
Ergodic Algorithmic Model (EAM), with Water as Implicit Solvent, in Chemical, Biochemical, and Biological Processes



Abstract
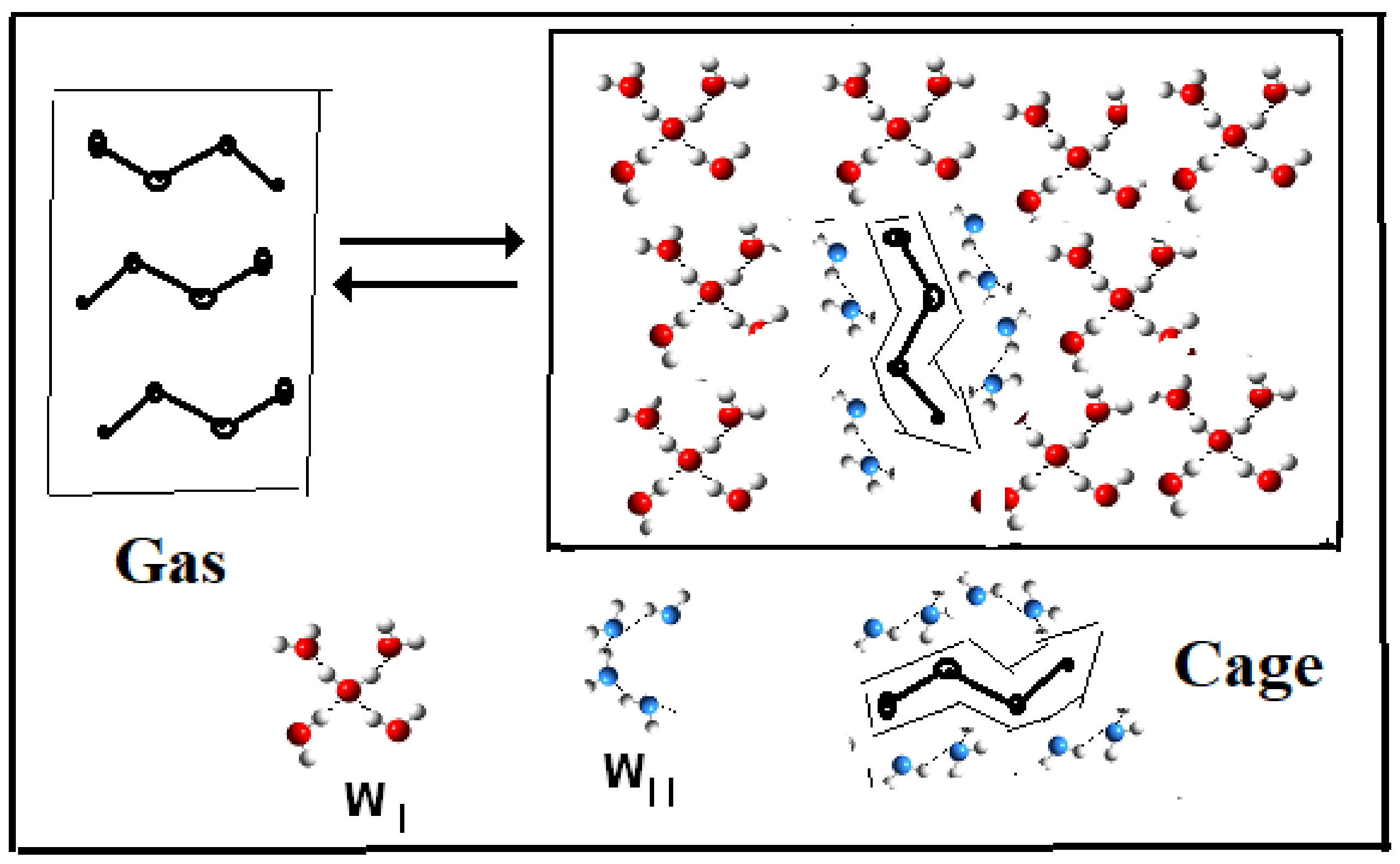
:1. Introduction
Thermal Equivalent Dilution (TED): Ergodicity
