

Multiple-Molecule Drug Repositioning for Disrupting Progression of SARS-CoV-2 Infection by Utilizing the Systems Biology Method through Host-Pathogen-Interactive Time Profile Data and DNN-Based DTI Model with Drug Design Specifications

Abstract
1. Introduction
2. Results
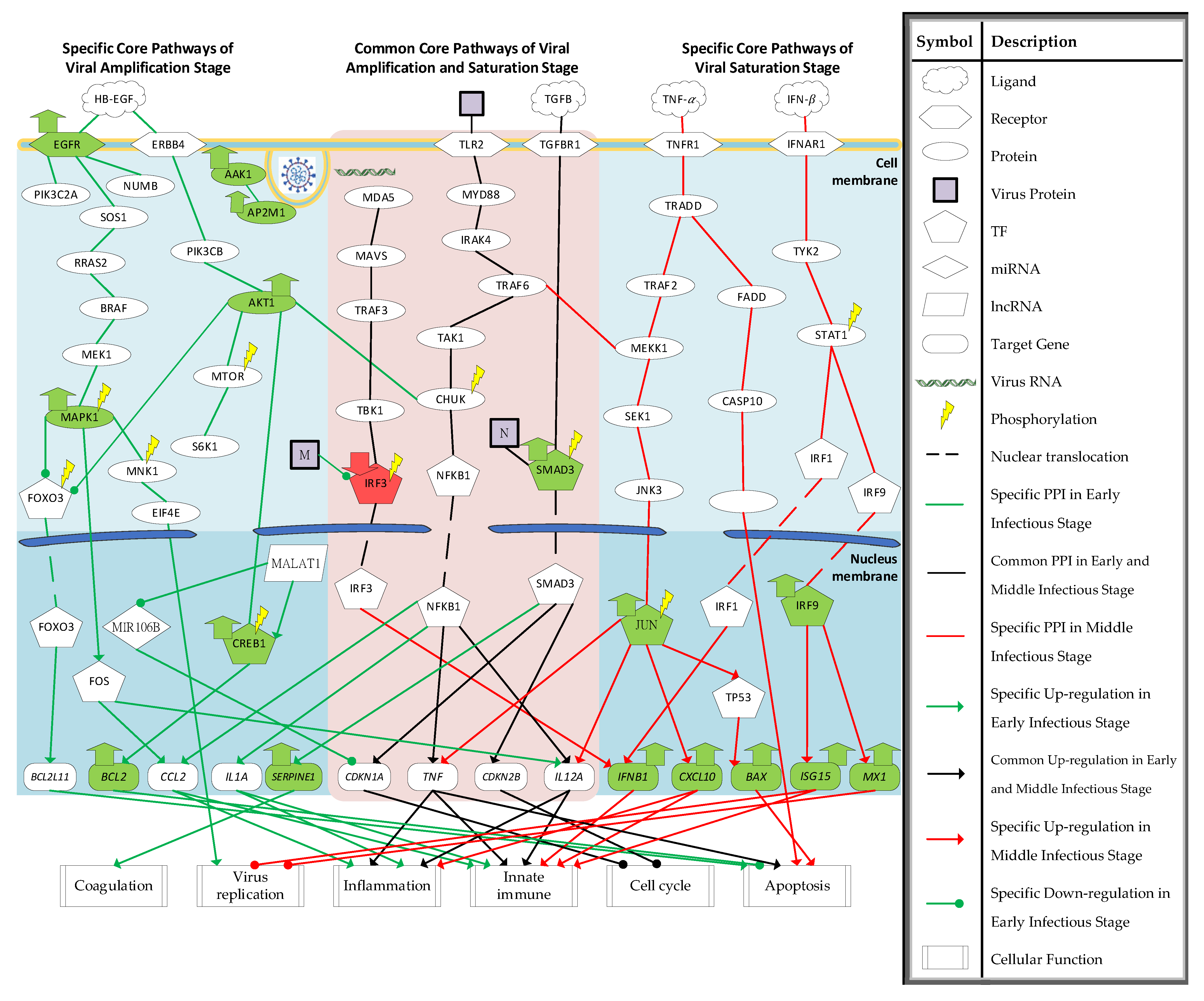
2.1. Core HPI Signaling Pathways during Amplification and Saturation Stage of SARS-CoV-2 Infection by the Systems Biology Method
2.2. Investigation of Specific Core HPI Signaling Pathways and Their Downstream Abnormal Cellular Functions during SARS-CoV-2 Infection
2.2.1. Investigation of Specific Core HPI Signaling Pathways in Amplification Infectious Stage
2.2.2. Investigation of Common Core HPI Signaling Pathways of Amplification and Saturation Infectious Stages
2.2.3. Investigation of Specific Core HPI Signaling Pathways at Saturation Infectious Stage
2.3. Multiple-Molecule Drug Discovery and Design by DNN-Based DTI Model with Drug Design Specifications
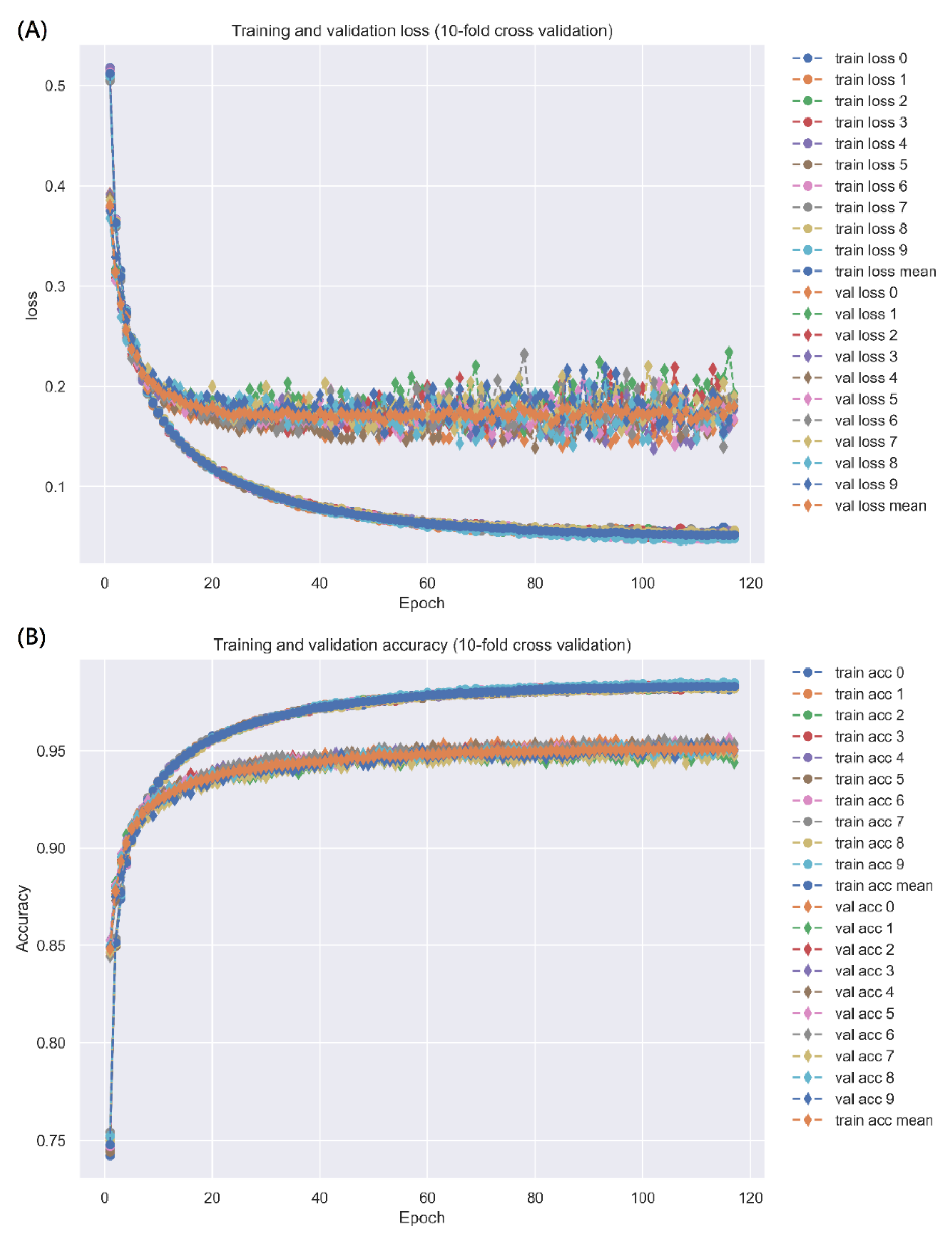
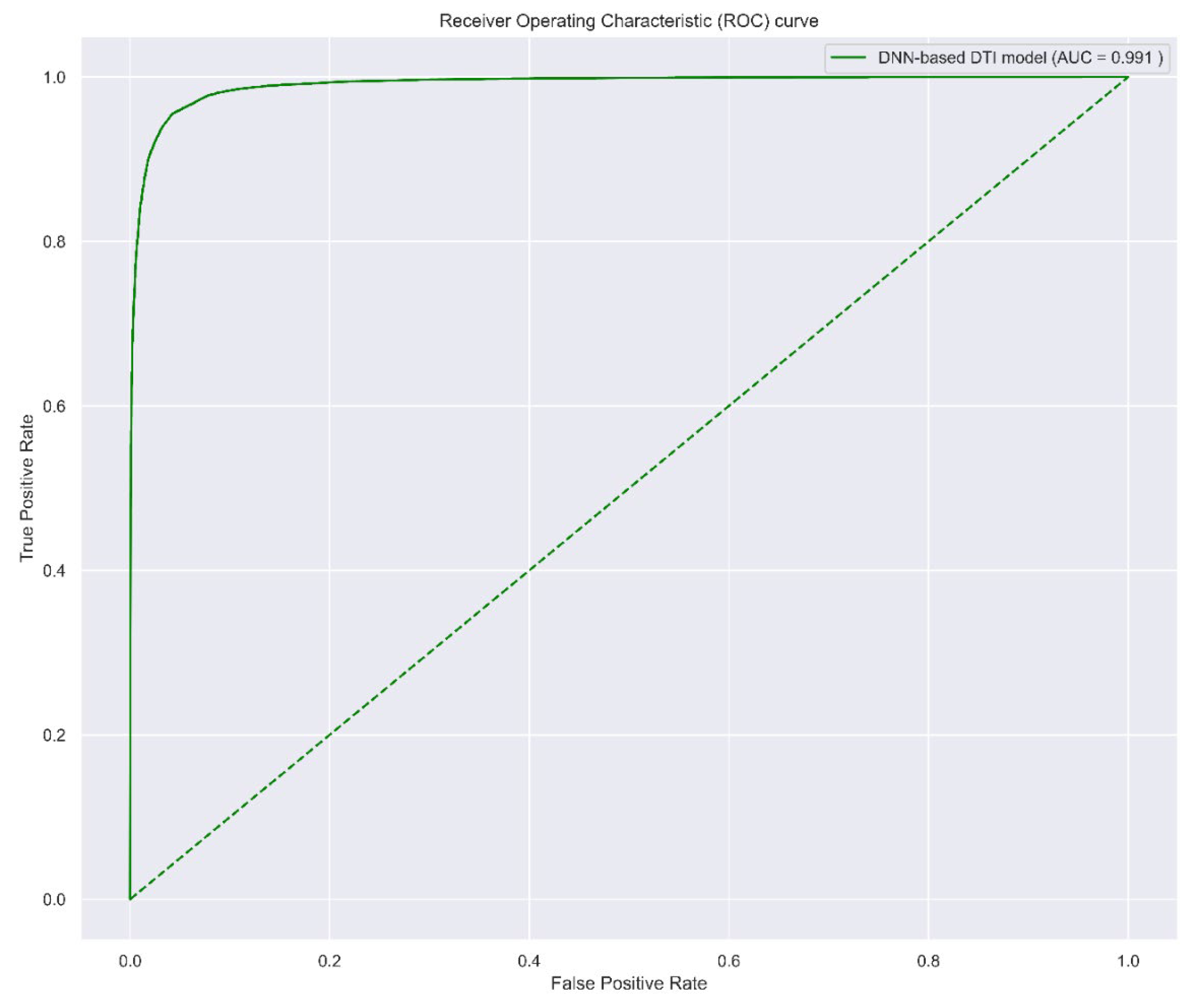
2.3.1. Prediction Performance of DNN-Based DTI Model
2.3.2. Multiple-Molecule Drug Repositioning for Disrupting the Progression of SARS-CoV-2 Infection
3. Discussion
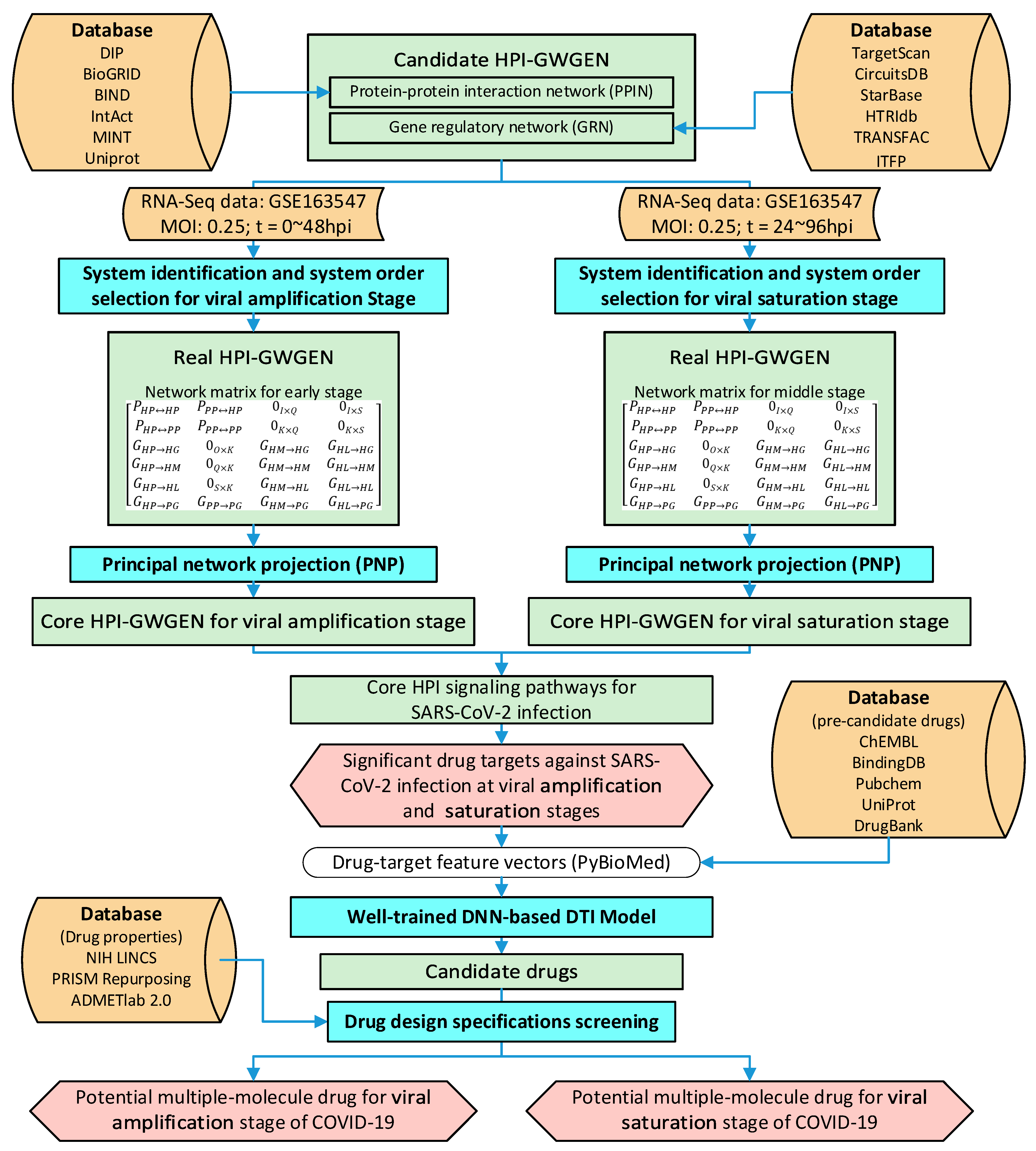
4. Materials and Methods
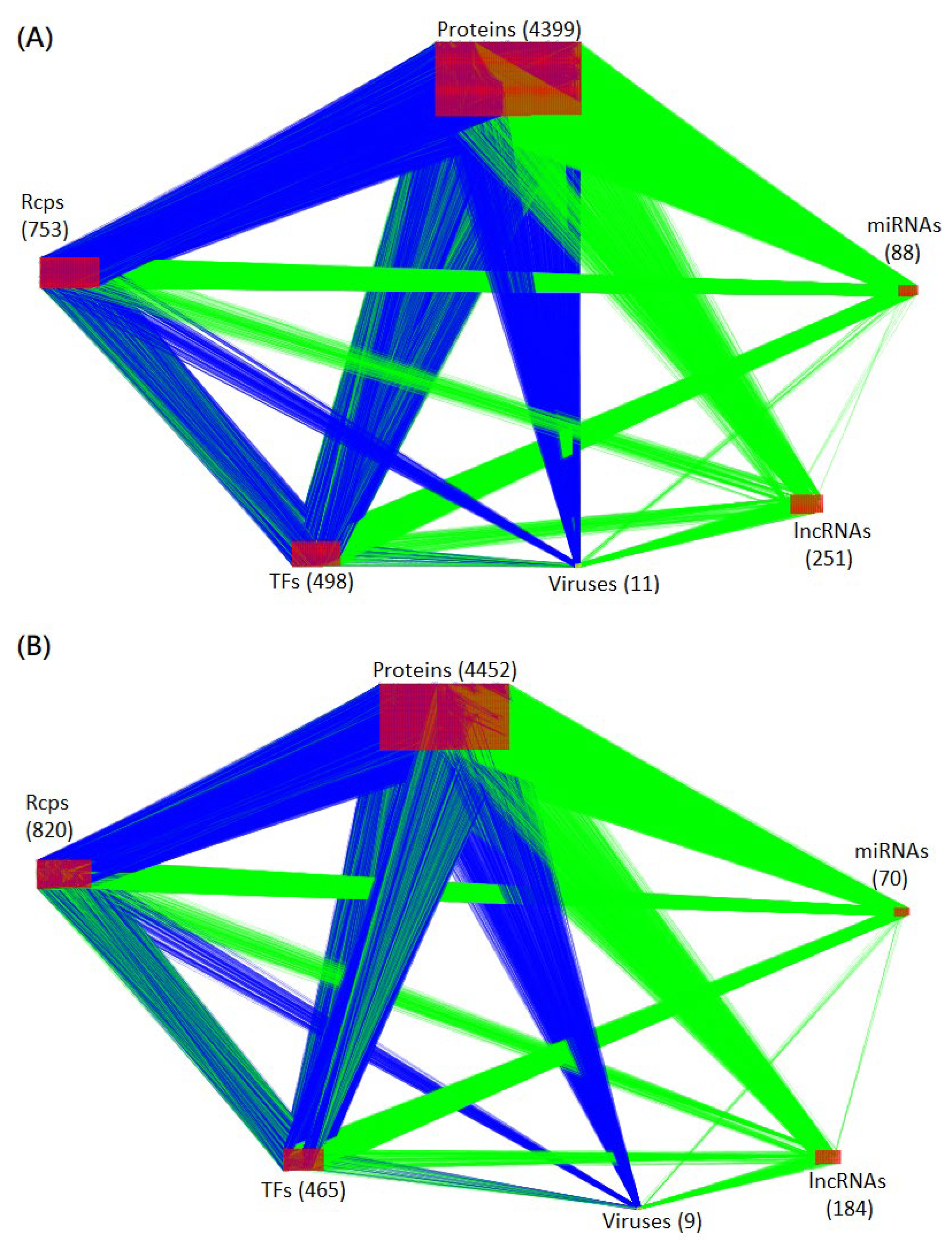
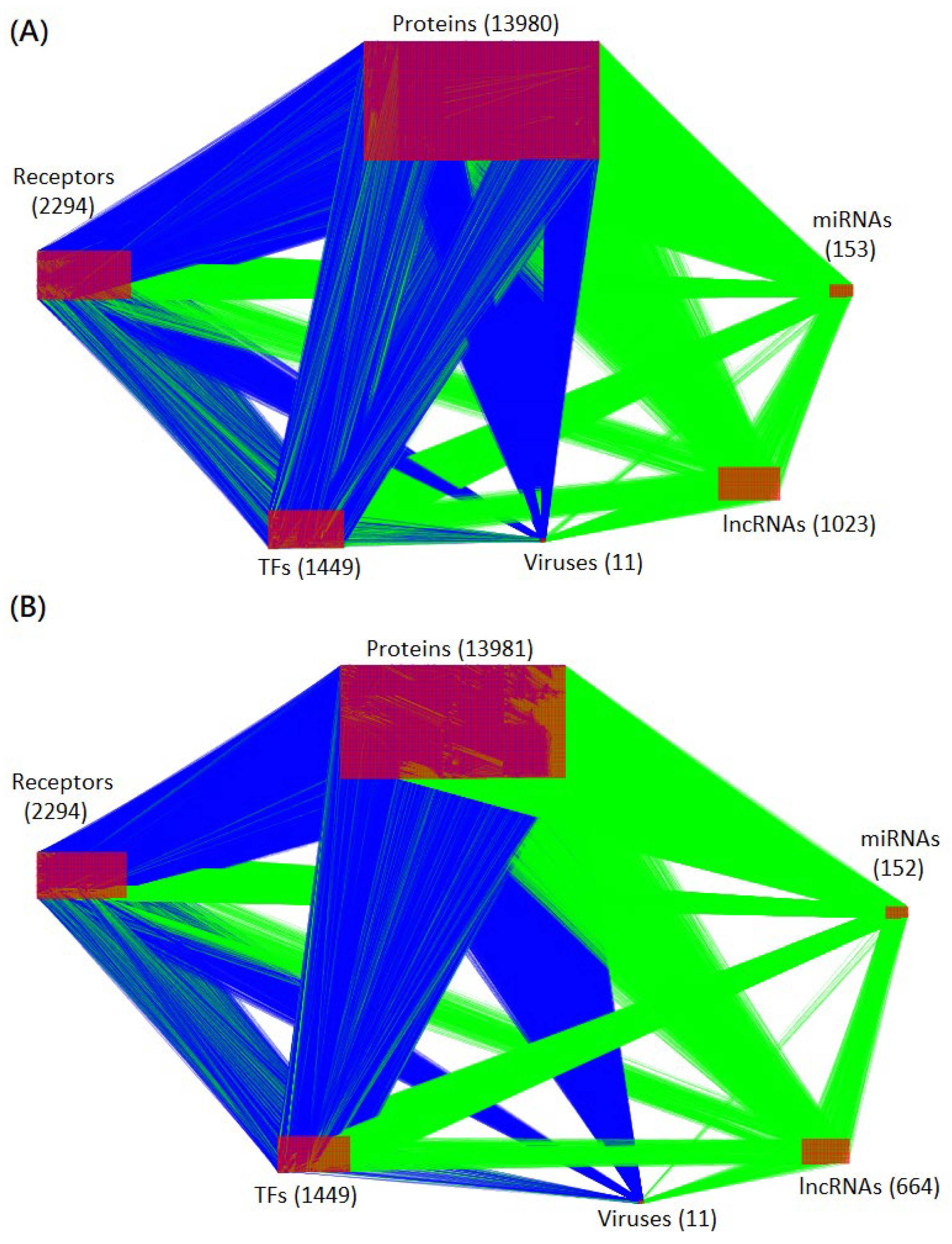
4.1. Construction of the Candidate HPI-GWGEN Using Big Data Mining
4.2. System Identification of HPI-GWGEN Using HPI RNA-Seq Time-Profile Data
4.2.1. HPI RNA-Seq Time-Profile Data
4.2.2. Dynamic Models for HPI-GWGEN
4.2.3. System Identification and System Order Selection for HPI-GWGEN
