Observation of Light-Induced Reactions of Olefin–Ozone Complexes in Cryogenic Matrices Using Fourier-Transform Infrared Spectroscopy


Abstract
:1. Introduction
2. Materials and Methods
2.1. Experiment
2.2. Calculation
3. Results
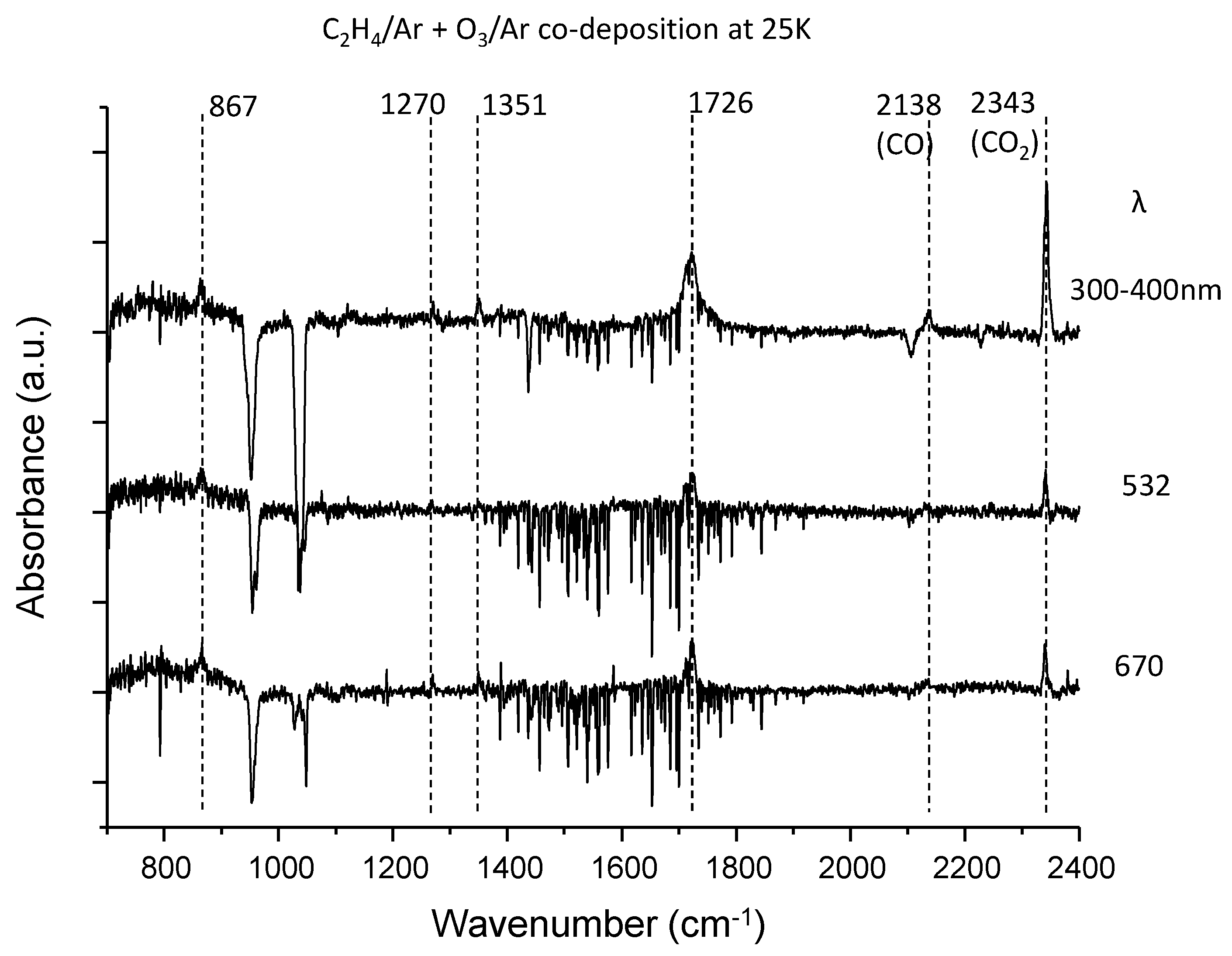
3.1. C2H4–O3 System
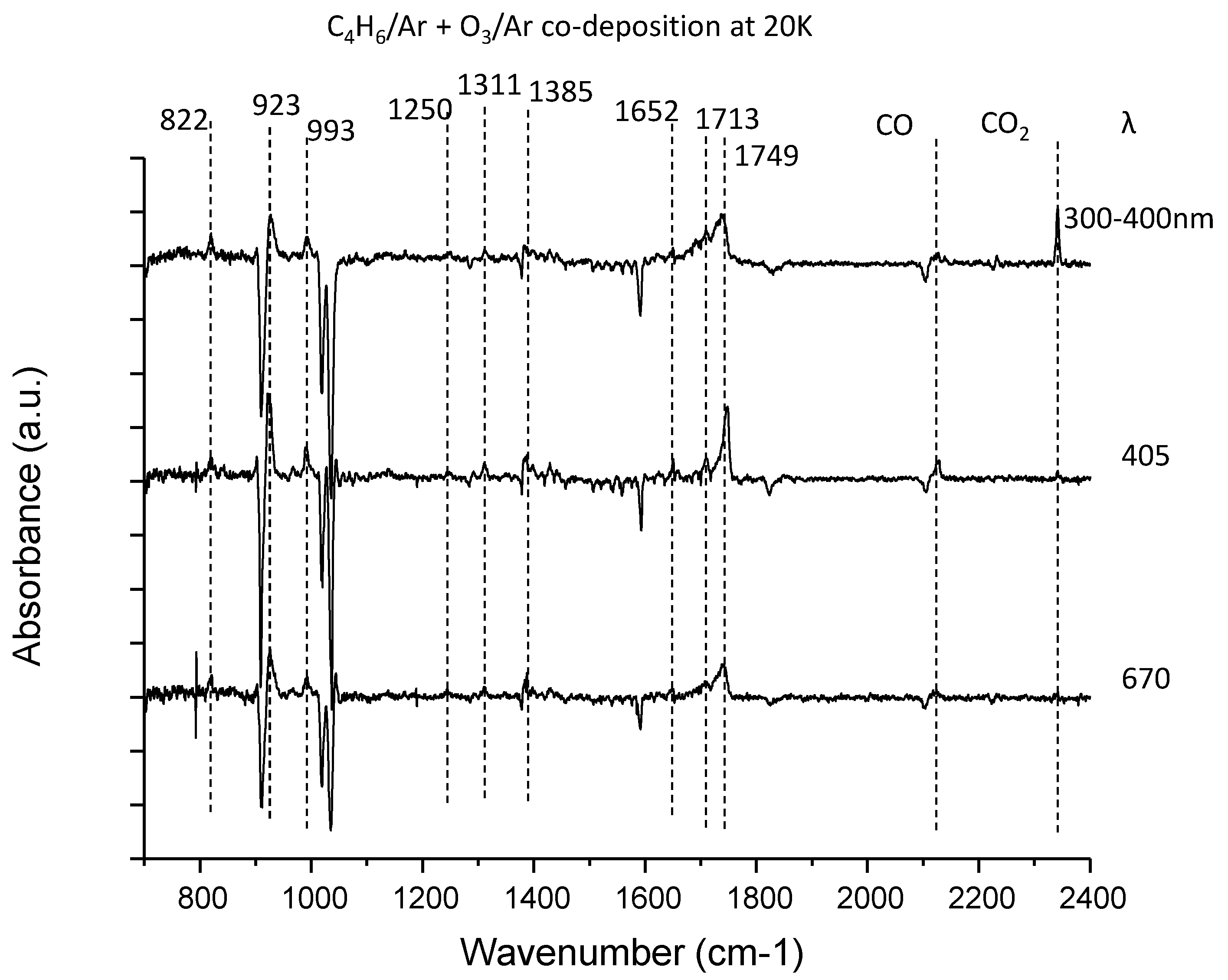
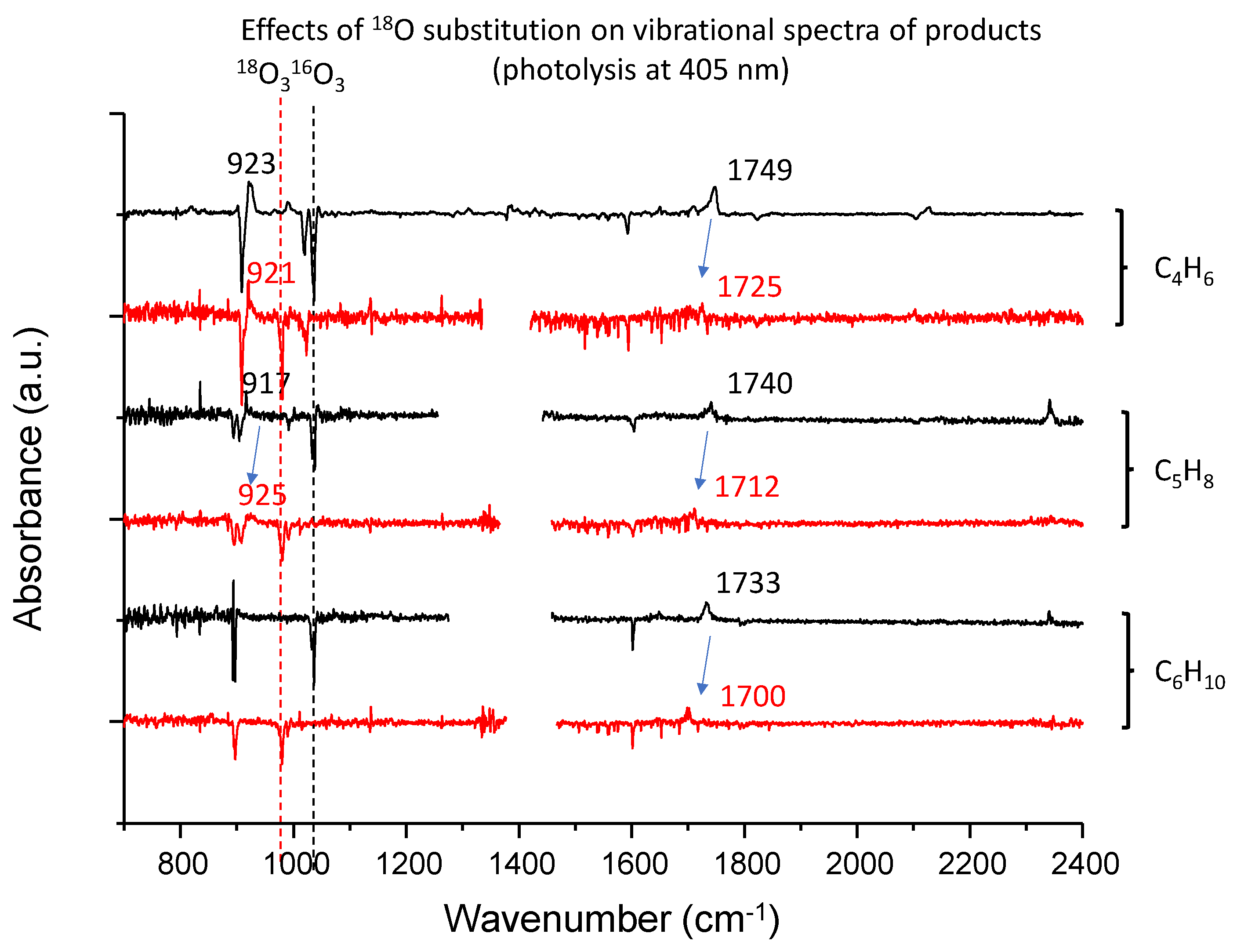
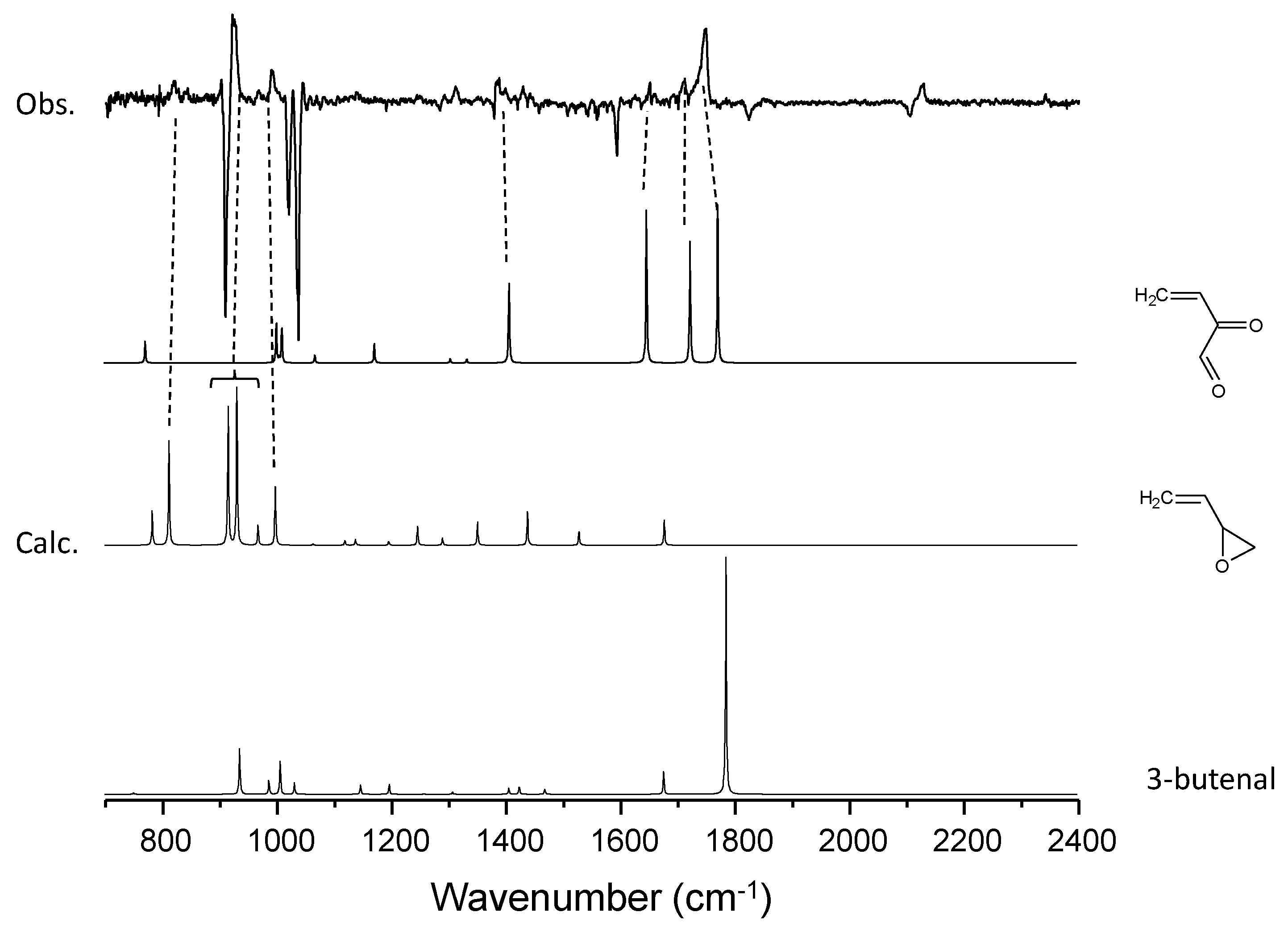
3.2. C4H6–O3 System
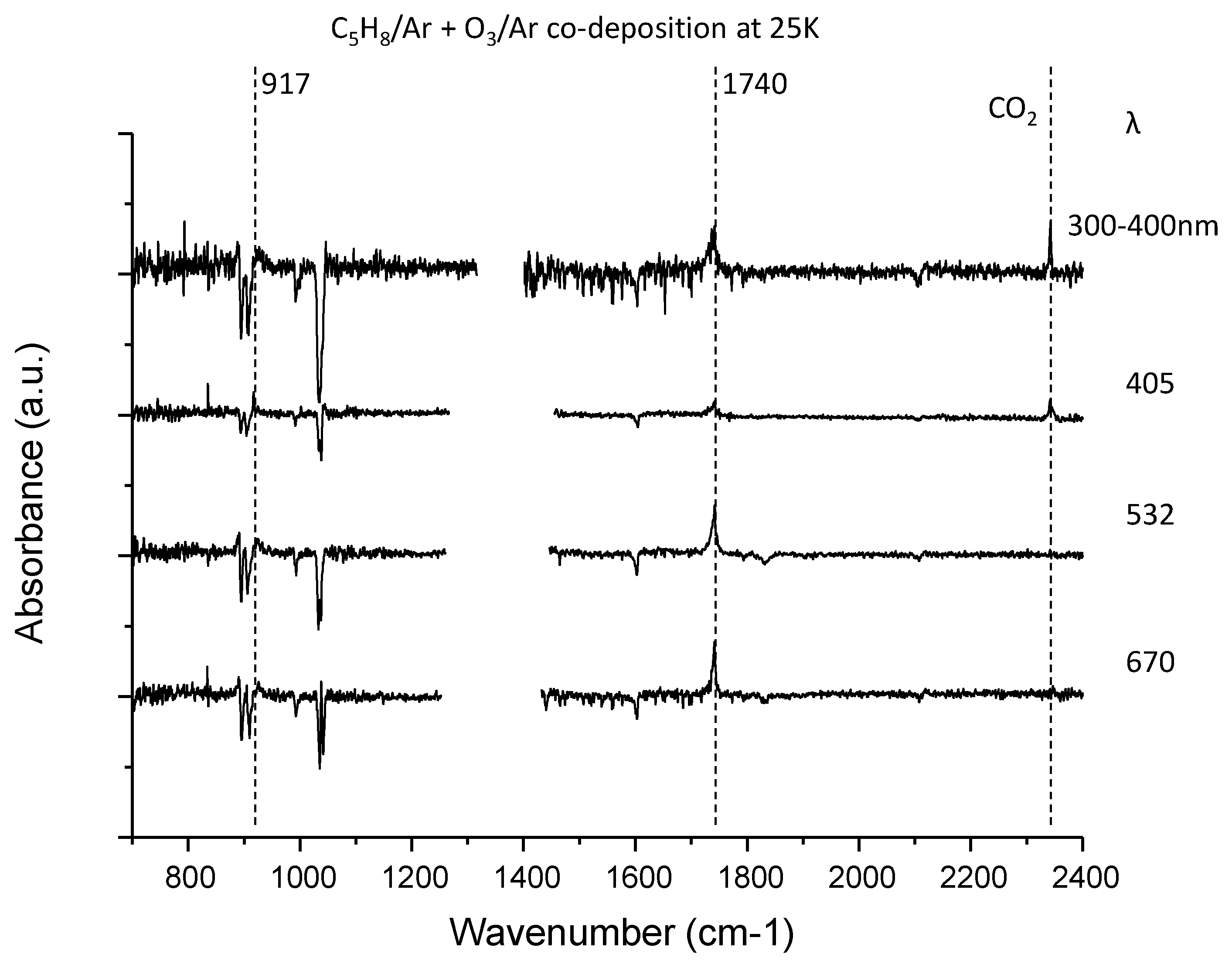
3.3. C5H8–O3 System
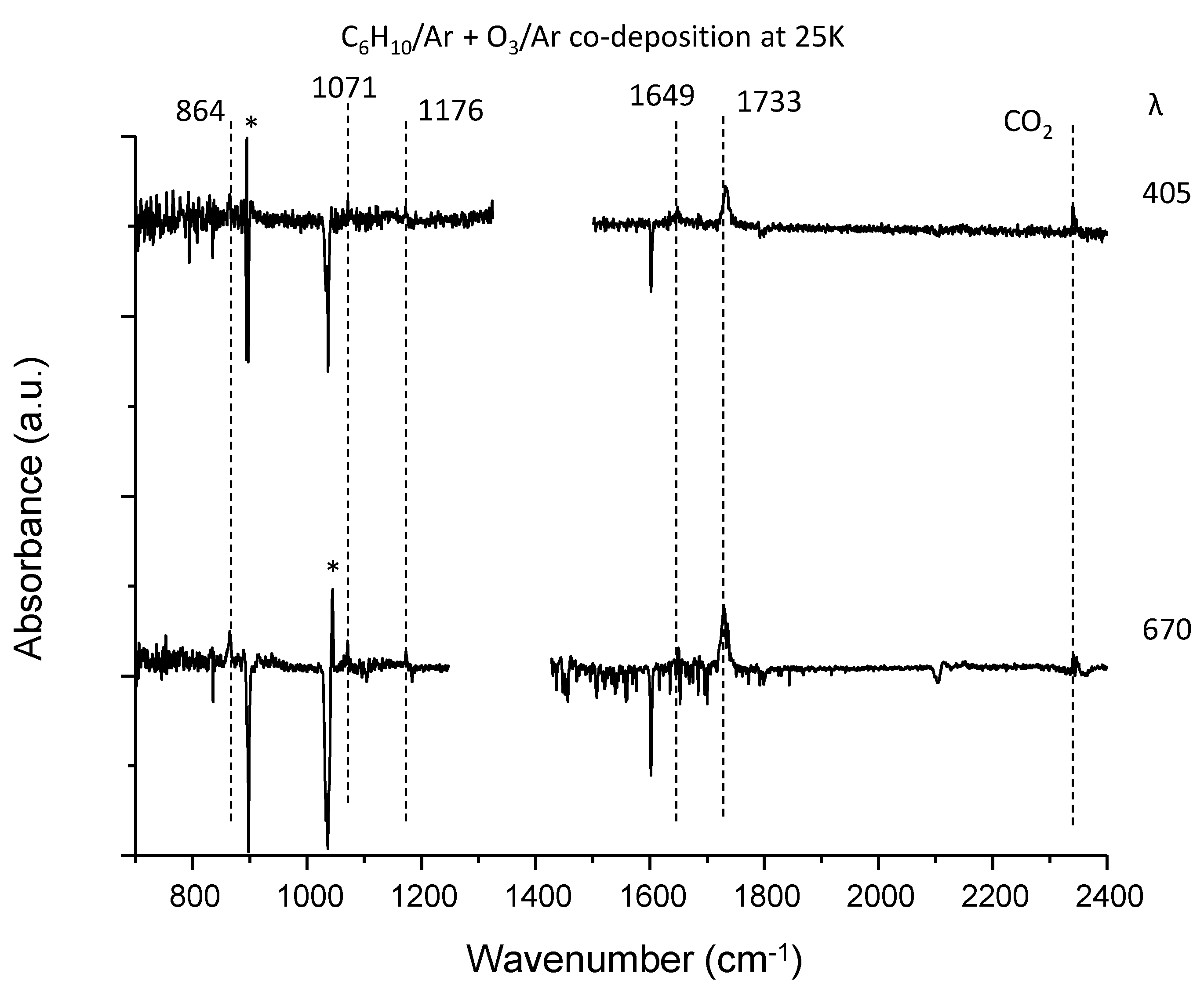
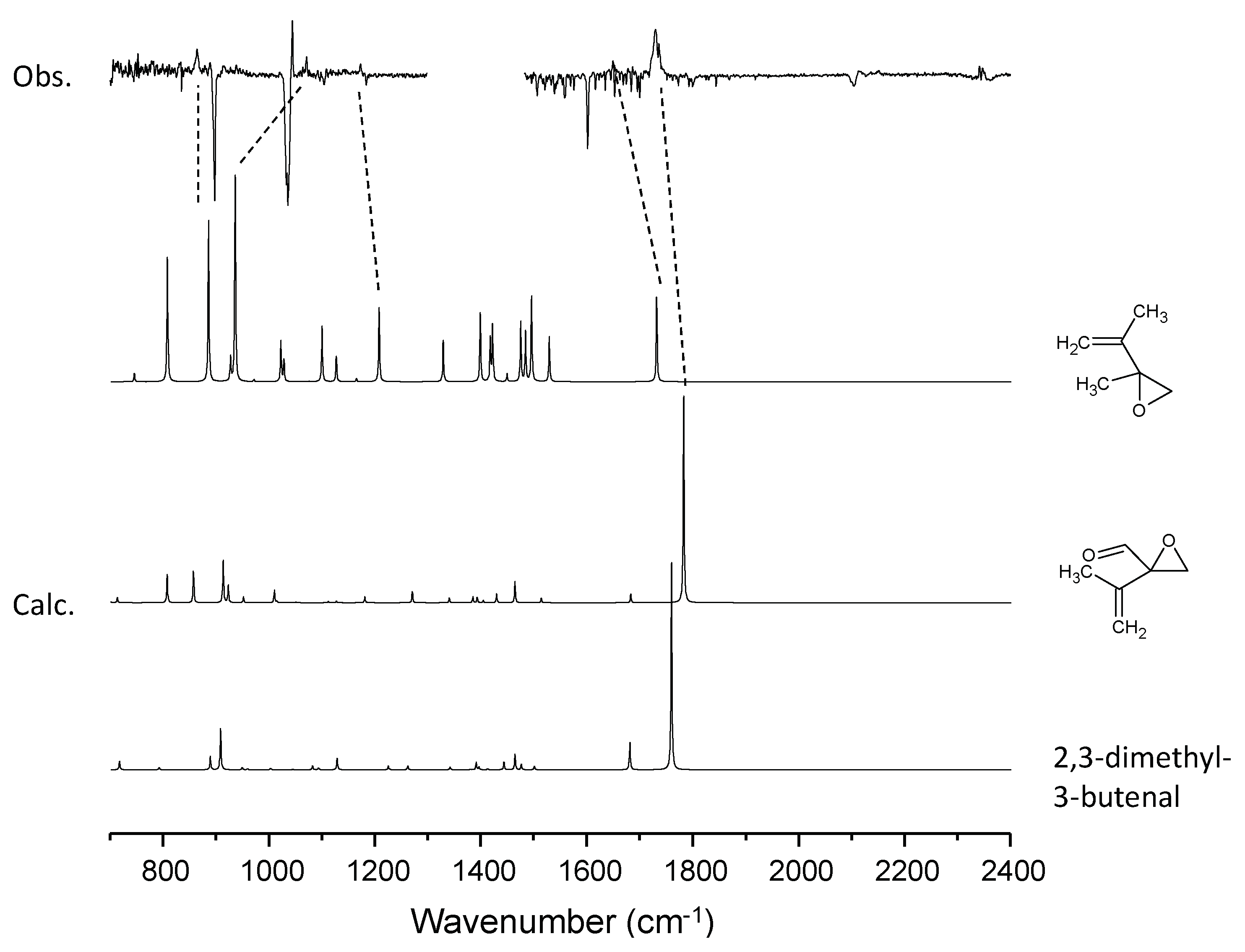
3.4. C6H10–O3 System
4. Discussions
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change; Wiley: New York, NY, USA, 1998. [Google Scholar]
- Johnson, D.; Marston, G. The gas-phase ozonolysis of unsaturated volatile organic compounds in the troposphere. Chem. Soc. Rev. 2008, 37, 699–716. [Google Scholar] [CrossRef] [PubMed]
- Wennberg, P.O.; Bates, K.H.; Crounse, J.D.; Dodson, L.G.; McVay, R.C.; Mertens, L.A.; Nguyen, T.B.; Praske, E.; Schwantes, R.H.; Smarte, M.D.; et al. Gas-Phase Reactions of Isoprene and Its Major Oxidation Products. Chem. Rev. 2018, 118, 3337–3390. [Google Scholar] [CrossRef] [Green Version]
- Singmaster, K.A.; Pimentel, G.C. Photolysis of Allene-Ozone Mixtures at 647 nm in Crygenic Matrices Part 1. Formation of allene oxide. J. Mol. Struct. 1989, 194, 215–238. [Google Scholar] [CrossRef]