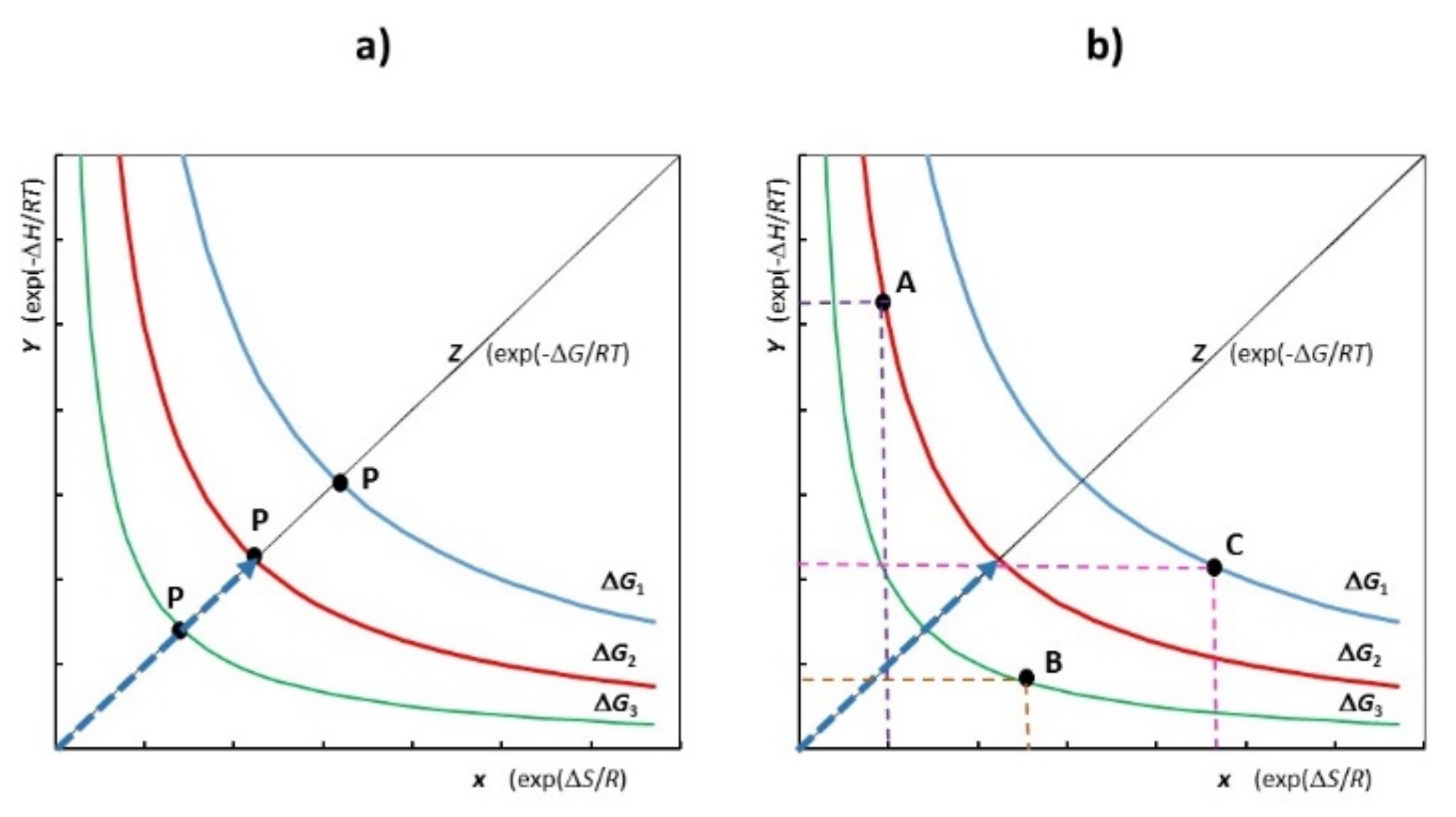
2. Free Energy
2.1. Thermodynamic Free Energy
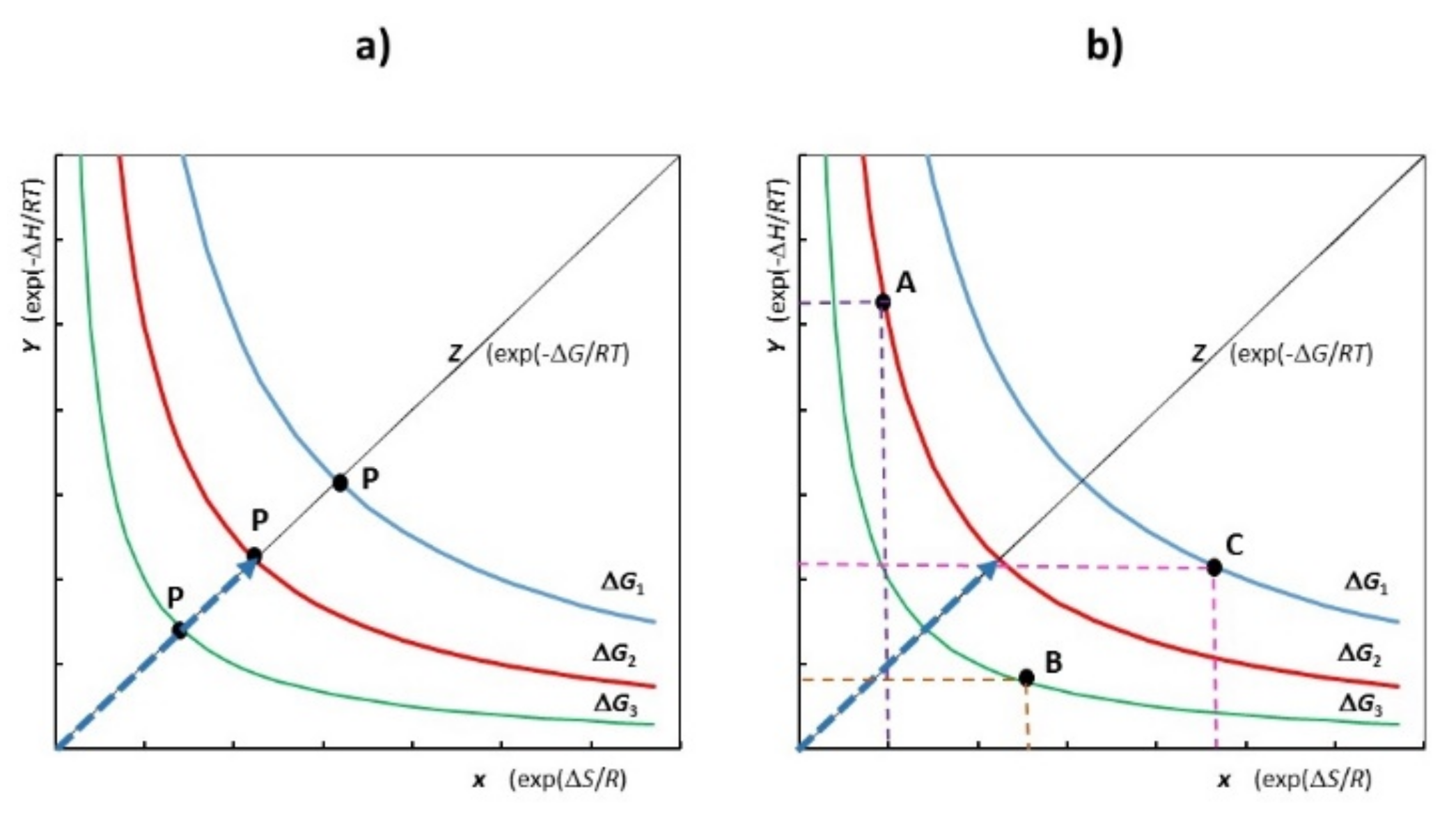
2.2. Pseudo-Free Energy Function
- (i)
- (ii)
- (iii)
- (iv)
3. Explicit Solvent and Implicit Solvent
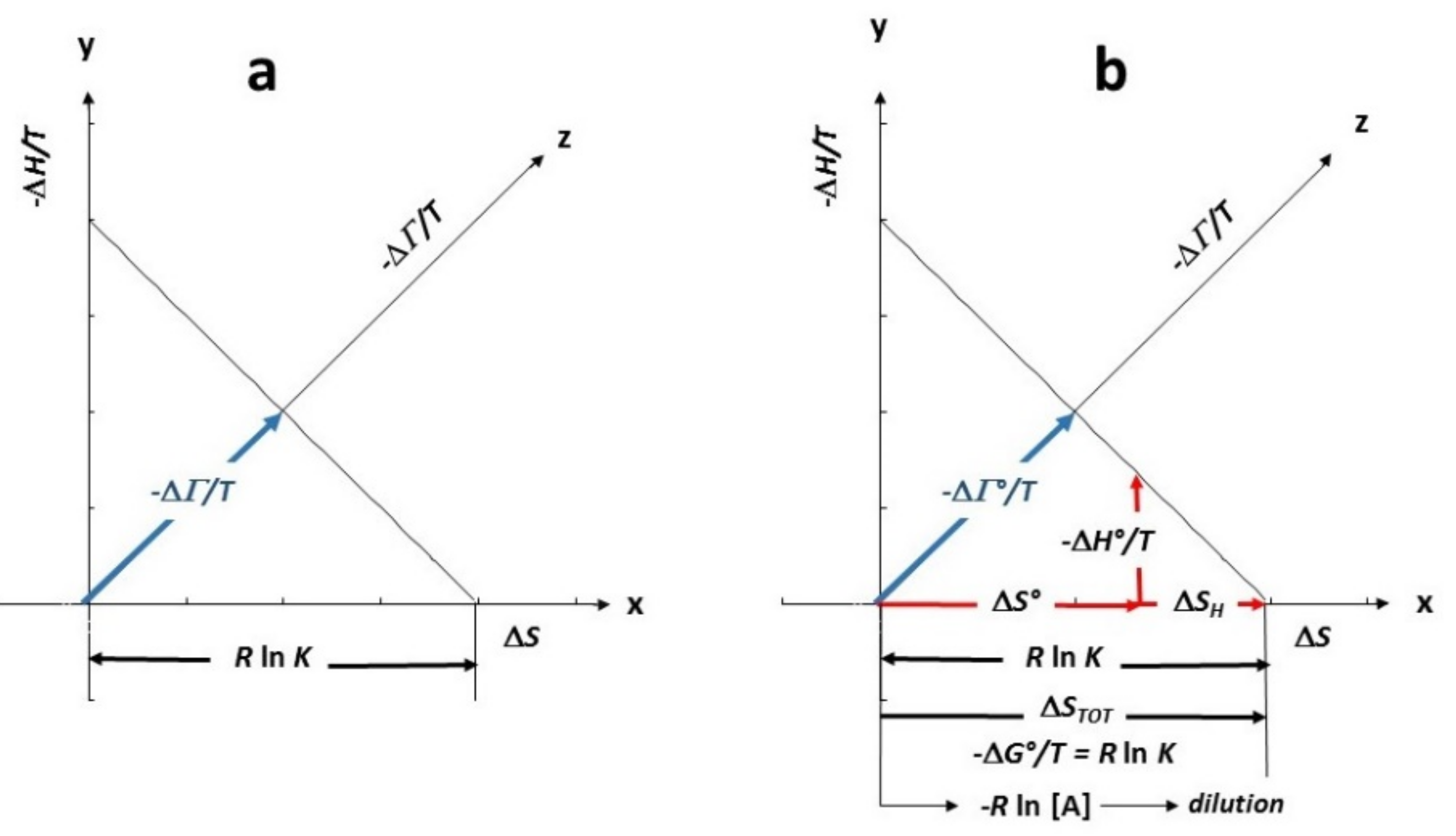
