2021, 2, 106–134. [Google Scholar] [CrossRef]
- Carreras, J.; Kikuti, Y.Y.; Miyaoka, M.; Hiraiwa, S.; Tomita, S.; Ikoma, H.; Kondo, Y.; Ito, A.; Shiraiwa, S.; Hamoudi, R.; et al. A Single Gene Expression Set Derived from Artificial Intelligence Predicted the Prognosis of Several Lymphoma Subtypes; and High Immunohistochemical Expression of TNFAIP8 Associated with Poor Prognosis in Diffuse Large B-Cell Lymphoma. AI 2020, 1, 342–360. [Google Scholar] [CrossRef]
- Carreras, J.; Kikuti, Y.Y.; Roncador, G.; Miyaoka, M.; Hiraiwa, S.; Tomita, S.; Ikoma, H.; Kondo, Y.; Ito, A.; Shiraiwa, S.; et al. High Expression of Caspase-8 Associated with Improved Survival in Diffuse Large B-Cell Lymphoma: Machine Learning and Artificial Neural Networks Analyses. BioMedInformatics 2021, 1, 18–46. [Google Scholar] [CrossRef]
- Carreras, J.; Hiraiwa, S.; Kikuti, Y.Y.; Miyaoka, M.; Tomita, S.; Ikoma, H.; Ito, A.; Kondo, Y.; Roncador, G.; Garcia, J.F.; et al. Artificial Neural Networks Predicted the Overall Survival and Molecular Subtypes of Diffuse Large B-Cell Lymphoma Using a Pancancer Immune-Oncology Panel. Cancers 2021, 13, 6384. [Google Scholar] [CrossRef]
- Carreras, J.; Nakamura, N.; Hamoudi, R. Artificial Intelligence Analysis of Gene Expression Predicted the Overall Survival of Mantle Cell Lymphoma and a Large Pan-Cancer Series. Healthcare 2022, 10, 155. [Google Scholar] [CrossRef]
- Carreras, J.; Kikuti, Y.Y.; Miyaoka, M.; Roncador, G.; Garcia, J.F.; Hiraiwa, S.; Tomita, S.; Ikoma, H.; Kondo, Y.; Ito, A.; et al. Integrative Statistics, Machine Learning and Artificial Intelligence Neural Network Analysis Correlated CSF1R with the Prognosis of Diffuse Large B-Cell Lymphoma. Hemato 2021, 2, 182–206. [Google Scholar] [CrossRef]
- Carreras, J.; Hamoudi, R. Artificial Neural Network Analysis of Gene Expression Data Predicted Non-Hodgkin Lymphoma Subtypes with High Accuracy. Mach. Learn. Knowl. Extr. 2021, 3, 720–739. [Google Scholar] [CrossRef]
- Carreras, J.; Kikuti, Y.Y.; Miyaoka, M.; Hiraiwa, S.; Tomita, S.; Ikoma, H.; Kondo, Y.; Ito, A.; Nakamura, N.; Hamoudi, R. Artificial Intelligence Analysis of the Gene Expression of Follicular Lymphoma Predicted the Overall Survival and Correlated with the Immune Microenvironment Response Signatures. Mach. Learn. Knowl. Extr. 2020, 2, 647–671. [Google Scholar] [CrossRef]
- Brown, J.R.; Freedman, A.S.; Aster, J.C. Pathobiology of Follicular Lymphoma; UpToDate: Waltham, MA, USA, 2021. [Google Scholar]
- Ikoma, H.; Miyaoka, M.; Hiraiwa, S.; Yukie Kikuti, Y.; Shiraiwa, S.; Hara, R.; Kojima, M.; Ohmachi, K.; Ando, K.; Carreras, J.; et al. Clinicopathological analysis of follicular lymphoma with BCL2, BCL6, and MYC rearrangements. Pathol. Int. 2022; Online ahead of print. [Google Scholar] [CrossRef]