4.3. PNP Method to Extract the Core HPI-GWGEN from Network Matrix of Real HPI-GWGEN
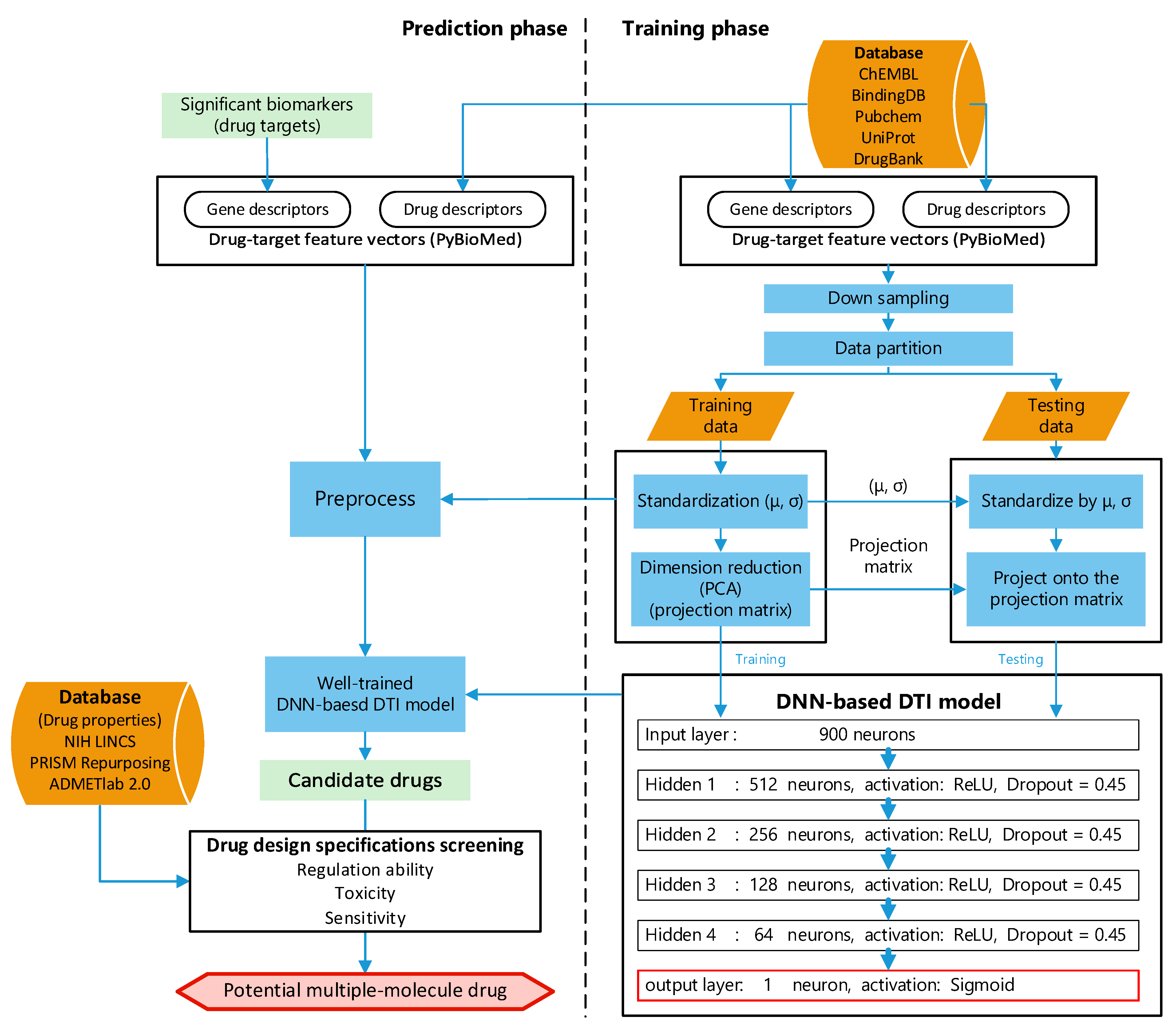
4.4. Systematic Discovery and Design of Multiple-Molecule Drug by UtilizingDNN-Based DTI Model with Drug Design Specifications
4.4.1. Preprocess of Targets and Drugs Data
4.4.2. Architecture of DNN-Based DTI Model
4.4.3. Drug Design Specifications
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A

References
- WHO. Available online: https://covid19.who.int/ (accessed on 22 June 2022).
- Lechien, J.R.; Chiesa-Estomba, C.M.; Place, S.; Van Laethem, Y.; Cabaraux, P.; Mat, Q.; Huet, K.; Plzak, J.; Horoi, M.; Hans, S.; et al. Clinical and epidemiological characteristics of 1420 European patients with mild-to-moderate coronavirus disease 2019. J. Intern. Med. 2020, 288, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Stokes, E.K.; Zambrano, L.D.; Anderson, K.N.; Marder, E.P.; Raz, K.M.; El Burai Felix, S.; Tie, Y.; Fullerton, K.E. Coronavirus Disease 2019 Case Surveillance—United States, 22 January–30 May 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Kim, D.; Lee, J.Y.; Yang, J.S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.A.; Belk, J.A.; Qi, Y.; Yasumoto, Y.; Wei, J.; Alfajaro, M.M.; Shi, Q.; Mumbach, M.R.; Limaye, A.; DeWeirdt, P.C.; et al. Discovery and functional interrogation of SARS-CoV-2 RNA-host protein interactions. Cell 2021, 184, 2394–2411.e16. [Google Scholar] [CrossRef] [PubMed]