- Singmaster, K.A.; Pimentel, G.C. Spectroscopic Detection of Ozone-Olefin Charge-Transfer Complexes in Cryogenic Matrices. J. Phys. Chem. 1990, 94, 5226–5229. [Google Scholar] [CrossRef]
- Jonnalagadda, S.; Chan, S.; Garrido, J.; Bond, J.; Singmaster, K.A. Detection of Ethylene-Ozone and Cyclohexene-Ozone Charge-Transfer Complexes in Cryogenic Matrices. J. Am. Chem. Soc. 1995, 117, 562–563. [Google Scholar] [CrossRef]
- Pinelo, L.F.; Kugel, R.W.; Ault, B.S. Charge-Transfer Complexes and Photochemistry of Ozone with Ferrocene and n-Butylferrocene: A UV−vis Matrix-Isolation Study. J. Phys. Chem. A 2015, 119, 10272–10278. [Google Scholar] [CrossRef]
- Herron, J.T.; Huie, R.E. Stopped-Flow Studies of the Mechanisms of Ozone-Alkene Reactions in the Gas Phase. Ethylene. J. Am. Chem. Soc. 1977, 99, 5430–5435. [Google Scholar] [CrossRef]
- Niki, H.; Maker, P.D.; Savage, C.M.; Breltenbach, L.P. A FT IR Study of a Transitory Product in the Gas-Phase Ozone-Ethylene Reaction. J. Phys. Chem. 1981, 85, 1024–1027. [Google Scholar] [CrossRef]
- Su, F.; Calverl, J.G.; Shaw, J.H. A FT 1R Spectroscopic Study of the Ozone-Ethene Reaction Mechanism in O2-Rich Mixtures. J. Phys. Chem. 1980, 84, 239–246. [Google Scholar] [CrossRef]
- Horie, O.; Moortgat, G.K. Decomposition pathways of the excited Criegee intermediates in the ozonolysis of simple alkenes. Atmos. Environ. A 1991, 25, 1881–1896. [Google Scholar] [CrossRef]
