
Exploring Stressors: Impact on Cellular Organelles and Implications for Cellular Functions

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Cellular Stressors
2.1. Environmental Stressors
2.2. Chemical Stressors
2.3. Biological Stressors
2.4. Physical Stressors
3. Cell Membrane
3.1. Mechanisms of Cell Membrane in Stress
3.2. Effects of Various Stressors on Cell Membrane
3.2.1. Oxidative Stress
3.2.2. Chemical Stress
3.2.3. Thermal Stress
3.3. Consequences of Cell Membrane Stress
4. Mitochondria
4.1. Mechanisms of Mitochondria in Stress
4.2. Effects of Various Stressors on Mitochondria
4.2.1. Oxidative Stress
4.2.2. Nutrient Stress
4.2.3. Hypoxia
4.2.4. Environmental Stress
4.2.5. Chemical Stress
4.2.6. Physical Stress
4.3. Consequences of Mitochondrial Stress
5. Endoplasmic Reticulum
5.1. Mechanisms of ER in Stress
5.2. Effects of Various Stressors on ER
5.2.1. Oxidative Stress
5.2.2. Nutrient Stress
5.2.3. Viral Infections
5.2.4. Chemical Stress
5.2.5. Protein Overloading
5.3. Consequences of ER Stress
6. Golgi Apparatus
6.1. Mechanisms of Golgi Apparatus in Stress
6.2. Effects of Various Stressors on Golgi Apparatus
6.2.1. Oxidative Stress
6.2.2. Nutrient Stress
6.2.3. Hypoxia
6.2.4. Viral Infections
6.2.5. Chemical Stress
6.2.6. Physical Stress
6.3. Consequences of Golgi Apparatus Stress
7. Lysosomes
7.1. Mechanisms of Lysosomes in Stress
7.2. Effects of Various Stressors on Lysosomes
7.2.1. Oxidative Stress
7.2.2. Nutrient Stress
7.2.3. Viral Infections
7.2.4. Environmental and Chemical Stress
7.3. Consequences of Lysosomes Stress
8. Membrane-Less Organelles
8.1. Stress Granules
8.2. Processing Bodies
8.3. Cajal Bodies
8.4. Consequences of MLOs Stress
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, Regulation and Functions of the Unfolded Protein Response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J. Hormesis: Path and Progression to Significance. Int. J. Mol. Sci. 2018, 19, 2871. [Google Scholar] [CrossRef] [PubMed]
- Evans, T.G.; Kültz, D. The Cellular Stress Response in Fish Exposed to Salinity Fluctuations. J. Exp. Zool. A Ecol. Integr. Physiol. 2020, 333, 421–435. [Google Scholar] [PubMed]
- Simmons, S.O.; Fan, C.-Y.; Ramabhadran, R. Cellular Stress Response Pathway System as a Sentinel Ensemble in Toxicological Screening. Toxicol. Sci. 2009, 111, 202–225. [Google Scholar]
- McKechnie, A.E.; Wolf, B.O. The Physiology of Heat Tolerance in Small Endotherms. Physiology 2019, 34, 302–313. [Google Scholar]
- Rastogi, R.P.; Richa; Kumar, A.; Tyagi, M.B.; Sinha, R.P. Molecular Mechanisms of Ultraviolet Radiation-induced DNA Damage and Repair. J. Nucleic Acids 2010, 2010, 592980. [Google Scholar]
- Balali-Mood, M.; Naseri, K.; Tahergorabi, Z.; Khazdair, M.R.; Sadeghi, M. Toxic Mechanisms of Five Heavy Metals: Mercury, Lead, Chromium, Cadmium, and Arsenic. Front. Pharmacol. 2021, 12, 643972. [Google Scholar] [CrossRef]
- Slotkin, T.A. Guidelines for Developmental Neurotoxicity and Their Impact on Organophosphate Pesticides: A Personal View from an Academic Perspective. Neurotoxicology 2004, 25, 631–640. [Google Scholar]
- Wang, L.; He, X.; Bi, Y.; Ma, Q. Stem Cell and Benzene-Induced Malignancy and Hematotoxicity. Chem. Res. Toxicol. 2012, 25, 1303–1315. [Google Scholar] [CrossRef]
- Stanhope, K.L. Sugar Consumption, Metabolic Disease and Obesity: The State of the Controversy. Crit. Rev. Clin. Lab. Sci. 2016, 53, 52–67. [Google Scholar]
- Wieczfinska, J.; Kleniewska, P.; Pawliczak, R. Oxidative Stress-related Mechanisms in SARS-CoV-2 Infections. Oxid. Med. Cell Longev. 2022, 2022, 5589089. [Google Scholar] [CrossRef] [PubMed]
- Verbrugghe, E.; Boyen, F.; Gaastra, W.; Bekhuis, L.; Leyman, B.; Van Parys, A.; Haesebrouck, F.; Pasmans, F. The Complex Interplay between Stress and Bacterial Infections in Animals. Vet. Microbiol. 2012, 155, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-S.J.; Haga, J.H.; Chien, S. Molecular Basis of the Effects of Shear Stress on Vascular Endothelial Cells. J. Biomech. 2005, 38, 1949–1971. [Google Scholar] [CrossRef] [PubMed]
- Burg, M.B.; Ferraris, J.D.; Dmitrieva, N.I. Cellular Response to Hyperosmotic Stresses. Physiol. Rev. 2007, 87, 1441–1474. [Google Scholar] [CrossRef]
- Sasaki, K.; Yoshida, H. Organelle Autoregulation—Stress Responses in the ER, Golgi, Mitochondria and Lysosome. J. Biochem. 2015, 157, 185–195. [Google Scholar] [CrossRef]
- Lindström, M.S.; Latonen, L. The Nucleolus as a Stress Response Organelle. In Proteins of the Nucleolus: Regulation, Translocation, & Biomedical Functions; Springer: Dordrecht, The Netherlands, 2013; pp. 251–273. [Google Scholar]
- Mehta, D.; Malik, A.B. Signaling Mechanisms Regulating Endothelial Permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef]
- Schirrmacher, V. Mitochondria at Work: New Insights into Regulation and Dysregulation of Cellular Energy Supply and Metabolism. Biomedicines 2020, 8, 526. [Google Scholar] [CrossRef]
- Mierke, C.T.; Mierke, C.T. Mechanical View on the Endoplasmatic Reticulum and Golgi. In Cellular Mechanics and Biophysics: Structure and Function of Basic Cellular Components Regulating Cell Mechanics; Springer: Cham, Switzerland, 2020; pp. 191–262. [Google Scholar]
- Jaishy, B.; Abel, E.D. Lipids, Lysosomes, and Autophagy. J. Lipid Res. 2016, 57, 1619–1635. [Google Scholar] [CrossRef]
- Rabouille, C.; Alberti, S. Cell Adaptation upon Stress: The Emerging Role of Membrane-Less Compartments. Curr. Opin. Cell Biol. 2017, 47, 34–42. [Google Scholar] [CrossRef]
- Sharifi-Rad, M.; Anil Kumar, N.V.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Tsouh Fokou, P.V.; Azzini, E.; Peluso, I. Lifestyle, Oxidative Stress, and Antioxidants: Back and Forth in the Pathophysiology of Chronic Diseases. Front. Physiol. 2020, 11, 694. [Google Scholar] [CrossRef]
- Halliwell, B.; Adhikary, A.; Dingfelder, M.; Dizdaroglu, M. Hydroxyl Radical Is a Significant Player in Oxidative DNA Damage in Vivo. Chem. Soc. Rev. 2021, 50, 8355–8360. [Google Scholar] [CrossRef] [PubMed]
- van Leeuwen, W.; Rabouille, C. Cellular Stress Leads to the Formation of Membraneless Stress Assemblies in Eukaryotic Cells. Traffic 2019, 20, 623–638. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, R.L.; Falcinelli, M.; Flint, M.S. Stress and Drug Resistance in Cancer. Cancer Drug Resist. 2019, 2, 773. [Google Scholar] [CrossRef] [PubMed]
- Labrie, M.; Brugge, J.S.; Mills, G.B.; Zervantonakis, I.K. Therapy Resistance: Opportunities Created by Adaptive Responses to Targeted Therapies in Cancer. Nat. Rev. Cancer 2022, 22, 323–339. [Google Scholar] [CrossRef]
- Eckerling, A.; Ricon-Becker, I.; Sorski, L.; Sandbank, E.; Ben-Eliyahu, S. Stress and Cancer: Mechanisms, Significance and Future Directions. Nat. Rev. Cancer 2021, 21, 767–785. [Google Scholar] [CrossRef]
- Teixeira, A.P.; Dias, J.M.L.; Carinhas, N.; Sousa, M.; Clemente, J.J.; Cunha, A.E.; von Stosch, M.; Alves, P.M.; Carrondo, M.J.T.; Oliveira, R. Cell Functional Enviromics: Unravelling the Function of Environmental Factors. BMC Syst. Biol. 2011, 5, 92. [Google Scholar] [CrossRef]
- Farhad, H.A. An Overview of Stress in Cellular and Molecular Levels and the Importance of Studying Responses to Stresses in Biology. Res. J. Biotechnol. Vol. 2021, 16, 4. [Google Scholar]
- McConkey, D.J. The Integrated Stress Response and Proteotoxicity in Cancer Therapy. Biochem. Biophys. Res. Commun. 2017, 482, 450–453. [Google Scholar] [CrossRef]
- Yan, Z.; Xu, L.; Zhang, W.; Yang, G.; Zhao, Z.; Wang, Y.; Li, X. Comparative Toxic Effects of Microplastics and Nanoplastics on Chlamydomonas Reinhardtii: Growth Inhibition, Oxidative Stress, and Cell Morphology. J. Water Process Eng. 2021, 43, 102291. [Google Scholar] [CrossRef]
- Banerjee, A.; Shelver, W.L. Micro-and Nanoplastic Induced Cellular Toxicity in Mammals: A Review. Sci. Total Environ. 2021, 755, 142518. [Google Scholar] [CrossRef]
- Tavakolpournegari, A.; Annangi, B.; Villacorta, A.; Banaei, G.; Martin, J.; Pastor, S.; Marcos, R.; Hernández, A. Hazard Assessment of Different-Sized Polystyrene Nanoplastics in Hematopoietic Human Cell Lines. Chemosphere 2023, 325, 138360. [Google Scholar] [PubMed]
- Annangi, B.; Villacorta, A.; Vela, L.; Tavakolpournegari, A.; Marcos, R.; Hernández, A. Effects of True-to-Life PET Nanoplastics Using Primary Human Nasal Epithelial Cells. Environ. Toxicol. Pharmacol. 2023, 100, 104140. [Google Scholar] [PubMed]
- Banaei, G.; García-Rodríguez, A.; Tavakolpournegari, A.; Martín-Pérez, J.; Villacorta, A.; Marcos, R.; Hernández, A. The Release of Polylactic Acid Nanoplastics (PLA-NPLs) from Commercial Teabags. Obtention, Characterization, and Hazard Effects of True-to-Life PLA-NPLs. J. Hazard. Mater. 2023, 458, 131899. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar]
- Sulmon, C.; Van Baaren, J.; Cabello-Hurtado, F.; Gouesbet, G.; Hennion, F.; Mony, C.; Renault, D.; Bormans, M.; El Amrani, A.; Wiegand, C. Abiotic Stressors and Stress Responses: What Commonalities Appear between Species across Biological Organization Levels? Environ. Pollut. 2015, 202, 66–77. [Google Scholar]
- Rider, C.V.; Boekelheide, K.; Catlin, N.; Gordon, C.J.; Morata, T.; Selgrade, M.K.; Sexton, K.; Simmons, J.E. Cumulative Risk: Toxicity and Interactions of Physical and Chemical Stressors. Toxicol. Sci. 2014, 137, 3–11. [Google Scholar]
- Holmstrup, M.; Bindesbøl, A.-M.; Oostingh, G.J.; Duschl, A.; Scheil, V.; Köhler, H.-R.; Loureiro, S.; Soares, A.M.V.M.; Ferreira, A.L.G.; Kienle, C. Interactions between Effects of Environmental Chemicals and Natural Stressors: A Review. Sci. Total Environ. 2010, 408, 3746–3762. [Google Scholar]
- Kruk, J.; Aboul-Enein, H.Y.; Kładna, A.; Bowser, J.E. Oxidative Stress in Biological Systems and Its Relation with Pathophysiological Functions: The Effect of Physical Activity on Cellular Redox Homeostasis. Free Radic. Res. 2019, 53, 497–521. [Google Scholar]