- Stukalov, A.; Girault, V.; Grass, V.; Karayel, O.; Bergant, V.; Urban, C.; Haas, D.A.; Huang, Y.; Oubraham, L.; Wang, A.; et al. Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature 2021, 594, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, L.; Milavetz, B. Epigenetic Regulation of Viral Biological Processes. Viruses 2017, 9, 346. [Google Scholar] [CrossRef]
- Zhang, Q.; Cao, X. Epigenetic regulation of the innate immune response to infection. Nat. Rev. Immunol. 2019, 19, 417–432. [Google Scholar] [CrossRef]
- Leong, M.M.L.; Lung, M.L. The Impact of Epstein-Barr Virus Infection on Epigenetic Regulation of Host Cell Gene Expression in Epithelial and Lymphocytic Malignancies. Front. Oncol. 2021, 11, 629780. [Google Scholar] [CrossRef]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, A.; Thakur, N.; Monga, I.; Thakur, A.; Kumar, M. VIRmiRNA: A comprehensive resource for experimentally validated viral miRNAs and their targets. Database 2014, 2014, bau103. [Google Scholar] [CrossRef] [PubMed]
- Saliminejad, K.; Khorram Khorshid, H.R.; Soleymani Fard, S.; Ghaffari, S.H. An overview of microRNAs: Biology, functions, therapeutics, and analysis methods. J. Cell. Physiol. 2019, 234, 5451–5465. [Google Scholar] [CrossRef] [PubMed]
- Dykes, I.M.; Emanueli, C. Transcriptional and Post-transcriptional Gene Regulation by Long Non-coding RNA. Genom. Proteom. Bioinform. 2017, 15, 177–186. [Google Scholar] [CrossRef]
- Ma, L.; Cao, J.; Liu, L.; Du, Q.; Li, Z.; Zou, D.; Bajic, V.B.; Zhang, Z. LncBook: A curated knowledgebase of human long non-coding RNAs. Nucleic Acids Res. 2019, 47, D128–D134. [Google Scholar] [CrossRef]
- Chen, L.; Zhou, Y.; Li, H. LncRNA, miRNA and lncRNA-miRNA interaction in viral infection. Virus Res. 2018, 257, 25–32. [Google Scholar] [CrossRef]
