
Numerical Integration of Weight Loss Curves for Kinetic Analysis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Numerical Methods for Simple Materials Based on One Process Only
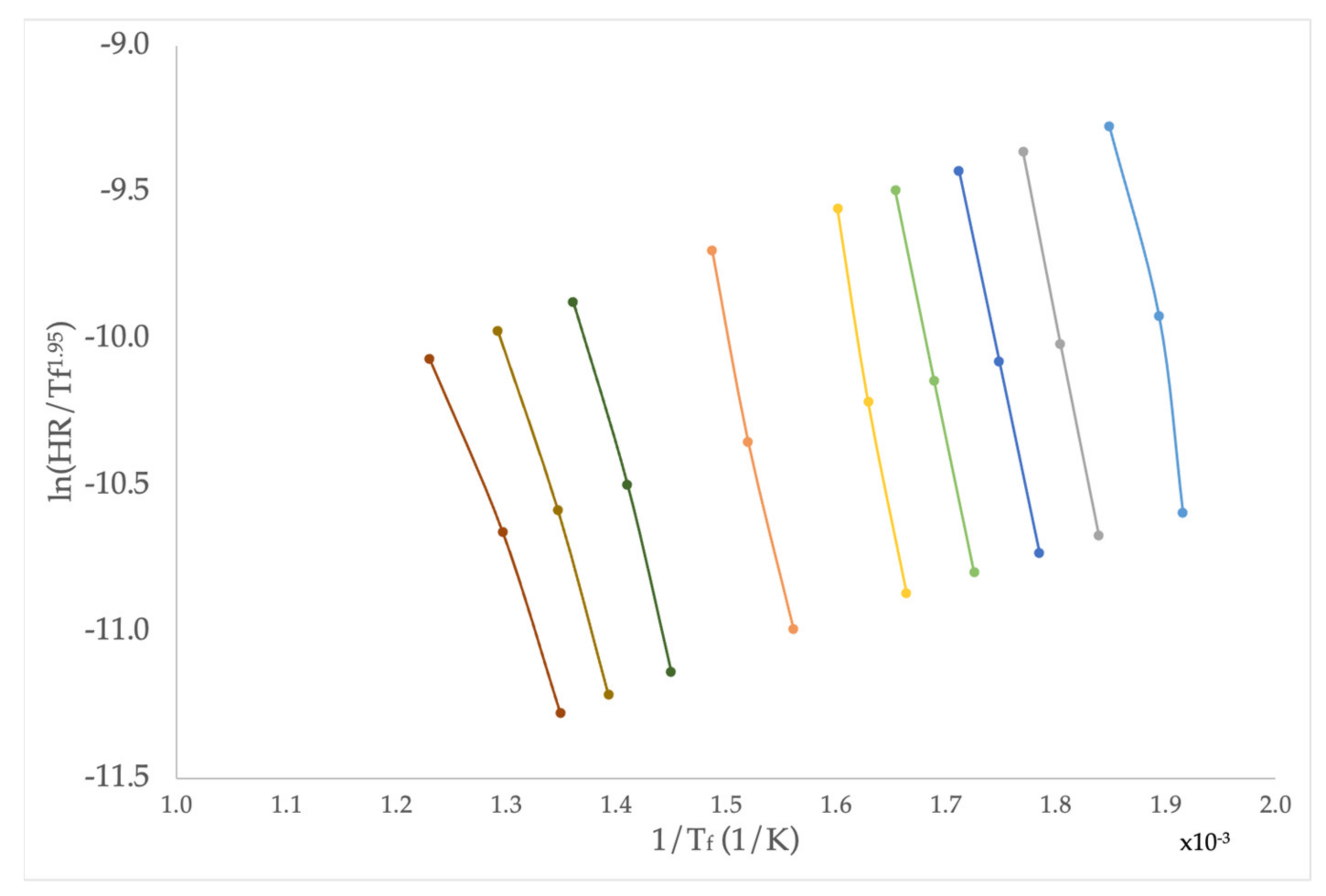
2.1. Theoretical Background
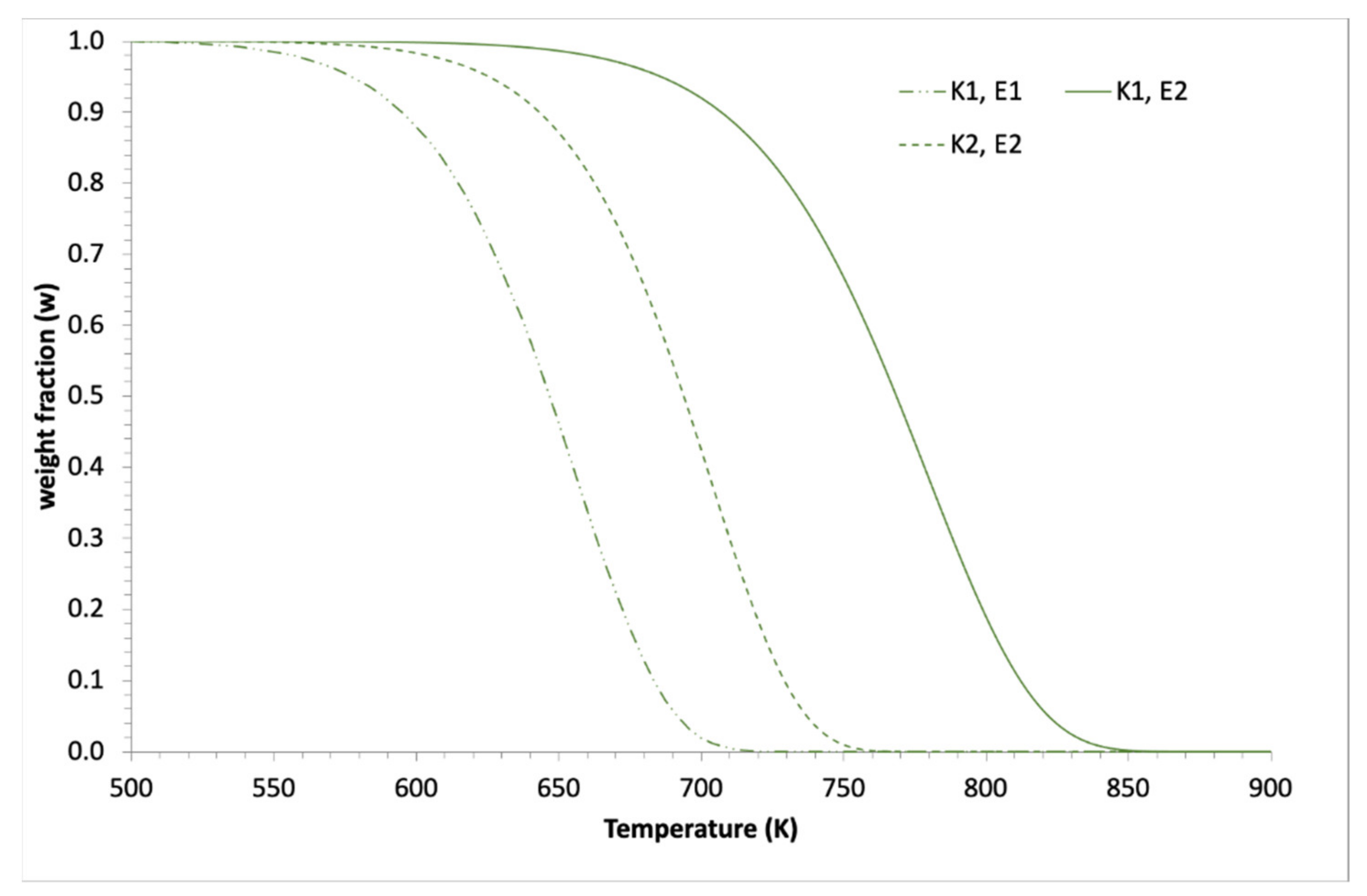
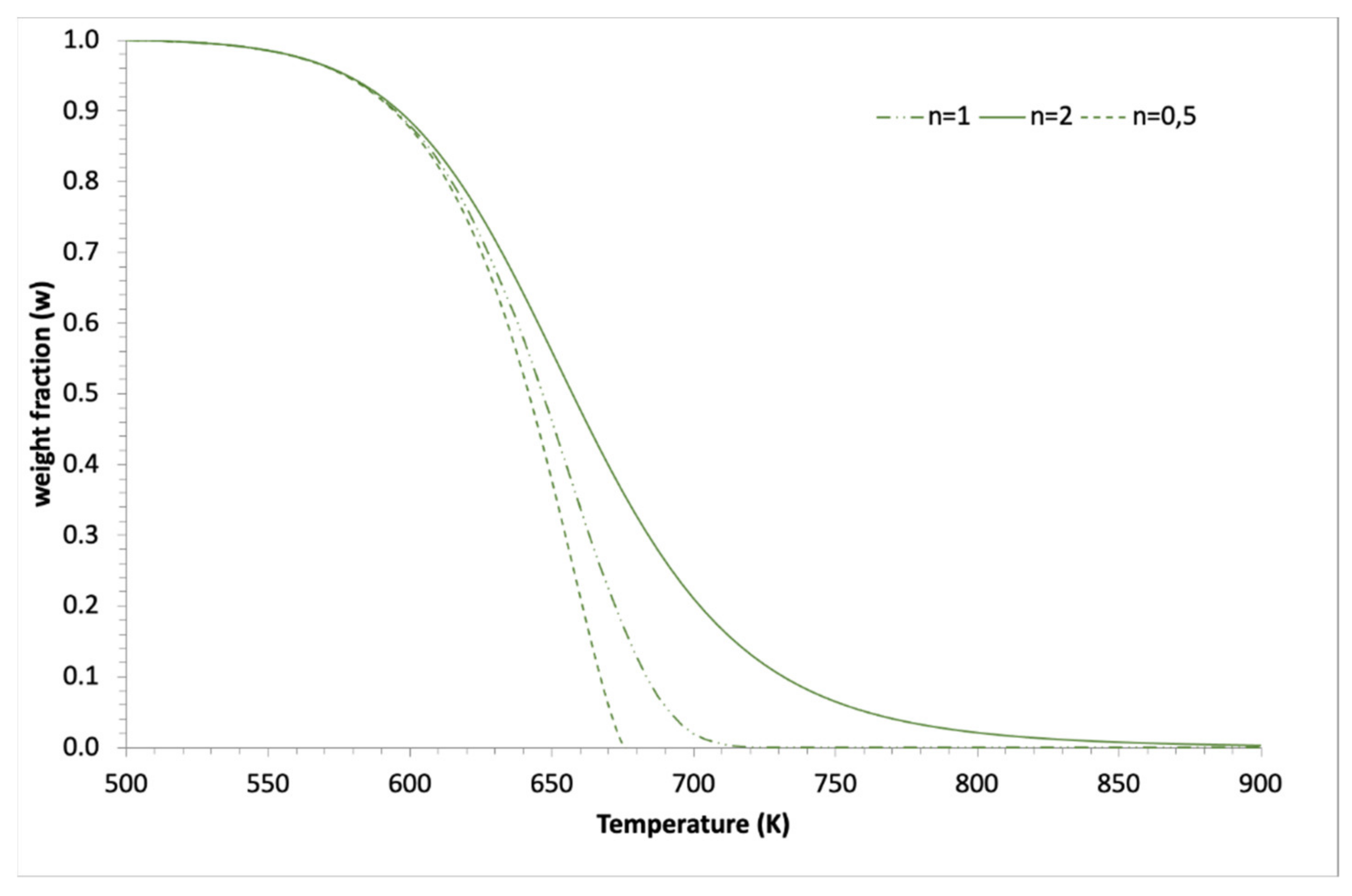
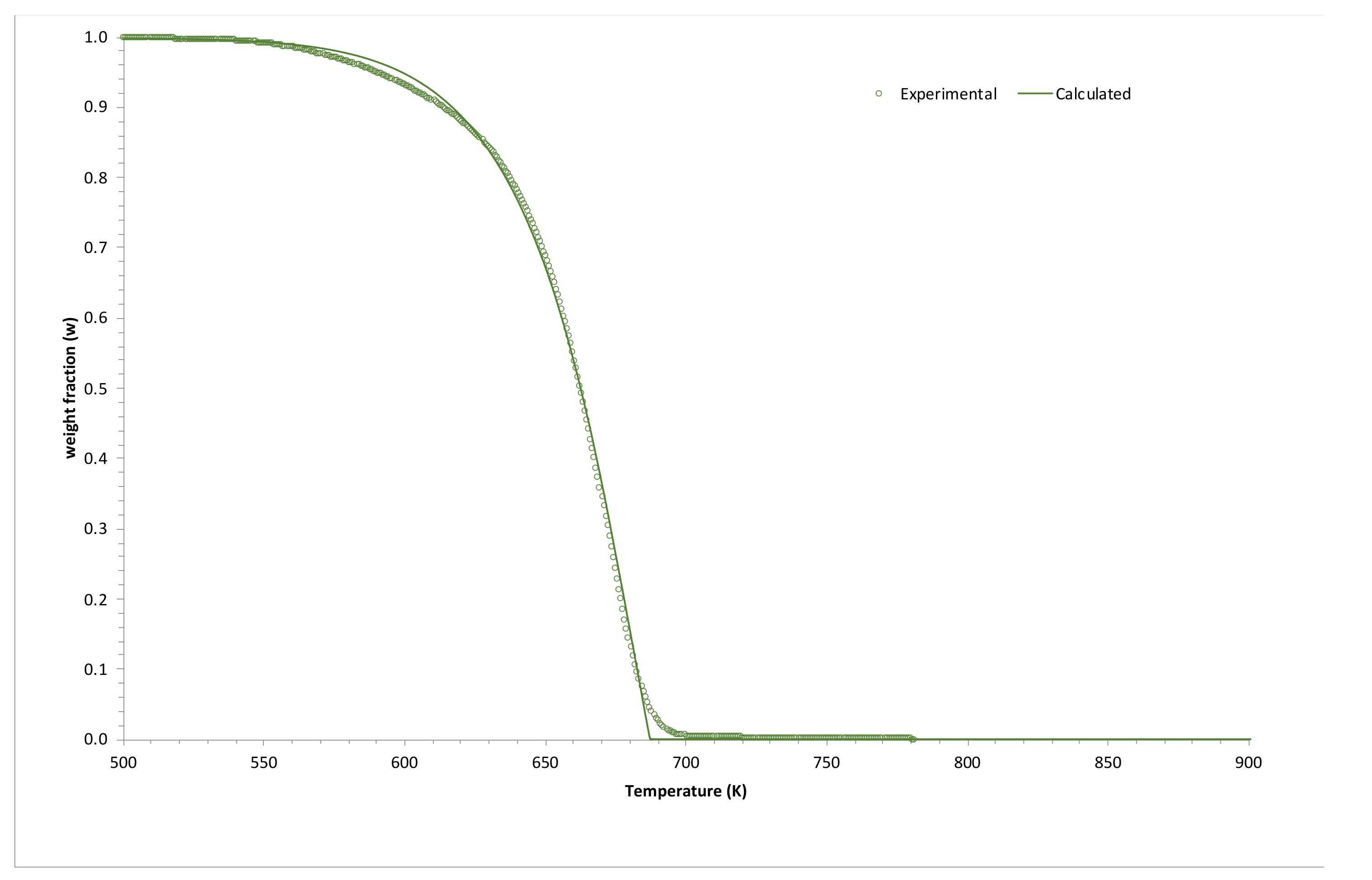
2.2. Application to Some Examples
3. Numerical Methods for Complex Materials
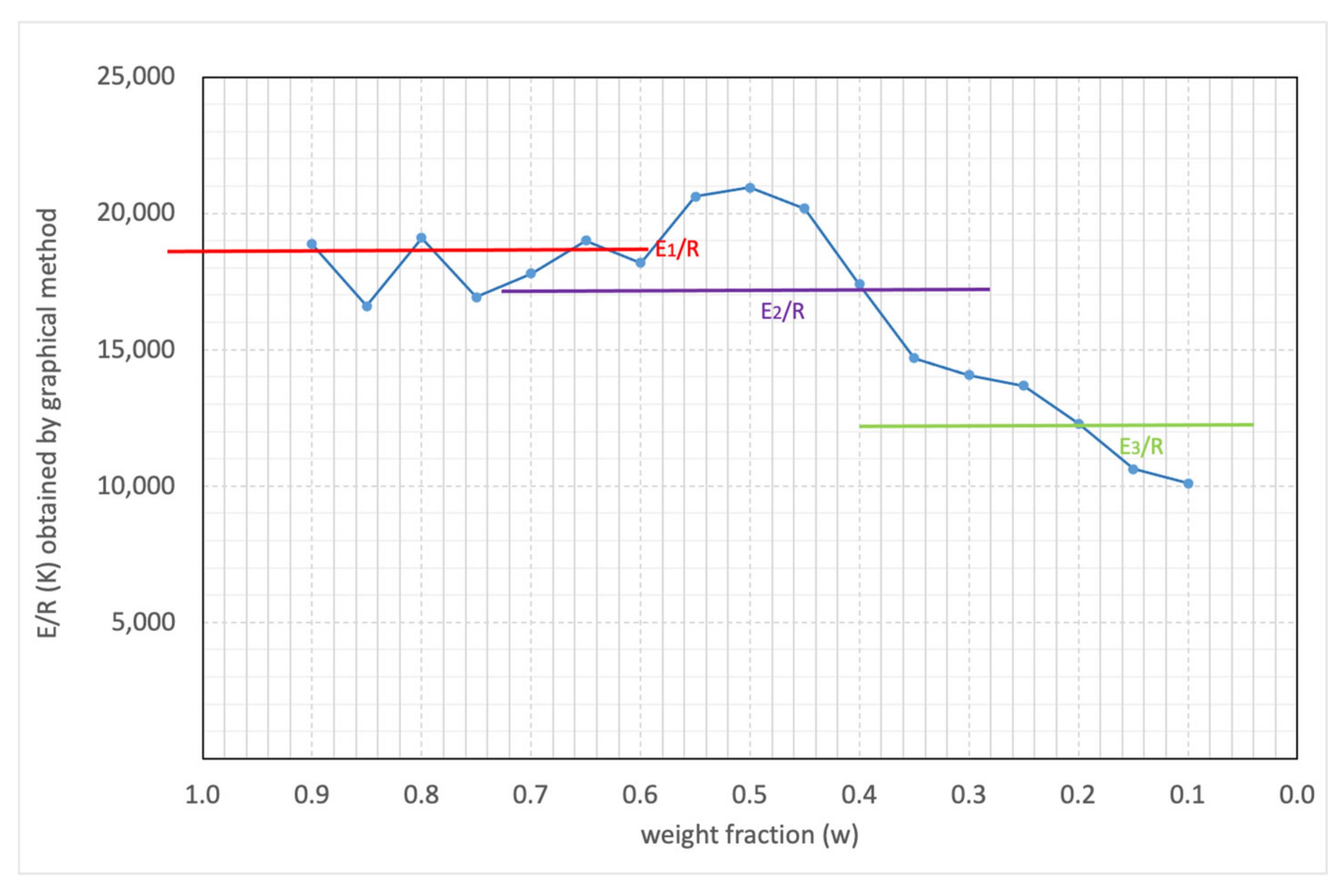
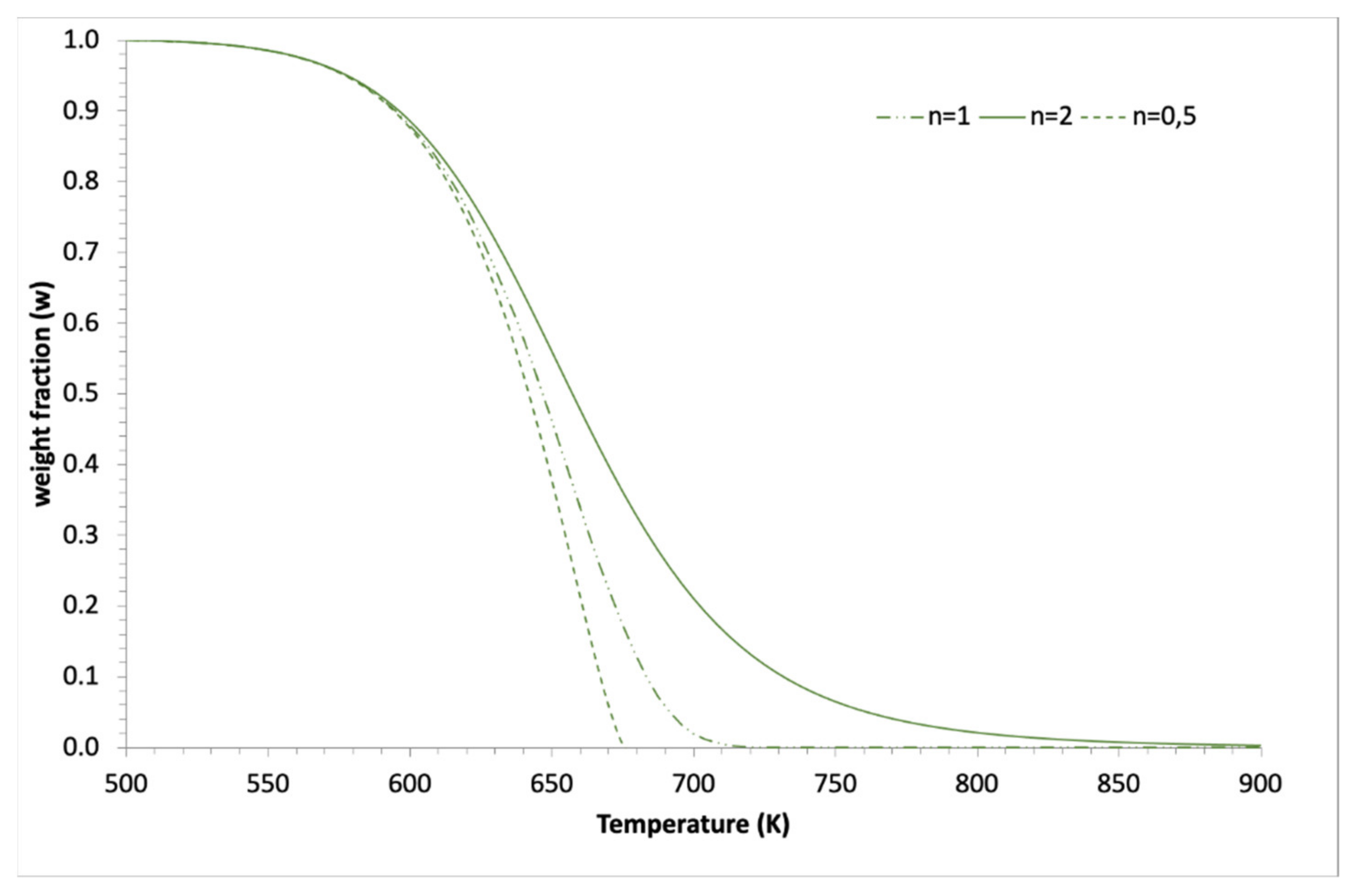
3.1. Theoretical Background
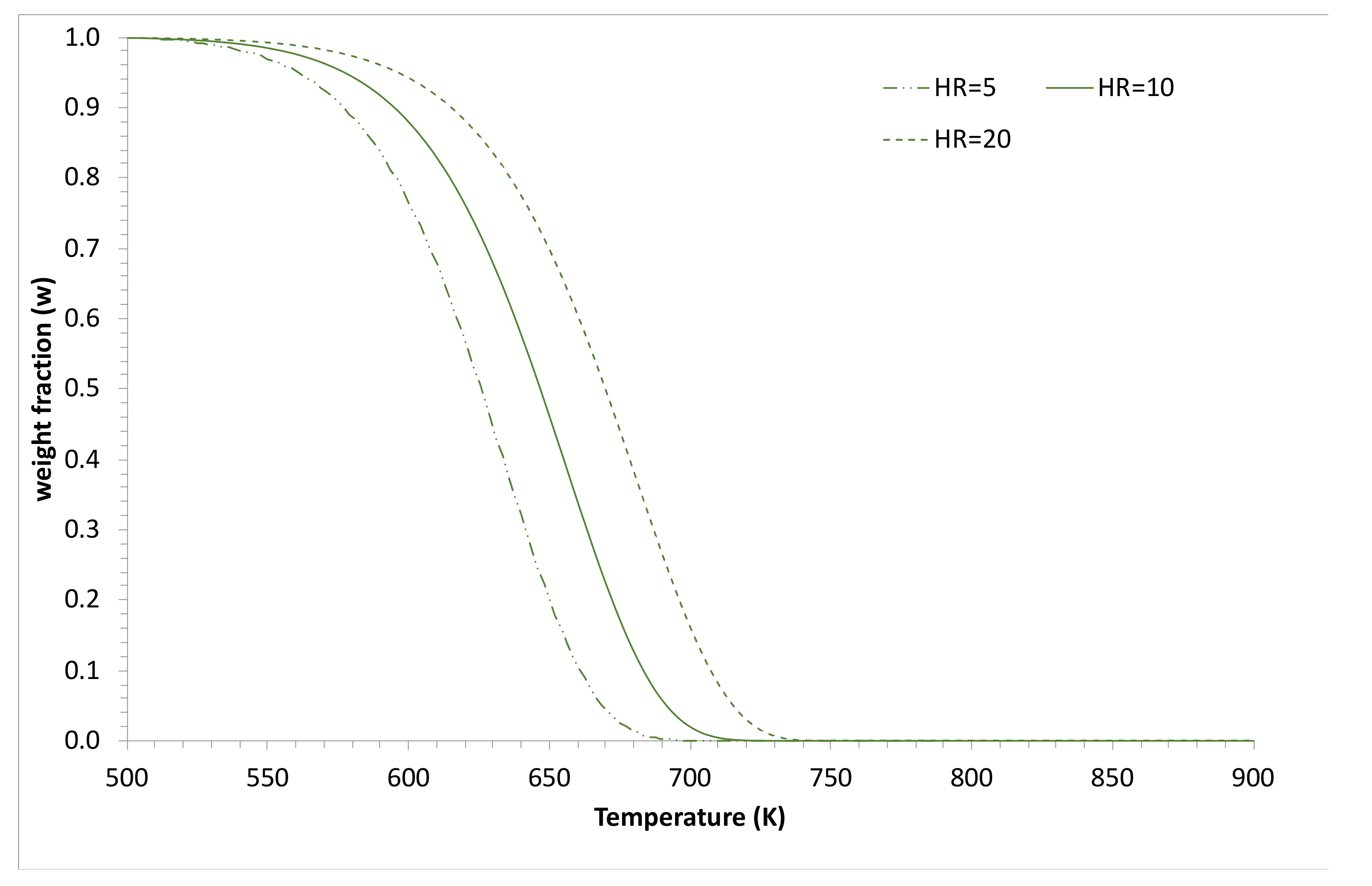
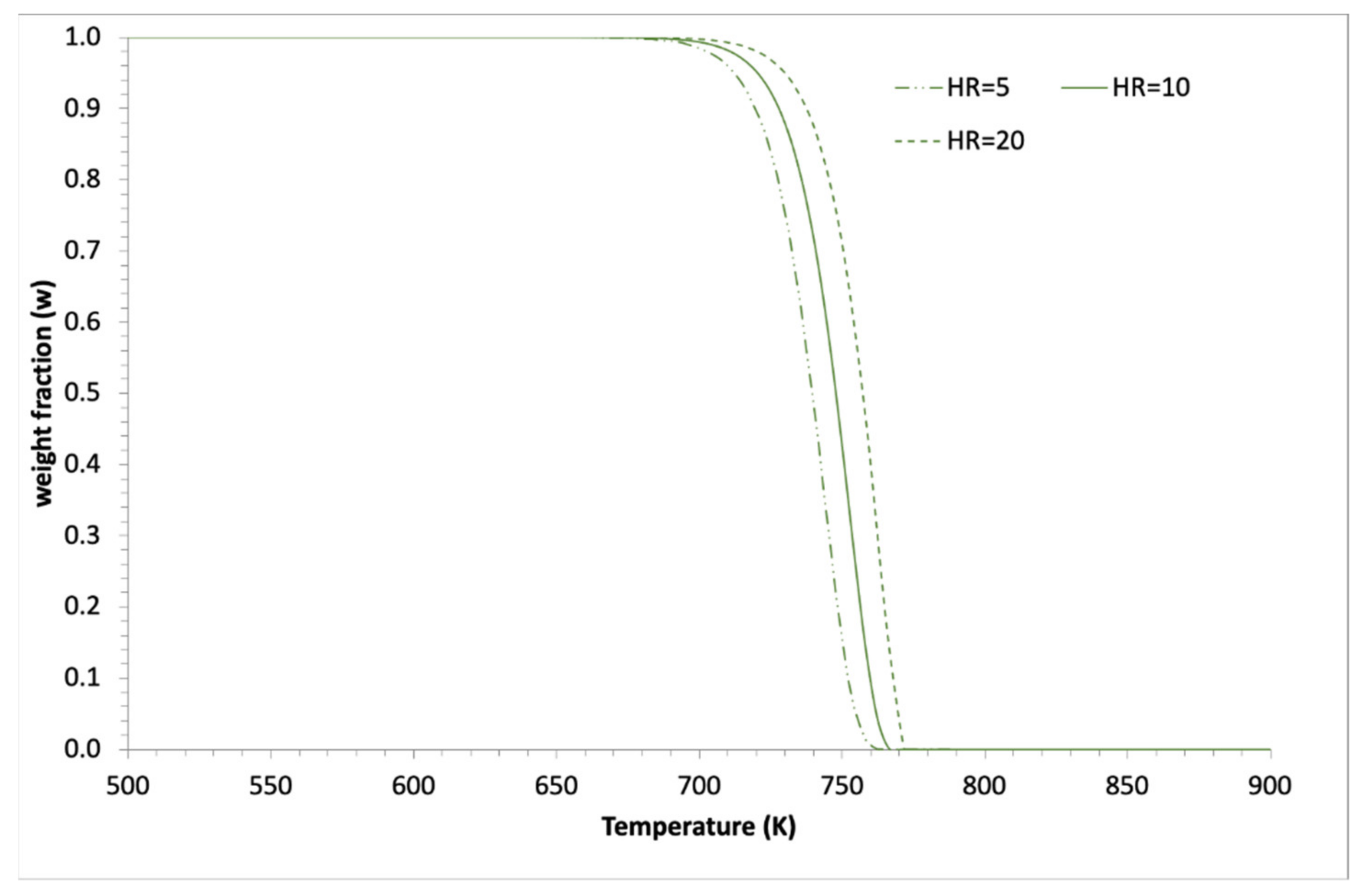
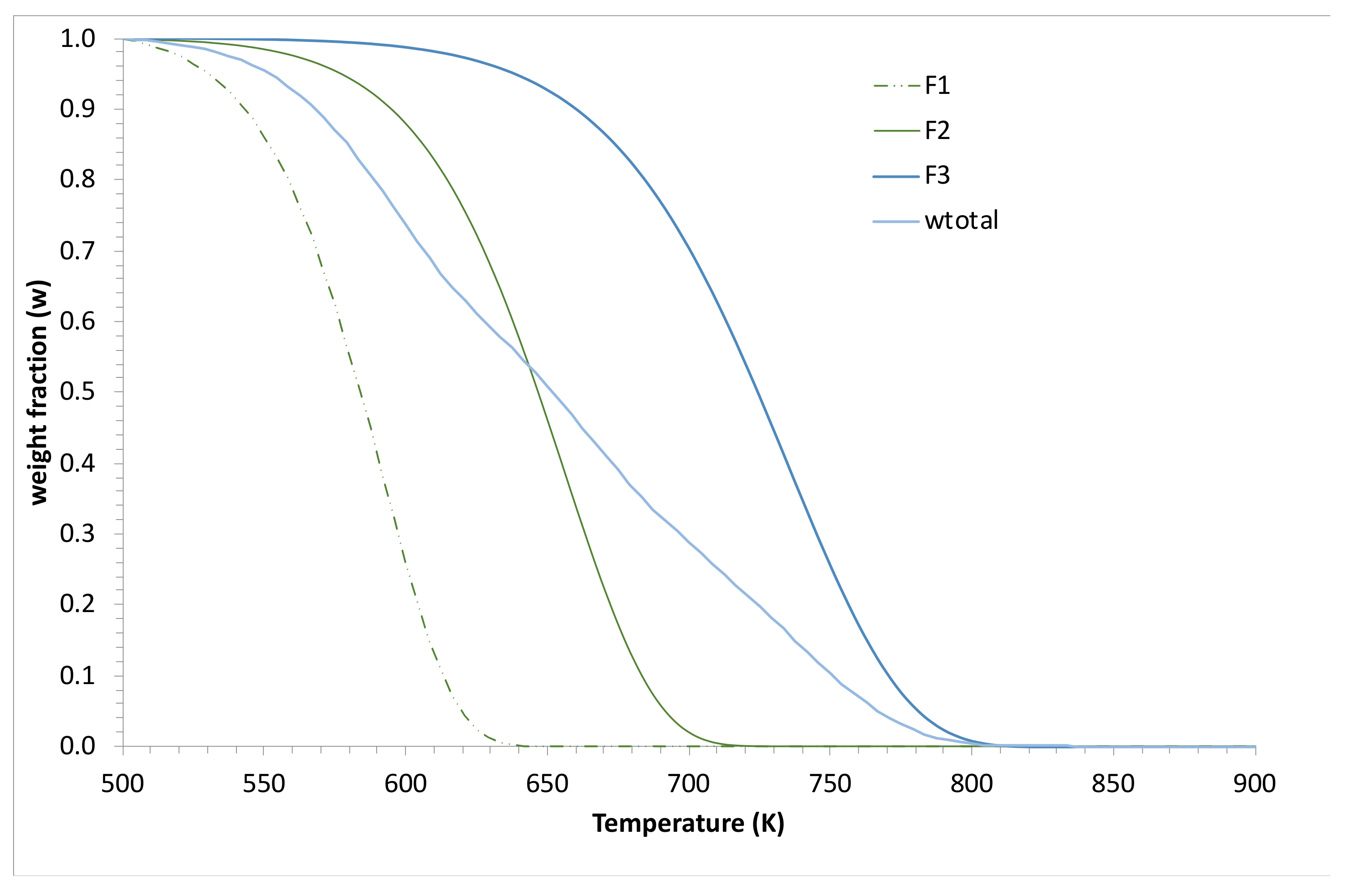
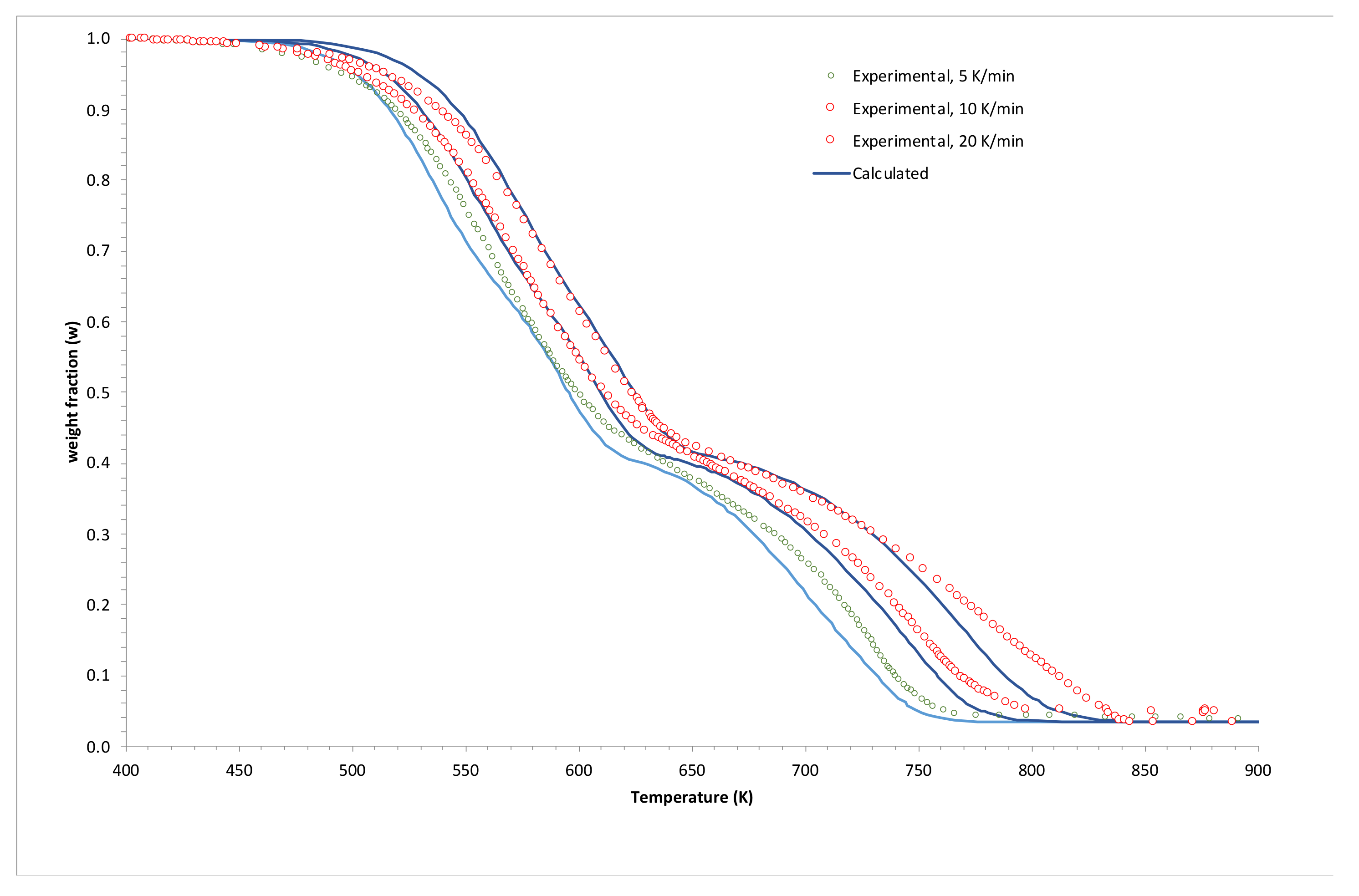
3.2. Simulations and Application to Some Materials
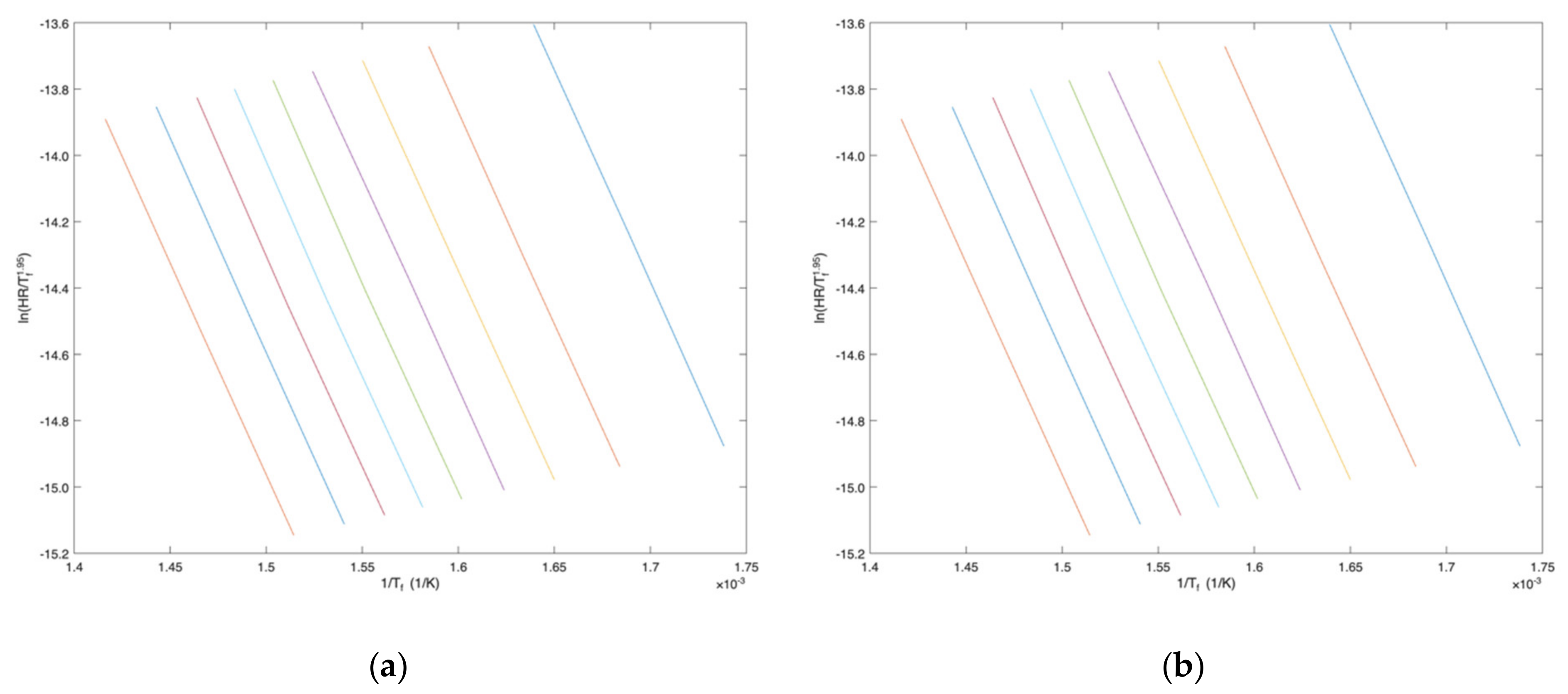
4. Comparison with Other Methods
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Freeman, E.S.; Carroll, B. The application of thermoanalytical techniques to reaction kinetics. The thermogravimetric evaluation of the kinetics of the decomposition of calcium oxalate monohydrate. J. Phys. Chem. 1958, 62, 394–397. [Google Scholar] [CrossRef]