- Neeb, P.; Horie, O.; Moortgat, G.K. The nature of the transitory product in the gas-phase ozonolysis of ethene. Chem. Phys. Lett. 1995, 246, 150–156. [Google Scholar] [CrossRef]
- Neeb, P.; Horie, O.; Moortgat, G.K. The Ethene—Ozone Reaction in the Gas Phase. J. Phys. Chem. A 1998, 102, 6778–6785. [Google Scholar] [CrossRef]
- Porterfield, J.P.; Eibenberger, S.; Patterson, D.; McCarthy, M.C. The ozonolysis of isoprene in a cryogenic buffer gas cell by high resolution microwave spectroscopy. Phys. Chem. Chem. Phys. 2018, 20, 16828–16834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousso, A.C.; Hansen, N.; Jasper, A.W.; Ju, Y. Low-Temperature Oxidation of Ethylene by Ozone in a Jet-Stirred Reactor. J. Phys. Chem. A 2018, 122, 8674–8685. [Google Scholar] [CrossRef]
- Kühne, H.; Vaccani, S.; Ha, T.-K.; Bauder, A.; Günthard, H.H. Infrared-matrix and microwave spectroscopy of the ethylene-ozone gas-phase reaction. Chem. Phys. Lett. 1976, 38, 449–455. [Google Scholar] [CrossRef]
- Kühne, H.; Günthard, H.H. Spectroscopic Study of the Ozone-Ethylene Reaction. Matrix-Infrared Spectra of Three Isotopic Ethylene Ozonides. J. Phys. Chem. 1976, 80, 1238–1247. [Google Scholar] [CrossRef]
- Nelander, B.; Nord, L. The reaction between ethylene and ozone. A matrix study. Tetrahedron Lett. 1977, 32, 2821–2822. [Google Scholar] [CrossRef]
- Hawkins, M.; Kohlmlller, C.K.; Andrews, L. Matrix Infrared Spectra and Photolysis and Pyrolysis of Isotopic Secondary Ozonides of Ethylene. J. Phys. Chem. 1982, 86, 3154–3166. [Google Scholar] [CrossRef]