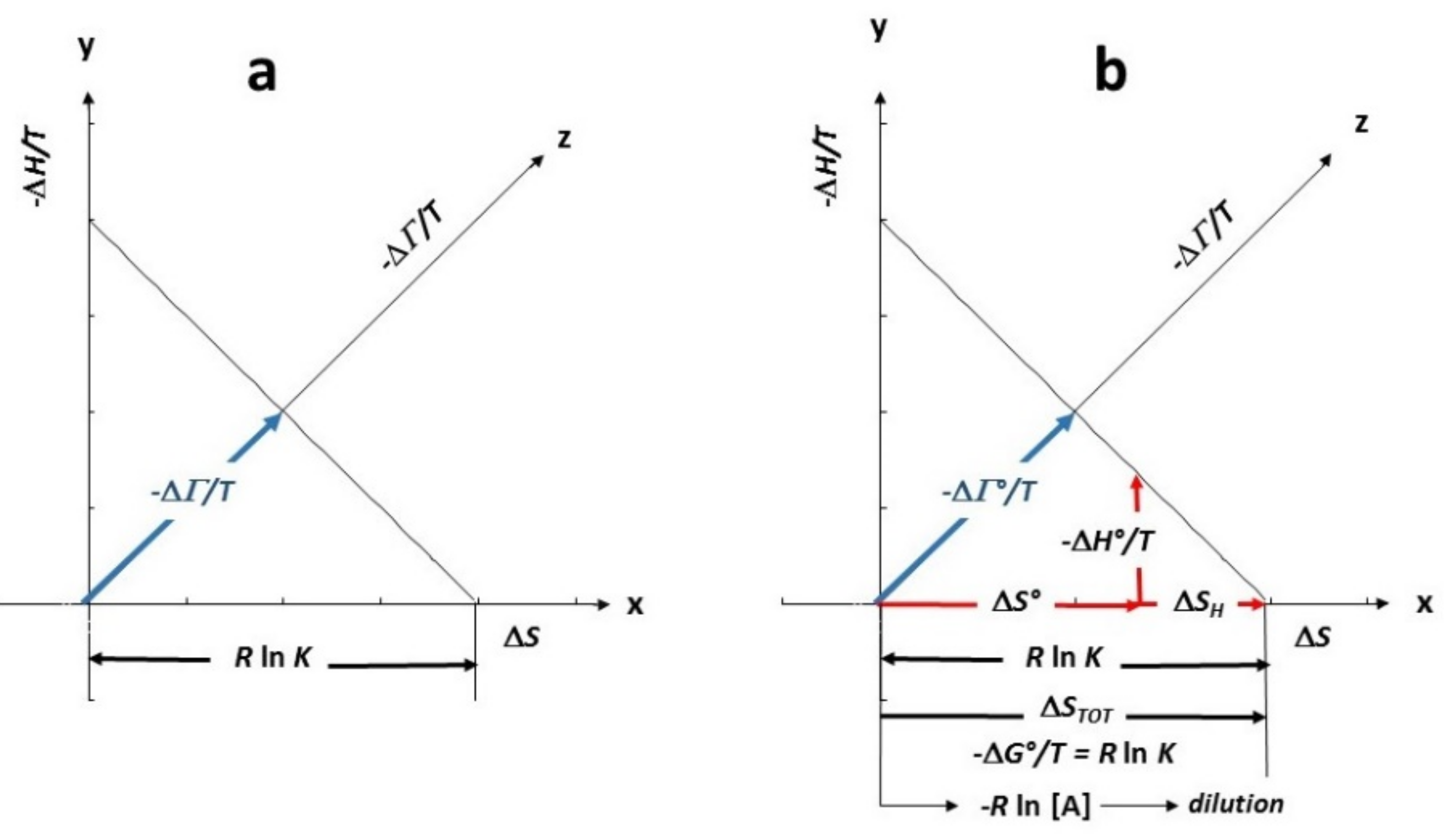
4. Free Energy: Intensity Entropy and Density Entropy Components
5. Procedure According to Ergodic Algorithmic Model (EAM)
- (a)
- Calculation of convoluted binding functions from experimental