- Bodor, C.; Grossmann, V.; Popov, N.; Okosun, J.; O’Riain, C.; Tan, K.; Marzec, J.; Araf, S.; Wang, J.; Lee, A.M.; et al. EZH2 mutations are frequent and represent an early event in follicular lymphoma. Blood 2013, 122, 3165–3168. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R. Chromatin modifying gene mutations in follicular lymphoma. Blood 2018, 131, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Gentles, A.J.; Nair, R.V.; Irish, J.M.; Kihira, S.; Liu, C.L.; Kela, I.; Hopmans, E.S.; Myklebust, J.H.; Ji, H.; et al. Hierarchy in somatic mutations arising during genomic evolution and progression of follicular lymphoma. Blood 2013, 121, 1604–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, M.R.; Kihira, S.; Liu, C.L.; Nair, R.V.; Salari, R.; Gentles, A.J.; Irish, J.; Stehr, H.; Vicente-Duenas, C.; Romero-Camarero, I.; et al. Mutations in early follicular lymphoma progenitors are associated with suppressed antigen presentation. Proc. Natl. Acad. Sci. USA 2015, 112, E1116–E1125. [Google Scholar] [CrossRef] [Green Version]
- Kridel, R.; Chan, F.C.; Mottok, A.; Boyle, M.; Farinha, P.; Tan, K.; Meissner, B.; Bashashati, A.; McPherson, A.; Roth, A.; et al. Histological Transformation and Progression in Follicular Lymphoma: A Clonal Evolution Study. PLoS Med. 2016, 13, e1002197. [Google Scholar] [CrossRef]
- Okosun, J.; Bodor, C.; Wang, J.; Araf, S.; Yang, C.Y.; Pan, C.; Boller, S.; Cittaro, D.; Bozek, M.; Iqbal, S.; et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat. Genet. 2014, 46, 176–181. [Google Scholar] [CrossRef]
- Pasqualucci, L.; Dominguez-Sola, D.; Chiarenza, A.; Fabbri, G.; Grunn, A.; Trifonov, V.; Kasper, L.H.; Lerach, S.; Tang, H.; Ma, J.; et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 2011, 471, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Pasqualucci, L.; Khiabanian, H.; Fangazio, M.; Vasishtha, M.; Messina, M.; Holmes, A.B.; Ouillette, P.; Trifonov, V.; Rossi, D.; Tabbo, F.; et al. Genetics of follicular lymphoma transformation. Cell Rep. 2014, 6, 130–140. [Google Scholar] [CrossRef] [Green Version]
- Carreras, J.; Lopez-Guillermo, A.; Kikuti, Y.Y.; Itoh, J.; Masashi, M.; Ikoma, H.; Tomita, S.; Hiraiwa, S.; Hamoudi, R.; Rosenwald, A.; et al. High TNFRSF14 and low BTLA are associated with poor prognosis in Follicular Lymphoma and in Diffuse Large B-cell Lymphoma transformation. J. Clin. Exp. Hematop 2019, 59, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Valero, J.G.; Matas-Cespedes, A.; Arenas, F.; Rodriguez, V.; Carreras, J.; Serrat, N.; Guerrero-Hernandez, M.; Yahiaoui, A.; Balague, O.; Martin, S.; et al. The receptor of the colony-stimulating factor-1 (CSF-1R) is a novel prognostic factor and therapeutic target in follicular lymphoma. Leukemia 2021, 35, 2635–2649. [Google Scholar] [CrossRef]
- Carreras, J.; Kikuti, Y.Y.; Hiraiwa, S.; Miyaoka, M.; Tomita, S.; Ikoma, H.; Ito, A.; Kondo, Y.; Itoh, J.; Roncador, G.; et al. High PTX3 expression is associated with a poor prognosis in diffuse large B-cell lymphoma. Cancer Sci. 2022, 113, 334–348. [Google Scholar] [CrossRef] [PubMed]
- Carreras, J.; Hamoudi, R.; Nakamura, N. Artificial Intelligence Analysis of Gene Expression Data Predicted the Prognosis of Patients with Diffuse Large B-Cell Lymphoma. Tokai J. Exp. Clin. Med. 2020, 45, 37–48. [Google Scholar] [PubMed]
- IBM. Troubleshooting. Available online: https://www.ibm.com/support/pages/can-i-make-same-model-neural-networks-spss (accessed on 23 February 2022).




