Excitation Wavelength-Dependent Photochemistry


Abstract
1. Introduction
2. The Historical Kasha’s Rule
3. The Unfounded Extension of Kasha’s Rule Application to Photochemistry
4. Anti-Kasha Emission
5. Distribution of the Literature in Photochemistry
6. Early Observations
7. Photoreactions of Species in Equilibrium

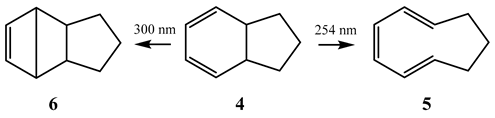
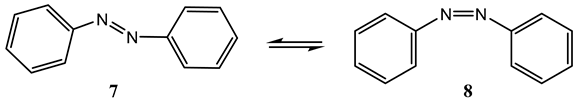
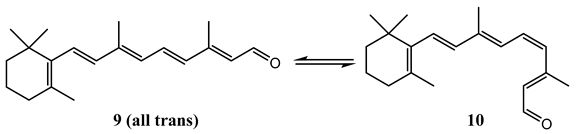
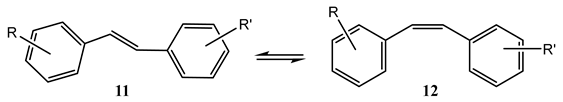
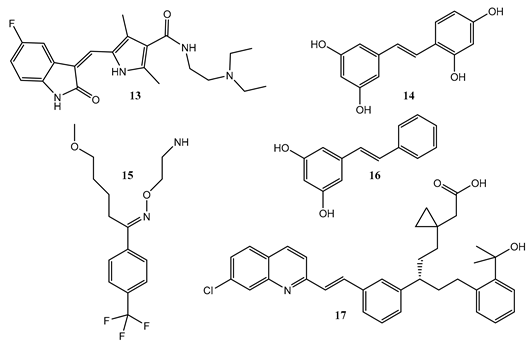
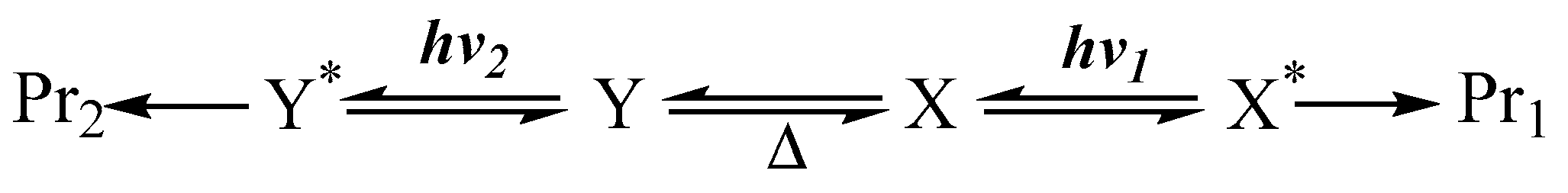
8. Photoreactions of Multi-Chromophoric Species Yielding Different End-Products
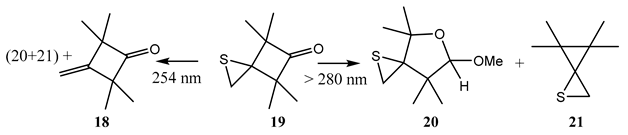
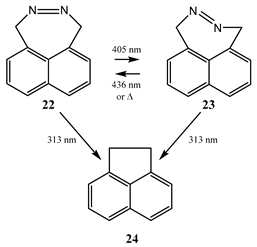
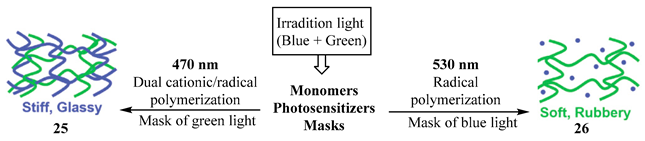
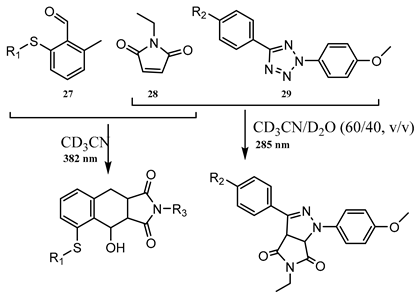
9. Photoreactions of Multi-Chromophoric Species Yielding a Unique Product Irrespective of the Excitation Wavelength

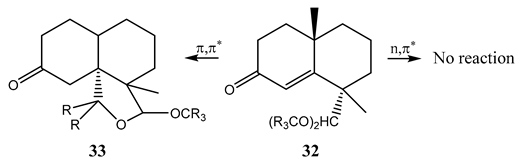
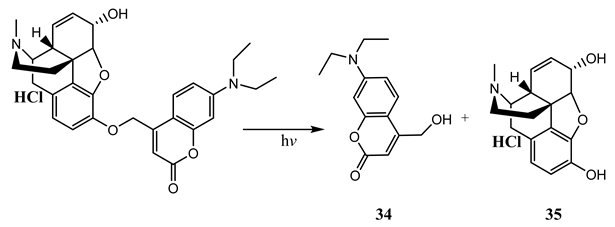
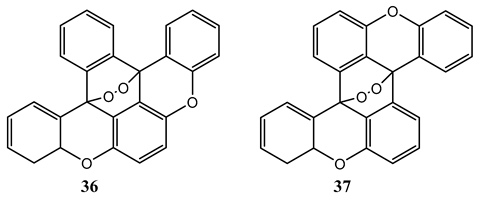
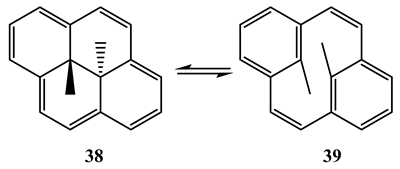
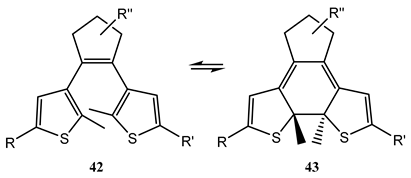
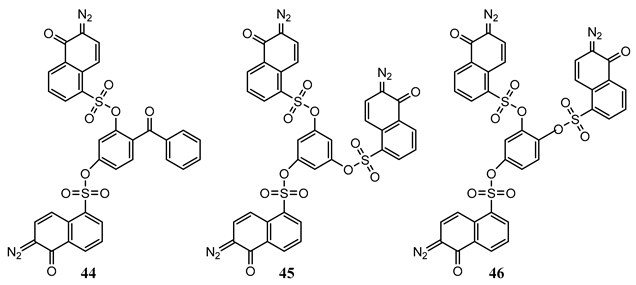
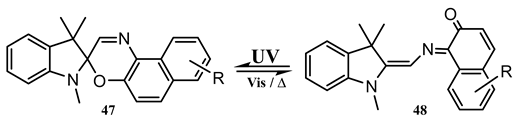
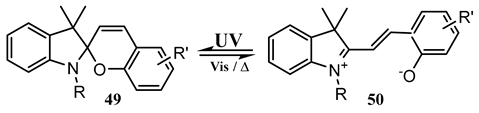
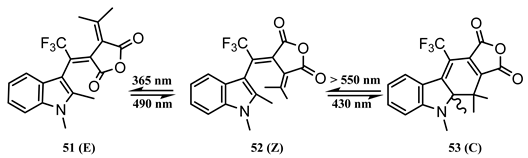
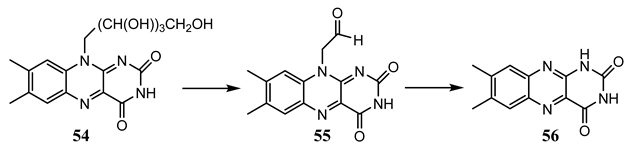
10. Vibronic States Photochemistry
11. Some Inorganic Actinometers
12. Challenges/Limitations
13. Concluding Remarks
Funding
Data Availability Statement
Conflicts of Interest
References
- Kislyak, A.; Frisch, H.; Gernhardt, M.; Van Steenberge, P.H.M.; D’Hooge, D.R.; Barner-Kowollik, C. Time-dependent differential and integral quantum yields for wavelength-dependent [4+4] photocycloadditions. Chem. Eur. J. 2020, 26, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Ermolaev, V.L. Ultrafast nonradiative transitions between higher excited states in organic molecules. Russ. Chem. Rev. 2001, 70, 471–490. [Google Scholar] [CrossRef]
- Tseng, H.-W.; Shen, J.-Y.; Kuo, T.-Y.; Tu, T.-S.; Chen, Y.-A.; Demchenko, A.P.; Chou, P.-T. Excited-state intramolecular proton-transfer reaction demonstrating anti-Kasha behavior. Chem. Sci. 2016, 7, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Bogdanova, A.; Popik, V.V. Wavelength-Dependent Photochemistry of Diazo Meldrum’s Acid and Its Spirocyclic Isomer, Diazirino Meldrum’s Acid: Wolff Rearrangement versus Isomerization. J. Am. Chem. Soc. 2003, 125, 1456–1457. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, A.P.; Tomin, V.I.; Chou, P.-T. Breaking the Kasha Rule for More Efficient Photochemistry. Chem. Rev. 2017, 117, 13353–13381. [Google Scholar] [CrossRef] [PubMed]
- Kasha, M. Characterization of electronic transitions in complex molecules. Discuss. Faraday Soc. 1950, 9, 14–19. [Google Scholar] [CrossRef]
- Turro, N.J.; Ramamurthy, V.; Cherry, W.; Farneth, W. The effect of wavelength on organic photoreactions in solution. Reactions from upper excited states. Chem. Rev. 1978, 78, 125–145. [Google Scholar] [CrossRef]
- Shi, L.; Xie, X.; Troisia, A. Rapid calculation of internal conversion and intersystem crossing rate for organic materials discovery. J. Chem. Phys. 2022, 157, 134106. [Google Scholar] [CrossRef]
- Jabłoński, A. Efficiency of Anti-Stokes Fluorescence in Dyes. Nature 1933, 131, 839–840. [Google Scholar] [CrossRef]
- Atkins, P.; de Paula, J. Atkins’ Physical Chemistry; Oxford University Press: Oxford, UK, 2006; ISBN 0-7167-8759-8. [Google Scholar]
- Itoh, T. Fluorescence and phosphorescence from higher excited states of organic molecules. Chem. Rev. 2012, 112, 4541–4568. [Google Scholar] [CrossRef]
- Del Valle, J.C.; Catalán, J. A Reappraisal of Kasha’s Rule. Phys. Chem. Chem. Phys. 2019, 21, 10061–10069. [Google Scholar] [CrossRef] [PubMed]
- Vavilov, S.I. The dependence of the intensity of the fluorescence of dyes upon the wave-length of the exciting light. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1922, 43, 307–320. [Google Scholar] [CrossRef]
- Braslavsky, S.E. Glossary of terms used in photochemistry (IUPAC). Pure Appl. Chem. 2007, 79, 293–465. [Google Scholar] [CrossRef]
- Lewis, G.N.; Kasha, M. Phosphorescence and the Triplet State. J. Am. Chem. Soc. 1944, 66, 2100–2116. [Google Scholar] [CrossRef]
- Klan, P.; Wirz, J. Photochemistry of Organic Compounds: From Concepts to Practice; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar] [CrossRef]
- Pianowski, Z.L. (Ed.) Molecular Photoswitches: Chemistry, Properties, and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2022; ISBN 978-3-527-35104-6. [Google Scholar]
- Wayne, R.P. Principles and Applications of Photochemistry; Oxford University Press: Cary, NC, USA, 1988; ISBN 9780198552345/0198552343. [Google Scholar]
- Rosspeintner, A.; Lang, B.; Vauthey, E. Ultrafast Photochemistry in Liquids. Annu. Rev. Phys. Chem. 2013, 64, 247–271. [Google Scholar] [CrossRef] [PubMed]
- Kumpulainen, T.; Lang, B.; Rosspeintner, A.; Vauthey, E. Ultrafast elementary photochemical processes of organic molecules in liquid solution. Chem. Rev. 2017, 117, 10826–10939. [Google Scholar] [CrossRef] [PubMed]
- Livshits, M.Y.; Wang, L.; Vittardi, S.B.; Ruetzel, S.; King, A.; Brixner, T.; Rack, J.J. An excited state dynamics driven reaction: Wavelength-dependent photoisomerization quantum yields in [Ru(bpy)2(dmso)2]2+. Chem. Sci. 2020, 11, 5797–5807. [Google Scholar] [CrossRef]
- Turro, N.J.; Ramamurthy, V.; Scaiano, J.C. Modern Molecular Photochemistry of Organic Molecules; University Science Books: Sausalito, CA, USA, 2010; ISBN 1-891389-25-4. [Google Scholar]