determinations; then, by applying EAM we derive Rln Kmot.
- (b)
- Calculation of simulation functions according to HEP.
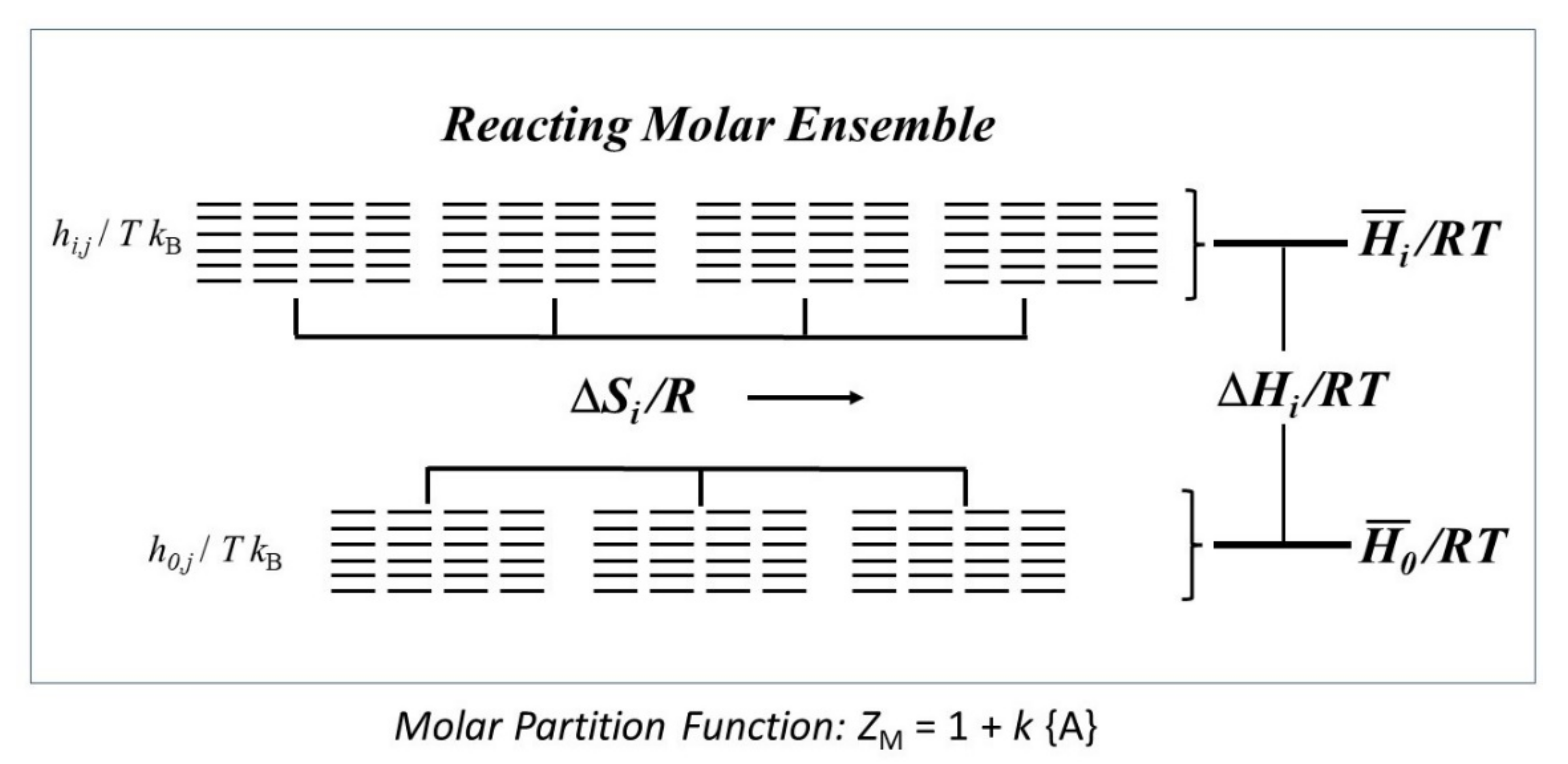
6. Thermodynamic Functions and Information Elements
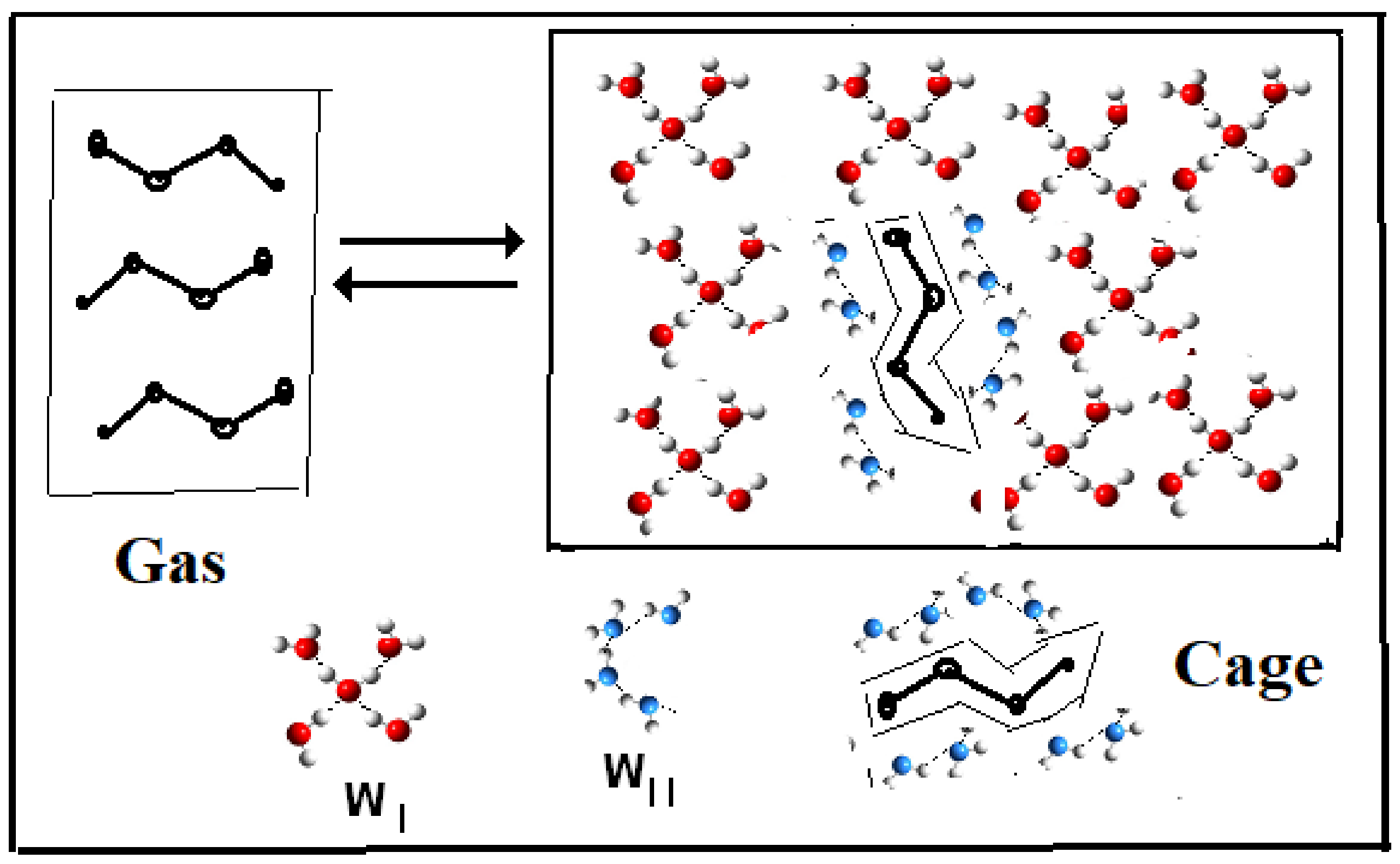