- Simons, E.L.; Newkirk, A.E.; Aliferis, I. Performance of a pen-recording chevenard thermobalance. Anal. Chem. 1957, 29, 48–54. [Google Scholar] [CrossRef]
- Horowitz, H.H.; Metzger, G. A new analysis of thermogravimetric traces. Anal. Chem. 1963, 35, 1464–1468. [Google Scholar] [CrossRef]
- Coats, A.W.; Redfern, J.P. Kinetic parameters from thermogravimetric data. Nature 1964, 201, 68–69. [Google Scholar] [CrossRef]
- Friedman, H.L. Kinetics of thermal degradation of char-forming plastics from thermogravimetry. Application to a phenolic plastic. J. Polym. Sci. Part C 2007, 6, 183–195. [Google Scholar] [CrossRef]
- Conesa, J.A.A.; Marcilla, A.; Font, R.; Caballero, J.A.A. Thermogravimetric studies on the thermal decomposition of polyethylene. J. Anal. Appl. Pyrolysis 1996, 36, 1–15. [Google Scholar] [CrossRef]
- Conesa, J.A.A.; Caballero, J.A.; Marcilla, A.; Font, R. Analysis of different kinetic models in the dynamic pyrolysis of cellulose. Thermochim. Acta 1995, 254, 175–192. [Google Scholar] [CrossRef]
- Agrawal, R.K. Analysis of irreversible complex chemical reactions and some observations on their overall activation energy. Thermochim. Acta 1988, 128, 185–208. [Google Scholar] [CrossRef]