- Yu, J.; Ma, H.; Huang, W.; Liang, Z.; Zhou, K.; Lv, A.; Li, X.-G.; He, Z. Purely Organic Room-Temperature Phosphorescence Endowing Fast Intersystem Crossing from Through-Space Spin−Orbit Coupling. J. Am. Chem. Soc. Au. 2021, 1, 1694–1699. [Google Scholar] [CrossRef]
- Levine, S.Z.; Knight, A.R.; Steer, R.P. Fluorescence from the Second Excited Singlet State of Thiophosgene Vapour. Chem. Phys. Lett. 1974, 29, 73–76. [Google Scholar] [CrossRef]
- Oka, T.; Knight, A.R.; Steer, R.P. A spectroscopic study of fluorescence from the second excited singlet state of thiophosgene vapor. J. Chem. Phys. 1975, 63, 2414–2420. [Google Scholar] [CrossRef]
- Steer, R.P. Structure and decay dynamics of electronic excited states of thiocarbonyl compounds. Rev. Chem. Intermed. 1981, 4, 1–41. [Google Scholar] [CrossRef]
- Acuña, A.U.; Catalán, J.; Toribio, F. Photon energy relaxation and thermal effects on gas-phase electronically excited methyl salicylate. J. Phys. Chem. 1981, 85, 241–245. [Google Scholar] [CrossRef]
- Catalán, J.; Del Valle, J.C.; Palomar, J.; Díaz, C.; De Paz, J.L.G. The six-membered intramolecular hydrogen bond position as a switch for inducing an excited state intramolecular proton transfer (ESIPT) in esters of o-Hydroxynaphthoic Acids. J. Phys. Chem. A 1999, 50, 10921–10934. [Google Scholar] [CrossRef]
- Viswanath, G.; Kasha, M. Confirmation of the Anomalous Fluorescence of Azulene. J. Chem. Phys. 1956, 24, 574–577. [Google Scholar] [CrossRef]
- Binsch, G.; Heilbronner, E.; Jankow, R.; Schmidt, D. On the fluorescence anomaly of azulene. Chem. Phys. Lett. 1967, 1, 135–138. [Google Scholar] [CrossRef]
- Birks, J.B. The photophysics of azulene. Chem. Phys. Lett. 1972, 17, 370–372. [Google Scholar] [CrossRef]
- Prlj, A.; Begušić, T.; Zhang, Z.T.; Fish, G.C.; Wehrle, M.; Zimmermann, T.; Choi, S.; Roulet, J.; Moser, J.-E.; Vaníček, J. Semiclassical approach to photophysics beyond Kasha’s rule and vibronic spectroscopy beyond the condon approximation. The case of azulene. J. Chem. Theory Comput. 2020, 16, 2617–2626. [Google Scholar] [CrossRef] [PubMed]
- Geldof, P.A.; Rettschnick, R.P.H.; Hoytink, G.J. Fluorescence from the second excited singlets of pyrene and 3,4-benzpyrene. Chem. Phys. Lett. 1969, 4, 59–61. [Google Scholar] [CrossRef]
- Peterson, D.L.; Lytle, F.E.; Laurendeau, N.M. Determination of flame temperature using the anomalous fluorescence of pyrene. Opt. Lett. 1986, 11, 345–347. [Google Scholar] [CrossRef]
- Easterly, C.E.; Christophorou, L.G.; Carter, J.G. Fluorescence from the second excited π-singlet state of aromatic hydrocarbons in solution. J. Chem. Soc. Faraday Trans. 1973, 69, 471–483. [Google Scholar] [CrossRef]
- Nakajima, A.; Baba, H. Fluorescence spectrum of pyrene vapor: Emission from the second excited singlet state. Bull. Chem. Soc. Jpn. 1970, 43, 967. [Google Scholar] [CrossRef]
- Baba, H.; Nakajima, A.; Aoi, M.; Chihara, K.J. Fluorescence from the second excited singlet state and radiationless processes in pyrene vapor. J. Chem. Phys. 1971, 55, 2433–2438. [Google Scholar] [CrossRef]
- Choi, I.S.; Lee, S.K. Analysis of Rotationally Cooled Vibronic Emission Spectra () of m-Xylyl Radical. Bull. Korean Chem. Soc. 1996, 17, 749–753. [Google Scholar]
- Uttamlal, M.; Holmes-Smith, A.S. The excitation wavelength dependent fluorescence of porphyrins. Chem. Phys. Lett. 2008, 454, 223–228. [Google Scholar] [CrossRef]
- Malpicci, D.; Lucenti, E.; Giannini, C.; Forni, A.; Botta, C.; Cariati, E. Prompt and long-lived anti-Kasha emission from organic dyes. Molecules 2021, 26, 6999. [Google Scholar] [CrossRef] [PubMed]
- Pavlovich, V.S. Dependence of excitation spectra of solutions of dipolar molecules on registration wavelength. Zh. Prikl. Spektr. 1976, 25, 480–487. [Google Scholar]
- Cushing, S.K.; Li, M.; Huang, F.; Wu, N. Origin of strong excitation wavelength dependent fluorescence of graphene oxide. ACS Nano 2014, 8, 1002–1013. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, A.P. The red-edge effects: 30 years of exploration. Luminescence 2002, 17, 19–42. [Google Scholar] [CrossRef] [PubMed]
- Samanta, A. Dynamic Stokes shift and excitation wavelength dependent fluorescence of dipolar molecules in room temperature ionic liquids. J. Phys. Chem. B 2006, 110, 13704–13716. [Google Scholar] [CrossRef]
- Khara, D.C.; Samanta, A. Solvation dynamics and red-edge effect of two electrically charged solutes in an imidazolium ionic liquid. Indian. J. Chem. 2010, 49, 714–720. [Google Scholar]
- Józefowicz, M.; Heldt, J.R. Excitation-wavelength dependent fluorescence of ethyl 5-(4-aminophenyl)-3-amino2,4-dicyanobenzoate. J. Fluoresc. 2011, 21, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Galley, W.C.; Purkey, R.M. Role of heterogeneity of the solvation site in electronic spectra in solution. Proc. Natl. Acad. Sci. USA 1970, 67, 1116–1121. [Google Scholar] [CrossRef] [PubMed]
- Milton, J.C.; Purkey, R.M.; Galley, W.C. The kinetics of solvent reorientation in hydroxylated solvents from the exciting-wavelength dependence of chromophore emission spectra. J. Chem. Phys. 1978, 68, 5396–5404. [Google Scholar] [CrossRef]
- Pavlovich, V.S.; Pershukevich, P.P.; Pikulik, L.G. Instantanous phosphorescence spectra of glassy polar solutions of acetyl phthalamide derivatives. Zh. Prikl. Spektr. 1983, 39, 779–785. [Google Scholar]