- Kohlmiller, C.K.; Andrews, L. Infrared Spectrum of the Primary Ozonide of Ethylene in Solid Xenon. J. Am. Chem. Soc. 1981, 103, 2578–2583. [Google Scholar] [CrossRef]
- Andrews, L.; Kohlmiller, C.K. Infrared Spectra and Photochemistry of the Primary and Secondary Ozonides of Propene, trans-2-Butene, and Methylpropene in Solid Argon. J. Phys. Chem. 1982, 86, 4548–4557. [Google Scholar] [CrossRef]
- Samuni, U.; Haas, Y.; Fajgar, R.; Pola, J. Matrix effects in the low-temperature oxonation of ethylene, tetramethylethylene and 1-hexene. J. Mol. Struct. 1998, 449, 177–201. [Google Scholar] [CrossRef]
- Clark, R.J.H.; Foley, L.J. Photochemically Induced Reactions of Ozone with 1,2-Dibromoethene and 1,2-Dichloroethene: An FT-IR Matrix Isolation Study. J. Phys. Chem. A 2002, 106, 3356–3364. [Google Scholar] [CrossRef]
- Clay, M.; Ault, B.S. Infrared matrix isolation and theoretical study of the initial intermediates in the reaction of ozone with cis-2-butene. J. Phys. Chem. A 2010, 114, 2799–2805. [Google Scholar] [CrossRef] [PubMed]
- Hoops, M.D.; Ault, B.S. Matrix isolation study of the early intermediates in the ozonolysis of cyclopentene and cyclopentadiene: Observation of two criegee intermediates. J. Am. Chem. Soc. 2009, 131, 2853–2863. [Google Scholar] [CrossRef]