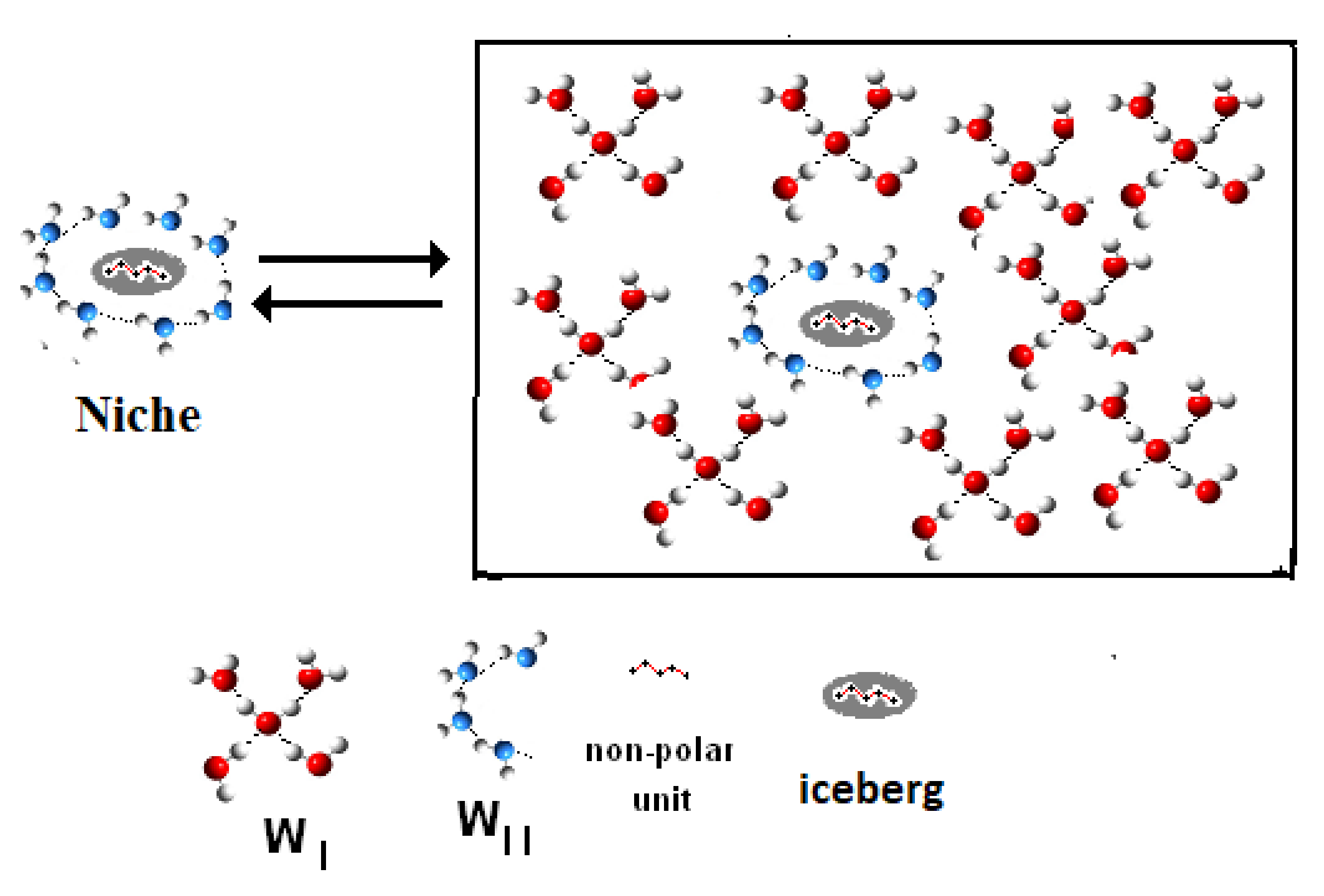
7. Dual-Structure Partition Function. Molecular Structures
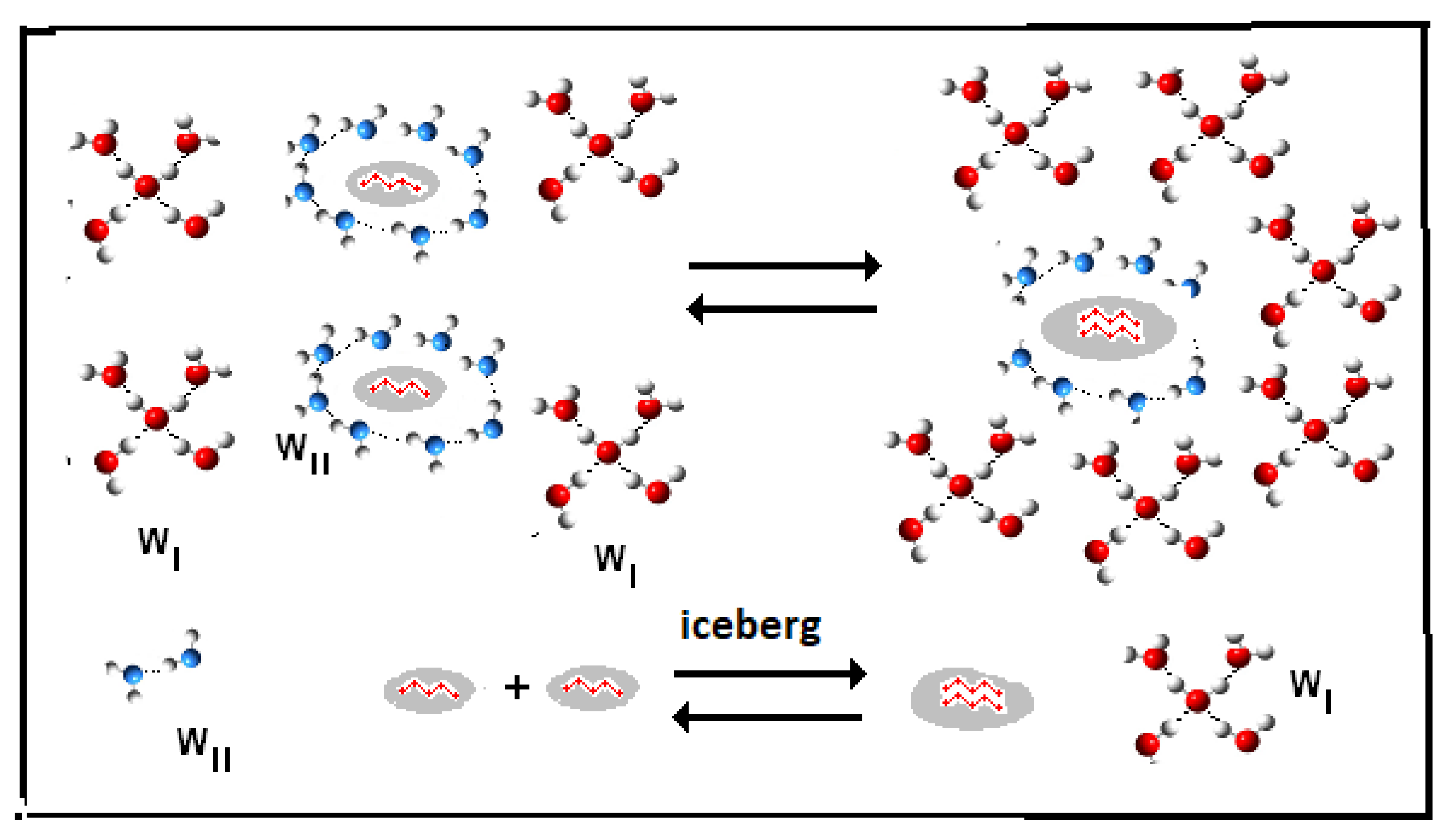
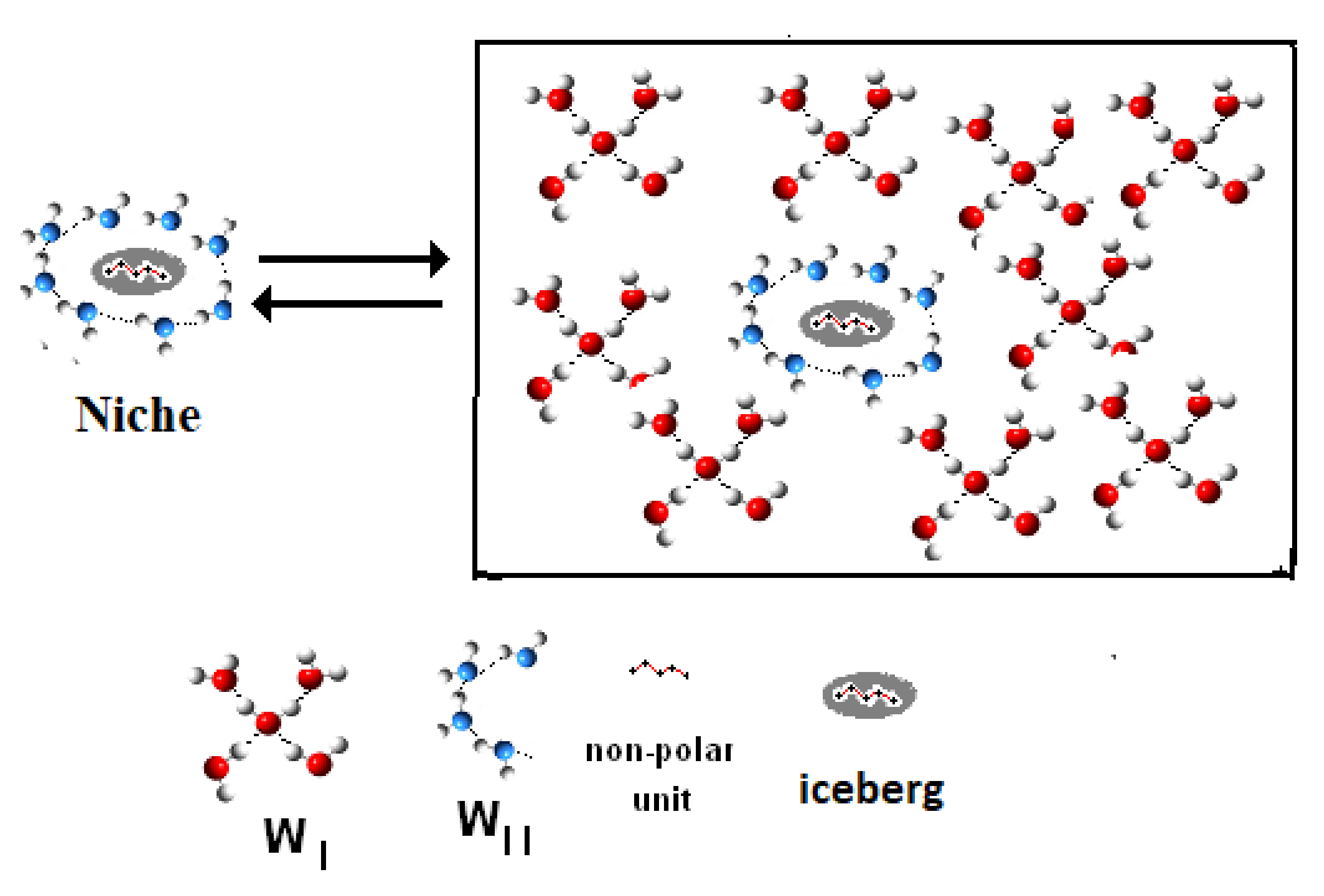
- (a)
- (Reaction A {–ξw WI (solvent) → ξw WII (iceberg, solute)}).