- Khisamov, R.M.; Ryadun, A.A.; Sukhikh, T.S.; Konchenko, S.N. Excitation wavelength-dependent room-temperature phosphorescence: Unusual properties of novel phosphinoamines. Mol. Syst. Des. Eng. 2021, 6, 1056–1065. [Google Scholar] [CrossRef]
- Xiao, G.; Ma, Y.-J.; Qi, Z.; Fang Chena, T.; Yan, D. A flexible ligand and halogen engineering enable one phosphor-based full-colour persistent luminescence in hybrid perovskitoids. Chem. Sci. 2024, 15, 3625–3632. [Google Scholar] [CrossRef] [PubMed]
- Holovka, J.M.; Gardner, P.D. Photolysis and photoisomerization of the benzene oxide-oxepin system. J. Am. Chem. Soc. 1967, 89, 6390–6391. [Google Scholar] [CrossRef]
- Dauben, W.G.; Kellog, M.S. Wavelength dependency in the photochemistry of 1,3-cyclohexadienes. cis-Bicyclo [4.3.0]nona-2,4-diene to cis,cis,trans-1,3,5-cyclononatriene valence tautomerism. J. Am. Chem. Soc. 1971, 93, 3805–3807. [Google Scholar] [CrossRef]
- Zimmerman, G.; Chow, L.-Y.; Paik, U.-J. The photochemical isomerization of azobenzene. J. Am. Chem. Soc. 1958, 80, 3528–3531. [Google Scholar] [CrossRef]
- Rau, H. Azo Compounds. In Advances in Photochemistry; Neckers, D.C., Volman, D.H., von Bunau, G., Eds.; Wliley & Sons: New York, NY, USA, 1995; Volume 19, ISBN 0-471-04912-3. [Google Scholar]
- Montali, M.; Credi, A.; Prodi, L.; Gandolfi, M.T. Handbook of Photochemistry, 3rd ed.; Taylor and Francis: Boca Raton, FL, USA, 2006. [Google Scholar]
- Rau, H. Photochromism: Molecules and Systems; Durr, H., Bounas-Laurent, H., Eds.; Elsevier: Amsterdam, The Netherland, 2003; Volume 1, ISBN 9780444513229. [Google Scholar]
- Gagliardi, L.; Orlandi, G.; Bernardi, F.; Cembran, A.; Garavelli, M. A theoretical study of the lowest electronic states of azobenzene: The role of torsion coordinate in the cis-trans photoisomerization. Theor. Chem. Acc. 2004, 111, 363–372. [Google Scholar] [CrossRef]
- Ladányi, V.; Dvořák, P.; Al-Anshori, J.; Vetráková, L.; Wirz, J.; Heger, D. Azobenzene photoisomerization quantum yields in methanol redetermined. Photochem. Photobiol. Sci. 2017, 16, 1757–1761. [Google Scholar] [CrossRef] [PubMed]
- Mauser, H.; Gauglitz, G.; Compton, R.G.; Hancock, G. (Eds.) Comprehensive Chemical Kinetics, Photokinetics: Theoretical Fundamentals and Applications; Elsevier: Amesterdam, The Netherland, 1998; Volume 36, ISBN 978-0444825360. [Google Scholar]
- Stranius, K.; Börjesson, K. Determining the Photoisomerization Quantum Yield of Photoswitchable Molecules in Solution and in the Solid State. Sci. Rep. 2017, 7, 41145. [Google Scholar] [CrossRef] [PubMed]
- Gade, R.; Porada, T. Determination of quantum yields and product spectra by studying reversible photoisomerizations in solution. J. Photochem. Photobiol. A Chem. 1997, 107, 27–34. [Google Scholar] [CrossRef]
- Deniel, M.H.; Lavabre, D.; Micheau, J.C. Photokinetics under continuous irradiation. In Organic Photochromic and Thermochromic Compounds; Crano, J.C., Guglielmetti, R.J., Eds.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 1999; ISBN 978-0-306-45882-8. [Google Scholar] [CrossRef]
- Delbaere, S.; Vermeersh, G.; Micheau, J.C. Quantitative analysis of the dynamic behaviour of photochromic systems. J. Photochem. Photobiol. C Chem. Photochem. Rev. 2011, 12, 74–105. [Google Scholar] [CrossRef]
- Lewis, G.N.; Milgel, T.T.; Lipkin, D. The Absorption and Re-emission of Light by cis- and trans-Stilbenes and the Efficiency of their Photochemical Isomerization. J. Am. Chem. Soc. 1940, 62, 2973–2980. [Google Scholar] [CrossRef]
- Olson, A.R. The study of chemical reactions from potential energy diagrams. Trans. Faraday Soc. 1931, 27, 69–76. [Google Scholar] [CrossRef]
- Hartley, G.S. The cis-form of azobenzene and the velocity of the thermal cis→trans-conversion of azobenzene and some derivatives. J. Chem. Soc. 1938, 633–642. [Google Scholar] [CrossRef]
- Dhammika Bandara, H.M.; Burdette, S.C. Photoisomerization in Different Classes of Azobenzene. Chem. Soc. Rev. 2012, 41, 1809–1825. [Google Scholar] [CrossRef]
- Diau, E.W.-G. A new trans-to-cis photoisomerization mechanism of azobenzene on the S1(n, π*) surface. J. Phys. Chem. A 2004, 108, 950–956. [Google Scholar] [CrossRef]
- Samai, S.; Bradley, D.J.; Choi, T.L.Y.; Yan, Y.; Ginger, D.S. Temperature dependent photoisomerization quantum yield for azobenzene-modified DNA. J. Phys. Chem. C 2017, 121, 6997–7004. [Google Scholar] [CrossRef]
- Shi, Y.-B.; Hearst, J.E. Wavelength dependence for the photoreactions of DNA-psoralen monoadducts. 2: Photo-cross-linking of monoadducts. Biochemistry 1987, 26, 3792–9798. [Google Scholar] [CrossRef] [PubMed]
- Schneider, E.E.; Goodeve, C.F.; Lythgoe, R.J. The spectral variation of the photosensitivity of visual purple. Proc. Natl. Acad. Sci. USA 1939, 170, 102–112. [Google Scholar]
- Kim, J.E.; Tauber, M.J.; Mathies, R.A. Wavelength-dependent cis-trans isomerization in vision. Biochemistry 2001, 40, 13774–13778. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Tauber, M.J.; Mathies, R.A. Analysis of the mode specific excited-state energy distribution and wavelength-dependent photoreaction quantum yield in rhodopsin. Biophys. J. 2003, 84, 2492–2501. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.; Stock, G. Quantum mechanical modelling of the femtosecond isomerization of rhodopsin. J. Phys. Chem. 2000, 104, 1146–1149. [Google Scholar] [CrossRef]
- Tscherbul, T.V.; Brumer, P. Escitation of biomolecules with incoherent light: Quantum yield for the photoisomerization of model retinal. J. Phys. Chem. 2014, 118, 3100–3111. [Google Scholar] [CrossRef]