- Waring, M.J.; Arrowsmith, J.; Leach, A.R.; Leeson, P.D.; Mandrell, S.; Owen, R.M.; Pairaudeau, G.; Pennie, W.D.; Pickett, S.D.; Wang, J.; et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 2015, 14, 475–486. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef]
- Maenza, J.; Flexner, C. Combination antiretroviral therapy for HIV infection. Am. Fam. Physician 1998, 57, 2789–2798. [Google Scholar] [PubMed]
- Fang, J.; Li, H.; Du, W.; Yu, P.; Guan, Y.-Y.; Ma, S.-Y.; Liu, D.; Chen, W.; Shi, G.-C.; Bian, X.-L. Efficacy of Early Combination Therapy With Lianhuaqingwen and Arbidol in Moderate and Severe COVID-19 Patients: A Retrospective Cohort Study. Front. Pharmacol. 2020, 11, 560209. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Zhou, F.; Hou, W.; Heybati, K.; Ali, S.; Chang, O.; Silver, Z.; Dhivagaran, T.; Ramaraju, H.B.; Wong, C.Y.; et al. Efficacy of lopinavir–ritonavir combination therapy for the treatment of hospitalized COVID-19 patients: A meta-analysis. Future Virol. 2022, 17, 169–189. [Google Scholar] [CrossRef]
- Roshanshad, A.; Kamalipour, A.; Ashraf, M.A.; Roshanshad, R.; Jafari, S.; Nazemi, P.; Akbari, M. The efficacy of remdesivir in coronavirus disease 2019 (COVID-19): A systematic review. Iran. J. Microbiol. 2020, 12, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Ansems, K.; Grundeis, F.; Dahms, K.; Mikolajewska, A.; Thieme, V.; Piechotta, V.; Metzendorf, M.I.; Stegemann, M.; Benstoem, C.; Fichtner, F. Remdesivir for the treatment of COVID-19. Cochrane Database Syst. Rev. 2021, 8, Cd014962. [Google Scholar]