| Num. | Model | Overall Accuracy (%) | No. Fields Used | Variables Used in the Final Model |
|---|---|---|---|---|
| 1 | XGBoost tree | 100 | 16 | 16 |
| 2 | Random trees | 97.2 | 16 | 13 (predictor importance: PRB1, CSF1R, IL10, L1CAM, BTD, MOCOS, FOXP3, PDCD1, EPB41L4B, and IPI score; top 10 inputs) |
| 3 | Random forest | 86.9 | 16 | Predictor importance: IPI score 1, CEP57, L1CAM, EPB41L4B, TNFRSF14, BTD, SPIN2A, PDCD1, CTNS, SRGAP3, IL10, MOCOS, PRB1, FOXP3, CD163, CSF1R, and IPI score 2 |
| 4 | LSVM | 74.4 | 16 | Predictor importance: SRGAP3, IPI score, L1CAM, CD163, EPB41L4B, PDCD1, CSF1R, IL10, MOCOS, BTD; top 10 inputs |
| 5 | Neural network | 69.4 (79.9% of overall percent correct) | 16 | Predictor importance: SRGAP3, IL10, TNFRSF14, EPB41L4B, CD163, SPIN2A, IPI score, CEP57, MOCOS, PDCD1, PRB1, L1CAM, CTNS, FOXP3, CSF1R, BTD |
| 6 | SVM | 68.9 | 16 | 16 |
| 7 | C&R tree | 68.9 | 6 | IPI score |
| 8 | C5 tree | 67.8 | 1 | IPI score |
| 9 | XGBoost linear | 67.8 | 16 | 16 |
| 10 | Quest | 67.8 | 6 | IPI score |
| 11 | Tree-AS | 67.2 | 1 | IPI score |
| 12 | CHAID | 67.2 | 1 | IPI score |
| 13 | Logistic regression | 66.7 | 16 | Equation for dead outcome: −1.8*SRGAP3 + −0.9*CEP57 + 1.0*EPB41L4B + 0.8*SPIN2A + 0.7*IL10 + 0.15*MOCOS + 0.3*CD163 + 0.9*CTNS + 0.1*BTD + −0.01*PDCD1 + −0.4*L1CAM + −0.1*PRB1 + 0.5*TNFRSF14 + −0.4*CSF1R + 0.03*FOXP3 + −1.1*IPI score = 1 + 0.7*IPI score = 2 + −3.8. |
| 14 | Discriminant analysis | 66.7 | 15 | 16, except IPI score |
| 15 | KNN algorithm | 63.4 | 16 | 16 |
| 16 | Bayesian network | 57.4 | 16 | 16 |
| Gene | Protein | Function |
|---|---|---|
| EPB41L4B | Band 4.1-like protein 4B | Promotes cellular adhesion, migration, and motility |
| MOCOS | Molybdenum cofactor sulfurase | Molybdopterin cofactor metabolic process |
| SPIN2A | Spindlin-2A | Regulation of cell cycle progression |
| BTD | Biotinidase | Biotin metabolic process |
| SRGAP3 | SLIT-ROBO Rho GTPase-activating protein 3 | Negative regulation of cell migration |
| CTNS | Cystinosin | Positive regulation of TORC1 signaling |
| PRB1 | Basic salivary proline-rich protein 1 | Glycoprotein |
| L1CAM | Neural cell adhesion molecule L1 | Cell adhesion and in the generation of transmembrane signals at tyrosine kinase receptors |
| CEP57 | Centrosomal protein CEP57L1 | Centrosomal protein, which may be required for microtubule attachment to centrosomes. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carreras, J.; Kikuti, Y.Y.; Miyaoka, M.; Hiraiwa, S.; Tomita, S.; Ikoma, H.; Kondo, Y.; Ito, A.; Hamoudi, R.; Nakamura, N. The Use of the Random Number Generator and Artificial Intelligence Analysis for Dimensionality Reduction of Follicular Lymphoma Transcriptomic Data. BioMedInformatics 2022, 2, 268-280. https://doi.org/10.3390/biomedinformatics2020017
Carreras J, Kikuti YY, Miyaoka M, Hiraiwa S, Tomita S, Ikoma H, Kondo Y, Ito A, Hamoudi R, Nakamura N. The Use of the Random Number Generator and Artificial Intelligence Analysis for Dimensionality Reduction of Follicular Lymphoma Transcriptomic Data. BioMedInformatics. 2022; 2(2):268-280. https://doi.org/10.3390/biomedinformatics2020017
Chicago/Turabian StyleCarreras, Joaquim, Yara Yukie Kikuti, Masashi Miyaoka, Shinichiro Hiraiwa, Sakura Tomita, Haruka Ikoma, Yusuke Kondo, Atsushi Ito, Rifat Hamoudi, and Naoya Nakamura. 2022. "The Use of the Random Number Generator and Artificial Intelligence Analysis for Dimensionality Reduction of Follicular Lymphoma Transcriptomic Data" BioMedInformatics 2, no. 2: 268-280. https://doi.org/10.3390/biomedinformatics2020017
APA StyleCarreras, J., Kikuti, Y. Y., Miyaoka, M., Hiraiwa, S., Tomita, S., Ikoma, H., Kondo, Y., Ito, A., Hamoudi, R., & Nakamura, N. (2022). The Use of the Random Number Generator and Artificial Intelligence Analysis for Dimensionality Reduction of Follicular Lymphoma Transcriptomic Data. BioMedInformatics, 2(2), 268-280. https://doi.org/10.3390/biomedinformatics2020017