- Balke, D.E.; Becker, R.S. Relationship between the absorption and excitation spectra and relative quantum yields of fluorescence of all-trans-retinal. J. Am. Chem. Soc. 1968, 90, 6710–6711. [Google Scholar] [CrossRef]
- Dartnall, H.J.A. The photosensitivities of visual pigments in the presence of hydroxylamine. Vision. Res. 1968, 8, 339–358. [Google Scholar] [CrossRef]
- Stensitzki, T.; Muders, V.; Schlesinger, R.; Heberle, J.; Heyne, K. The primary photoreaction of channelrhodopsin-1: Wavelength dependent photoreactions induced by ground-state heterogeneity. Front. Mol. Biosci. 2015, 2, 41. [Google Scholar] [CrossRef]
- Schulte-Frohlinde, D.; Blume, H.; Gusten, H. Photochemical cis-trans-isomerization of substituted stilbene. J. Phys. Chem. 1962, 66, 2486–2491. [Google Scholar] [CrossRef]
- Gorner, H.; Kuhn, H.J. Cis-trans isomerization of stilbenes and stilbene-like molecules. In Advances in Photochemistry; Neckers, D.C., Volman, D.H., von Bunau, G., Eds.; Wliley & Sons: New York, NY, USA, 1995; Volume 19, ISBN 0-471-04912-3. [Google Scholar]
- Maafi, M.; Maafi, W. Φ-Order kinetics of photoreversible-drug reactions. Int. J. Pharm. 2014, 471, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Maafi, M.; Maafi, W. Quantitative assessment of photostability and photostabilisation of fluvoxamine and its design for actinometry. Photochem. Photobiol. Sci. 2015, 14, 982–994. [Google Scholar] [CrossRef] [PubMed]
- Maafi, M.; Lee, L.Y. Actinometric and Φ-order photodegradation of anti-cancer Sunitinib. J. Pharm. Biomed. Anal. 2015, 110, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Maafi, M.; Maafi, W. Montelukast photodegradation: Elucidation of Φ-order kinetics, determination of quantum yields and application to actinometry. Int. J. Pharm. 2014, 471, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Maafi, M.; Al-Qarni, M.A. Φ-order spectrophotokinetic characterisation and quantification of trans-cis oxyresveratrol reactivity, photodegradation and actinometry. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 188, 64–71. [Google Scholar] [CrossRef]
- Maafi, M.; Al-Qarni, M.A. Dependence of pinosylvin photochemical quantum yield on excitation wavelength. Trends Photochem. Photobiol. 2019, 18, 45–58. [Google Scholar]
- Maafi, M. On photokinetics under monochromatic light. Front. Chem. 2023, 11, 1233151. [Google Scholar] [CrossRef]
- Maafi, M. On photokinetics under polychromatic light. Front. Chem. 2024, 12, 1367276. [Google Scholar] [CrossRef] [PubMed]
- Gardner, C.; Moer, J.F. Photochemische Reaktionen. 50. Mitteilung [1]. Zur Photochemie von gesättigten β-Ketosulfiden II. Synthese von 6-Hydroxy-2-thia-7-oxa-isotwistan. Helv. Chim. Acta 1969, 52, 725–737. [Google Scholar] [CrossRef]
- Pacifici, J.G.; Diebert, C. Wavelength effect in photolysis of 4,4,6,6-tetramethyl-1-thiaspiro [2.3]hexan-5-one. J. Am. Chem. Soc. 1969, 91, 4595. [Google Scholar] [CrossRef]
- Gisin, M.; Wirz, J. Photolysis of the azo-precursors of 2,3- and 1,s-naphtoquinodimethane. Helv. Chim. Acta 1976, 59, 2273–2277. [Google Scholar] [CrossRef]
- Pagni, R.M.; Burnett, M.N.; Dodd, J.R. Photochemistry of 1,4-Dihydronaphtho [1,8-de] [1,2] diazepine. Preparation and electron spin resonance observation of the uinsubstituted 1,8-Naphthoquinodimethane. J. Am. Chem. Soc. 1977, 99, 1972–1973. [Google Scholar] [CrossRef]
- Bochet, C.G. Chromic orthogonality in organic synthesis. Synlett 2004, 13, 2268–2274. [Google Scholar] [CrossRef]
- Bochet, C.G. Photochemical release of various functional groups. Pure Appl. Chem. 2006, 78, 241–247. [Google Scholar] [CrossRef]
- Protti, S.; Ravelli, D.; Fagnoni, M. Wavelength-dependence and wavelength-selectivity in photochemical reactions. Photochem. Photobiol. Sci. 2019, 18, 2094–2101. [Google Scholar] [CrossRef] [PubMed]
- Bochet, C.G. Wavelength-selective cleavage of photolabile protecting groups. Tetrahedron Lett. 2000, 41, 6341–6346. [Google Scholar] [CrossRef]
- Bochet, C.G. Orthogonal photolysis of protecting Groups. Angew. Int. Ed. 2001, 40, 2071–2073. [Google Scholar] [CrossRef]
- Stegmaier, P.; Alonso, J.M.; del Campo, A. Photoresponsive Surfaces with Two Independent Wavelength-Selective Functional Levels. Langmuir 2008, 24, 11872–11879. [Google Scholar] [CrossRef] [PubMed]
- Kotzur, N.; Briand, B.; Beyermann, M.; Hagen, V. Wavelength-selective photoactivatable protecting groups for thiols. J. Am. Chem. Soc. 2009, 25, 16927–16931. [Google Scholar] [CrossRef]
- Kantevari, S.; Matsuzaki, M.; Kanemoto, Y.; Kasai, H.; Ellis-Davies, G.C.R. Two-color, two-photon uncaging of glutamate and GABA. Nat. Methods 2010, 7, 123–125. [Google Scholar] [CrossRef]
- San Miguel, V.; Bochet, C.G.; del Campo, A. Wavelength-selective caged surfaces: How many functional levels are possible? J. Am. Chem. Soc. 2011, 133, 5380–5388. [Google Scholar] [CrossRef] [PubMed]
- Schafer, F.; Joshi, K.B.; Fichte, M.A.H.; Mack, T.; Wachtveitl, J.; Heckel, A. Wavelength-selective uncaging of dA and dC residues. Org. Lett. 2011, 13, 1450–1453. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; San Miguel, V.; del Campo, A. Light-triggered multifunctionality at surfaces mediated by photolabile protecting groups. Macromol. Rapid. Commun. 2013, 34, 310–329. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Fernández, L.; Herbivo, C.; San Miguel Arranz, V.; Warther, D.; Donato, L.; Specht, A.; del Campo, A. Dual photosensitive polymers with wavelength-selective photoresponse. Adv. Mater. 2014, 26, 5012–5017. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.J.; Velema, W.A.; Lerch, M.M.; Szymanski, W.; Feringa, B.L. Wavelength-selective cleavage of photoprotecting groups: Strategies and applications in dynamic systems. Chem. Soc. Rev. 2015, 44, 3358–3377. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, N.; Ciftci, M.; Jung, K.; Boyer, C. Mediating Reaction Orthogonality in Polymer and Materials Science. Angew. Chem. Int. Ed. 2021, 60, 1748–1781. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, J.; Sinsel, F.; Heckel, A. Chromatic Selectivity of Coumarin-Caged Oligonucleotides. Chem. Eur. J. 2023, 29, e202204014. [Google Scholar] [CrossRef] [PubMed]