- Ferrer, J.A.C. Curso Básico de Ánalisis Térmico: Termogravimetría, Cinética de Reacciones y Análisis Térmico Diferencial; Editorial Club Universitario: Alicante, Spain, 2000; ISBN 84-8454-018-9. [Google Scholar]
- Islamova, S.I.; Timofeeva, S.S.; Khamatgalimov, A.R.; Ermolaev, D.V. Kinetic analysis of the thermal decomposition of lowland and high-moor peats. Solid Fuel Chem. 2020, 54, 154–162. [Google Scholar] [CrossRef]
- Reinehr, T.O.; Ohara, M.A.; de Oliveira Santos, M.P.; Barros, J.L.M.; Bittencourt, P.R.S.; Baraldi, I.J.; da Silva, E.A.; Zanatta, E.R. Study of pyrolysis kinetic of green corn husk. J. Therm. Anal. Calorim. 2020, 1–12. [Google Scholar] [CrossRef]
- Salin, I.M.; Seferis, J.C. Kinetic analysis of high-resolution TGA variable heating rate data. J. Appl. Polym. Sci. 1993, 47, 847–856. [Google Scholar] [CrossRef]
- Fetisova, O.Y.; Kuznetsov, P.N.; Chesnokov, N.V. Kinetic study of the thermal decomposition of low-ranked coal of Mongolia and Tuva. Chem. Sustain. Dev. 2019. [Google Scholar] [CrossRef]