- Münzel, T.; Daiber, A. Environmental Stressors and Their Impact on Health and Disease with Focus on Oxidative Stress. Antioxid. Redox Signal 2018, 28, 735–740. [Google Scholar]
- Poljšak, B.; Milisav, I. Clinical Implications of Cellular Stress Responses. Bosn. J. Basic. Med. Sci. 2012, 12, 122. [Google Scholar]
- Harrison, R. Biological Membranes: Their Structure and Function; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2013; ISBN 9401168571. [Google Scholar]
- Hughes, R.C. Membrane Glycoproteins: A Review of Structure and Function; Elsevier: Amsterdam, The Netherlands, 2014; ISBN 1483163962. [Google Scholar]
- Mohandas, N.; Gallagher, P.G. Red Cell Membrane: Past, Present, and Future. Blood J. Am. Soc. Hematol. 2008, 112, 3939–3948. [Google Scholar]
- Lee, M.J.; Yaffe, M.B. Protein Regulation in Signal Transduction. Cold Spring Harb. Perspect. Biol. 2016, 8, a005918. [Google Scholar]
- Sezgin, E.; Levental, I.; Mayor, S.; Eggeling, C. The Mystery of Membrane Organization: Composition, Regulation and Roles of Lipid Rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361–374. [Google Scholar] [PubMed]
- Ouweneel, A.B.; Thomas, M.J.; Sorci-Thomas, M.G. The Ins and Outs of Lipid Rafts: Functions in Intracellular Cholesterol Homeostasis, Microparticles, and Cell Membranes: Thematic Review Series: Biology of Lipid Rafts. J. Lipid Res. 2020, 61, 676–686. [Google Scholar]
- Mencarelli, C.; Martinez–Martinez, P. Ceramide Function in the Brain: When a Slight Tilt Is Enough. Cell. Mol. Life Sci. 2013, 70, 181–203. [Google Scholar]
- Hashiguchi, A.; Komatsu, S. Impact of Post-Translational Modifications of Crop Proteins under Abiotic Stress. Proteomes 2016, 4, 42. [Google Scholar] [CrossRef]
- Snead, W.T.; Gladfelter, A.S. The Control Centers of Biomolecular Phase Separation: How Membrane Surfaces, PTMs, and Active Processes Regulate Condensation. Mol. Cell 2019, 76, 295–305. [Google Scholar]
- Penke, B.; Bogár, F.; Crul, T.; Sántha, M.; Tóth, M.E.; Vígh, L. Heat Shock Proteins and Autophagy Pathways in Neuroprotection: From Molecular Bases to Pharmacological Interventions. Int. J. Mol. Sci. 2018, 19, 325. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Bergink, S. Heat Shock Proteins as Potential Targets for Protective Strategies in Neurodegeneration. Lancet Neurol. 2016, 15, 748–759. [Google Scholar] [CrossRef]
- Ernst, R.; Ejsing, C.S.; Antonny, B. Homeoviscous Adaptation and the Regulation of Membrane Lipids. J. Mol. Biol. 2016, 428, 4776–4791. [Google Scholar] [CrossRef]
- Murata, N.; Los, D.A. Membrane Fluidity and Temperature Perception. Plant Physiol. 1997, 115, 875. [Google Scholar] [CrossRef] [PubMed]
- McNeil, P.L.; Steinhardt, R.A. Plasma Membrane Disruption: Repair, Prevention, Adaptation. Annu. Rev. Cell Dev. Biol. 2003, 19, 697–731. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan, S.; Mandrekar, P. Cellular Stress Response and Innate Immune Signaling: Integrating Pathways in Host Defense and Inflammation. J. Leukoc. Biol. 2013, 94, 1167–1184. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Catalá, A.; Díaz, M. Impact of Lipid Peroxidation on the Physiology and Pathophysiology of Cell Membranes. Front. Physiol. 2016, 7, 423. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, J.; Yu, Y.; Liu, X.; Shang, X.; Du, Z.; Xu, M.L.; Zhang, T. Recent Advances in the Inhibition of Membrane Lipid Peroxidation by Food-Borne Plant Polyphenols via the Nrf2/GPx4 Pathway. J. Agric. Food Chem. 2024, 72, 12340–12355. [Google Scholar] [CrossRef]
- Sadiq, I.Z. Free Radicals and Oxidative Stress: Signaling Mechanisms, Redox Basis for Human Diseases, and Cell Cycle Regulation. Curr. Mol. Med. 2023, 23, 13–35. [Google Scholar] [CrossRef]
- Song, K.; Li, Y.; Zhang, H.; An, N.; Wei, Y.; Wang, L.; Tian, C.; Yuan, M.; Sun, Y.; Xing, Y. Oxidative Stress-Mediated Blood-Brain Barrier (BBB) Disruption in Neurological Diseases. Oxid. Med. Cell Longev. 2020, 2020, 4356386. [Google Scholar] [CrossRef]
- Lemma, T.; Ruiz, G.C.M.; Oliveira, O.N., Jr.; Constantino, C.J.L. Disruption of Giant Unilamellar Vesicles Mimicking Cell Membranes Induced by the Pesticides Glyphosate and Picloram. Biophys. Chem. 2019, 250, 106176. [Google Scholar] [CrossRef]
- Renu, K.; Chakraborty, R.; Myakala, H.; Koti, R.; Famurewa, A.C.; Madhyastha, H.; Vellingiri, B.; George, A.; Gopalakrishnan, A.V. Molecular Mechanism of Heavy Metals (Lead, Chromium, Arsenic, Mercury, Nickel and Cadmium)-Induced Hepatotoxicity–A Review. Chemosphere 2021, 271, 129735. [Google Scholar] [PubMed]
- Russo, G.; Capuozzo, A.; Barbato, F.; Irace, C.; Santamaria, R.; Grumetto, L. Cytotoxicity of Seven Bisphenol Analogues Compared to Bisphenol A and Relationships with Membrane Affinity Data. Chemosphere 2018, 201, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Gnauck, A.; Lentle, R.G.; Kruger, M.C. The Characteristics and Function of Bacterial Lipopolysaccharides and Their Endotoxic Potential in Humans. Int. Rev. Immunol. 2016, 35, 189–218. [Google Scholar] [CrossRef] [PubMed]
- Joyce, H.; McCann, A.; Clynes, M.; Larkin, A. Influence of Multidrug Resistance and Drug Transport Proteins on Chemotherapy Drug Metabolism. Expert. Opin. Drug Metab. Toxicol. 2015, 11, 795–809. [Google Scholar]
- Liu, X.; Yang, H.; Liu, Z. Signaling Pathways Involved in Paraquat-Induced Pulmonary Toxicity: Molecular Mechanisms and Potential Therapeutic Drugs. Int. Immunopharmacol. 2022, 113, 109301. [Google Scholar]
- Wong, F.; Stokes, J.M.; Cervantes, B.; Penkov, S.; Friedrichs, J.; Renner, L.D.; Collins, J.J. Cytoplasmic Condensation Induced by Membrane Damage Is Associated with Antibiotic Lethality. Nat. Commun. 2021, 12, 2321. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, X.; Su, Y.; Xu, W.; Liu, H.; Liu, Z.; Chen, W.; Wang, J. Dimethyl Phthalate Damaged the Cell Membrane of Escherichia Coli K12. Ecotoxicol. Environ. Saf. 2019, 180, 208–214. [Google Scholar]
- Delpire, E.; Gagnon, K.B. Water Homeostasis and Cell Volume Maintenance and Regulation. Curr. Top. Membr. 2018, 81, 3–52. [Google Scholar]
- Török, Z.; Crul, T.; Maresca, B.; Schütz, G.J.; Viana, F.; Dindia, L.; Piotto, S.; Brameshuber, M.; Balogh, G.; Péter, M. Plasma Membranes as Heat Stress Sensors: From Lipid-Controlled Molecular Switches to Therapeutic Applications. Biochim. et Biophys. Acta (BBA)-Biomembr. 2014, 1838, 1594–1618. [Google Scholar] [CrossRef]
- Padmini, E. Physiological Adaptations of Stressed Fish to Polluted Environments: Role of Heat Shock Proteins. In Reviews of Environmental Contamination and Toxicology; Springer: New York, NY, USA, 2010; Volume 206, pp. 1–27. [Google Scholar]
- Kalmar, B.; Greensmith, L. Cellular Chaperones as Therapeutic Targets in ALS to Restore Protein Homeostasis and Improve Cellular Function. Front. Mol. Neurosci. 2017, 10, 251. [Google Scholar]
- Pockley, A.G.; Henderson, B. Extracellular Cell Stress (Heat Shock) Proteins—Immune Responses and Disease: An Overview. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20160522. [Google Scholar]
- Kelty, J.D.; Noseworthy, P.A.; Feder, M.E.; Robertson, R.M.; Ramirez, J.-M. Thermal Preconditioning and Heat-Shock Protein 72 Preserve Synaptic Transmission during Thermal Stress. J. Neurosci. 2002, 22, RC193. [Google Scholar] [PubMed]
- Penke, B.; Paragi, G.; Gera, J.; Berkecz, R.; Kovács, Z.; Crul, T.; Vígh, L. The Role of Lipids and Membranes in the Pathogenesis of Alzheimer’s Disease: A Comprehensive View. Curr. Alzheimer Res. 2018, 15, 1191–1212. [Google Scholar] [PubMed]
- Calì, T.; Ottolini, D.; Brini, M. Calcium Signaling in Parkinson’s Disease. Cell Tissue Res. 2014, 357, 439–454. [Google Scholar]
- Poznyak, A.V.; Grechko, A.V.; Orekhova, V.A.; Chegodaev, Y.S.; Wu, W.-K.; Orekhov, A.N. Oxidative Stress and Antioxidants in Atherosclerosis Development and Treatment. Biology 2020, 9, 60. [Google Scholar] [CrossRef]
- Soares, R.O.S.; Losada, D.M.; Jordani, M.C.; Évora, P.; Castro-e-Silva, O. Ischemia/Reperfusion Injury Revisited: An Overview of the Latest Pharmacological Strategies. Int. J. Mol. Sci. 2019, 20, 5034. [Google Scholar] [CrossRef]
- Römer, A.; Linn, T.; Petry, S.F. Lipotoxic Impairment of Mitochondrial Function in β-Cells: A Review. Antioxidants 2021, 10, 293. [Google Scholar] [CrossRef]
- Ray, S.; Das, S.; Suar, M. Molecular Mechanism of Drug Resistance. In Drug Resistance in Bacteria, Fungi, Malaria, and Cancer; Springer: Berlin/Heidelberg, Germany, 2017; pp. 47–110. [Google Scholar]
- Clemente-Suárez, V.J.; Redondo-Flórez, L.; Beltrán-Velasco, A.I.; Ramos-Campo, D.J.; Belinchón-deMiguel, P.; Martinez-Guardado, I.; Dalamitros, A.A.; Yáñez-Sepúlveda, R.; Martín-Rodríguez, A.; Tornero-Aguilera, J.F. Mitochondria and Brain Disease: A Comprehensive Review of Pathological Mechanisms and Therapeutic Opportunities. Biomedicines 2023, 11, 2488. [Google Scholar] [CrossRef]
- Dai, D.-F.; Chiao, Y.A.; Marcinek, D.J.; Szeto, H.H.; Rabinovitch, P.S. Mitochondrial Oxidative Stress in Aging and Healthspan. Longev. Heal. 2014, 3, 6. [Google Scholar]
- Fernandez-Vizarra, E.; Zeviani, M. Mitochondrial Disorders of the OXPHOS System. FEBS Lett. 2021, 595, 1062–1106. [Google Scholar]
- Ackerman, S.H.; Tzagoloff, A. Function, Structure, and Biogenesis of Mitochondrial ATP Synthase. Prog. Nucleic Acid. Res. Mol. Biol. 2005, 80, 95–133. [Google Scholar] [PubMed]
- Martín-Pérez, J.; Villacorta, A.; Banaei, G.; Morataya-Reyes, M.; Tavakolpournegari, A.; Marcos, R.; Hernández, A.; García-Rodriguez, A. Hazard Assessment of Nanoplastics Is Driven by Their Surface-Functionalization. Effects in Human-Derived Primary Endothelial Cells. Sci. Total Environ. 2024, 934, 173236. [Google Scholar] [CrossRef] [PubMed]
- Pacheu-Grau, D.; Rucktäschel, R.; Deckers, M. Mitochondrial Dysfunction and Its Role in Tissue-Specific Cellular Stress. Cell Stress 2018, 2, 184. [Google Scholar] [CrossRef] [PubMed]