- Gil-Sierra, M.D.; Briceño-Casado, M.P.; Alegre-Del Rey, E.J.; Sánchez-Hidalgo, M. Efficacy of early use of remdesivir: A systematic review of subgroup analysis. Rev. Esp. Quimioter. 2022, 35, 249–259. [Google Scholar] [CrossRef]
- Chang, S.; Wang, L.H.; Chen, B.-S. Investigating Core Signaling Pathways of Hepatitis B Virus Pathogenesis for Biomarkers Identification and Drug Discovery via Systems Biology and Deep Learning Method. Biomedicines 2020, 8, 320. [Google Scholar] [CrossRef]
- Chang, S.; Chen, J.-Y.; Chuang, Y.-J.; Chen, B.-S. Systems Approach to Pathogenic Mechanism of Type 2 Diabetes and Drug Discovery Design Based on Deep Learning and Drug Design Specifications. Int. J. Mol. Sci. 2021, 22, 166. [Google Scholar] [CrossRef]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [PubMed]
- Knox, C.; Law, V.; Jewison, T.; Liu, P.; Ly, S.; Frolkis, A.; Pon, A.; Banco, K.; Mak, C.; Neveu, V.; et al. DrugBank 3.0: A comprehensive resource for “omics” research on drugs. Nucleic Acids Res. 2011, 39, D1035–D1041. [Google Scholar] [CrossRef] [PubMed]
- Bovolenta, L.; Acencio, M.; Lemke, N. HTRIdb: An open-access database for experimentally verified human transcriptional regulation interactions. BMC Genom. 2012, 13, 405. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Tu, K.; Yang, Q.; Xiong, Y.; Wei, C.; Xie, L.; Zhu, Y.; Li, Y. ITFP: An integrated platform of mammalian transcription factors. Bioinformatics 2008, 24, 2416–2417. [Google Scholar] [CrossRef]
- Wingender, E.; Chen, X.; Hehl, R.; Karas, H.; Liebich, I.; Matys, V.; Meinhardt, T.; Prüss, M.; Reuter, I.; Schacherer, F. TRANSFAC: An integrated system for gene expression regulation. Nucleic Acids Res. 2000, 28, 316–319. [Google Scholar] [CrossRef]