- Kugel, R.W.; Ault, B.S. Infrared matrix isolation and theoretical studies of reactions of ozone with bicyclic alkenes: α-pinene, norbornene, and norbornadiene. J. Phys. Chem. A 2015, 119, 312–322. [Google Scholar] [CrossRef]
- Ault, B.S. Matrix isolation study of the reaction of O (3P) with 1,3 butadiene: Unexpected formation of ethylketone. J. Mol. Struct. 2019, 1176, 47–53. [Google Scholar] [CrossRef]
- Ito, F. Infrared and quantum chemical studies of isoprene-methanol complexes in noble gas matrices. J. Mol. Spectrosc. 2019, 362, 90–95. [Google Scholar] [CrossRef]
- Ito, F. Infrared and density functional theory studies of isoprene-water complexes in noble gas matrices. J. Mol. Spectrosc. 2017, 341, 27–34. [Google Scholar] [CrossRef]
- Torres, E.; DiLabio, G.A. A (Nearly) Universally Applicable Method for Modeling Noncovalent Interactions Using B3LYP. J. Phys. Chem. Lett. 2012, 3, 1738–1744. [Google Scholar] [CrossRef]
- Barone, V. Vibrational zero-point energies and thermodynamic functions beyond the harmonic approximation. J. Chem. Phys. 2004, 120, 3059–3065. [Google Scholar] [CrossRef]
- Barone, V. Anharmonic vibrational properties by a fully automated second-order perturbative approach. J. Chem. Phys. 2005, 122, 014108. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Ault, B.S. Matrix Isolation Study of the Complexes of Small-ring Heterocycles. J. Mol. Struct. 1985, 127, 343–356. [Google Scholar] [CrossRef]