- Bidooki, S.H.; Quero, J.; Sánchez-Marco, J.; Herrero-Continente, T.; Marmol, I.; Lasheras, R.; Sebastian, V.; Arruebo, M.; Osada, J.; Rodriguez-Yoldi, M.J. Squalene in Nanoparticles Improves Antiproliferative Effect on Human Colon Carcinoma Cells Through Apoptosis by Disturbances in Redox Balance. Int. J. Mol. Sci. 2024, 25, 13048. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The Multifaceted Contributions of Mitochondria to Cellular Metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS Signaling in Organismal Homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef]
- Ristow, M.; Schmeisser, K. Mitohormesis: Promoting Health and Lifespan by Increased Levels of Reactive Oxygen Species (ROS). Dose-Response 2014, 12, 288–341. [Google Scholar] [CrossRef]
- Yun, J.; Finkel, T. Mitohormesis. Cell Metab. 2014, 19, 757–766. [Google Scholar] [CrossRef]
- Hood, D.A.; Memme, J.M.; Oliveira, A.N.; Triolo, M. Maintenance of Skeletal Muscle Mitochondria in Health, Exercise, and Aging. Annu. Rev. Physiol. 2019, 81, 19–41. [Google Scholar] [CrossRef]
- Radak, Z.; Zhao, Z.; Koltai, E.; Ohno, H.; Atalay, M. Oxygen Consumption and Usage during Physical Exercise: The Balance between Oxidative Stress and ROS-Dependent Adaptive Signaling. Antioxid. Redox Signal 2013, 18, 1208–1246. [Google Scholar] [CrossRef] [PubMed]
- Palabiyik, A.A.; Palabiyik, E. Pharmacological Approaches to Enhance Mitochondrial Biogenesis: Focus on PGC-1A, AMPK, and SIRT1 in Cellular Health. Mol. Biol. Rep. 2025, 52, 270. [Google Scholar] [CrossRef] [PubMed]
- Jamwal, S.; Blackburn, J.K.; Elsworth, J.D. PPARγ/PGC1α Signaling as a Potential Therapeutic Target for Mitochondrial Biogenesis in Neurodegenerative Disorders. Pharmacol. Ther. 2021, 219, 107705. [Google Scholar] [CrossRef]
- Assaf, L.; Eid, A.A.; Nassif, J. Role of AMPK/MTOR, Mitochondria, and ROS in the Pathogenesis of Endometriosis. Life Sci. 2022, 306, 120805. [Google Scholar] [CrossRef]
- Xia, M.; Zhang, Y.; Jin, K.; Lu, Z.; Zeng, Z.; Xiong, W. Communication between Mitochondria and Other Organelles: A Brand-New Perspective on Mitochondria in Cancer. Cell Biosci. 2019, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Velmurugan, G.V.; Vekaria, H.J.; Hartz, A.M.S.; Bauer, B.; Hubbard, W.B. Oxidative Stress Alters Mitochondrial Homeostasis in Isolated Brain Capillaries. Fluids Barriers CNS 2024, 21, 81. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Poole, A.C.; Thomas, R.E.; Andrews, L.A.; McBride, H.M.; Whitworth, A.J.; Pallanck, L.J. The PINK1/Parkin Pathway Regulates Mitochondrial Morphology. Proc. Natl. Acad. Sci. USA 2008, 105, 1638–1643. [Google Scholar] [CrossRef]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial Metabolism of Reactive Oxygen Species. Biochemistry 2005, 70, 200–214. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, Oxidants, and Aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef]
- Tavakolpournegari, A.; Villacorta, A.; Morataya-Reyes, M.; Arranz, J.A.; Banaei, G.; Pastor, S.; Velázquez, A.; Marcos, R.; Hernández, A.; Annangi, B. Harmful Effects of True-to-Life Nanoplastics Derived from PET Water Bottles in Human Alveolar Macrophages. Environ. Pollut. 2024, 348, 123823. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Melov, S. SOD2 in Mitochondrial Dysfunction and Neurodegeneration. Free Radic. Biol. Med. 2013, 62, 4–12. [Google Scholar] [PubMed]
- Zhao, R.; Jiang, S.; Zhang, L.; Yu, Z. Mitochondrial Electron Transport Chain, ROS Generation and Uncoupling. Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [PubMed]
- Rodrigues, T.; Ferraz, L.S. Therapeutic Potential of Targeting Mitochondrial Dynamics in Cancer. Biochem. Pharmacol. 2020, 182, 114282. [Google Scholar]
- Kowalczyk, P.; Sulejczak, D.; Kleczkowska, P.; Bukowska-Ośko, I.; Kucia, M.; Popiel, M.; Wietrak, E.; Kramkowski, K.; Wrzosek, K.; Kaczyńska, K. Mitochondrial Oxidative Stress—A Causative Factor and Therapeutic Target in Many Diseases. Int. J. Mol. Sci. 2021, 22, 13384. [Google Scholar] [CrossRef]
- Hu, Q.; Ren, J.; Li, G.; Wu, J.; Wu, X.; Wang, G.; Gu, G.; Ren, H.; Hong, Z.; Li, J. The Mitochondrially Targeted Antioxidant MitoQ Protects the Intestinal Barrier by Ameliorating Mitochondrial DNA Damage via the Nrf2/ARE Signaling Pathway. Cell Death Dis. 2018, 9, 403. [Google Scholar] [CrossRef]
- Vargas-Mendoza, N.; Angeles-Valencia, M.; Morales-González, Á.; Madrigal-Santillán, E.O.; Morales-Martínez, M.; Madrigal-Bujaidar, E.; Álvarez-González, I.; Gutiérrez-Salinas, J.; Esquivel-Chirino, C.; Chamorro-Cevallos, G. Oxidative Stress, Mitochondrial Function and Adaptation to Exercise: New Perspectives in Nutrition. Life 2021, 11, 1269. [Google Scholar] [CrossRef]
- Kuppuswami, J.; Senthilkumar, G.P. Nutri-Stress, Mitochondrial Dysfunction, and Insulin Resistance—Role of Heat Shock Proteins. Cell Stress Chaperones 2023, 28, 35–48. [Google Scholar] [CrossRef]
- Valdecantos, M.P.; Pérez-Matute, P.; González-Muniesa, P.; Prieto-Hontoria, P.L.; Moreno-Aliaga, M.J.; Martínez, J.A. Lipoic Acid Improves Mitochondrial Function in Nonalcoholic Steatosis through the Stimulation of Sirtuin 1 and Sirtuin 3. Obesity 2012, 20, 1974–1983. [Google Scholar]
- Halling, J.F.; Pilegaard, H. PGC-1α-Mediated Regulation of Mitochondrial Function and Physiological Implications. Appl. Physiol. Nutr. Metab. 2020, 45, 927–936. [Google Scholar]
- Miotto, P.M.; LeBlanc, P.J.; Holloway, G.P. High-Fat Diet Causes Mitochondrial Dysfunction as a Result of Impaired ADP Sensitivity. Diabetes 2018, 67, 2199–2205. [Google Scholar]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of Metabolism and Mitochondrial Homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [PubMed]
- Balsa, E.; Soustek, M.S.; Thomas, A.; Cogliati, S.; García-Poyatos, C.; Martín-García, E.; Jedrychowski, M.; Gygi, S.P.; Enriquez, J.A.; Puigserver, P. ER and Nutrient Stress Promote Assembly of Respiratory Chain Supercomplexes through the PERK-EIF2α Axis. Mol. Cell 2019, 74, 877–890. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhu, M.; Bao, H.; Li, B.; Dong, Y.; Xiao, C.; Zhang, G.Y.; Henter, I.; Rudorfer, M.; Vitiello, B. The Role of Nutrients in Protecting Mitochondrial Function and Neurotransmitter Signaling: Implications for the Treatment of Depression, PTSD, and Suicidal Behaviors. Crit. Rev. Food Sci. Nutr. 2016, 56, 2560–2578. [Google Scholar] [PubMed]
- Kierans, S.J.; Taylor, C.T. Regulation of Glycolysis by the Hypoxia-inducible Factor (HIF): Implications for Cellular Physiology. J. Physiol. 2021, 599, 23–37. [Google Scholar] [CrossRef]
- Emelyanova, L.; Ashary, Z.; Cosic, M.; Negmadjanov, U.; Ross, G.; Rizvi, F.; Olet, S.; Kress, D.; Sra, J.; Tajik, A.J. Selective Downregulation of Mitochondrial Electron Transport Chain Activity and Increased Oxidative Stress in Human Atrial Fibrillation. Am. J. Physiol.-Heart Circ. Physiol. 2016, 311, H54–H63. [Google Scholar]
- Deng, X.; Wang, Q.; Cheng, M.; Chen, Y.; Yan, X.; Guo, R.; Sun, L.; Li, Y.; Liu, Y. Pyruvate Dehydrogenase Kinase 1 Interferes with Glucose Metabolism Reprogramming and Mitochondrial Quality Control to Aggravate Stress Damage in Cancer. J. Cancer 2020, 11, 962. [Google Scholar]
- Orang, A.V.; Petersen, J.; McKinnon, R.A.; Michael, M.Z. Micromanaging Aerobic Respiration and Glycolysis in Cancer Cells. Mol. Metab. 2019, 23, 98–126. [Google Scholar]
- Zhou, B.; Tian, R. Mitochondrial Dysfunction in Pathophysiology of Heart Failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar]
- Jimi, S.; Uchiyama, M.; Takaki, A.Y.A.; Suzumiya, J.; Hara, S. Mechanisms of Cell Death Induced by Cadmium and Arsenic. In Mitochondrial Pathogenesis: From Genes and Apoptosis to Aging and Disease; Springer: Berlin/Heidelberg, Germany, 2004; pp. 325–331. [Google Scholar]
- Lee, H.K.; Pak, Y.K. Persistent Organic Pollutants, Mitochondrial Dysfunction, and Metabolic Syndrome. In Mitochondrial Dysfunction Caused by Drugs and Environmental Toxicants; Wiley Online Library: Hoboken, NJ, USA, 2018; pp. 691–707. [Google Scholar]
- Meyer, J.N.; Leung, M.C.K.; Rooney, J.P.; Sendoel, A.; Hengartner, M.O.; Kisby, G.E.; Bess, A.S. Mitochondria as a Target of Environmental Toxicants. Toxicol. Sci. 2013, 134, 1–17. [Google Scholar]
- Martinez, T.N.; Greenamyre, J.T. Toxin Models of Mitochondrial Dysfunction in Parkinson’s Disease. Antioxid. Redox Signal 2012, 16, 920–934. [Google Scholar] [PubMed]
- Inigo, J.R.; Chandra, D. The Mitochondrial Unfolded Protein Response (UPRmt): Shielding against Toxicity to Mitochondria in Cancer. J. Hematol. Oncol. 2022, 15, 98. [Google Scholar] [PubMed]
- Adeva-Andany, M.M.; Carneiro-Freire, N.; Seco-Filgueira, M.; Fernández-Fernández, C.; Mouriño-Bayolo, D. Mitochondrial β-Oxidation of Saturated Fatty Acids in Humans. Mitochondrion 2019, 46, 73–90. [Google Scholar] [PubMed]
- Jang, Y.J.; Won, J.H.; Back, M.J.; Fu, Z.; Jang, J.M.; Ha, H.C.; Hong, S.; Chang, M.; Kim, D.K. Paraquat Induces Apoptosis through a Mitochondria-Dependent Pathway in RAW264. 7 Cells. Biomol. Ther. 2015, 23, 407. [Google Scholar]
- Montanarí, C.; Franco-Campos, F.; Taroncher, M.; Rodríguez-Carrasco, Y.; Zingales, V.; Ruiz, M.J. Chlorpyrifos Induces Cytotoxicity via Oxidative Stress and Mitochondrial Dysfunction in HepG2 Cells. Food Chem. Toxicol. 2024, 192, 114933. [Google Scholar]
- Nair, S.; Sobotka, K.S.; Joshi, P.; Gressens, P.; Fleiss, B.; Thornton, C.; Mallard, C.; Hagberg, H. Lipopolysaccharide-induced Alteration of Mitochondrial Morphology Induces a Metabolic Shift in Microglia Modulating the Inflammatory Response in Vitro and in Vivo. Glia 2019, 67, 1047–1061. [Google Scholar]
- Lomeli, N.; Di, K.; Czerniawski, J.; Guzowski, J.F.; Bota, D.A. Cisplatin-Induced Mitochondrial Dysfunction Is Associated with Impaired Cognitive Function in Rats. Free Radic. Biol. Med. 2017, 102, 274–286. [Google Scholar]
- Zuhra, K.; Szabo, C. The Two Faces of Cyanide: An Environmental Toxin and a Potential Novel Mammalian Gasotransmitter. FEBS J. 2022, 289, 2481–2515. [Google Scholar]