- Conesa, J.A.; Marcilla, A. Kinetic study of the thermogravimetric behavior of different rubbers. J. Anal. Appl. Pyrolysis 1996, 37, 95–110. [Google Scholar] [CrossRef]
- Conesa, J.A.; Font, R.; Marcilla, A. Gas from the pyrolysis of scrap tires in a fluidized bed reactor. Energy Fuels 1996, 10, 134–140. [Google Scholar] [CrossRef]
- Várhegyi, G.; Czégény, Z.; Jakab, E.; McAdam, K.; Liu, C. Tobacco pyrolysis. Kinetic evaluation of thermogravimetric-mass spectrometric experiments. J. Anal. Appl. Pyrolysis 2009, 86, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Grønli, M.G.; Várhegyi, G.; Di Blasi, C. Thermogravimetric analysis and devolatilization kinetics of wood. Ind. Eng. Chem. Res. 2002, 41, 4201–4208. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Wight, C.A. Model-free and model-fitting approaches to kinetic analysis of isothermal and nonisothermal data. Thermochim. Acta 1999, 340–341, 53–68. [Google Scholar] [CrossRef]
- Font, R.; García, A.N. Application of the transition state theory to the pyrolysis of biomass and tars. J. Anal. Appl. Pyrolysis 1995, 35, 249–258. [Google Scholar] [CrossRef]
- Di Blasi, C. Linear pyrolysis of cellulosic and plastic waste. J. Anal. Appl. Pyrolysis 1997, 40–41, 463–479. [Google Scholar] [CrossRef]