- Khoshkhoo, H.; Nixon, E.R. Infrared and Raman spectra of formaldehyde in argon and nitrogen matrices. Spectrochim. Acta A 1973, 29, 603–612. [Google Scholar] [CrossRef]
- Wiberg, K.B.; Thiel, Y.; Goodman, L.; Leszczynski, J. Acetaldehyde: Harmonic Frequencies, Force Field, and Infrared Intensities. J. Phys. Chem. 1995, 99, 13850–13864. [Google Scholar] [CrossRef]
- Maçôas, E.M.S.; Lundell, J.; Pettersson, M.; Khriachtchev, L.; Fausto, R.; Räsänen, M. Vibrational spectroscopy of cis- and trans-formic acid in solid argon. J. Mol. Spectrosc. 2003, 219, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Macoas, E.M.S.; Khriachtchev, L.; Pettersson, M.; Fausto, R.; Rasanen, M. Rotational isomerism of acetic acid isolated in rare-gas matrices: Effect of medium and isotopic substitution on IR-induced isomerization quantum yield and cis→trans tunneling rate. J. Chem. Phys. 2004, 121, 1331–1338. [Google Scholar] [CrossRef] [Green Version]
- Diem, M.; MacDonald, B.G.; Lee, E.K.C. Photolysis and Laser-Excited Fluorescence and Phosphorescence Emission of trans-Glyoxal in an Argon Matrix at 13 K. J. Phys. Chem. 1981, 85, 2227–2232. [Google Scholar] [CrossRef]
- Pugh, L.A.; Rao, K.N. Intensities from infrared spectra. In Molecular Spectroscopy: Modern Research; Rao, K.N., Ed.; Academic Press: Cambridge, MA, USA, 1976; Volume 2, pp. 165–226. [Google Scholar]