- Moloi, T.P.; Ziqubu, K.; Mazibuko-Mbeje, S.E.; Mabaso, N.H.; Ndlovu, Z. Aflatoxin B1-Induced Hepatotoxicity through Mitochondrial Dysfunction, Oxidative Stress, and Inflammation as Central Pathological Mechanisms: A Review of Experimental Evidence. Toxicology 2024, 509, 153983. [Google Scholar]
- Songbo, M.; Lang, H.; Xinyong, C.; Bin, X.; Ping, Z.; Liang, S. Oxidative Stress Injury in Doxorubicin-Induced Cardiotoxicity. Toxicol. Lett. 2019, 307, 41–48. [Google Scholar]
- Marchi, S.; Guilbaud, E.; Tait, S.W.G.; Yamazaki, T.; Galluzzi, L. Mitochondrial Control of Inflammation. Nat. Rev. Immunol. 2023, 23, 159–173. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in Innate Immune Responses and Inflammatory Pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R.F.; Jonker, M.R.; Brandenburg, S.M.; De Bruin, H.G.; Ten Hacken, N.H.T.; Van Oosterhout, A.J.M.; Heijink, I.H. Mitochondrial Dysfunction Increases Pro-Inflammatory Cytokine Production and Impairs Repair and Corticosteroid Responsiveness in Lung Epithelium. Sci. Rep. 2019, 9, 15047. [Google Scholar] [CrossRef] [PubMed]
- Ziolkowska, S.; Binienda, A.; Jabłkowski, M.; Szemraj, J.; Czarny, P. The Interplay between Insulin Resistance, Inflammation, Oxidative Stress, Base Excision Repair and Metabolic Syndrome in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2021, 22, 11128. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The Role of Mitochondria in NLRP3 Inflammasome Activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef]
- Neufer, P.D.; Bamman, M.M.; Muoio, D.M.; Bouchard, C.; Cooper, D.M.; Goodpaster, B.H.; Booth, F.W.; Kohrt, W.M.; Gerszten, R.E.; Mattson, M.P. Understanding the Cellular and Molecular Mechanisms of Physical Activity-Induced Health Benefits. Cell Metab. 2015, 22, 4–11. [Google Scholar] [CrossRef]
- Simpson, R.J.; Campbell, J.P.; Gleeson, M.; Krüger, K.; Nieman, D.C.; Pyne, D.B.; Turner, J.E.; Walsh, N.P. Can Exercise Affect Immune Function to Increase Susceptibility to Infection? Exerc. Immunol. Rev. 2020, 26, 8–22. [Google Scholar]
- Chastin, S.F.M.; Abaraogu, U.; Bourgois, J.G.; Dall, P.M.; Darnborough, J.; Duncan, E.; Dumortier, J.; Pavón, D.J.; McParland, J.; Roberts, N.J. Effects of Regular Physical Activity on the Immune System, Vaccination and Risk of Community-Acquired Infectious Disease in the General Population: Systematic Review and Meta-Analysis. Sports Med. 2021, 51, 1673–1686. [Google Scholar] [CrossRef]
- Abu Shelbayeh, O.; Arroum, T.; Morris, S.; Busch, K.B. PGC-1α Is a Master Regulator of Mitochondrial Lifecycle and ROS Stress Response. Antioxidants 2023, 12, 1075. [Google Scholar] [CrossRef]
- Wu, J.; Li, J.; Feng, B.; Bi, Z.; Zhu, G.; Zhang, Y.; Li, X. Activation of AMPK-PGC-1α Pathway Ameliorates Peritoneal Dialysis Related Peritoneal Fibrosis in Mice by Enhancing Mitochondrial Biogenesis. Ren. Fail. 2022, 44, 1546–1558. [Google Scholar] [CrossRef]
- Dawson, T.M.; Dawson, V.L. Mitochondrial Mechanisms of Neuronal Cell Death: Potential Therapeutics. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 437–454. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Kosiol, M.; Mayr, M.; Schulz, R.; Rohrbach, S. Mitochondria and Ageing: Role in Heart, Skeletal Muscle and Adipose Tissue. J. Cachexia Sarcopenia Muscle 2017, 8, 349–369. [Google Scholar] [CrossRef] [PubMed]
- Panov, A.; Mayorov, V.I.; Dikalov, S. Metabolic Syndrome and β-Oxidation of Long-Chain Fatty Acids in the Brain, Heart, and Kidney Mitochondria. Int. J. Mol. Sci. 2022, 23, 4047. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; McEwen, B.S.; Epel, E.S.; Sandi, C. An Energetic View of Stress: Focus on Mitochondria. Front. Neuroendocr. 2018, 49, 72–85. [Google Scholar]
- Memme, J.M.; Erlich, A.T.; Phukan, G.; Hood, D.A. Exercise and Mitochondrial Health. J. Physiol. 2021, 599, 803–817. [Google Scholar] [CrossRef]
- Monzio Compagnoni, G.; Di Fonzo, A.; Corti, S.; Comi, G.P.; Bresolin, N.; Masliah, E. The Role of Mitochondria in Neurodegenerative Diseases: The Lesson from Alzheimer’s Disease and Parkinson’s Disease. Mol. Neurobiol. 2020, 57, 2959–2980. [Google Scholar]
- Carmo, C.; Naia, L.; Lopes, C.; Rego, A.C. Mitochondrial Dysfunction in Huntington’s Disease. In Polyglutamine Disorders; Springer: Berlin/Heidelberg, Germany, 2018; pp. 59–83. [Google Scholar]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The Role of Mitochondria in Amyotrophic Lateral Sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar]
- Hu, C.; Huang, Y.; Li, L. Drp1-Dependent Mitochondrial Fission Plays Critical Roles in Physiological and Pathological Progresses in Mammals. Int. J. Mol. Sci. 2017, 18, 144. [Google Scholar] [CrossRef]
- Kim, Y.; Vadodaria, K.C.; Lenkei, Z.; Kato, T.; Gage, F.H.; Marchetto, M.C.; Santos, R. Mitochondria, Metabolism, and Redox Mechanisms in Psychiatric Disorders. Antioxid. Redox Signal 2019, 31, 275–317. [Google Scholar] [CrossRef]
- Rovira-Llopis, S.; Bañuls, C.; Diaz-Morales, N.; Hernandez-Mijares, A.; Rocha, M.; Victor, V.M. Mitochondrial Dynamics in Type 2 Diabetes: Pathophysiological Implications. Redox Biol. 2017, 11, 637–645. [Google Scholar] [CrossRef]
- Burgos-Morón, E.; Abad-Jiménez, Z.; Martinez de Maranon, A.; Iannantuoni, F.; Escribano-López, I.; López-Domènech, S.; Salom, C.; Jover, A.; Mora, V.; Roldan, I. Relationship between Oxidative Stress, ER Stress, and Inflammation in Type 2 Diabetes: The Battle Continues. J. Clin. Med. 2019, 8, 1385. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zou, M.-H. AMPK, Mitochondrial Function, and Cardiovascular Disease. Int. J. Mol. Sci. 2020, 21, 4987. [Google Scholar] [CrossRef] [PubMed]
- Forte, M.; Schirone, L.; Ameri, P.; Basso, C.; Catalucci, D.; Modica, J.; Chimenti, C.; Crotti, L.; Frati, G.; Rubattu, S. The Role of Mitochondrial Dynamics in Cardiovascular Diseases. Br. J. Pharmacol. 2021, 178, 2060–2076. [Google Scholar] [CrossRef] [PubMed]
- Bertero, E.; Maack, C. Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circ. Res. 2018, 122, 1460–1478. [Google Scholar] [CrossRef]
- Kopinski, P.K.; Singh, L.N.; Zhang, S.; Lott, M.T.; Wallace, D.C. Mitochondrial DNA Variation and Cancer. Nat. Rev. Cancer 2021, 21, 431–445. [Google Scholar] [CrossRef]
- Minarrieta, L.; Annis, M.G.; Audet-Delage, Y.; Kuasne, H.; Pacis, A.; St-Louis, C.; Nowakowski, A.; Biondini, M.; Khacho, M.; Park, M. Mitochondrial Elongation Impairs Breast Cancer Metastasis. Sci. Adv. 2024, 10, eadm8212. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, W.; Li, Z.; Lin, S.; Zheng, T.; Hao, B.; Hou, Y.; Zhang, Y.; Wang, K.; Qin, C. Mitochondria Dysfunction in CD8+ T Cells as an Important Contributing Factor for Cancer Development and a Potential Target for Cancer Treatment: A Review. J. Exp. Clin. Cancer Res. 2022, 41, 227. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Kaplowitz, N.; Lebeaupin, C.; Kroemer, G.; Kaufman, R.J.; Malhi, H.; Ren, J. Endoplasmic Reticulum Stress in Liver Diseases. Hepatology 2023, 77, 619–639. [Google Scholar] [CrossRef]
- Krshnan, L.; van de Weijer, M.L.; Carvalho, P. Endoplasmic Reticulum–Associated Protein Degradation. Cold Spring Harb. Perspect. Biol. 2022, 14, a041247. [Google Scholar] [CrossRef]
- Bidooki, S.H.; Barranquero, C.; Sánchez-Marco, J.; Martínez-Beamonte, R.; Rodríguez-Yoldi, M.J.; Navarro, M.A.; Fernandes, S.C.M.; Osada, J. TXNDC5 Plays a Crucial Role in Regulating Endoplasmic Reticulum Activity through Different ER Stress Signaling Pathways in Hepatic Cells. Int. J. Mol. Sci. 2024, 25, 7128. [Google Scholar] [CrossRef]
- Bidooki, S.H.; Sánchez-Marco, J.; Martínez-Beamonte, R.; Herrero-Continente, T.; Navarro, M.A.; Rodríguez-Yoldi, M.J.; Osada, J. Endoplasmic Reticulum Protein TXNDC5 Interacts with PRDX6 and HSPA9 to Regulate Glutathione Metabolism and Lipid Peroxidation in the Hepatic AML12 Cell Line. Int. J. Mol. Sci. 2023, 24, 17131. [Google Scholar] [CrossRef] [PubMed]
- Ekundayo, B.E.; Obafemi, T.O.; Adewale, O.B.; Obafemi, B.A.; Oyinloye, B.E.; Ekundayo, S.K. Oxidative Stress, Endoplasmic Reticulum Stress and Apoptosis in the Pathology of Alzheimer’s Disease. Cell Biochem. Biophys. 2024, 82, 457–477. [Google Scholar] [PubMed]
- Chen, X.; Shi, C.; He, M.; Xiong, S.; Xia, X. Endoplasmic Reticulum Stress: Molecular Mechanism and Therapeutic Targets. Signal Transduct. Target. Ther. 2023, 8, 352. [Google Scholar] [CrossRef] [PubMed]
- Gariballa, N.; Mohamed, F.; Badawi, S.; Ali, B.R. The Double Whammy of ER-Retention and Dominant-Negative Effects in Numerous Autosomal Dominant Diseases: Significance in Disease Mechanisms and Therapy. J. Biomed. Sci. 2024, 31, 64. [Google Scholar] [CrossRef]
- Karna, K.K.; Choi, N.Y.; Kim, C.Y.; Kim, H.K.; Shin, Y.S.; Park, J.K. Gui-a-Gra Attenuates Testicular Dysfunction in Varicocele-Induced Rats via Oxidative Stress, Er Stress and Mitochondrial Apoptosis Pathway. Int. J. Mol. Sci. 2020, 21, 9231. [Google Scholar] [CrossRef]
- Awasthi, D.; Chopra, S.; Cho, B.A.; Emmanuelli, A.; Sandoval, T.A.; Hwang, S.-M.; Chae, C.-S.; Salvagno, C.; Tan, C.; Vasquez-Urbina, L. Inflammatory ER Stress Responses Dictate the Immunopathogenic Progression of Systemic Candidiasis. J. Clin. Investig. 2023, 133, e167359. [Google Scholar]
- Bidooki, S.H.; Navarro, M.A.; Fernandes, S.C.M.; Osada, J. Thioredoxin Domain Containing 5 (TXNDC5): Friend or Foe? Curr. Issues Mol. Biol. 2024, 46, 3134–3163. [Google Scholar] [CrossRef]
- Jin, J.-K.; Blackwood, E.A.; Azizi, K.; Thuerauf, D.J.; Fahem, A.G.; Hofmann, C.; Kaufman, R.J.; Doroudgar, S.; Glembotski, C.C. ATF6 Decreases Myocardial Ischemia/Reperfusion Damage and Links ER Stress and Oxidative Stress Signaling Pathways in the Heart. Circ. Res. 2017, 120, 862–875. [Google Scholar]
- Estébanez, B.; De Paz, J.A.; Cuevas, M.J.; González-Gallego, J. Endoplasmic Reticulum Unfolded Protein Response, Aging and Exercise: An Update. Front. Physiol. 2018, 9, 1744. [Google Scholar] [CrossRef]
- Sprenkle, N.T.; Sims, S.G.; Sánchez, C.L.; Meares, G.P. Endoplasmic Reticulum Stress and Inflammation in the Central Nervous System. Mol. Neurodegener. 2017, 12, 42. [Google Scholar] [CrossRef]