- Baker, R.R. Kinetic mechanism of the thermal decomposition of tobacco. Thermochim. Acta 1979, 28, 45–57. [Google Scholar] [CrossRef]
- Ortega, A. The kinetics of solid-state reactions toward consensus, Part 2: Fitting kinetics data in dynamic conventional thermal analysis. Int. J. Chem. Kinet. 2002, 34, 193–208. [Google Scholar] [CrossRef]
- Gao, X.; Chen, D.; Dollimore, D. The correlation between the value of α at the maximum reaction rate and the reaction mechanisms. A theoretical study. Thermochim. Acta 1993, 223, 75–82. [Google Scholar] [CrossRef]
- Agrawal, R.K. Compensation effect in the pyrolysis of cellulosic materials. Thermochim. Acta 1985, 90, 347–351. [Google Scholar] [CrossRef]
- Chornet, E.; Roy, C. Compensation effect in the thermal decomposition of cellulosic materials. Thermochim. Acta 1980, 35, 389–393. [Google Scholar] [CrossRef]
- Caballero, J.A.; Conesa, J.A. Mathematical considerations for nonisothermal kinetics in thermal decomposition. J. Anal. Appl. Pyrolysis 2005, 73, 85–100. [Google Scholar] [CrossRef]
- Edwin, K.P.; Chong, S.H.Z. An Introduction to Optimization, 4th ed.; Wiley and Sons: Hoboken, NJ, USA, 2013; Available online: https://www.wiley.com/en-us/An+Introduction+to+Optimization%2C+4th+Edition-p-9781118515150 (accessed on 18 January 2021).
- Conesa, J.A.A.; Font, R.; Fullana, A.; Caballero, J.A.A. Kinetic model for the combustion of tyre wastes. Fuel 1998, 77, 1469–1475. [Google Scholar] [CrossRef]
- Font, R.; Marcilla, A.; Garcia, A.N.; Caballero, J.A.; Conesa, J.A. Kinetic models for the thermal degradation of heterogeneous materials. J. Anal. Appl. Pyrolysis 1995, 32, 29–39. [Google Scholar] [CrossRef]
- Conesa, J.A.A.; Marcilla, A.; Prats, D.; Rodriguez-Pastor, M. Kinetic study of the pyrolysis of sewage sludge. Waste Manag. Res. 1997, 15, 293–305. [Google Scholar] [CrossRef]
- Conesa, J.A.; Font, R.; Marcilla, A. Mass spectrometry validation of a kinetic model for the thermal decomposition of tyre wastes. J. Anal. Appl. Pyrolysis 1997, 43, 83–96. [Google Scholar] [CrossRef]
- Koga, N.; Šesták, J.; Málek, J. Distortion of the Arrhenius parameters by the inappropriate kinetic model function. Thermochim. Acta 1991, 188, 333–336. [Google Scholar] [CrossRef]