- Dolinski, N.D.; Page, Z.A.; Callaway, E.B.; Eisenreich, F.; Garcia, R.V.; Chavez, R.; Bothman, D.P.; Hecht, S.; Zok, F.W.; Hawker, C.J. Solution mask liquid lithography (SMaLL) for one-step, multimaterial 3D printing. Adv. Mater. 2018, 30, 1800364. [Google Scholar] [CrossRef] [PubMed]
- Menzel, J.P.; Feist, F.; Tuten, B.; Weil, T.; Blinco, J.P.; Barner-Kowollik, C. Light-Controlled Orthogonal Covalent Bond Formation at Two Different Wavelengths. Angew. Chem. Int. Ed. 2019, 58, 7470–7474. [Google Scholar] [CrossRef]
- Birks, J.B. (Ed.) Organic Molecular Photophysics; Wiley-Interscience: New York, NY, USA, 1973; Volume I, ISBN 10.0471074152. [Google Scholar]
- Nobs, F.; Burger, U.; Schaffner, K. Specifically (π → π*)-Induced Cyclohexenone Reactions: 4a-(Z-1-Propenyl)-bicyclo [4.4.0]dec-1(8a)-en-2-one and 4a-Z-1-Propenyl-bicyclo [4.4.0]deca-1(8a), 7-dien-2-one. Helv. Chim. Acta 1977, 60, 1607–1628. [Google Scholar] [CrossRef]
- Adriano, N.; Ahearn, C.; Black, C.; Cracchiolo, M.; Ghere, D.; Nuñez, A.; Olivan, L.; Patel, R.; Saner, S.; Smith, K.R.; et al. Solvent- and Wavelength-Dependent Photolysis of Estrone. Photochem. Photobiol. 2022, 98, 783–797. [Google Scholar] [CrossRef] [PubMed]
- Bridge, N.K. Effect of wavelength on the flash photolysis of quinones. Trans. Faraday Soc. 1960, 56, 1001–1007. [Google Scholar] [CrossRef]
- Gloor, J.; Schaffner, K. Spezifisch π→π*-induzierte Reaktionen von γ-Dimethoxymethylcyclo hexen-2-onen: 1,3-Umlagerung und Wasserstoffabstraktion durch das α-Kohlenstoffatom. Helv. Chim. Acta 1974, 57, 1815–1845. [Google Scholar] [CrossRef]
- Dunkel, P. Photoremovable Protecting Groups. Encyclopedia 2022, 2, 1225–1236. [Google Scholar] [CrossRef]
- Saracini, C.; Liakos, D.G.; Zapata Rivera, J.E.; Neese, F.; Meyer, G.J.; Karlin, K.D. Excitation wavelength dependent O2 release from copper(II)-superoxide compounds: Laser flash-photolysis experiments and theoretical studies. J. Am. Chem. Soc. 2014, 136, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Ziani, Z.; Loiseau, F.; Lognon, E.; Boggio-Pasqua, M.; Philouze, C.; Cobo, S.; Royal, G.C. Synthesis of a negative photochrome with high switching quantum yields and capable of singlet-oxygen production and storage. Chem. Eur. J. 2021, 27, 16642–16653. [Google Scholar] [CrossRef] [PubMed]
- Thoma, K.; Klimek, R. Photostabilisation of drugs in dosage forms without protection from packaging materials. Int. J. Pharm. 1991, 67, 169–175. [Google Scholar] [CrossRef]
- Maafi, W.; Maafi, M. Modelling nifedipine photodegradation, photostability and actinometry properties. Int. J. Pharm. 2013, 456, 153–164. [Google Scholar] [CrossRef]
- Maafi, M.; Maafi, W. Quantification of Unimolecular Photoreaction Kinetics: Determination of Quantum Yields and Development of Actinometers-The Photodegradation Case of Cardiovascular Drug Nisoldipine. Int. J. Photoenergy 2015, 2015, 1–12. [Google Scholar] [CrossRef]
- Maafi, M.; Lee, L.Y. Determination of dacarbazine Φ-order photokinetics, quantum yields, and potential for actinometry. J. Pharm. Sci. 2015, 104, 3501–3509. [Google Scholar] [CrossRef]
- El-Hendawy, M.M.; Fayed, T.A.; Awad, M.K.; English, N.J.; Etaiw, S.E.H.; Zaki, A.B. Photophysics, photochemistry and thermal stability of diarylethene-containing benzothiazolium species. J. Photochem. Photochem. Chem. A 2015, 301, 20–31. [Google Scholar] [CrossRef]
- Ribeiro, A.; Ballardini, R.; Belser, P.; Gandolfi, M.T.; Iyer, V.M.; Moggi, L. Photochemical investigation of photochromic diaryethene compound that can be used as a wide range actinometer. Photochem. Photobiol. Sci. 2009, 8, 1734–1742. [Google Scholar] [CrossRef] [PubMed]
- Ern, J.; Bens, A.T.; Martin, H.-D.; Kuldova, K.; Trommsdorff, H.P.; Kryschi, C. Ring-opening and -closure reaction dynamics of a photochromic dithienylethene derivative. J. Phys. Chem. A 2002, 106, 1654–1660. [Google Scholar] [CrossRef]
- Herder, M.; Schmidt, B.M.; Grubert, L.; Patzel, M.; Schwarz, J.; Hecht, S. Improving the Fatigue Resistance of Diarylethene Switches. J. Am. Chem. Soc. 2015, 137, 2738–2747. [Google Scholar] [CrossRef] [PubMed]
- Maafi, M.; Brown, R.G. Kinetic analysis and elucidation options for AB(1k,2F) systems. New spectrokinetic methods for photochromes. Photochem. Photobiol. Sci. 2008, 7, 1360–1372. [Google Scholar] [CrossRef]
- Maafi, M.; Brown, R.G. The kinetic model for AB(1F) systems. A closed-form integration of the differential equation with a variable photokinetic factor. J. Photochem. Photobiol. A Chem. 2007, 187, 319–324. [Google Scholar] [CrossRef]
- Maafi, M.; Al-Qarni, M.A. Mono- and polychromatic light diarylethene-actinometer for the visible range. Dye. Pigment. 2022, 198, 109942. [Google Scholar] [CrossRef]
- Sumi, T.; Takagi, Y.; Yagi, A.; Morimoto, M.; Irie, M. Photoirradiation wavelength dependence of cycloreversion quantum yields of diarylethenes. Chem. Commun. 2014, 50, 3928–3930. [Google Scholar] [CrossRef]