- Yang, Y.; Lu, D.; Wang, M.; Liu, G.; Feng, Y.; Ren, Y.; Sun, X.; Chen, Z.; Wang, Z. Endoplasmic Reticulum Stress and the Unfolded Protein Response: Emerging Regulators in Progression of Traumatic Brain Injury. Cell Death Dis. 2024, 15, 156. [Google Scholar] [CrossRef] [PubMed]
- Read, A.; Schröder, M. The Unfolded Protein Response: An Overview. Biology 2021, 10, 384. [Google Scholar] [CrossRef]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic Reticulum Stress, Unfolded Protein Response, and Cancer Cell Fate. Front. Oncol. 2017, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, D.; Korhonen, L.; Eriksson, O.; Kõks, S. Recent Insights into the Role of Unfolded Protein Response in ER Stress in Health and Disease. Front. Cell Dev. Biol. 2017, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Resende, R.; Fernandes, T.; Pereira, A.C.; Marques, A.P.; Pereira, C.F. Endoplasmic Reticulum-Mitochondria Contacts Modulate Reactive Oxygen Species-Mediated Signaling and Oxidative Stress in Brain Disorders: The Key Role of Sigma-1 Receptor. Antioxid. Redox Signal 2022, 37, 758–780. [Google Scholar]
- Ong, G.; Logue, S.E. Unfolding the Interactions between Endoplasmic Reticulum Stress and Oxidative Stress. Antioxidants 2023, 12, 981. [Google Scholar] [CrossRef]
- Ren, J.; Bi, Y.; Sowers, J.R.; Hetz, C.; Zhang, Y. Endoplasmic Reticulum Stress and Unfolded Protein Response in Cardiovascular Diseases. Nat. Rev. Cardiol. 2021, 18, 499–521. [Google Scholar]
- Oakes, S.A. Endoplasmic Reticulum Stress Signaling in Cancer Cells. Am. J. Pathol. 2020, 190, 934–946. [Google Scholar]
- Roy, A.; Kumar, A. ER Stress and Unfolded Protein Response in Cancer Cachexia. Cancers 2019, 11, 1929. [Google Scholar] [CrossRef]
- Lucas, D.; Sarkar, T.; Niemeyer, C.Y.; Harnoss, J.C.; Schneider, M.; Strowitzki, M.J.; Harnoss, J.M. IRE1 Is a Promising Therapeutic Target in Pancreatic Cancer. Am. J. Physiol.-Cell Physiol. 2025, 328, 806–824. [Google Scholar] [CrossRef]
- Brito, M.L.; Coutinho-Wolino, K.S.; Almeida, P.P.; de Castro Trigueira, P.; de Paula Alves, A.P.; Magliano, D.C.; Stockler-Pinto, M.B. Unstressing the Reticulum: Nutritional Strategies for Modulating Endoplasmic Reticulum Stress in Obesity. Mol. Nutr. Food Res. 2024, 68, 2400361. [Google Scholar]
- Zhou, X.; Fouda, S.; Li, D.; Zhang, K.; Ye, J.-M. Involvement of the Autophagy-ER Stress Axis in High Fat/Carbohydrate Diet-Induced Nonalcoholic Fatty Liver Disease. Nutrients 2020, 12, 2626. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, D.; Stabile, A.M.; Vacca, C.; Pistilli, A.; Rende, M.; Gioiello, A.; Cruciani, G.; Galli, F. Endoplasmic Reticulum Stress and NF-kB Activation in SARS-CoV-2 Infected Cells and Their Response to Antiviral Therapy. IUBMB Life 2022, 74, 93–100. [Google Scholar]
- Cirone, M. ER Stress, UPR Activation and the Inflammatory Response to Viral Infection. Viruses 2021, 13, 798. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Qin, C.; Ramatchandirin, B.; Pearah, A.; Guo, S.; Hussain, M.; Yu, L.; Wondisford, F.E.; He, L. Activation of the Canonical ER Stress IRE1–XBP1 Pathway by Insulin Regulates Glucose and Lipid Metabolism. J. Biol. Chem. 2022, 298, 102283. [Google Scholar]
- Choi, J.-A.; Song, C.-H. Insights into the Role of Endoplasmic Reticulum Stress in Infectious Diseases. Front. Immunol. 2020, 10, 3147. [Google Scholar] [CrossRef]
- Lim, D.; Tapella, L.; Dematteis, G.; Genazzani, A.A.; Corazzari, M.; Verkhratsky, A. The Endoplasmic Reticulum Stress and Unfolded Protein Response in Alzheimer’s Disease: A Calcium Dyshomeostasis Perspective. Ageing Res. Rev. 2023, 87, 101914. [Google Scholar] [CrossRef]
- Gao, P.-C.; Wang, A.-Q.; Chen, X.-W.; Cui, H.; Li, Y.; Fan, R.-F. Selenium Alleviates ER Calcium Depletion-Induced Endoplasmic Reticulum Stress Dependent Apoptosis via PERK/ATF4/CHOP Pathway in Chicken Myocardium after Mercuric Chloride Exposure. Environ. Sci. Pollut. Res. 2023, 30, 51531–51541. [Google Scholar]
- Afroze, D.; Kumar, A. ER Stress in Skeletal Muscle Remodeling and Myopathies. FEBS J. 2019, 286, 379–398. [Google Scholar]
- Sun, Z.; Brodsky, J.L. Protein Quality Control in the Secretory Pathway. J. Cell Biol. 2019, 218, 3171–3187. [Google Scholar]
- Liu, X.; Green, R.M. Endoplasmic Reticulum Stress and Liver Diseases. Liver Res. 2019, 3, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Ghemrawi, R.; Khair, M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 6127. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Liu, Z.; Zhou, J.; Ke, C.; Li, D. Significance of Programmed Cell Death Pathways in Neurodegenerative Diseases. Int. J. Mol. Sci. 2024, 25, 9947. [Google Scholar] [CrossRef] [PubMed]
- Eleftheriadis, T.; Pissas, G.; Golfinopoulos, S.; Efthymiadi, M.; Poulianiti, C.; Polyzou Konsta, M.A.; Liakopoulos, V.; Stefanidis, I. Routes of Albumin Overload Toxicity in Renal Tubular Epithelial Cells. Int. J. Mol. Sci. 2023, 24, 9640. [Google Scholar] [CrossRef]
- Rowland, M.B.; Moore, P.E.; Correll, R.N. Regulation of Cardiac Fibroblast Cell Death by Unfolded Protein Response Signaling. Front. Physiol. 2024, 14, 1304669. [Google Scholar]
- Hashemi, M.; Hosseini, A.S.; Monjezi, S.; Hasany, S.; Binaei, S.; Nejat, M.; Melyani, H.; Bashandeh, N.; Matinahmadi, A.; Zayani, Z. Prostate Cancer, Apoptosis, Autophagy and Ferroptosis: Cell Death Mechanisms and Their Cross-Talk. In Prostate Cancer: Molecular Events and Therapeutic Modalities; Springer: Berlin/Heidelberg, Germany, 2024; pp. 71–107. [Google Scholar]
- Liu, Q.; Körner, H.; Wu, H.; Wei, W. Endoplasmic Reticulum Stress in Autoimmune Diseases. Immunobiology 2020, 225, 151881. [Google Scholar] [CrossRef]
- Ni, L.; Yang, L.; Lin, Y. Recent Progress of Endoplasmic Reticulum Stress in the Mechanism of Atherosclerosis. Front. Cardiovasc. Med. 2024, 11, 1413441. [Google Scholar]
- Yuan, S.; She, D.; Jiang, S.; Deng, N.; Peng, J.; Ma, L. Endoplasmic Reticulum Stress and Therapeutic Strategies in Metabolic, Neurodegenerative Diseases and Cancer. Mol. Med. 2024, 30, 40. [Google Scholar]
- Whaley, W.G. The Golgi Apparatus; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012; ISBN 3709176700. [Google Scholar]
- Zhang, X.; Wang, Y. Glycosylation Quality Control by the Golgi Structure. J. Mol. Biol. 2016, 428, 3183–3193. [Google Scholar]
- Luini, A.; Parashuraman, S. Golgi and TGN. Encycl. Cell Biol. 2016, 2, 183–191. [Google Scholar]
- Pavelka, M. Functional Morphology of the Golgi Apparatus; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2013; ISBN 364272826X. [Google Scholar]
- Eşrefoğlu, M. The Golgi Apparatus: Morphology and Function with Recent Facts. Bezmialem Sci. 2019, 7, 331–338. [Google Scholar] [CrossRef]
- Machamer, C.E. The Golgi Complex in Stress and Death. Front. Neurosci. 2015, 9, 421. [Google Scholar] [CrossRef] [PubMed]
- Hicks, S.W.; Machamer, C.E. Golgi Structure in Stress Sensing and Apoptosis. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2005, 1744, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Oku, M.; Tanakura, S.; Uemura, A.; Sohda, M.; Misumi, Y.; Taniguchi, M.; Wakabayashi, S.; Yoshida, H. Novel Cis-Acting Element GASE Regulates Transcriptional Induction by the Golgi Stress Response. Cell Struct. Funct. 2011, 36, 1–12. [Google Scholar] [CrossRef]
- Yin, Y.; Kan, X.; Miao, X.; Sun, Y.; Chen, S.; Qin, T.; Ding, C.; Peng, D.; Liu, X. H5 Subtype Avian Influenza Virus Induces Golgi Apparatus Stress Response via TFE3 Pathway to Promote Virus Replication. PLoS Pathog. 2024, 20, e1012748. [Google Scholar] [CrossRef]
- Gao, J.; Gao, A.; Liu, W.; Chen, L. Golgi Stress Response: A Regulatory Mechanism of Golgi Function. Biofactors 2021, 47, 964–974. [Google Scholar] [CrossRef]
- Li, J.; Ahat, E.; Wang, Y. Golgi Structure and Function in Health, Stress, and Diseases. In The Golgi Apparatus and Centriole: Functions, Interactions and Role in Disease; Springer: Berlin/Heidelberg, Germany, 2019; pp. 441–485. [Google Scholar]
- Eisenberg-Lerner, A.; Benyair, R.; Hizkiahou, N.; Nudel, N.; Maor, R.; Kramer, M.P.; Shmueli, M.D.; Zigdon, I.; Cherniavsky Lev, M.; Ulman, A. Golgi Organization Is Regulated by Proteasomal Degradation. Nat. Commun. 2020, 11, 409. [Google Scholar] [CrossRef]
- Chang, H.; Yang, W.Y. Golgi Quality Control and Autophagy. IUBMB Life 2022, 74, 361–370. [Google Scholar] [CrossRef]
- Kim, W.K.; Choi, W.; Deshar, B.; Kang, S.; Kim, J. Golgi Stress Response: New Insights into the Pathogenesis and Therapeutic Targets of Human Diseases. Mol. Cells 2023, 46, 191–199. [Google Scholar] [CrossRef]
- He, Q.; Liu, H.; Deng, S.; Chen, X.; Li, D.; Jiang, X.; Zeng, W.; Lu, W. The Golgi Apparatus May Be a Potential Therapeutic Target for Apoptosis-Related Neurological Diseases. Front. Cell Dev. Biol. 2020, 8, 830. [Google Scholar] [CrossRef]
- Kumari, N.; Reabroi, S.; North, B.J. Unraveling the Molecular Nexus between GPCRs, ERS, and EMT. Mediat. Inflamm. 2021, 2021, 6655417. [Google Scholar] [CrossRef] [PubMed]
- Raja, R.; Fonseka, O.; Ganenthiran, H.; Liu, W. The Multifaceted Roles of ER and Golgi in Metabolic Cardiomyopathy. Front. Cardiovasc. Med. 2022, 9, 999044. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Duan, Y.; Dong, J.; Zhang, K.; Jin, X.; Gao, M.; Jia, H.; Chen, J.; Liu, M.; Wei, M. Early Signs of Neurodegenerative Diseases: Possible Mechanisms and Targets for Golgi Stress. Biomed. Pharmacother. 2024, 175, 116646. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Yoshida, H. TFE3, HSP47, and CREB3 Pathways of the Mammalian Golgi Stress Response. Cell Struct. Funct. 2017, 42, 27–36. [Google Scholar] [CrossRef]
- Harapas, C.R.; Idiiatullina, E.; Al-Azab, M.; Hrovat-Schaale, K.; Reygaerts, T.; Steiner, A.; Laohamonthonkul, P.; Davidson, S.; Yu, C.-H.; Booty, L. Organellar Homeostasis and Innate Immune Sensing. Nat. Rev. Immunol. 2022, 22, 535–549. [Google Scholar] [CrossRef]