- Li, J.-H.; Liu, S.; Zhou, H.; Qu, L.-H.; Yang, J.-H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2013, 42, D92–D97. [Google Scholar] [CrossRef]
- Friard, O.; Re, A.; Taverna, D.; De Bortoli, M.; Corá, D. CircuitsDB: A database of mixed microRNA/transcription factor feed-forward regulatory circuits in human and mouse. BMC Bioinform. 2010, 11, 435. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Licata, L.; Briganti, L.; Peluso, D.; Perfetto, L.; Iannuccelli, M.; Galeota, E.; Sacco, F.; Palma, A.; Nardozza, A.P.; Santonico, E.; et al. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012, 40, D857–D861. [Google Scholar] [CrossRef]
- UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2020, 49, D480–D489. [Google Scholar]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; del-Toro, N.; et al. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2013, 42, D358–D363. [Google Scholar] [CrossRef] [PubMed]
- Bader, G.D.; Betel, D.; Hogue, C.W. BIND: The Biomolecular Interaction Network Database. Nucleic Acids Res. 2003, 31, 248–250. [Google Scholar] [CrossRef] [PubMed]
- Stark, C.; Breitkreutz, B.J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef] [PubMed]
- Salwinski, L.; Miller, C.S.; Smith, A.J.; Pettit, F.K.; Bowie, J.U.; Eisenberg, D. The Database of Interacting Proteins: 2004 update. Nucleic Acids Res. 2004, 32, D449–D451. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Agajanian, M.J.; Walker, M.P.; Axtman, A.D.; Ruela-de-Sousa, R.R.; Serafin, D.S.; Rabinowitz, A.D.; Graham, D.M.; Ryan, M.B.; Tamir, T.; Nakamichi, Y.; et al. WNT Activates the AAK1 Kinase to Promote Clathrin-Mediated Endocytosis of LRP6 and Establish a Negative Feedback Loop. Cell Rep. 2019, 26, 79–93.e8. [Google Scholar] [CrossRef]