- Cipolloni, M.; Gentili, P.L.; Ortica, F.; Becker, R.S.; Favaro, G. Effects of solvent, excitation wavelength, and concentration on the photobehavior of some diazonaphthoquinones. Arkivoc 2011, 9, 205–220. [Google Scholar] [CrossRef]
- Di Nunzio, M.R.; Gentili, P.L.; Romani, A.; Favaro, G. Photochromic, thermochromic, and fluorescent spirooxazines and naphthopyrans: A spectrokinetic and thermodynamic study. Chemphyschem 2008, 9, 768–775. [Google Scholar] [CrossRef]
- Wilkinson, F.; Hobley, J.; Naftaly, M. Photochromism of spiro-naphthoxazines: Molar absorption coefficients and quantum efficiencies. J. Chem. Soc. Faraday Trans. 1992, 88, 1511–1517. [Google Scholar] [CrossRef]
- Fischer, E. Calculation of photostationary states in systems A.dblarw. B when only A is known. J. Phys. Chem. 1967, 71, 3704–3706. [Google Scholar] [CrossRef]
- Kaiser, C.; Halbritter, T.; Heckel, A.; Wachtveitl, J. Thermal, Photochromic and Dynamic Properties of Water-Soluble Spiropyrans. ChemistrySelect 2017, 2, 4111–4123. [Google Scholar] [CrossRef]
- Ortica, F.; Smimmo, P.; Favaro, G.; Mazzucato, U.; Delbaere, S.; Venec, D.; Vermeersch, G.; Frigoli, M.; Moustrou, C.; Samat, A. Effect of oligothiophene substituents on the photophysical and photochemical properties of a naphthopyran. Photochem. Photobiol. Sci. 2004, 3, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Ortica, F.; Moustrou, C.; Berthet, J.; Favaro, G.; Samat, A.; Guglielmetti, R.; Vermeersch, G.; Mazzucato, U. Comprehensive photokinetic and NMR study of a biphotochromic supermolecule involving two naphthopyrans linked to a central thiophene unit through acetylenic bonds. Photochem. Photobiol. 2003, 78, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, Y.; Tekahashi, Y. Trifluoromethyl-substituted photochromic indolylfulgide. A remarkably durable fulgide towards photochemical and thermal treatments. Chem. Lett. 1996, 25, 1037–1038. [Google Scholar] [CrossRef]
- Wolak, M.A.; Gillespie, N.B.; Thomas, C.J.; Birge, R.R.; Lees, W.J. Optical properties of photochromic fluorinated indolylfulgides. J. Photochem. Photobiol. A Chem. 2001, 144, 83–91. [Google Scholar] [CrossRef]
- Wolak, M.A.; Gillespie, N.B.; Thomas, C.J.; Birge, R.R.; Lees, W.J. Thermolysis of fluorinated cycloalkylidene fulgides yields a new class of photochromic compounds. Chem. Commun. 2003, 992–993. [Google Scholar] [CrossRef]
- Cordes, T.; Malkmus, S.; DiGirolamo, J.A.; Lees, W.J.; Nenov, A.; de Vivie-Riedle, R.; Braun, M.; Zinth, W. Accelerated and efficient photochemistry from higher excited electronic states in fulgide molecules. J. Phys. Chem. A 2008, 112, 13364–13371. [Google Scholar] [CrossRef]
- Reinfelds, M.; Hermanns, V.; Halbritter, T.; Wachtveitlz, J.; Braun, M.; Slanina, T.; Heckel, A. A robust, broad-absorbing fulgide derivative as a universal chemical actinometer for UV to NIR region. ChemPhotoChem 2019, 3, 441–449. [Google Scholar] [CrossRef]
- Volarić, J.; Szymanski, W.; Simeth, N.A.; Feringa, B.L. Molecular photoswitches in aqueous environments. Chem. Soc. Rev. 2021, 50, 12377–12449. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Zhang, J. (Eds.) Photochromic Materials: Preparation, Properties and Applications; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2016; ISBN 978-3-527-68372-7. [Google Scholar]
- Maafi, M.; Maafi, W. Modelling and elucidation of the kinetics of multiple consecutive photoreactions AB4(4Φ) with Φ-order kinetics. Application to the photodegradation of ribofavin. J. Pharm. Sci. 2016, 105, 3537–3548. [Google Scholar] [CrossRef] [PubMed]
- Becker, R.S.; Dolan, E.; Balke, D.E. Vibronic effects in photochemistry. Competition between internal conversion and photochemistry. J. Chem. Phys. 1969, 50, 239–245. [Google Scholar] [CrossRef]
- Becker, R.S.; Michl, J. Photochromism of synthetic and naturally occurring 2H-chromenes and 2H-pyrans. J. Am. Chem. Soc. 1966, 88, 5931–5933. [Google Scholar] [CrossRef]
- Lenoble, C.; Becker, R.S. Photophysics, photochemistry and kinetics of photochromic 2H-pyrans and chromenes. J. Photochem. 1986, 33, 187–197. [Google Scholar] [CrossRef]
- Becker, R.S.; Favaro, G.; Romani, A.; Gentili, P.L.; Dias, F.M.B. Vibronic effects in pathways of photochemistry and vibrational relaxation. Chem. Phys. 2005, 316, 108–116. [Google Scholar] [CrossRef]
- Becker, R.S.; Pelliccioli, A.P.; Romani, A.; Favaro, G. Vibronic quantum effects in fluorescence and photochemistry. Competition between vibrational relaxation and photochemistry and consequences on photochemical control. J. Am. Chem. Soc. 1999, 121, 2104–2109. [Google Scholar] [CrossRef]
- Becker, R.S.; Favaro, G. New concepts in photochemistry and photophysics: Photochromic and other type molecules. J. Photochem. Photobiol. C Photochem. Rev. 2011, 12, 167–176. [Google Scholar] [CrossRef]
- Zewail, A.H. Femtochemistry: Atomic-scale dynamics of the chemical bond using ultrafast lasers (Nobel Lecture). Angew. Chem. Int. Ed. 2000, 39, 2586–2631. [Google Scholar] [CrossRef]
- Tamai, N.; Miyasaka, H. Ultrafast dynamics of photochromic systems. Chem. Rev. 2000, 100, 1875–1890. [Google Scholar] [CrossRef]
- Briand, J.; Bram, O.; Rehault, J.; Leonard, J.; Cannizzo, A.; Chergui, M.; Zanirato, V.; Olivucci, M.; Helbing, J.; Haacke, S. Coherent ultrafast torsional motion and isomerization of a biomimetic dipolar photoswitch. Phys. Chem. Chem. Phys. 2010, 12, 3178–3187. [Google Scholar] [CrossRef] [PubMed]