- (b)
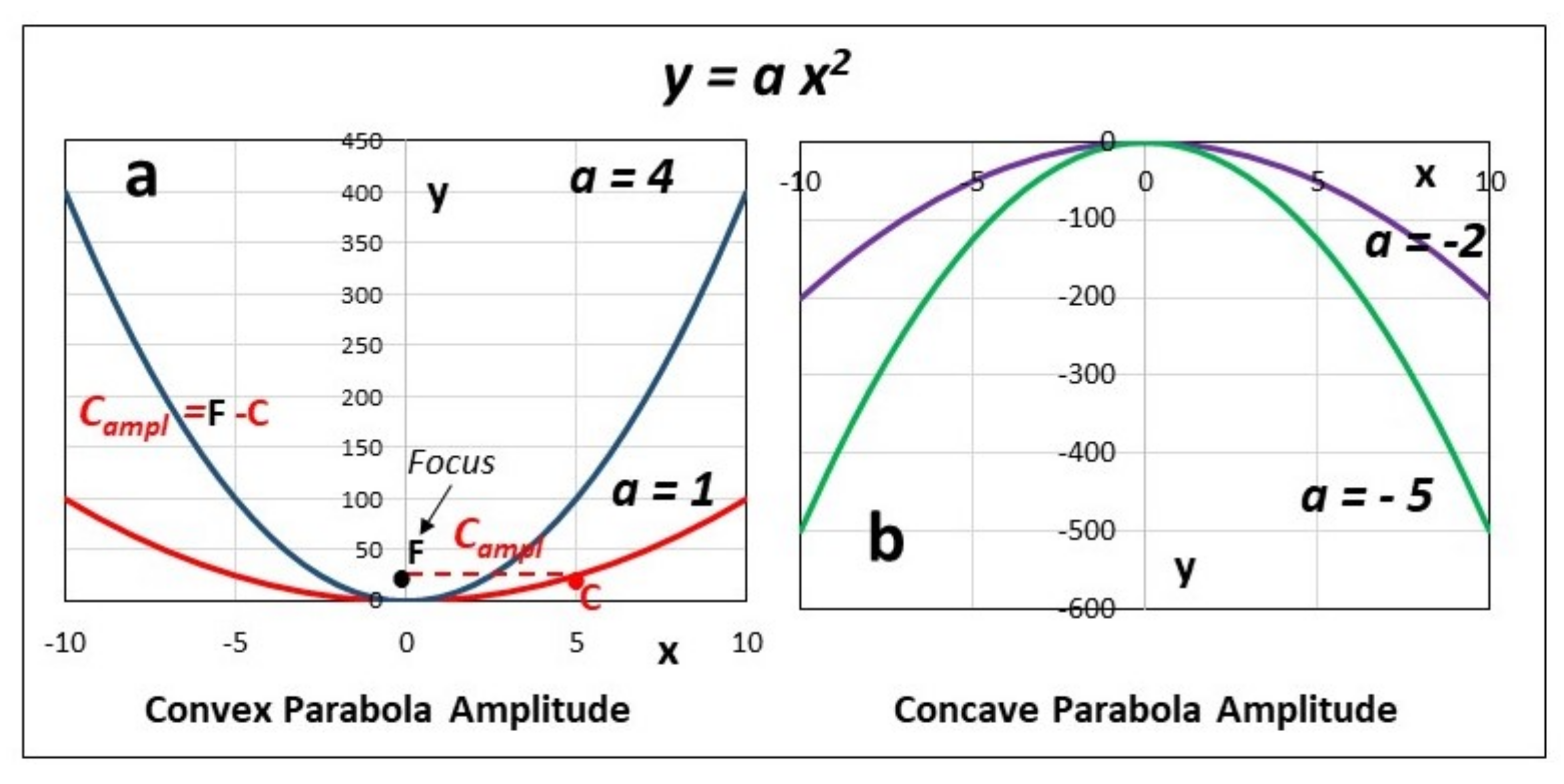
- Convexity (ΔCp,hydr = ξw Cp,w > 0).
- (c)
- Iceberg size and water stoichiometry: ξw.
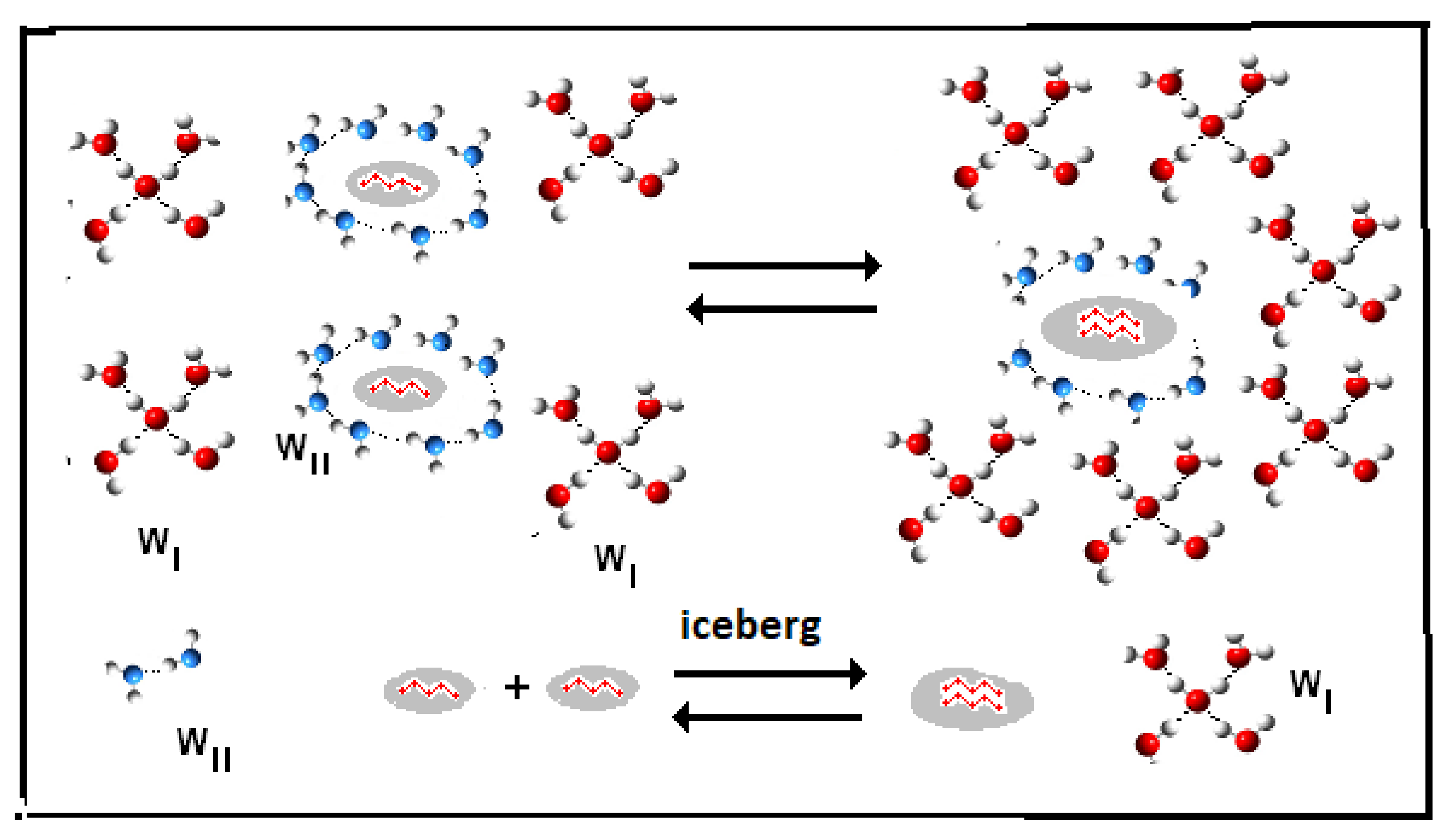
- (a)
- (reaction: B {–ξw WII (iceberg, solute)} → ξw WI (solvent).