- Xu, Y.; Jin, Y.; Huang, Y.; Wen, Y.; Gu, Z.; Zhu, Y. Targeted Drug Delivery System for Golgi Apparatus’s Diseases. Eng. Regen. 2025, 6, 17–33. [Google Scholar] [CrossRef]
- Liu, Y.-N.; Chen, Y.-L.; Zhang, Z.-J.; Wu, F.-Y.; Wang, H.-J.; Wang, X.-L.; Liu, G.-Q. Phosphatidic Acid Directly Activates MTOR and Then Regulates SREBP to Promote Ganoderic Acid Biosynthesis under Heat Stress in Ganoderma Lingzhi. Commun. Biol. 2024, 7, 1503. [Google Scholar] [CrossRef]
- Fernandes-da-Silva, A.; Miranda, C.S.; Santana-Oliveira, D.A.; Oliveira-Cordeiro, B.; Rangel-Azevedo, C.; Silva-Veiga, F.M.; Martins, F.F.; Souza-Mello, V. Endoplasmic Reticulum Stress as the Basis of Obesity and Metabolic Diseases: Focus on Adipose Tissue, Liver, and Pancreas. Eur. J. Nutr. 2021, 60, 2949–2960. [Google Scholar] [CrossRef]
- Liu, J.; Huang, Y.; Li, T.; Jiang, Z.; Zeng, L.; Hu, Z. The Role of the Golgi Apparatus in Disease. Int. J. Mol. Med. 2021, 47, 38. [Google Scholar] [CrossRef]
- Khoder-Agha, F.; Kietzmann, T. The Glyco-Redox Interplay: Principles and Consequences on the Role of Reactive Oxygen Species during Protein Glycosylation. Redox Biol. 2021, 42, 101888. [Google Scholar] [CrossRef]
- Lee, P.; Chandel, N.S.; Simon, M.C. Cellular Adaptation to Hypoxia through Hypoxia Inducible Factors and Beyond. Nat. Rev. Mol. Cell Biol. 2020, 21, 268–283. [Google Scholar] [PubMed]
- Mennerich, D.; Kellokumpu, S.; Kietzmann, T. Hypoxia and Reactive Oxygen Species as Modulators of Endoplasmic Reticulum and Golgi Homeostasis. Antioxid. Redox Signal 2019, 30, 113–137. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yu, J.; Gong, L.; Zhang, Y.; Dong, S.; Shi, J.; Li, C.; Li, Y.; Zhang, Y.; Li, H. Heme Oxygenase-1 (HO-1) Regulates Golgi Stress and Attenuates Endotoxin-Induced Acute Lung Injury through Hypoxia Inducible Factor-1α (HIF-1α)/HO-1 Signaling Pathway. Free Radic. Biol. Med. 2021, 165, 243–253. [Google Scholar] [PubMed]
- Huang, X.; Akgün, E.E.; Mehmood, K.; Zhang, H.; Tang, Z.; Li, Y. Mechanism of Hypoxia-Mediated Smooth Muscle Cell Proliferation Leading to Vascular Remodeling. Biomed. Res. Int. 2022, 2022, 3959845. [Google Scholar] [CrossRef]
- Gonzalez-Garcia, P.; Fiorillo Moreno, O.; Zarate Peñata, E.; Calderon-Villalba, A.; Pacheco Lugo, L.; Acosta Hoyos, A.; Villarreal Camacho, J.L.; Navarro Quiroz, R.; Pacheco Londoño, L.; Aroca Martinez, G. From Cell to Symptoms: The Role of SARS-CoV-2 Cytopathic Effects in the Pathogenesis of COVID-19 and Long COVID. Int. J. Mol. Sci. 2023, 24, 8290. [Google Scholar] [CrossRef]
- Pandey, K.P.; Zhou, Y. Influenza a Virus Infection Activates NLRP3 Inflammasome through Trans-Golgi Network Dispersion. Viruses 2022, 14, 88. [Google Scholar] [CrossRef]
- Mărunţelu, I.; Constantinescu, A.-E.; Covache-Busuioc, R.-A.; Constantinescu, I. The Golgi Apparatus: A Key Player in Innate Immunity. Int. J. Mol. Sci. 2024, 25, 4120. [Google Scholar] [CrossRef]
- Kaminska, P.; Tempes, A.; Scholz, E.; Malik, A.R. Cytokines on the Way to Secretion. Cytokine Growth Factor. Rev. 2024, 79, 52–65. [Google Scholar]
- Daussy, C.F.; Wodrich, H. “Repair Me If You Can”: Membrane Damage, Response, and Control from the Viral Perspective. Cells 2020, 9, 2042. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Read, E.; Fu, M.; Pei, Y.; Wu, L.; Wang, R.; Yang, G. Golgi Stress Response, Hydrogen Sulfide Metabolism, and Intracellular Calcium Homeostasis. Antioxid. Redox Signal 2020, 32, 583–601. [Google Scholar]
- Suga, K.; Saito, A.; Mishima, T.; Akagawa, K. Data for the Effects of ER and Golgi Stresses on the ER–Golgi SNARE Syntaxin5 Expression and on the ΒAPP Processing in Cultured Hippocampal Neurons. Data Brief. 2015, 5, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.-H.; Lee, L.; Chen, J.; Li, W.-S. Lithocholic Acid Analogues, New and Potent α-2, 3-Sialyltransferase Inhibitors. Chem. Commun. 2006, 629–631. [Google Scholar] [CrossRef] [PubMed]
- Saenz, J.B.; Sun, W.J.; Chang, J.W.; Li, J.; Bursulaya, B.; Gray, N.S.; Haslam, D.B. Golgicide A Reveals Essential Roles for GBF1 in Golgi Assembly and Function. Nat. Chem. Biol. 2009, 5, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Robineau, S.; Chabre, M.; Antonny, B. Binding Site of Brefeldin A at the Interface between the Small G Protein ADP-Ribosylation Factor 1 (ARF1) and the Nucleotide-Exchange Factor Sec7 Domain. Proc. Natl. Acad. Sci. USA 2000, 97, 9913–9918. [Google Scholar]
- Feng, Y.; Jadhav, A.P.; Rodighiero, C.; Fujinaga, Y.; Kirchhausen, T.; Lencer, W.I. Retrograde Transport of Cholera Toxin from the Plasma Membrane to the Endoplasmic Reticulum Requires the Trans-Golgi Network but Not the Golgi Apparatus in Exo2-treated Cells. EMBO Rep. 2004, 5, 596–601. [Google Scholar] [CrossRef]
- Farber-Katz, S.E.; Dippold, H.C.; Buschman, M.D.; Peterman, M.C.; Xing, M.; Noakes, C.J.; Tat, J.; Ng, M.M.; Rahajeng, J.; Cowan, D.M. DNA Damage Triggers Golgi Dispersal via DNA-PK and GOLPH3. Cell 2014, 156, 413–427. [Google Scholar]
- Tak, J.; Kim, S.G. Effects of Toxicants on Endoplasmic Reticulum Stress and Hepatic Cell Fate Determination. Toxicol. Res. 2023, 39, 533–547. [Google Scholar]
- Mohan, A.G.; Calenic, B.; Ghiurau, N.A.; Duncea-Borca, R.-M.; Constantinescu, A.-E.; Constantinescu, I. The Golgi Apparatus: A Voyage through Time, Structure, Function and Implication in Neurodegenerative Disorders. Cells 2023, 12, 1972. [Google Scholar] [CrossRef]
- Heng, B.C.; Zhang, X.; Aubel, D.; Bai, Y.; Li, X.; Wei, Y.; Fussenegger, M.; Deng, X. Role of YAP/TAZ in Cell Lineage Fate Determination and Related Signaling Pathways. Front. Cell Dev. Biol. 2020, 8, 735. [Google Scholar]
- Tortorella, I.; Argentati, C.; Emiliani, C.; Morena, F.; Martino, S. Biochemical Pathways of Cellular Mechanosensing/Mechanotransduction and Their Role in Neurodegenerative Diseases Pathogenesis. Cells 2022, 11, 3093. [Google Scholar] [CrossRef]
- Meng, S.; Liu, J.; Wang, Z.; Fan, Y.; Pei, S.; Wang, E.; Song, Y.; Cui, Y.; Xie, K. Inhibition of Golgi Stress Alleviates Sepsis-Induced Cardiomyopathy by Reducing Inflammation and Apoptosis. Int. Immunopharmacol. 2024, 133, 112103. [Google Scholar] [CrossRef] [PubMed]
- Mohamed Asik, R.; Suganthy, N.; Aarifa, M.A.; Kumar, A.; Szigeti, K.; Mathe, D.; Gulyás, B.; Archunan, G.; Padmanabhan, P. Alzheimer’s Disease: A Molecular View of β-Amyloid Induced Morbific Events. Biomedicines 2021, 9, 1126. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.K.; Muqit, M.M.K. Parkinson’s: A Disease of Aberrant Vesicle Trafficking. Annu. Rev. Cell Dev. Biol. 2020, 36, 237–264. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, L.; Anakor, E.; Connolly, O.; Vijayakumar, U.G.; Duddy, W.J.; Duguez, S. Molecular and Cellular Mechanisms Affected in ALS. J. Pers. Med. 2020, 10, 101. [Google Scholar] [CrossRef]
- Vlad, D.-B.; Dumitrascu, D.-I.; Dumitrascu, A.-L. Golgi’s Role in the Development of Possible New Therapies in Cancer. Cells 2023, 12, 1499. [Google Scholar] [CrossRef]
- Bone, R.N.; Oyebamiji, O.; Talware, S.; Selvaraj, S.; Krishnan, P.; Syed, F.; Wu, H.; Evans-Molina, C. A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes. Diabetes 2020, 69, 2364–2376. [Google Scholar] [CrossRef]
- Khan, M.S.; Lee, C.; Kim, S.G. Non-Alcoholic Fatty Liver Disease and Liver Secretome. Arch. Pharm. Res. 2022, 45, 938–963. [Google Scholar] [CrossRef]
- Daussy, C.F.; Monard, S.C.; Guy, C.; Muñoz-González, S.; Chazal, M.; Anthonsen, M.W.; Jouvenet, N.; Henry, T.; Dreux, M.; Meurs, E.F. The Inflammasome Components NLRP3 and ASC Act in Concert with IRGM to Rearrange the Golgi Apparatus during Hepatitis c Virus Infection. J. Virol. 2021, 95, 10–1128. [Google Scholar] [CrossRef]
- Pais, S.V.; Key, C.E.; Borges, V.; Pereira, I.S.; Gomes, J.P.; Fisher, D.J.; Mota, L.J. CteG Is a Chlamydia Trachomatis Effector Protein That Associates with the Golgi Complex of Infected Host Cells. Sci. Rep. 2019, 9, 6133. [Google Scholar] [CrossRef]
- Bonam, S.R.; Wang, F.; Muller, S. Lysosomes as a Therapeutic Target. Nat. Rev. Drug Discov. 2019, 18, 923–948. [Google Scholar] [CrossRef]
- Xu, H.; Ren, D. Lysosomal Physiology. Annu. Rev. Physiol. 2015, 77, 57–80. [Google Scholar] [PubMed]
- Gu, J.; Geng, M.; Qi, M.; Wang, L.; Zhang, Y.; Gao, J. The Role of Lysosomal Membrane Proteins in Glucose and Lipid Metabolism. FASEB J. 2021, 35, e21848. [Google Scholar] [CrossRef] [PubMed]
- Kononenko, N.L. Lysosomes Convene to Keep the Synapse Clean. J. Cell Biol. 2017, 216, 2251. [Google Scholar]
- Braulke, T.; Carette, J.E.; Palm, W. Lysosomal Enzyme Trafficking: From Molecular Mechanisms to Human Diseases. Trends Cell Biol. 2024, 34, 198–210. [Google Scholar]
- Yim, W.W.-Y.; Mizushima, N. Lysosome Biology in Autophagy. Cell Discov. 2020, 6, 6. [Google Scholar]
- Mahapatra, K.K.; Mishra, S.R.; Behera, B.P.; Patil, S.; Gewirtz, D.A.; Bhutia, S.K. The Lysosome as an Imperative Regulator of Autophagy and Cell Death. Cell. Mol. Life Sci. 2021, 78, 7435–7449. [Google Scholar] [CrossRef]
- Nixon, R.A.; Rubinsztein, D.C. Mechanisms of Autophagy–Lysosome Dysfunction in Neurodegenerative Diseases. Nat. Rev. Mol. Cell Biol. 2024, 25, 926–946. [Google Scholar]
- Rudinskiy, M.; Morone, D.; Molinari, M. Fluorescent Reporters, Imaging, and Artificial Intelligence Toolkits to Monitor and Quantify Autophagy, Heterophagy, and Lysosomal Trafficking Fluxes. Traffic 2024, 25, e12957. [Google Scholar]
- Platonova, N.; Manzo, T.; Mirandola, L.; Colombo, M.; Calzavara, E.; Vigolo, E.; Cermisoni, G.C.; De Simone, D.; Garavelli, S.; Cecchinato, V. PI3K/AKT Signaling Inhibits NOTCH1 Lysosome-mediated Degradation. Genes. Chromosomes Cancer 2015, 54, 516–526. [Google Scholar]
- Ploper, D.; De Robertis, E.M. The MITF Family of Transcription Factors: Role in Endolysosomal Biogenesis, Wnt Signaling, and Oncogenesis. Pharmacol. Res. 2015, 99, 36–43. [Google Scholar] [CrossRef]
- Raben, N.; Puertollano, R. TFEB and TFE3: Linking Lysosomes to Cellular Adaptation to Stress. Annu. Rev. Cell Dev. Biol. 2016, 32, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.L. Lysosomal Calcium and Autophagy. Int. Rev. Cell Mol. Biol. 2021, 362, 141–170. [Google Scholar] [PubMed]