- Venkataraman, T.; Coleman, C.M.; Frieman, M.B. Overactive Epidermal Growth Factor Receptor Signaling Leads to Increased Fibrosis after Severe Acute Respiratory Syndrome Coronavirus Infection. J. Virol. 2017, 91, e00182-17. [Google Scholar] [CrossRef]
- Luo, S.; Rubinsztein, D.C. BCL2L11/BIM. Autophagy 2013, 9, 104–105. [Google Scholar] [CrossRef] [PubMed]
- Häcker, G. Apoptosis in infection. Microbes Infect. 2018, 20, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Afsar, M.; Khandelwal, N.; Chander, Y.; Riyesh, T.; Dedar, R.K.; Gulati, B.R.; Pal, Y.; Barua, S.; Tripathi, B.N.; et al. Emetine suppresses SARS-CoV-2 replication by inhibiting interaction of viral mRNA with eIF4E. Antivir. Res. 2021, 189, 105056. [Google Scholar] [CrossRef] [PubMed]
- Campa, C.C.; Franco, I.; Hirsch, E. PI3K-C2α: One enzyme for two products coupling vesicle trafficking and signal transduction. FEBS Lett. 2015, 589, 1552–1558. [Google Scholar] [CrossRef]
- Arcaro, A.; Zvelebil Marketa, J.; Wallasch, C.; Ullrich, A.; Waterfield Michael, D.; Domin, J. Class II Phosphoinositide 3-Kinases Are Downstream Targets of Activated Polypeptide Growth Factor Receptors. Mol. Cell. Biol. 2000, 20, 3817–3830. [Google Scholar] [CrossRef]
- Abdi, K.; Neves, G.; Pyun, J.; Kiziltug, E.; Ahrens, A.; Kuo, C.T. EGFR Signaling Termination via Numb Trafficking in Ependymal Progenitors Controls Postnatal Neurogenic Niche Differentiation. Cell Rep. 2019, 28, 2012–2022.e4. [Google Scholar] [CrossRef]