- (b)
- Concavity (ΔCp,hydr = ξw Cp,w < 0).
- (c)
- Iceberg size and water stoichiometry: ξw.
8. Structures of Thermodynamic Functions
- (1)
- The simulated free energy functions RlnKsimul, as calculated by any simulation functions, either FEP, or TI, MV, MC, etc., and applied to hydrophobic hydration processes are inadequate to represent the properties of biphasic systems with implicit solvent. Many important thermodynamic functions are ignored by these calculations and all the important information elements carried by these functions are completely lost.
- (2)
- The hydrophobic hydration systems have a biphasic composition, constituted by a “non-reacting” molecule ensemble, at constant potential formed by the solvent and by a “reacting” mole ensemble formed by diluted solutes. The distribution of molecules over the sublevels hi,j of each macrolevel Hi follows a Boltzmann distribution, whereas the distribution of moles of reactants over macrolevels Hi follows a binomial distribution. The Ergodic Algorithmic Model (EAM) is adequate to the biphasic composition of aqueous solutions and suited to evaluate correctly the many essential information elements.
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Probability Space |
| Dual-Structure Partition Function (for Biphasic Systems) |
| {DS-PF} = {T-PF} · {M-PF}{ζw}} = Kmot·1 |
| {M-PF} = exp(ΔSmot/R), {T-PF} = exp(−ΔHmot/RT), exp(−ΔGmot/RT) = Kmot |
| exp(−ΔHmot/RT) exp(ΔSmot/R) = exp(−ΔGmot/RT) = Kdual |
| exp(Sdens/R) = aA−1 = xA−1 Φ−1 = xA−1· TCp,A/R |
| exp(Sint/Cp,A) = T |
| Kdual = Kmot · ζth |
| ζth = 1 (Implicit Solvent, at constant Thermodynamic Potential μs) |
| {M-PF} = Kmot → (Solute, Density Entropy parameter) |
| {T-PF} = ζth = 1 → (Solute, Intensity Entropy parameter) |
| aA = xA · Φ (aA = activity of A, Φ = T−Cp,A/R: Lambert thermal factor) |
| Ergodicity → (Dilution → Temperature) |
| (1/xA) = did(A) → dilution thermal factor TCp,A/R = 1/Φ |
| Thermodynamic Space |
| (A) Binding Function RlnKdual = (−ΔGdual/T) = {f(1/T)·g(T)} |
| 1.Curved convoluted function ({f(1/T)*g(T)}) |
| Rln Kdual = {(−ΔHdual/T) + (ΔSdual)} = {(−ΔHmot/T) + (−ΔHth/T)} + {(ΔSmot) + (ΔSth)} |
| ∂(RlnKdual)/∂(1/T) = −ΔHdual = −ΔHmot − ΔHth = −ΔHmot − ΔCp.hydrT (J mole−1) |
| 2. Linear function (f(1/T)) |
| RlnKmot = RlnKdual ={(−ΔHdual/T) + (ΔHth/T)} + {(ΔSdual) − (ΔSth)} = −ΔHmot/T + ΔSmot |
| ∂(RlnKmot)/∂(1/T) = −ΔHmot (J mole−1) |
| (B) Binding Function RTlnKdual = (−ΔGdual) = {f(T)·g(lnT)} |
| 1.Curved convoluted function ({f(T)·g(lnT)}) |
| RTln Kdual = {−ΔHdual + T ΔSdual} = {−ΔHmot − ΔHth} + T {ΔSmot + ΔSth} |
| ∂(RTlnKdual)/∂T = ΔSdual = ΔSmot + ΔSth = ΔSmot + ΔCp,hydrlnT (J K−1 mole−1) |
| 2.linear function (f(T)) |
| RTlnKmot = {−ΔHdual + ΔHth} + T {ΔSdual − ΔSth} = −ΔHmot + T ΔSmot |
| ∂(RTlnKmot)/∂T = ΔSmot |
| (C) Activity: aA = f(xA,T) |
| dSDens = −Rdln aA = (−Rdln xA)T + (Cp,A dlnT)x |
| (D) Ergodicity |
| Density Entropy (ΔS/R) and Intensity Entropy (ΔH/RT) |
| (dSDens)T = (−Rdln xA)T = (Rdlndid,A)T → (Changing Density Entropy) |
| (dSInt)xA = (Cp,A dlnT)xA → (Changing Intensity Entropy) |
| TED (Thermal Equivalent Dilution) |
| (dSDen)T = (dSInt)xA |
| nw (Rdln did,A)T = nw (Cp,A dlnT) xA |
| (E) Hydrophobic Heat Capacity |
| ΔCp,hydr = ±ξw Cp,w; Cp,w = 75.36 J K−1mol−1 (molar heat capacity for liquid water) |
Appendix B
| (a) Analysis within Classes | ||
| Class A: iceberg formation | Unit | Relative error |
| <Δhfor>A= −22.7 ± 0.07 | kJ⋅mol−1 ⋅ξw−1 | ±3.1% |
| <Δsfor>A = −445 ± 3 | J⋅K−1⋅mol−1⋅ξw−1 | ±0.7% |
| Class B: iceberg reduction | Unit | Relative error |
| <Δhred>B = +23.7 ± 0.6 | kJ⋅mol−1 ⋅ξw−1 | ±2.51% |
| <Δsred>B = +432 ± 4 | J⋅K−1⋅mol−1⋅ξw−1 | ±0.9% |
| (b) Comparison among Classes | ||
| Enthalpy | Entropy | |
| <Δhfor>A = −22.7 ± 0.7 kJ mol−1 ξw−1 | <Δsfor>A = −445 ± 3J K−1 mol−1 ξw−1 | |
| <Δhred>B = +23.7 ± 0.6 Kj mol−1 ξw−1 | <Δsred> B = +432 ± 4 J K−1 mol−1 ξw−1 | |
| mean abs.value <Δh>A,B = 22.95 ± 0.75 | mean abs.value <Δs>A,B = 438.5 ± 6.5 | |
| Student’s ratio: 0.75/0.92 = 0.815 | Student’s ratio: 6.5/5 = 1.3 | |
References
- Lambert, F.L. Configurational Entropy Revisited. J. Chem. Educ. 2007, 84, 1548–1550. [Google Scholar] [CrossRef]
- Lambert, F.L. The Misinterpretation of Entropy as “Disorder”. J. Chem. Educ. 2012, 89, 310. [Google Scholar] [CrossRef]
- Fisicaro, E.; Compari, C.; Braibanti, A. Entropy/enthalpy compensation: Hydrophobic effect, micelles and protein complexes. Phys. Chem. Chem. Phys. 2004, 6, 4156–4166. [Google Scholar] [CrossRef]
- Fisicaro, E.; Compari, C.; Duce, E.; Biemmi, M.; Peroni, M.; Braibanti, A. Thermodynamics of micelle formation in water, hydrophobic processes and surfactant self-assemblies. Phys. Chem. Chem. Phys. 2008, 10, 3903–3914. [Google Scholar] [CrossRef] [PubMed]
- Fisicaro, E.; Compari, C.; Braibanti, A. Hydrophobic hydration processes. General thermodynamic model by thermal equivalent dilution determinations. Biophys. Chem. 2010, 151, 119–138. [Google Scholar] [CrossRef] [PubMed]
- Fisicaro, E.; Compari, C.; Braibanti, A. Hydrophobic hydration processes. Biophys. Chem. 2011, 156, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Fisicaro, E.; Compari, C.; Braibanti, A. Hydrophobic Hydration Processes. I: Dual-Structure Partition Function for Biphasic Aqueous Systems. ACS Omega 2018, 3, 15043–15065. [Google Scholar] [CrossRef] [PubMed]