- Várhegyi, G.; Antal, M.J.; Jakab, E.; Szabó, P. Kinetic modeling of biomass pyrolysis. J. Anal. Appl. Pyrolysis 1997, 42, 73–87. [Google Scholar] [CrossRef] [Green Version]
- Nelder, J.A.; Mead, R. A simple method for function minimization. Comput. J. 1965, 7, 308–313. [Google Scholar] [CrossRef]
- Caballero, J.A.A.; Conesa, J.A.A.; Font, R.; Marcilla, A. Pyrolysis kinetics of almond shells and olive stones considering their organic fractions. J. Anal. Appl. Pyrolysis 1997, 42, 159–175. [Google Scholar] [CrossRef]
- Varhegyi, G.; Antal Jr, M.J.; Szekely, T.; Szabo, P. Kinetics of the thermal decomposition of cellulose, hemicellulose, and sugar cane bagasse. Energy Fuels 1989, 3, 329–335. [Google Scholar] [CrossRef]
- Iñiguez, M.E.; Conesa, J.A.; Fullana, A. Pollutant content in marine debris and characterization by thermal decomposition. Mar. Pollut. Bull. 2017. [Google Scholar] [CrossRef] [Green Version]
- Conesa, J.A.; Moltó, J.; Font, R.; Egea, S. Polyvinyl chloride and halogen-free electric wires thermal decomposition. Ind. Eng. Chem. Res. 2010, 49, 11841–11847. [Google Scholar] [CrossRef]