- Profeta, L.T.M.; Sams, R.L.; Johnson, T.J. Quantitative Infrared Intensity Studies of Vapor-Phase Glyoxal, Methylglyoxal, and 2,3-Butanedione (Diacetyl) with Vibrational Assignments. J. Phys. Chem. A 2011, 115, 9886–9900. [Google Scholar] [CrossRef]
- Adler-Golden, S.M.; Langhoff, S.R.; Bauschlicher, C.W., Jr.; Carney, G.D. Theoretical calculation of ozone vibrational infrared intensities. J. Chem. Phys. 1985, 83, 255–264. [Google Scholar] [CrossRef]
- Cvetanovic, R.J. Reaction of Oxygen Atoms with Ethylene. J. Chem. Phys. 1955, 23, 1375–1380. [Google Scholar] [CrossRef]
- Hirokami, S.; Cvetanovic, R.J. Reaction of Oxygen Atoms, O(3P), with Olefins in Liquid Nitrogen Solution at 77 K. J. Am. Chem. Soc. 1974, 96, 3738–3746. [Google Scholar] [CrossRef]
- Yang, X.; Maeda, S.; Ohno, K. Insight into Global Reaction Mechanism of [C2, H4, O] System from ab initio Calculations by the Scaled Hypersphere Search Method. J. Phys. Chem. A 2007, 111, 5099–5110. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, R.; Carter, W.P.L. Kinetics and Mechanisms of the Gas-Phase Reactions of Ozone with Organic Compounds under Atmospheric Conditions. Chem. Rev. 1984, 84, 437–470. [Google Scholar] [CrossRef]
- Tanaka, N.; Kajii, Y.; Shibuya, K.; Nakata, M. Visible Light Induced Reactions of NO2 with Conjugated Dienes in a Low-Temperature Ar Matrix. J. Phys. Chem. 1993, 97, 7048–7053. [Google Scholar] [CrossRef]
- 2-Methyl-2-vinyloxirane | C5H8O - PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/92166 (accessed on 23 February 2022).