- Fisicaro, E.; Compari, C.; Braibanti, A. Statistical Inference for Ergodic Algorithmic Model (EAM), Applied to Hydrophobic Hydration Processes. Entropy 2021, 23, 700–721. [Google Scholar] [CrossRef]
- Fisicaro, E.; Compari, C.; Braibanti, A. Intensity Entropy and Null Thermal Free Energy and Density Entropy and Motive Free Energy. ACS Omega 2019, 4, 19526–19547. [Google Scholar] [CrossRef]
- Chipot, C.; Pohorille, A. Free Energy Calculations. Theory and Application in Chemistry and Biology; Sperling: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Pohorille, A.; Jarzynski, C.; Chipot, C. Good Practice in Free-Energy Calculation. J. Phys. Chem. B 2010, 114, 10235–10253. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Dehez, F.; Cai, W.; Chipot, C. A Toolkit for the Analysis of Free-Energy Perturbation Calculations. J. Chem. Theory Comput. 2012, 8, 2606–2616. [Google Scholar] [CrossRef]
- Born, M. Volumes and heats of hydration of ions. Z. Phys. 1920, 1, 45–48. [Google Scholar] [CrossRef]
- Landau, L.D.; Lifshitz, E.M. Statistical Physics; ThriftBooks-Dallas: Seattle, WA, USA; The Claredon Press: Oxford, UK, 1938. [Google Scholar]
- Pearlman, D.A.; Charifson, P.S. Are Free Energy Calculations Useful in Practice? A Comparison with Rapid Scoring Functions for the p38 MAP Kinase Protein System. J. Med. Chem. 2001, 44, 3417–3423. [Google Scholar] [CrossRef]
- Zwanzig, R.W. High-temperature equation of state by a perturbation method. I. Nonpolar gases. J. Chem. Phys. 1954, 22, 1420–1426. [Google Scholar] [CrossRef]
- Chipot, C.; Rozanska, X.; Dixit, S.B. Can free energy calculations be fast and accurate at the same time? Binding of low-affinity, non-peptide inhibitors to the SH2 domain of the src protein. J. Comput.-Aided Mol. Des. 2005, 19, 765–770. [Google Scholar]
- Kirkwood, J.G. Statistical mechanics of fluid mixtures. J. Chem. Phys. 1935, 3, 300–313. [Google Scholar] [CrossRef]
- Chodera, J.D. Alchemical free energy methods for drug discovery: Progress and challenges. Curr. Opin. Struct. Biol. 2011, 21, 150–160. [Google Scholar] [CrossRef] [Green Version]
- Bennett, C.H. Efficient Estimation of Free Energy Differences from Monte Carlo Data. J. Comput. Phys. 1976, 22, 245–268. [Google Scholar] [CrossRef]
- Ferrenberg, A.M.; Swendsen, R.H. New Monte Carlo technique for studying phase transitions. Phys. Rev. Lett. 1988, 61, 2635–2638. [Google Scholar] [CrossRef] [Green Version]
- Jarzynski, C. Non equilibrium Equality for Free Energy Differences. Phys. Rev. Lett. 1997, 78, 2690. [Google Scholar] [CrossRef] [Green Version]
- Jarzynski, C. Equilibrium free-energy differences from nonequilibrium measurements: A master-equation approach. Phys. Rev. E 1997, 56, 5018–5035. [Google Scholar] [CrossRef] [Green Version]
- Abel, R.; Wang, L.; Harder, E.D.; Berne, B.J.; Friesner, R.A. Advancing Drug Discovery through Enhanced Free Energy Calculations. Acc. Chem. Res. 2017, 50, 1625–1632. [Google Scholar] [CrossRef]
- Knight, J.L.; Brooks, C.L., III. λ-dynamics free energy simulation methods. J. Comput. Chem. 2009, 30, 1692–1700. [Google Scholar] [CrossRef]
- Talhout, R.; Villa, A.; Mark, A.E.; Engberts, J.B.F.N. Understanding Binding Affinity: A Combined Isothermal Titration Calorimetry/Molecular Dynamics Study of the Binding of a Series of Hydrophobically Modified Benzamidinium Chloride Inhibitors to Trypsin. J. Am. Chem. Soc. 2003, 125, 10570–10579. [Google Scholar] [CrossRef]
- Freire, E. The binding thermodynamics of drug candidates. Methods Princ. Med. Chem. 2015, 65, 1–13. [Google Scholar]
- Schön, A.; Freire, E. Enthalpy screen of drug candidates. Anal. Biochem. 2016, 513, 1–6. [Google Scholar] [CrossRef] [Green Version]



| ∂(RlnKmot)/∂(1/T) = −ΔH°mot (J mol−1) |
| ∂(RTlnKmot)/∂T = ΔS°mot (J K−1 mol−1) |
| ΔCp,hydr = ±ξw Cp,w |
| with Cp,w = 75.36 J·K−1mol−1 (molar heat capacity for liquid water) |
| ΔCp,hydr = +ξw Cp,w (Class A, convex binding function) |
| ΔCp,hydr = −ξw Cp,w (Class B, concave binding function) |
| ENTHALPY | ENTROPY |
|---|---|
| RlnKdual = −ΔG°/T = −ΔHdual/T + Sdual. = −{ΔHmot+ΔCp,hydr,T}/T + ΔS’ where ΔCp,hydr = 2202(ΔHdual)/∂T ΔC p,hydr = isobaric heat capacity(*) | RT lnKdual = −ΔG° = −ΔHdual + T ΔSdual = −ΔH’+T{ΔSmot + ΔCp,hydr lnT} where ΔCp,hyd = ∂(ΔSdual)∂lnT ΔCp,hydr = isobaric heat capacity(*) |
| Motive Function from primary function f(1/T) in RlnKapp = {f((1/T)*g(T)} | Motive Function from primary function f(T) in RTlnKapp = {f((T)*g(lnT)} |
| R lnKmot = −(ΔHmot/T) + ΔS | RT lnKmot = −ΔHmot +T ΔS° |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fisicaro, E.; Compari, C.; Braibanti, A. Ergodic Algorithmic Model (EAM), with Water as Implicit Solvent, in Chemical, Biochemical, and Biological Processes. Thermo 2021, 1, 361-375. https://doi.org/10.3390/thermo1030022
Fisicaro E, Compari C, Braibanti A. Ergodic Algorithmic Model (EAM), with Water as Implicit Solvent, in Chemical, Biochemical, and Biological Processes. Thermo. 2021; 1(3):361-375. https://doi.org/10.3390/thermo1030022
Chicago/Turabian StyleFisicaro, Emilia, Carlotta Compari, and Antonio Braibanti. 2021. "Ergodic Algorithmic Model (EAM), with Water as Implicit Solvent, in Chemical, Biochemical, and Biological Processes" Thermo 1, no. 3: 361-375. https://doi.org/10.3390/thermo1030022
APA StyleFisicaro, E., Compari, C., & Braibanti, A. (2021). Ergodic Algorithmic Model (EAM), with Water as Implicit Solvent, in Chemical, Biochemical, and Biological Processes. Thermo, 1(3), 361-375. https://doi.org/10.3390/thermo1030022