- Wynne, K.; Hochstrasser, R.M. Coherence and adiabaticity in ultrafast electron transfer. Adv. Chem. Phys. Electron Transf. Isol. Mol. Biomol. Part 2 1999, 107, 263–309. [Google Scholar] [CrossRef]
- Rather, S.R.; Scholes, G.D. From fundamental theories to quantum coherences in electron transfer. J. Am. Chem. Soc. 2019, 141, 708–722. [Google Scholar] [CrossRef]
- Chudoba, C.; Riedle, E.; Pfeiffer, M.; Elsaesser, T. Vibrational Coherence in Ultrafast Excited State Proton Transfer. Chem. Phys. Lett. 1996, 263, 622–628. [Google Scholar] [CrossRef]
- Myahkostupov, M.; Pagba, C.V.; Gundlach, L.; Piotrowiak, P. Vibrational state dependence of interfacial electron transfer: Hot electron injection from the S1 state of azulene into TiO2 nanoparticles. J. Phys. Chem. C 2013, 117, 20485–20493. [Google Scholar] [CrossRef]
- Bishenden, E.; Donaldson, D.J. Mode-specific chemical branching ratios in the photodissociation of OClO. J. Chem. Phys. 1993, 99, 3129–3132. [Google Scholar] [CrossRef]
- Fidder, H.; Tschirschwitz, F.; Dühr, O.; Nibbering, E.T.J. Vibrational mode-specific photochemical reaction dynamics of chlorine dioxide in solution. J. Chem. Phys. 2001, 114, 6781–6794. [Google Scholar] [CrossRef]
- Wallace, P.; Bollinger, C.; Hayes, S.; Reid, A. On the actinic wavelength dependence of OClO photochemistry in solution. J. Chem. Phys. 2003, 116, 1883–1890. [Google Scholar] [CrossRef]
- Vaida, V.; Feierabend, K.J.; Rontu, N.; Takahashi, K. Sunlight-initiated photochemistry: Excited vibrational states of atmospheric chromophores. Int. J. Photoenergy 2008, 138091. [Google Scholar] [CrossRef]
- Reynard, L.M.; Donaldson, D.J. Overtone-induced chemistry of trifluoroacetic acid: An experimental and theoretical study. J. Phys. Chem. A 2002, 106, 8651–8657. [Google Scholar] [CrossRef]
- Staikova, M.; Oh, M.; Donaldson, D.J. Overtone-induced decarboxylation: A potential sink for atmospheric diacids. J. Phys. Chem. A 2005, 109, 597–602. [Google Scholar] [CrossRef]
- Young, C.J.; Donaldson, D.J. Overtone-induced degradation of perfluorinated alcohols in the atmosphere. J. Phys. Chem. A 2007, 111, 13466–13471. [Google Scholar] [CrossRef] [PubMed]
- Manzanares, C.E.; Delgado, Y.P.; Sunuwar, S. Phase shift cavity ring down and FT-NIR spectra of CH vibrational transitions and band strengths of ethyl acetate. J. Quant. Spectr. Rad. Transf. 2024, 312, 108810. [Google Scholar] [CrossRef]
- Donaldson, D.J.; Tuck, A.F.; Vaida, V. Atmospheric photochemistry via vibrational overtone absorption. Chem. Rev. 2003, 103, 4717–4729. [Google Scholar] [CrossRef] [PubMed]
- Miller, Y.; Chaban, G.M.; Finlayson-Pitts, B.J.; Gerber, R.B. Photochemical processes induced by vibrational overtone excitations: Dynamics simulations for cis-HONO, trans-HONO, HNO3, and HNO3-H2O. J. Phys. Chem. A 2006, 110, 5342–5354. [Google Scholar] [CrossRef]
- Miller, Y.; Gerber, R.B. Dynamics of vibrational overtone excitations of H2SO4, H2SO4-H2O: Hydrogen-hopping and photodissociation processes. J. Am. Chem. Soc. 2006, 128, 9594–9595. [Google Scholar] [CrossRef] [PubMed]
- Goss, L.M.; Vaida, V.; Brault, J.W.; Skodje, R.T. Sequential two-photon dissociation of atmospheric water. J. Phys. Chem. A 2001, 105, 70–75. [Google Scholar] [CrossRef]
- Brown, S.S.; Metz, R.B.; Berghout, H.L.; Crim, F.F. Vibrationally mediated photodissociation of isocyanic acid (HNCO): Preferential N–H bond fission by excitation of the reaction coordinate. J. Chem. Phys. 1996, 105, 6293–9303. [Google Scholar] [CrossRef]
- Lai, H.Y.; Jhang, W.R.; Tseng, C.M. Mode-dependent excited-state lifetime of phenol under the S1/S2 conical intersection. J. Chem. Phys. 2018, 149, 031104. [Google Scholar] [CrossRef]
- Troe, J. Analysis of Quantum Yields for the Photolysis of Formaldehyde at λ > 310 nm. J. Phys. Chem. A 2007, 111, 3868–3874. [Google Scholar] [CrossRef]
- Röth, E.P.; Ehhalt, D.H. A simple formulation of the CH2O photolysis quantum yields. Atmos. Chem. Phys. 2015, 15, 7195–7202. [Google Scholar] [CrossRef]
- Jankowski, J.J.; Kieber, D.J.; Mopper, K.; Neale, P.J. Development and intercalibration of ultraviolet solar actinometer. Photochem. Photobiol. 2000, 71, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Hatchard, C.G.; Parker, C.A. A New Sensitive Chemical Actinometer. II. Potassium Ferrioxalate as a Standard Chemical Actinometer. Proc. R. Soc. London. Ser. A Math. Phys. Sci. 1956, 235, 518–536. [Google Scholar] [CrossRef]
- Favaro, G.; di Nunzio, M.R.; Gentili, P.L.; Romani, A.; Becker, R.S. Effects of the Exciting Wavelength and Viscosity on the Photobehavior of 9- and 9,10-Bromoanthracenes. J. Phys. Chem. A 2007, 111, 5948–5953. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.M.; Silva, E.; Reis, R.L. Light-triggered release of photocaged therapeutics—Where are we now? J. Contr. Release 2019, 298, 154–176. [Google Scholar] [CrossRef] [PubMed]
- Setlow, R.B. The wavelengths in sunlight effective in producing skin cancer: A theoretical analysis. Proc. Natl. Acad. Sci. USA 1974, 71, 3363–3366. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, E.; Abe, R.; Ohtani, B. Visible light-induced photocatalytic reaction of gold-modified titanium(iv) oxide particles: Action spectrum analysis. Chem. Commun. 2009, 2, 241–243. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-K.; Mills, A.; O’rourke, C. Action spectra in semiconductor photocatalysis. Chem. Soc. Rev. 2017, 46, 4877–4894. [Google Scholar] [CrossRef] [PubMed]