- Eriksson, I.; Wäster, P.; Öllinger, K. Restoration of Lysosomal Function after Damage Is Accompanied by Recycling of Lysosomal Membrane Proteins. Cell Death Dis. 2020, 11, 370. [Google Scholar] [CrossRef] [PubMed]
- Pascua-Maestro, R.; Diez-Hermano, S.; Lillo, C.; Ganfornina, M.D.; Sanchez, D. Protecting Cells by Protecting Their Vulnerable Lysosomes: Identification of a New Mechanism for Preserving Lysosomal Functional Integrity upon Oxidative Stress. PLoS Genet. 2017, 13, e1006603. [Google Scholar] [CrossRef]
- Wang, H.; Wang, N.; Xu, D.; Ma, Q.; Chen, Y.; Xu, S.; Xia, Q.; Zhang, Y.; Prehn, J.H.M.; Wang, G. Oxidation of Multiple MiT/TFE Transcription Factors Links Oxidative Stress to Transcriptional Control of Autophagy and Lysosome Biogenesis. Autophagy 2020, 16, 1683–1696. [Google Scholar] [CrossRef]
- Cao, M.; Luo, X.; Wu, K.; He, X. Targeting Lysosomes in Human Disease: From Basic Research to Clinical Applications. Signal Transduct. Target. Ther. 2021, 6, 379. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, W.; Zhang, S.; Iyaswamy, A.; Sun, J.; Wang, J.; Yang, C. Novel Insight into Functions of Transcription Factor EB (TFEB) in Alzheimer’s Disease and Parkinson’s Disease. Aging Dis. 2023, 14, 652. [Google Scholar] [CrossRef]
- Chen, X.; Li, Z.; Yang, C.; Tang, J.; Lan, H.; Liu, H. Lysosome Depletion-Triggered Autophagy Impairment in Progressive Kidney Injury. Kidney Dis. 2021, 7, 254–267. [Google Scholar] [CrossRef]
- Yamashita, G.; Takano, N.; Kazama, H.; Tsukahara, K.; Miyazawa, K. P53 Regulates Lysosomal Membrane Permeabilization as Well as Cytoprotective Autophagy in Response to DNA-Damaging Drugs. Cell Death Discov. 2022, 8, 502. [Google Scholar] [CrossRef]
- De Santis, M.C.; Gozzelino, L.; Margaria, J.P.; Costamagna, A.; Ratto, E.; Gulluni, F.; Di Gregorio, E.; Mina, E.; Lorito, N.; Bacci, M. Lysosomal Lipid Switch Sensitises to Nutrient Deprivation and MTOR Targeting in Pancreatic Cancer. Gut 2023, 72, 360–371. [Google Scholar] [CrossRef]
- Rabanal-Ruiz, Y.; Korolchuk, V.I. MTORC1 and Nutrient Homeostasis: The Central Role of the Lysosome. Int. J. Mol. Sci. 2018, 19, 818. [Google Scholar] [CrossRef] [PubMed]
- Leprivier, G.; Rotblat, B. How Does MTOR Sense Glucose Starvation? AMPK Is the Usual Suspect. Cell Death Discov. 2020, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Misra, S. Vacuolar ATPase (V-ATPase) Proton Pump and Its Significance in Human Health. In Ion Transporters-From Basic Properties to Medical Treatment; IntechOpen: London, UK, 2022; ISBN 1803555505. [Google Scholar]
- Seebacher, N.A.; Lane, D.J.R.; Jansson, P.J.; Richardson, D.R. Glucose Modulation Induces Lysosome Formation and Increases Lysosomotropic Drug Sequestration via the P-Glycoprotein Drug Transporter. J. Biol. Chem. 2016, 291, 3796–3820. [Google Scholar] [CrossRef] [PubMed]
- Mijanovic, O.; Petushkova, A.I.; Brankovic, A.; Turk, B.; Solovieva, A.B.; Nikitkina, A.I.; Bolevich, S.; Timashev, P.S.; Parodi, A.; Zamyatnin, A.A., Jr. Cathepsin D—Managing the Delicate Balance. Pharmaceutics 2021, 13, 837. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, Q.; Song, D.; Zen, R.; Zhang, L.; Wang, Y.; Yang, H.; Zhang, D.; Jia, J.; Zhang, J. Lysosomal Dysfunction and Autophagy Blockade Contribute to Autophagy-Related Cancer Suppressing Peptide-Induced Cytotoxic Death of Cervical Cancer Cells through the AMPK/MTOR Pathway. J. Exp. Clin. Cancer Res. 2020, 39, 197. [Google Scholar] [CrossRef]
- Saikia, R.; Joseph, J. AMPK: A Key Regulator of Energy Stress and Calcium-Induced Autophagy. J. Mol. Med. 2021, 99, 1539–1551. [Google Scholar] [CrossRef]
- Teixeira, S.C.; Teixeira, T.L.; Tavares, P.C.B.; Alves, R.N.; da Silva, A.A.; Borges, B.C.; Martins, F.A.; Dos Santos, M.A.; de Castilhos, P.; Notário, A.F.O. Subversion Strategies of Lysosomal Killing by Intracellular Pathogens. Microbiol. Res. 2023, 277, 127503. [Google Scholar] [CrossRef]
- Sachdeva, K.; Sundaramurthy, V. The Interplay of Host Lysosomes and Intracellular Pathogens. Front. Cell Infect. Microbiol. 2020, 10, 595502. [Google Scholar] [CrossRef]
- Blaess, M.; Kaiser, L.; Sauer, M.; Csuk, R.; Deigner, H.-P. COVID-19/SARS-CoV-2 Infection: Lysosomes and Lysosomotropism Implicate New Treatment Strategies and Personal Risks. Int. J. Mol. Sci. 2020, 21, 4953. [Google Scholar] [CrossRef]
- He, W.; Gao, Y.; Zhou, J.; Shi, Y.; Xia, D.; Shen, H.-M. Friend or Foe? Implication of the Autophagy-Lysosome Pathway in SARS-CoV-2 Infection and COVID-19. Int. J. Biol. Sci. 2022, 18, 4690. [Google Scholar] [CrossRef]
- Nie, B.; Liu, X.; Lei, C.; Liang, X.; Zhang, D.; Zhang, J. The Role of Lysosomes in Airborne Particulate Matter-Induced Pulmonary Toxicity. Sci. Total Environ. 2024, 919, 170893. [Google Scholar] [CrossRef] [PubMed]
- Lima, H., Jr.; Jacobson, L.; Goldberg, M.; Chandran, K.; Diaz-Griffero, F.; Lisanti, M.P.; Brojatsch, J. Role of Lysosome Rupture in Controlling Nlrp3 Signaling and Necrotic Cell Death. Cell Cycle 2013, 12, 1868–1878. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, J.; Dong, S.; Cai, X.; Simaiti, A.; Yang, X.; Zhu, X.; Luo, J.; Jiang, L.-H.; Du, B. Silica Nanoparticles Induce Lung Inflammation in Mice via ROS/PARP/TRPM2 Signaling-Mediated Lysosome Impairment and Autophagy Dysfunction. Part. Fibre Toxicol. 2020, 17, 23. [Google Scholar] [CrossRef]
- Dhakal, S.; Macreadie, I. Protein Homeostasis Networks and the Use of Yeast to Guide Interventions in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 8014. [Google Scholar] [CrossRef] [PubMed]
- Anfinogenova, N.D.; Quinn, M.T.; Schepetkin, I.A.; Atochin, D.N. Alarmins and C-Jun N-Terminal Kinase (JNK) Signaling in Neuroinflammation. Cells 2020, 9, 2350. [Google Scholar] [CrossRef]
- Pei, X.; Liu, D.; Li, J.; Li, L.; Ding, X.; Zhang, W.; Li, Z.; Xu, G.; Li, C.; Li, D. TFEB Coordinates Autophagy and Pyroptosis as Hepatotoxicity Responses to ZnO Nanoparticles. Sci. Total Environ. 2023, 865, 161242. [Google Scholar] [CrossRef]
- Wei, H.; Kim, S.-J.; Zhang, Z.; Tsai, P.-C.; Wisniewski, K.E.; Mukherjee, A.B. ER and Oxidative Stresses Are Common Mediators of Apoptosis in Both Neurodegenerative and Non-Neurodegenerative Lysosomal Storage Disorders and Are Alleviated by Chemical Chaperones. Hum. Mol. Genet. 2008, 17, 469–477. [Google Scholar] [CrossRef]
- Udayar, V.; Chen, Y.; Sidransky, E.; Jagasia, R. Lysosomal Dysfunction in Neurodegeneration: Emerging Concepts and Methods. Trends Neurosci. 2022, 45, 184–199. [Google Scholar] [CrossRef]
- Huang, W.J.; Zhang, X.; Chen, W.W. Gaucher Disease: A Lysosomal Neurodegenerative Disorder. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 1219–1226. [Google Scholar]
- Ferreira, C.R.; Gahl, W.A. Lysosomal Storage Diseases. Transl. Sci. Rare Dis. 2017, 2, 1–71. [Google Scholar] [CrossRef]
- Lie, P.P.Y.; Nixon, R.A. Lysosome Trafficking and Signaling in Health and Neurodegenerative Diseases. Neurobiol. Dis. 2019, 122, 94–105. [Google Scholar] [PubMed]
- Pitcairn, C.; Wani, W.Y.; Mazzulli, J.R. Dysregulation of the Autophagic-Lysosomal Pathway in Gaucher and Parkinson’s Disease. Neurobiol. Dis. 2019, 122, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Darios, F.; Stevanin, G. Impairment of Lysosome Function and Autophagy in Rare Neurodegenerative Diseases. J. Mol. Biol. 2020, 432, 2714–2734. [Google Scholar] [PubMed]
- Valionyte, E.; Yang, Y.; Roberts, S.L.; Kelly, J.; Lu, B.; Luo, S. Lowering Mutant Huntingtin Levels and Toxicity: Autophagy-Endolysosome Pathways in Huntington’s Disease. J. Mol. Biol. 2020, 432, 2673–2691. [Google Scholar]
- Monteith, A.J.; Kang, S.; Scott, E.; Hillman, K.; Rajfur, Z.; Jacobson, K.; Costello, M.J.; Vilen, B.J. Defects in Lysosomal Maturation Facilitate the Activation of Innate Sensors in Systemic Lupus Erythematosus. Proc. Natl. Acad. Sci. USA 2016, 113, E2142–E2151. [Google Scholar]
- Machado, E.R.; Annunziata, I.; van de Vlekkert, D.; Grosveld, G.C.; d’Azzo, A. Lysosomes and Cancer Progression: A Malignant Liaison. Front. Cell Dev. Biol. 2021, 9, 642494. [Google Scholar]
- Trybus, W.; Trybus, E.; Król, T. Lysosomes as a Target of Anticancer Therapy. Int. J. Mol. Sci. 2023, 24, 2176. [Google Scholar] [CrossRef]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomes as Mediators of Drug Resistance in Cancer. Drug Resist. Updates 2016, 24, 23–33. [Google Scholar]
- Scoca, V.; Di Nunzio, F. Membraneless Organelles Restructured and Built by Pandemic Viruses: HIV-1 and SARS-CoV-2. J. Mol. Cell Biol. 2021, 13, 259–268. [Google Scholar] [CrossRef]
- FERIC, M.; BRA, C. The Shape-Shifting Blobs That Rule Biology. Nature 2022, 611, 24–27. [Google Scholar]
- Anderson, P.; Kedersha, N.; Ivanov, P. Stress Granules, P-Bodies and Cancer. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2015, 1849, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Riggs, C.L.; Kedersha, N.; Ivanov, P.; Anderson, P. Mammalian Stress Granules and P Bodies at a Glance. J. Cell Sci. 2020, 133, jcs242487. [Google Scholar] [CrossRef]
- Campos-Melo, D.; Hawley, Z.C.E.; Droppelmann, C.A.; Strong, M.J. The Integral Role of RNA in Stress Granule Formation and Function. Front. Cell Dev. Biol. 2021, 9, 621779. [Google Scholar] [CrossRef] [PubMed]
- Hubstenberger, A.; Courel, M.; Bénard, M.; Souquere, S.; Ernoult-Lange, M.; Chouaib, R.; Yi, Z.; Morlot, J.-B.; Munier, A.; Fradet, M. P-Body Purification Reveals the Condensation of Repressed MRNA Regulons. Mol. Cell 2017, 68, 144–157. [Google Scholar] [PubMed]
- Millar, S.R.; Huang, J.Q.; Schreiber, K.J.; Tsai, Y.-C.; Won, J.; Zhang, J.; Moses, A.M.; Youn, J.-Y. A New Phase of Networking: The Molecular Composition and Regulatory Dynamics of Mammalian Stress Granules. Chem. Rev. 2023, 123, 9036–9064. [Google Scholar] [CrossRef]
- Nawaz, M.S.; Vik, E.S.; Berges, N.; Fladeby, C.; Bjørås, M.; Dalhus, B.; Alseth, I. Regulation of Human Endonuclease V Activity and Relocalization to Cytoplasmic Stress Granules. J. Biol. Chem. 2016, 291, 21786–21801. [Google Scholar] [CrossRef]