- Su, Y.-T.; Huang, Y.-H.; Witek, H.A.; Lee, Y.-P. Infrared Absorption Spectrum of the Simplest Criegee Intermediate CH2OO. Science 2013, 340, 174–176. [Google Scholar] [CrossRef] [Green Version]
- Dreuw, A.; Weisman, J.L.; Head-Gordon, M. Long-range charge-transfer excited states in time-dependent density functional theory require non-local exchange. J. Chem. Phys. 2003, 119, 2943–2946. [Google Scholar] [CrossRef]
- Adamo, C.; Jacquemin, D. The calculations of excited-state properties with Time-Dependent Density Functional Theory. Chem. Soc. Rev. 2013, 42, 845–856. [Google Scholar] [CrossRef]
- Campetella, M.; Maschietto, F.; Frisch, M.J.; Scalmani, G.; Ciofini, I.; Adamo, C. Charge Transfer Excitations in TDDFT: A Ghost-Hunter Index. J. Comput. Chem. 2017, 38, 2151–2156. [Google Scholar] [CrossRef]
- Kamata, K.; Yoshioka, R.; Akai, N.; Nakata, M. Visible-light-Induced Reaction of an Ozone–Trimethylamine Complex Studied by Matrix-Isolation IR and Visible Absorption Spectroscopies. J. Phys. Chem. A 2020, 124, 9973–9979. [Google Scholar] [CrossRef]
- Kon, A.; Inano, N.; Terada, N.; Kamata, K.; Akai, N.; Nakata, M. Photoreactions of Ozone–Tetrahydrothiophene, Ozone–Pyrrolidine, and Ozone–Thiazolidine Complexes Studied Using Matrix-Isolation IR and Visible Absorption Spectroscopies. J. Phys. Chem. A 2021, 125, 2114–2120. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Olefin | Peak Position (cm−1) | Assignment | Calc. |
|---|---|---|---|
| C2H4 | 867 | Ethyleneoxide ν5 (ring deform) | |
| 1270 | Ethyleneoxide ν3 (ring stretch) | ||
| 1351 | unidentified | ||
| 1726 | Glyoxal ν10 (C=O stretch) | ||
| C4H6 | 822 | butadiene monoxide (ring deform) | |
| 923 | butadiene monoxide (ring deform) | ||
| 993 | butadiene monoxide (CH2=C bend) | ||
| 1250 | unidentified | ||
| 1311 | unidentified | ||
| 1385 | 2-oxo-3-butenal (CH2 bend) | 1408 | |
| 1652 | 2-oxo-3-butenal (C=C stretch) | 1648 | |
| 1713 | 2-oxo-3-butenal (C=O stretch) | 1725 | |
| 1749 | 2-oxo-3-butenal (C=O stretch) | 1773 | |
| C5H8 | 917 | 3,4-epoxy-2-methyl-1-butene (ring deform) | 907 |
| 1740 | 3-methyl-2-oxobut-3-enal (C = O stretch) | 1696 | |
| C6H10 | 864 | 2-methyl-2-(1-methylethenyl) oxirane (ring deform) | 864 |
| 1071 | 2-methyl-2-(1-methylethenyl)oxirane (CH2=C bend) | 917 | |
| 1176 | 2-methyl-2-(1-methylethenyl)oxirane (CH2=C bend) | 1180 | |
| 1649 | 2-methyl-2-(1-methylethenyl)oxirane (C=C stretch) | 1690 | |
| 1733 | 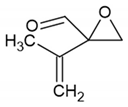 (C=O stretch) (C=O stretch)Or 2,3-dimethyl-3-butenal (C=O stretch) | 1782 | |
| 1759 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ito, F. Observation of Light-Induced Reactions of Olefin–Ozone Complexes in Cryogenic Matrices Using Fourier-Transform Infrared Spectroscopy. Photochem 2022, 2, 150-164. https://doi.org/10.3390/photochem2010012
Ito F. Observation of Light-Induced Reactions of Olefin–Ozone Complexes in Cryogenic Matrices Using Fourier-Transform Infrared Spectroscopy. Photochem. 2022; 2(1):150-164. https://doi.org/10.3390/photochem2010012
Chicago/Turabian StyleIto, Fumiyuki. 2022. "Observation of Light-Induced Reactions of Olefin–Ozone Complexes in Cryogenic Matrices Using Fourier-Transform Infrared Spectroscopy" Photochem 2, no. 1: 150-164. https://doi.org/10.3390/photochem2010012
APA StyleIto, F. (2022). Observation of Light-Induced Reactions of Olefin–Ozone Complexes in Cryogenic Matrices Using Fourier-Transform Infrared Spectroscopy. Photochem, 2(1), 150-164. https://doi.org/10.3390/photochem2010012