- Conesa, J.A.; Sánchez, N.E.; Garrido, M.A.; Casas, J.C. Semivolatile and volatile compound evolution during pyrolysis and combustion of Colombian coffee husk. Energy Fuels 2016, 30, 7827–7833. [Google Scholar] [CrossRef] [Green Version]
- Starink, M.J. The determination of activation energy from linear heating rate experiments: A comparison of the accuracy of isoconversion methods. Thermochim. Acta 2003, 404, 163–176. [Google Scholar] [CrossRef] [Green Version]
- Marcilla, A.; Beltrán, M. Thermogravimetric kinetic study of poly(vinyl chloride) pyrolysis. Polym. Degrad. Stab. 1995, 48, 219–229. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conesa, J.A. Numerical Integration of Weight Loss Curves for Kinetic Analysis. Thermo 2021, 1, 32-44. https://doi.org/10.3390/thermo1010003
Conesa JA. Numerical Integration of Weight Loss Curves for Kinetic Analysis. Thermo. 2021; 1(1):32-44. https://doi.org/10.3390/thermo1010003
Chicago/Turabian StyleConesa, Juan A. 2021. "Numerical Integration of Weight Loss Curves for Kinetic Analysis" Thermo 1, no. 1: 32-44. https://doi.org/10.3390/thermo1010003
APA StyleConesa, J. A. (2021). Numerical Integration of Weight Loss Curves for Kinetic Analysis. Thermo, 1(1), 32-44. https://doi.org/10.3390/thermo1010003