- Datcu, A.; Mendoza, M.L.; Pérez del Pino, A.; Logofatu, C.; Luculescu, C.; György, E. UV–vis light induced photocatalytic activity of TiO2/graphene oxide nanocomposite coatings. Catal. Today 2019, 321–322, 81–86. [Google Scholar] [CrossRef]
- Pettersson, L.A.A.; Roman, L.S.; Inganäs, O. Modeling photocurrent action spectra of photovoltaic devices based on organic thin films. J. Appl. Phys. 1999, 86, 487–496. [Google Scholar] [CrossRef]
- Langford, C.H.; Achari GIzadifard, M. Wavelength dependence of luminescence and quantum yield in dichlorination of PCBs. J. Photochem. Photobiol. A Chem. 2011, 222, 40–46. [Google Scholar] [CrossRef]
- Bhat, A.P.; Pomerantz, W.C.K.; Arnold, W.A. Wavelength-Dependent UV-LED Photolysis of Fluorinated Pesticides and Pharmaceuticals. Environ. Sci. Technol. 2023, 57, 5327–5336. [Google Scholar] [CrossRef] [PubMed]
- Kuldova, K.; Tsyganenko, K.; Corval, A.; Trommsdorff, H.P.; Bens, A.T.; Kryschi, C. Photo-switchable dithienylethenes: Threshold of the photoreactivity. Synth. Met. 2000, 115, 163–166. [Google Scholar] [CrossRef]
- Bens, A.T.; Frewert, D.; Kodatis, K.; Kryschi, C.; Martin, H.-D.; Trommsdorff, H.P. Coupling of Chromophores: Carotenoids and Photoactive Diarylethenes–Photoreactivity versus Radiationless Deactivation. Eur. J. Org. Chem. 1998, 11, 2333–2338. [Google Scholar] [CrossRef]
- Irie, M.; Eriguchi, T.; Takada, T.; Uchida, K. Photochromism of diarylethenes having thiophene oligomers as the aryl groups. Tetrahedron 1997, 53, 12263–12271. [Google Scholar] [CrossRef]
- Nagorny, S.; Schewe, M.; Weingartz, T.; Eitzeroth, A.; Adams, J.; Rembe, C.; Schmidt, A. Stabilities of bis(thienyl)ethenes in polymethyl methacrylate (PMMA) coatings as absorbance modulation layers for nanoscale imaging. Mater. Adv. 2024, 5, 159–170. [Google Scholar] [CrossRef]
- Maafi, M. Useful Spectrokinetic Methods for the Investigation of Photochromic and Thermo-Photochromic Spiropyrans. Molecules 2008, 13, 2260–2302. [Google Scholar] [CrossRef]
- Dulić, D.; Kudernac, T.; Pužys, A.; Feringa, B.L.; van Wees, B.J. Temperature gating of the ring-opening process in diarylethene molecular switches. Adv. Mater. 2007, 19, 2898–2902. [Google Scholar] [CrossRef]

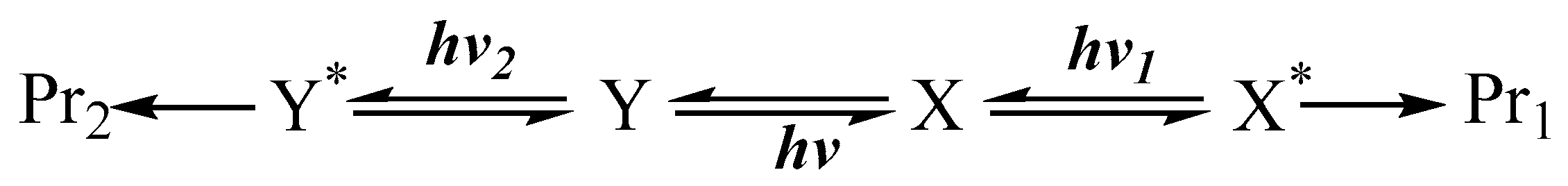
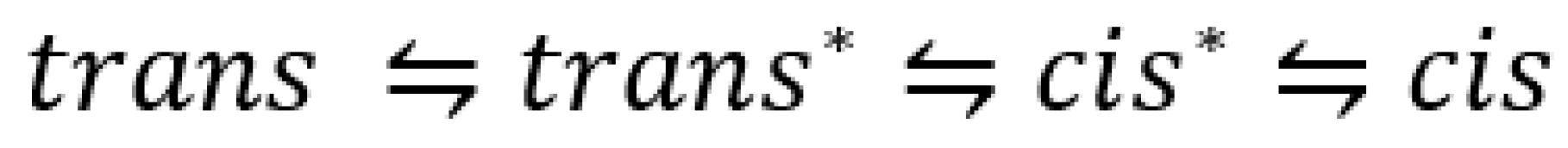

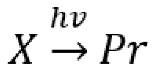
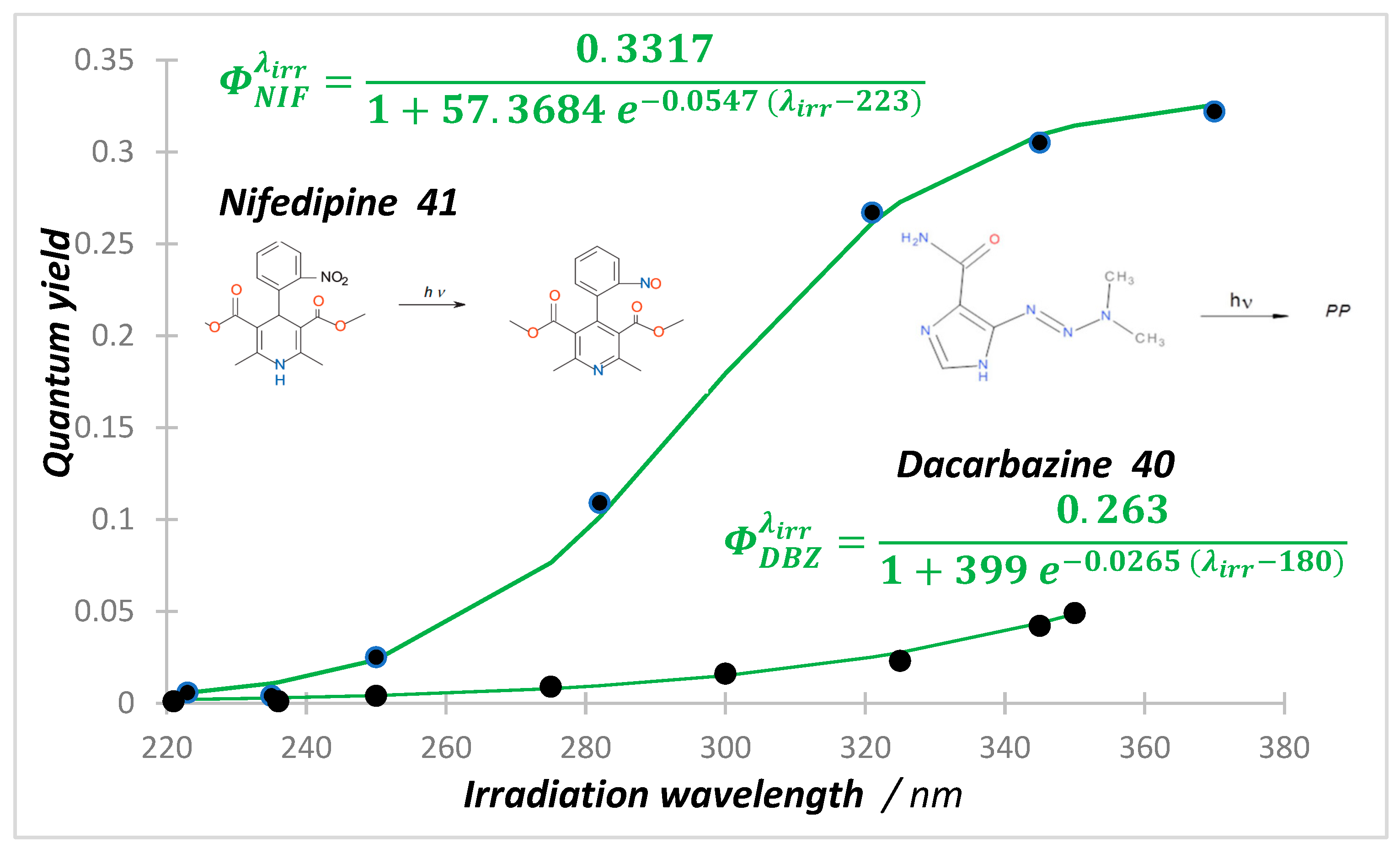
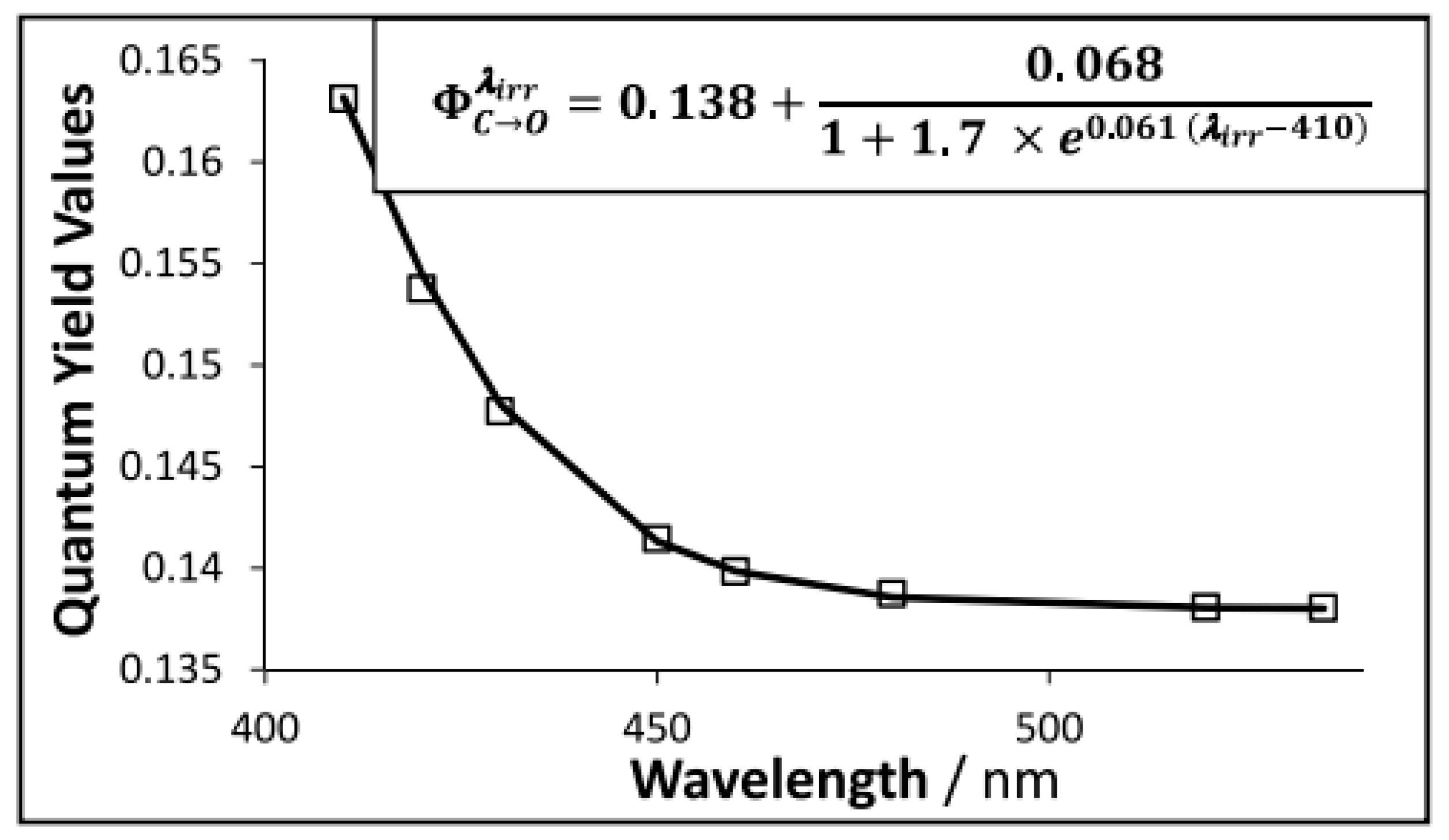
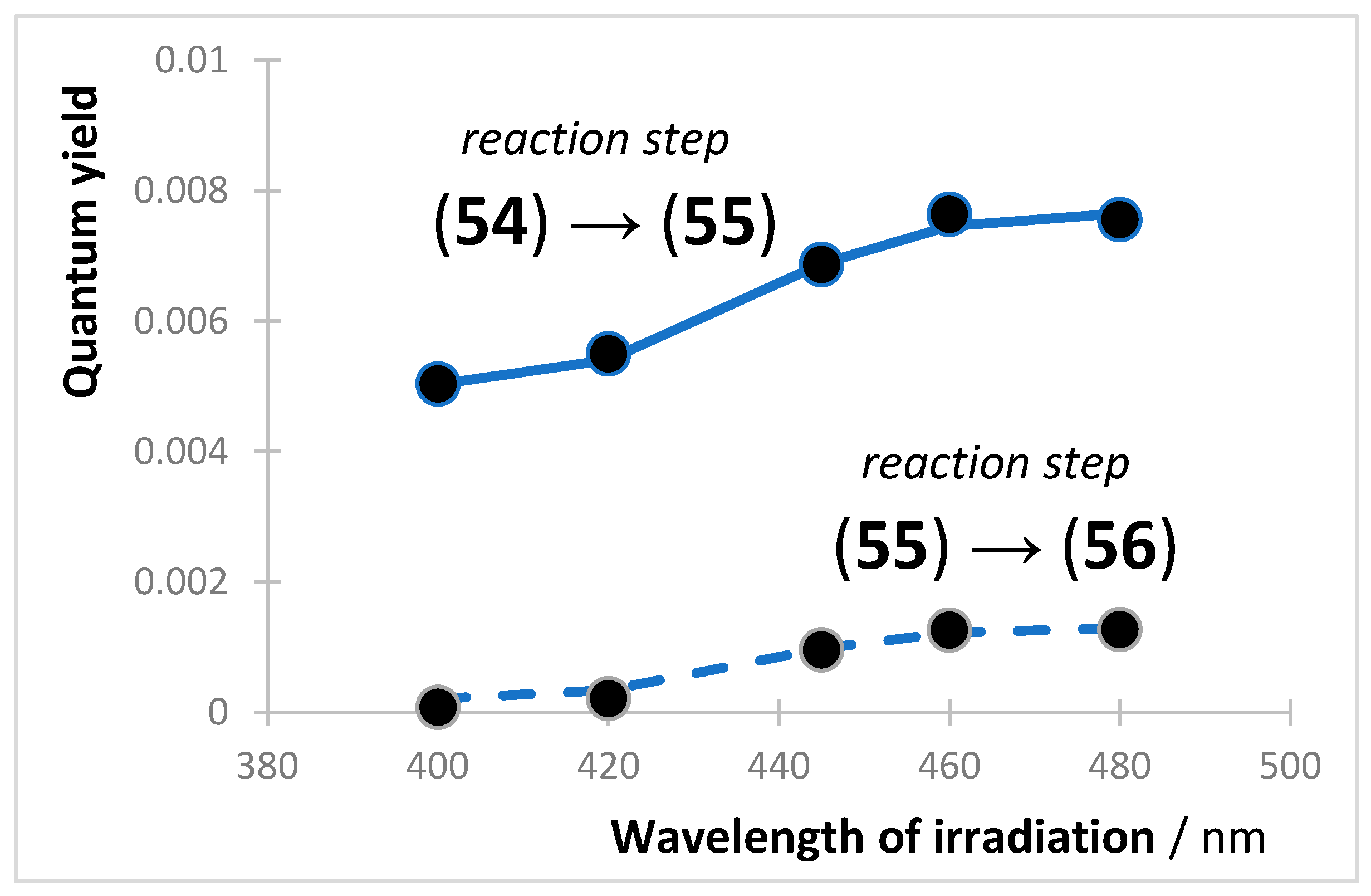
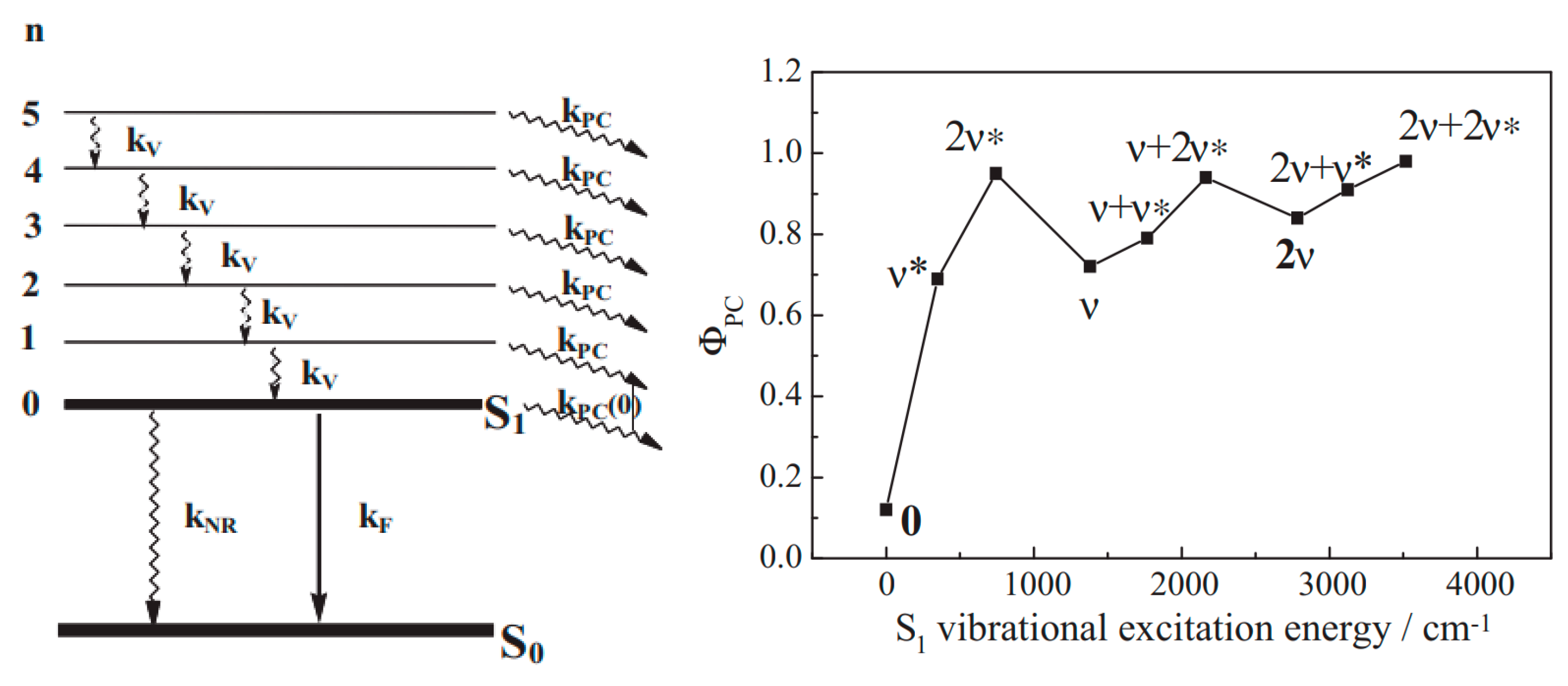
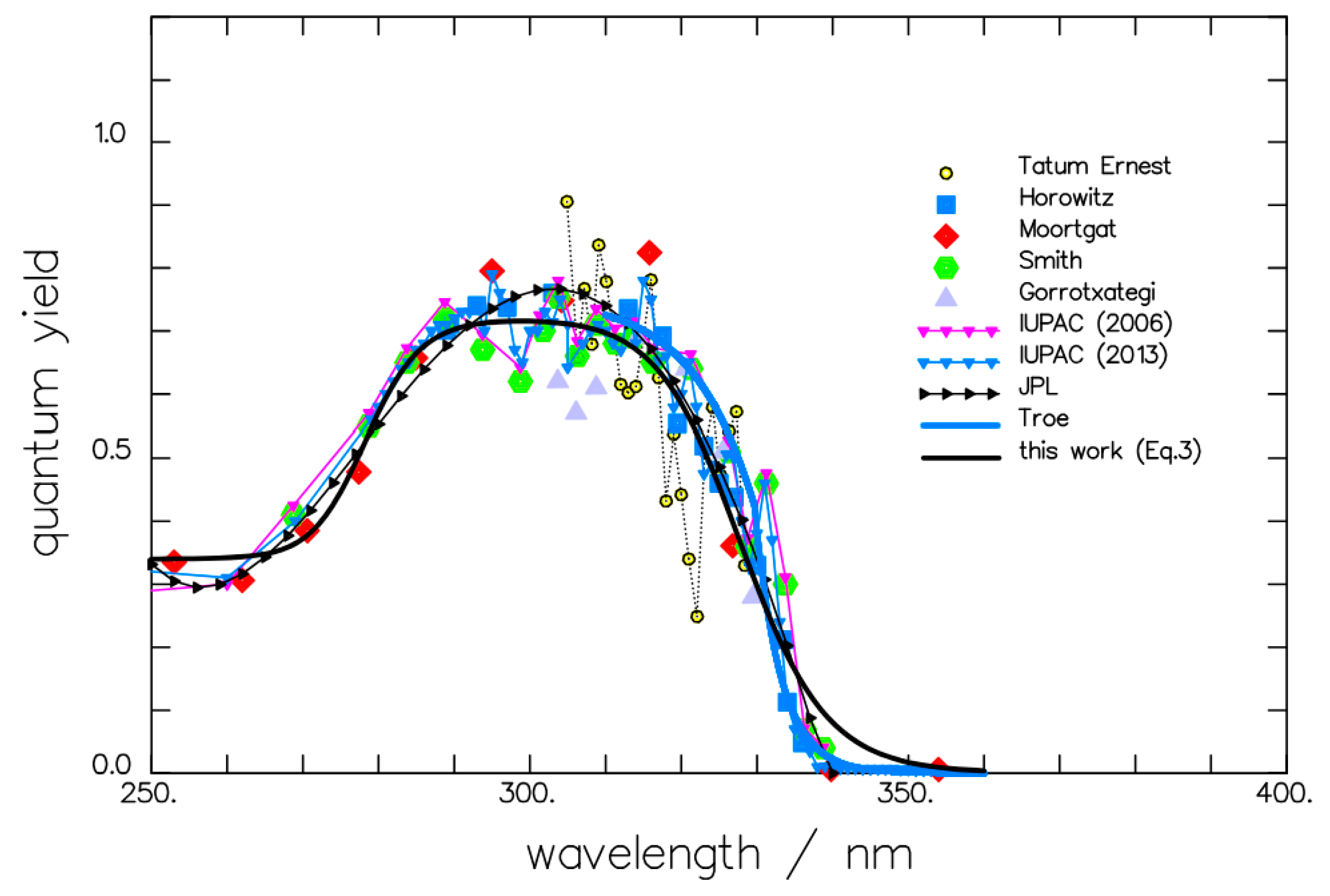

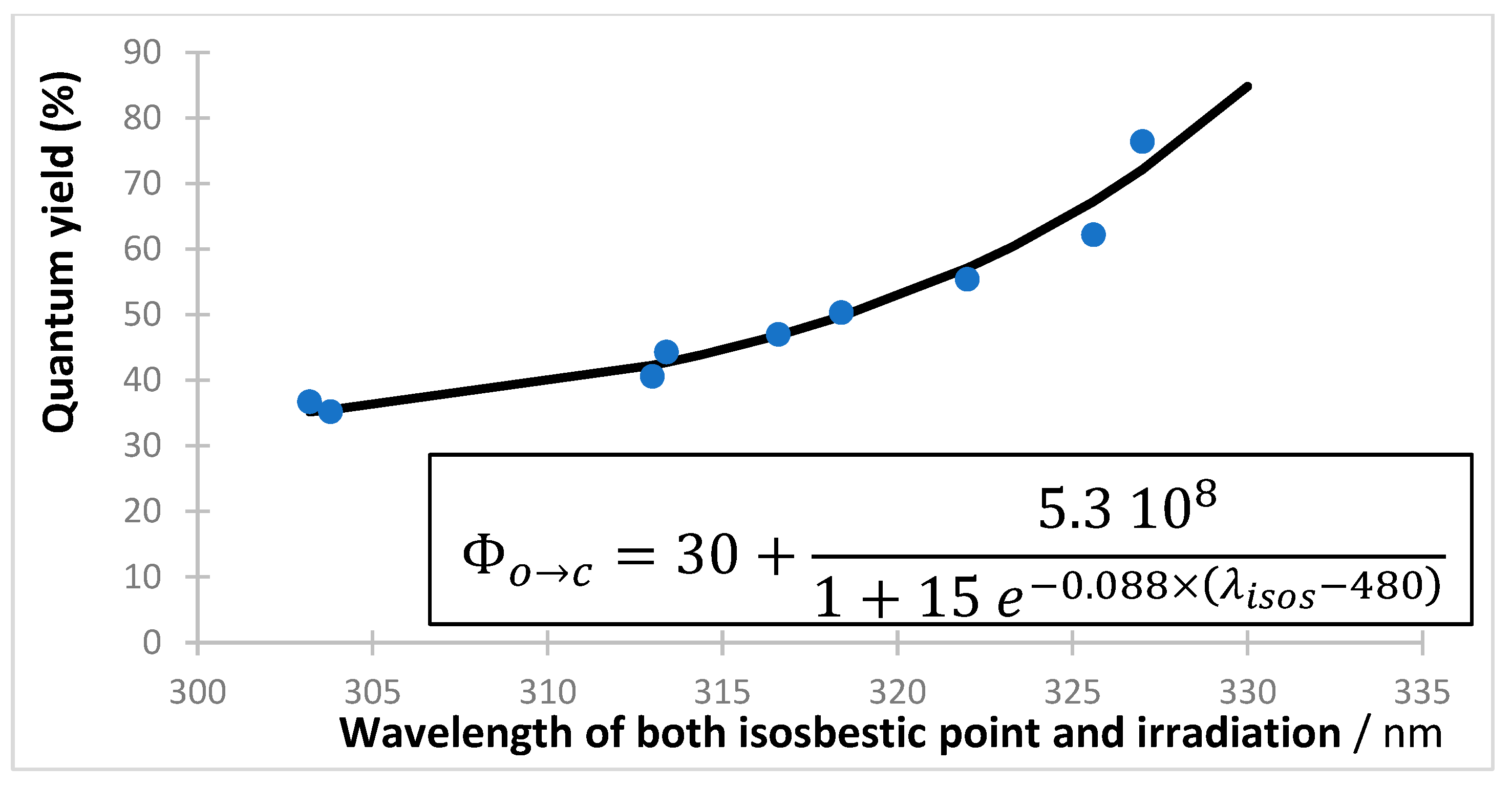
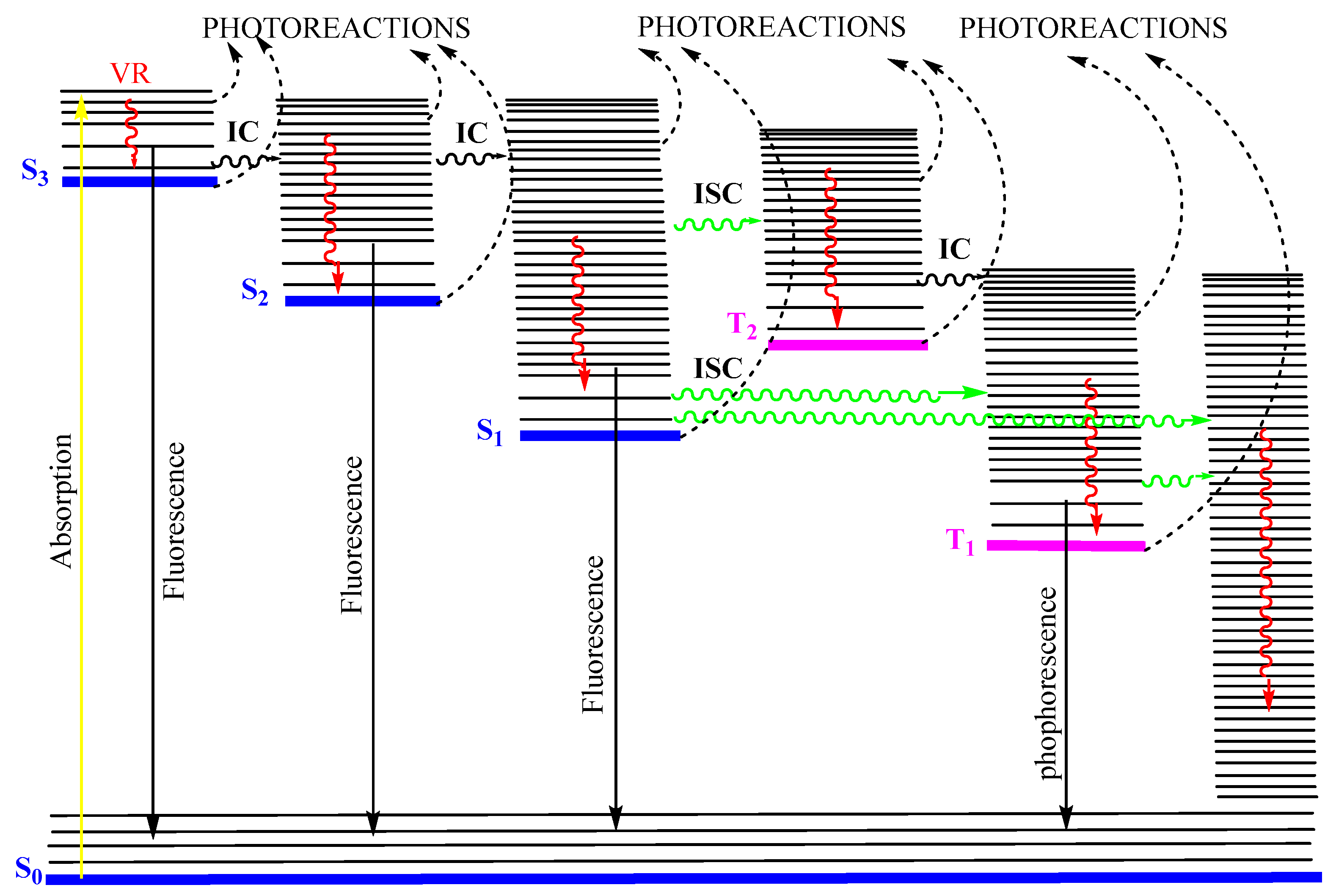

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Irradiation Wavelength/nm | Azobenzene Concentration/M | Medium Temperature/°C | Solvent | ||
|---|---|---|---|---|---|
| 254 | 2.5 × 10−5 | 25 | Isooctane | 0.13 | 0.14 |
| 254 | 6 × 10−4 | RT | Methanol | 0.26 | 0.31 |
| 280 | 6 × 10−4 | RT | Methanol | 0.12 | 0.34 |
| 313 | 6 × 10−4 | RT | Methanol | 0.13 | 0.3 |
| 313 | (0.04–2) × 10−3 | RT | Methanol | 0.14 | - |
| 313 | (0.2–5) × 10−5 | 20 | Methanol | 0.37 | - |
| 313 | 10−5–10−4 | RT | Ethanol | 0.12 | 0.31 |
| 313 | (0.01–2) × 10−3 | 15–25 | Ethanol | 0.22 | 0.69 |
| 313 | (0.04–2) × 10−3 | RT | Isopropanol | 0.1 | 0.5 |
| 313 | (0.2–5) × 10−4 | 20 | H2O-Ethanol | - | 0.4 |
| 313 | (0.2–5) × 10−4 | 20 | MeCN | - | 0.35 |
| 313 | (0.2–5) × 10−4 | 20 | THF | - | 0.4 |
| 313 | (0.025–1) × 10−3 | 25 | Isooctane | 0.1 | 0.41 |
| 313 | (0.04–2) × 10−3 | RT | Cyclohexane | 0.1 | 0.42 |
| 313 | (0.2–5) × 10−5 | 20 | Cyclohexane | 0.4 | - |
| 313 | (0.2–5) × 10−5 | 20 | n-hexane | - | 0.44 |
| 334 | 6 × 10−4 | RT | Methanol | 0.15 | 0.3 |
| 365 | 6 × 10−4 | RT | Methanol | 0.15 | 0.35 |
| 365 | (0.01–2) × 10−3 | 15–20 | Ethanol | 0.2 | 0.6 |
| 365 | (0.025–1) × 10−3 | 25 | Isooctane | 0.12 | 0.48 |
| 365 | (0.04–2) × 10−3 | RT | Cyclohexane | 0.14 | - |
| 405 | 6 × 10−4 | RT | Methanol | 0.2 | 0.57 |
| 405 | (0.025–1) × 10−3 | 25 | Isooctane | 0.23 | 0.55 |
| 436 | (0.04–2) × 10−3 | RT | Methanol | 0.28 | - |
| 436 | 6 × 10−4 | RT | Methanol | 0.22 | 0.63 |
| 436 | (0.01–2) × 10−3 | 15–20 | Ethanol | 0.36 | 0.45 |
| 436 | 10−5–10−4 | RT | Ethanol | 0.24 | 53 |
| 436 | (0.04–2) × 10−3 | RT | Isopropanol | 0.26 | 0.42 |
| 436 | (0.025–1) × 10−3 | 25 | Isooctane | 0.28 | 0.55 |
| 436 | (0.2–5) × 10−5 | 20 | Cyclohexane | 0.28 | 0.55 |
| 436 | 1 × 10−3 | 25 | Benzene | 0.26 | 0.46 |
| Molecule | Irradiation Wavelength Range | (Pattern) | (Pattern) |
|---|---|---|---|
| Fluvoxamine (15) | 226–290 | 0.0038–0.435 (Sigmoid) | 0.0015–0.0844 (Curved) |
| Pinosylvin (16) | 280–330 | 0.105–0.277 (Sigmoid) | 0.125–0.879 (Sigmoid) |
| Oxyresveratrol (14) | 260–360 | 0.069–0.180 (Sigmoid) | 0.023–0.26 (Sigmoid) |
| Montelukast (17) | 220–360 | 0.012–0.18 (Linear: positive slop) | 0.0033–0.061 (Triangular: reversed V-shape) |
| Sunitinib (13) | 240–480 | 0.03–0.026 (Flat line) | 0.06–0.175 (Sigmoid) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maafi, M. Excitation Wavelength-Dependent Photochemistry. Photochem 2024, 4, 233-270. https://doi.org/10.3390/photochem4020015
Maafi M. Excitation Wavelength-Dependent Photochemistry. Photochem. 2024; 4(2):233-270. https://doi.org/10.3390/photochem4020015
Chicago/Turabian StyleMaafi, Mounir. 2024. "Excitation Wavelength-Dependent Photochemistry" Photochem 4, no. 2: 233-270. https://doi.org/10.3390/photochem4020015
APA StyleMaafi, M. (2024). Excitation Wavelength-Dependent Photochemistry. Photochem, 4(2), 233-270. https://doi.org/10.3390/photochem4020015