- Mateju, D.; Franzmann, T.M.; Patel, A.; Kopach, A.; Boczek, E.E.; Maharana, S.; Lee, H.O.; Carra, S.; Hyman, A.A.; Alberti, S. An Aberrant Phase Transition of Stress Granules Triggered by Misfolded Protein and Prevented by Chaperone Function. EMBO J. 2017, 36, 1669–1687. [Google Scholar] [CrossRef]
- Yang, P.; Mathieu, C.; Kolaitis, R.-M.; Zhang, P.; Messing, J.; Yurtsever, U.; Yang, Z.; Wu, J.; Li, Y.; Pan, Q. G3BP1 Is a Tunable Switch That Triggers Phase Separation to Assemble Stress Granules. Cell 2020, 181, 325–345. [Google Scholar]
- Verma, A.; Sumi, S.; Seervi, M. Heat Shock Proteins-Driven Stress Granule Dynamics: Yet Another Avenue for Cell Survival. Apoptosis 2021, 26, 371–384. [Google Scholar] [CrossRef]
- Moon, S.L.; Parker, R. Analysis of EIF2B Bodies and Their Relationships with Stress Granules and P-Bodies. Sci. Rep. 2018, 8, 12264. [Google Scholar]
- Costa-Mattioli, M.; Walter, P. The Integrated Stress Response: From Mechanism to Disease. Science 2020, 368, eaat5314. [Google Scholar] [PubMed]
- Cadena Sandoval, M.; Heberle, A.M.; Rehbein, U.; Barile, C.; Ramos Pittol, J.M.; Thedieck, K. MTORC1 Crosstalk with Stress Granules in Aging and Age-Related Diseases. Front. Aging 2021, 2, 761333. [Google Scholar]
- Hodgson, R.E.; Varanda, B.A.; Ashe, M.P.; Allen, K.E.; Campbell, S.G. Cellular EIF2B Subunit Localization: Implications for the Integrated Stress Response and Its Control by Small Molecule Drugs. Mol. Biol. Cell 2019, 30, 942–958. [Google Scholar] [PubMed]
- Luo, Y.; Na, Z.; Slavoff, S.A. P-Bodies: Composition, Properties, and Functions. Biochemistry 2018, 57, 2424–2431. [Google Scholar] [CrossRef]
- Guzikowski, A.R.; Chen, Y.S.; Zid, B.M. Stress-induced MRNP Granules: Form and Function of Processing Bodies and Stress Granules. Wiley Interdiscip. Rev. RNA 2019, 10, e1524. [Google Scholar]
- Chu, C.; Geng, Y.; Zhou, Y.; Sicinski, P. Cyclin E in Normal Physiology and Disease States. Trends Cell Biol. 2021, 31, 732–746. [Google Scholar]
- Ansari Basir, S.; Adeli, K. MicroRNAs: Critical Regulators of MRNA Traffic and Translational Control with Promising Biotech and Therapeutic Applications. Iran. J. Biotechnol. 2013, 11, 147–155. [Google Scholar]
- Liu, L.; Weiss, E.; Panas, M.D.; Götte, B.; Sellberg, S.; Thaa, B.; McInerney, G.M. RNA Processing Bodies Are Disassembled during Old World Alphavirus Infection. J. Gen. Virol. 2019, 100, 1375–1389. [Google Scholar]
- Emara, M.M.; Brinton, M.A. Interaction of TIA-1/TIAR with West Nile and Dengue Virus Products in Infected Cells Interferes with Stress Granule Formation and Processing Body Assembly. Proc. Natl. Acad. Sci. USA 2007, 104, 9041–9046. [Google Scholar]
- Chahar, H.S.; Chen, S.; Manjunath, N. P-Body Components LSM1, GW182, DDX3, DDX6 and XRN1 Are Recruited to WNV Replication Sites and Positively Regulate Viral Replication. Virology 2013, 436, 1–7. [Google Scholar]
- Gall, J.G. Cajal Bodies: The First 100 Years. Annu. Rev. Cell Dev. Biol. 2000, 16, 273–300. [Google Scholar] [PubMed]
- Rudzka, M.; Wróblewska-Ankiewicz, P.; Majewska, K.; Hyjek-Składanowska, M.; Gołębiewski, M.; Sikora, M.; Smoliński, D.J.; Kołowerzo-Lubnau, A. Functional Nuclear Retention of Pre-MRNA Involving Cajal Bodies during Meiotic Prophase in European Larch (Larix decidua). Plant Cell 2022, 34, 2404–2423. [Google Scholar] [PubMed]
- Quinodoz, S.A.; Guttman, M. Essential Roles for RNA in Shaping Nuclear Organization. Cold Spring Harb. Perspect. Biol. 2022, 14, a039719. [Google Scholar]
- Morris, G.E. The Cajal Body. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2008, 1783, 2108–2115. [Google Scholar] [CrossRef]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular Condensates: Organizers of Cellular Biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar]
- Neugebauer, K.M. Special Focus on the Cajal Body. RNA Biol. 2017, 14, 669–670. [Google Scholar]
- Tichy, E.D.; Ma, N.; Sidibe, D.; Loro, E.; Kocan, J.; Chen, D.Z.; Khurana, T.S.; Hasty, P.; Mourkioti, F. Persistent NF-ΚB Activation in Muscle Stem Cells Induces Proliferation-Independent Telomere Shortening. Cell Rep. 2021, 35, 109098. [Google Scholar]
- Dominguez, C.E.; Cunningham, D.; Chandler, D.S. SMN Regulation in SMA and in Response to Stress: New Paradigms and Therapeutic Possibilities. Hum. Genet. 2017, 136, 1173–1191. [Google Scholar]
- Coucoravas, C.; Dhanjal, S.; Henriksson, S.; Böhm, S.; Farnebo, M. Phosphorylation of the Cajal Body Protein WRAP53β by ATM Promotes Its Involvement in the DNA Damage Response. RNA Biol. 2017, 14, 804–813. [Google Scholar]
- Yu, J.; Zhu, H.; Lape, R.; Greiner, T.; Du, J.; Lü, W.; Sivilotti, L.; Gouaux, E. Mechanism of Gating and Partial Agonist Action in the Glycine Receptor. Cell 2021, 184, 957–968. [Google Scholar] [CrossRef]
- Navascues, J.; Bengoechea, R.; Tapia, O.; Casafont, I.; Berciano, M.T.; Lafarga, M. SUMO-1 Transiently Localizes to Cajal Bodies in Mammalian Neurons. J. Struct. Biol. 2008, 163, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Young, P.J.; Day, P.M.; Zhou, J.; Androphy, E.J.; Morris, G.E.; Lorson, C.L. A Direct Interaction between the Survival Motor Neuron Protein and P53 and Its Relationship to Spinal Muscular Atrophy. J. Biol. Chem. 2002, 277, 2852–2859. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Levine, A.J. The P53 Functional Circuit. J. Cell Sci. 2001, 114, 4139–4140. [Google Scholar] [CrossRef] [PubMed]
- Rebelo, L.; Almeida, F.; Ramos, C.; Bohmann, K.; Lamond, A.I.; Carmo-Fonseca, M. The Dynamics of Coiled Bodies in the Nucleus of Adenovirus-Infected Cells. Mol. Biol. Cell 1996, 7, 1137–1151. [Google Scholar] [CrossRef]
- Rodrigues, S.H.; Silva, N.P.; Delício, L.R.; Granato, C.; Andrade, L.E.C. The Behavior of the Coiled Body in Cells Infected with Adenovirus in Vitro. Mol. Biol. Rep. 1996, 23, 183–189. [Google Scholar] [CrossRef]
- Zhou, Y.; Kok, K.H.; Chun, A.C.S.; Wong, C.-M.; Wu, H.W.; Lin, M.C.M.; Fung, P.C.W.; Kung, H.; Jin, D.-Y. Mouse Peroxiredoxin V Is a Thioredoxin Peroxidase That Inhibits P53-Induced Apoptosis. Biochem. Biophys. Res. Commun. 2000, 268, 921–927. [Google Scholar] [CrossRef]
- Kropotov, A.V.; Grudinkin, P.S.; Pleskach, N.M.; Gavrilov, B.A.; Tomilin, N.V.; Zhivotovsky, B. Downregulation of Peroxiredoxin V Stimulates Formation of Etoposide-Induced Double-Strand DNA Breaks. FEBS Lett. 2004, 572, 75–79. [Google Scholar] [CrossRef]
- Gao, X.; Jiang, L.; Gong, Y.; Chen, X.; Ying, M.; Zhu, H.; He, Q.; Yang, B.; Cao, J. Stress Granule: A Promising Target for Cancer Treatment. Br. J. Pharmacol. 2019, 176, 4421–4433. [Google Scholar] [CrossRef]
- Song, M.-S.; Grabocka, E. Stress Granules in Cancer. In Organelles in Disease; Springer: Berlin/Heidelberg, Germany, 2020; pp. 25–52. [Google Scholar]
- Curdy, N.; Lanvin, O.; Cerapio, J.-P.; Pont, F.; Tosolini, M.; Sarot, E.; Valle, C.; Saint-Laurent, N.; Lhuillier, E.; Laurent, C. The Proteome and Transcriptome of Stress Granules and P Bodies during Human T Lymphocyte Activation. Cell Rep. 2023, 42, 112211. [Google Scholar] [CrossRef]
- Chen, L.; Liu, B. Relationships between Stress Granules, Oxidative Stress, and Neurodegenerative Diseases. Oxid. Med. Cell Longev. 2017, 2017, 1809592. [Google Scholar] [CrossRef]
- Wolozin, B.; Ivanov, P. Stress Granules and Neurodegeneration. Nat. Rev. Neurosci. 2019, 20, 649–666. [Google Scholar] [CrossRef] [PubMed]
- Ryan, V.H.; Fawzi, N.L. Physiological, Pathological, and Targetable Membraneless Organelles in Neurons. Trends Neurosci. 2019, 42, 693–708. [Google Scholar] [CrossRef] [PubMed]
- Nsengimana, B.; Khan, F.A.; Ngowi, E.E.; Zhou, X.; Jin, Y.; Jia, Y.; Wei, W.; Ji, S. Processing Body (P-Body) and Its Mediators in Cancer. Mol. Cell Biochem. 2022, 477, 1217–1238. [Google Scholar] [CrossRef] [PubMed]
- Kodali, S.; Proietti, L.; Valcarcel, G.; López-Rubio, A.V.; Pessina, P.; Eder, T.; Shi, J.; Jen, A.; Lupión-Garcia, N.; Starner, A.C. RNA Sequestration in P-Bodies Sustains Myeloid Leukaemia. Nat. Cell Biol. 2024, 26, 1745–1758. [Google Scholar] [CrossRef]
- Johnson, M.E.; Grassetti, A.V.; Taroni, J.N.; Lyons, S.M.; Schweppe, D.; Gordon, J.K.; Spiera, R.F.; Lafyatis, R.; Anderson, P.J.; Gerber, S.A. Stress Granules and RNA Processing Bodies Are Novel Autoantibody Targets in Systemic Sclerosis. Arthritis Res. Ther. 2016, 18, 27. [Google Scholar] [CrossRef]
- Balak, C.; Benard, M.; Schaefer, E.; Iqbal, S.; Ramsey, K.; Ernoult-Lange, M.; Mattioli, F.; Llaci, L.; Geoffroy, V.; Courel, M. Rare de Novo Missense Variants in RNA Helicase DDX6 Cause Intellectual Disability and Dysmorphic Features and Lead to P-Body Defects and RNA Dysregulation. Am. J. Hum. Genet. 2019, 105, 509–525. [Google Scholar] [CrossRef]
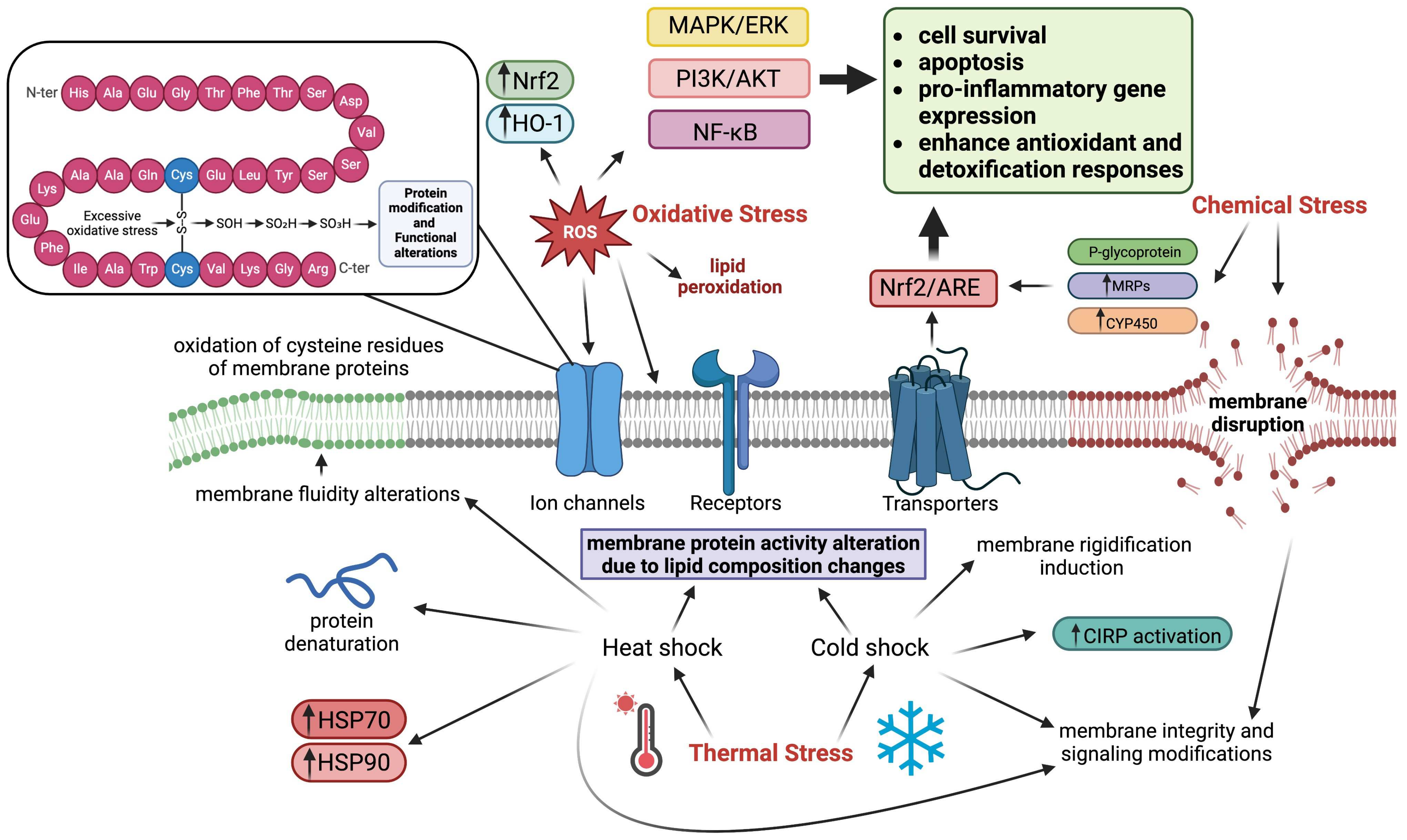
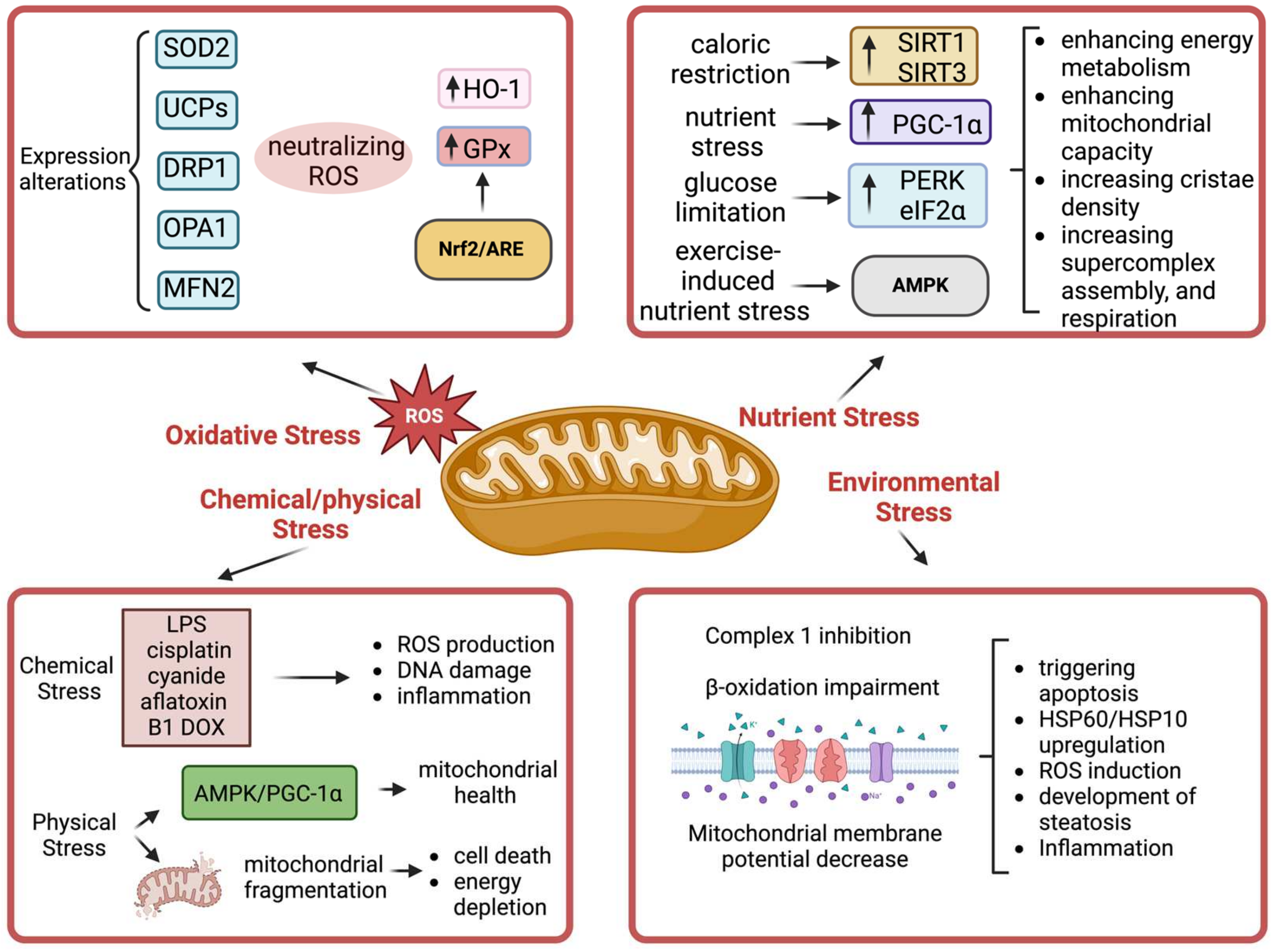

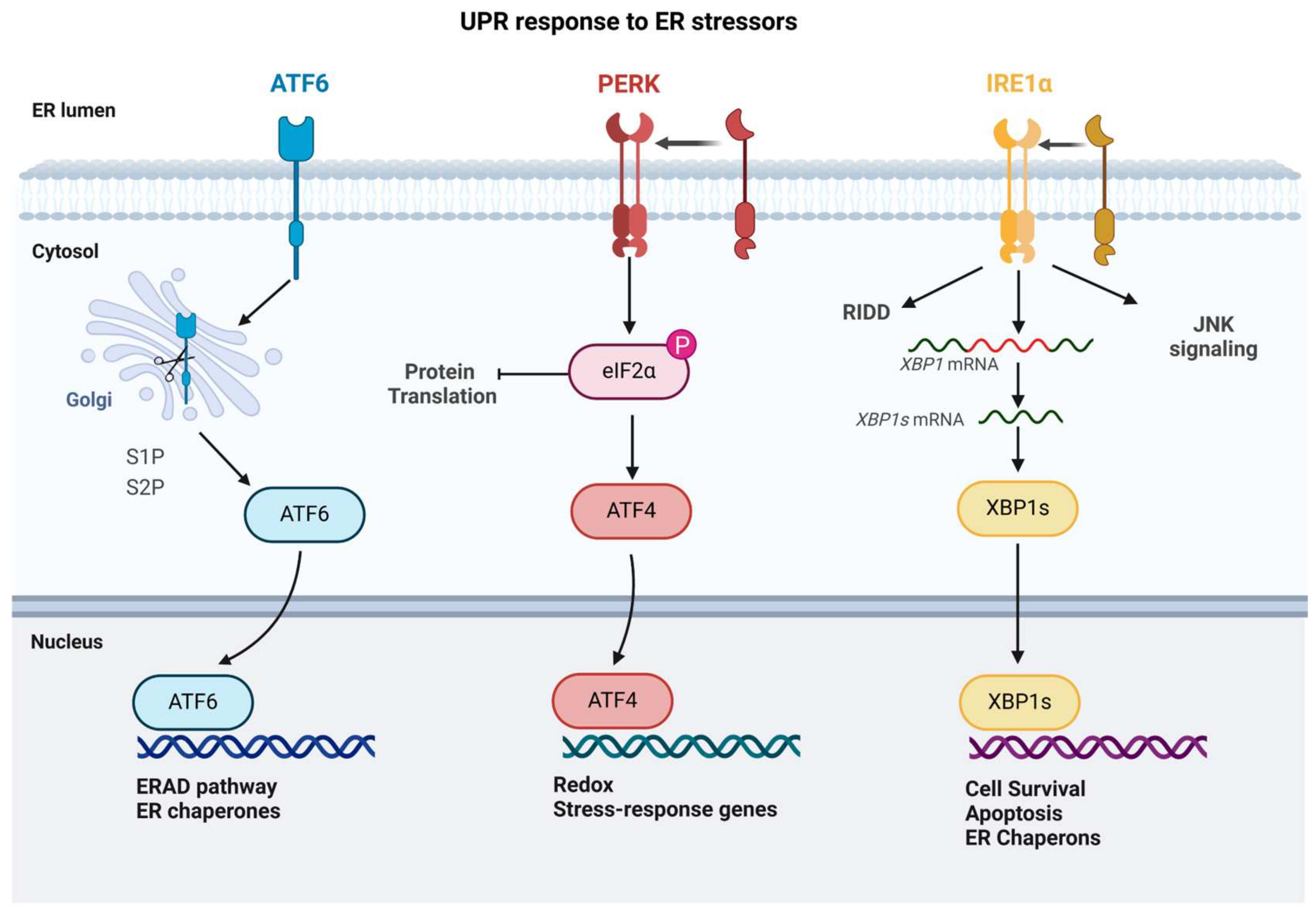
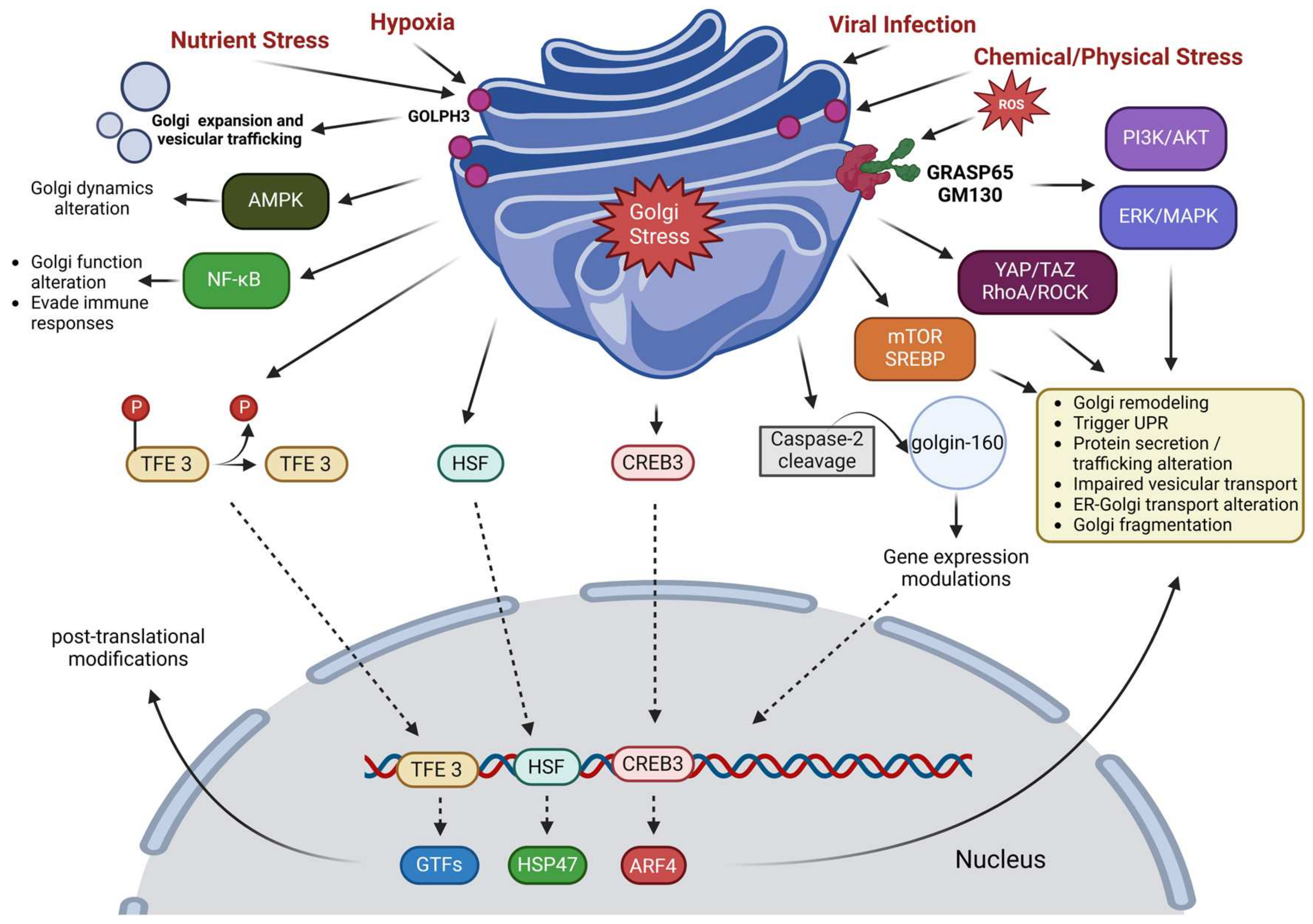



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zayani, Z.; Matinahmadi, A.; Tavakolpournegari, A.; Bidooki, S.H. Exploring Stressors: Impact on Cellular Organelles and Implications for Cellular Functions. Stresses 2025, 5, 26. https://doi.org/10.3390/stresses5020026
Zayani Z, Matinahmadi A, Tavakolpournegari A, Bidooki SH. Exploring Stressors: Impact on Cellular Organelles and Implications for Cellular Functions. Stresses. 2025; 5(2):26. https://doi.org/10.3390/stresses5020026
Chicago/Turabian StyleZayani, Zoofa, Arash Matinahmadi, Alireza Tavakolpournegari, and Seyed Hesamoddin Bidooki. 2025. "Exploring Stressors: Impact on Cellular Organelles and Implications for Cellular Functions" Stresses 5, no. 2: 26. https://doi.org/10.3390/stresses5020026
APA StyleZayani, Z., Matinahmadi, A., Tavakolpournegari, A., & Bidooki, S. H. (2025). Exploring Stressors: Impact on Cellular Organelles and Implications for Cellular Functions. Stresses, 5(2), 26. https://doi.org/10.3390/stresses5020026