- Ramaiah, M.J. mTOR inhibition and p53 activation, microRNAs: The possible therapy against pandemic COVID-19. Gene Rep. 2020, 20, 100765. [Google Scholar] [CrossRef]
- Huang, K.; Wang, C.; Vagts, C.; Raguveer, V.; Finn, P.W.; Perkins, D.L. Long non-coding RNAs (lncRNAs) NEAT1 and MALAT1 are differentially expressed in severe COVID-19 patients: An integrated single cell analysis. medRxiv 2021. [Google Scholar] [CrossRef]
- Yao, J.; Wang, X.Q.; Li, Y.J.; Shan, K.; Yang, H.; Wang, Y.N.; Yao, M.D.; Liu, C.; Li, X.M.; Shen, Y.; et al. Long non-coding RNA MALAT1 regulates retinal neurodegeneration through CREB signaling. EMBO Mol. Med. 2016, 8, 346–362. [Google Scholar] [CrossRef]
- Zhuang, M.; Zhao, S.; Jiang, Z.; Wang, S.; Sun, P.; Quan, J.; Yan, D.; Wang, X. MALAT1 sponges miR-106b-5p to promote the invasion and metastasis of colorectal cancer via SLAIN2 enhanced microtubules mobility. EBioMedicine 2019, 41, 286–298. [Google Scholar] [CrossRef]
- Ivanovska, I.; Ball, A.S.; Diaz, R.L.; Magnus, J.F.; Kibukawa, M.; Schelter, J.M.; Kobayashi, S.V.; Lim, L.; Burchard, J.; Jackson, A.L.; et al. MicroRNAs in the miR-106b family regulate p21/CDKN1A and promote cell cycle progression. Mol. Cell. Biol. 2008, 28, 2167–2174. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Chen, Y.; Qi, S.; Shi, D.; Feng, L.; Sun, D. A Mini-Review on Cell Cycle Regulation of Coronavirus Infection. Front. Vet. Sci. 2020, 7, 586826. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, M.; Saso, W.; Sugiyama, R.; Ishii, K.; Ohki, M.; Nagamori, S.; Suzuki, R.; Aizaki, H.; Ryo, A.; Yun, J.-H.; et al. Epidermal growth factor receptor is a host-entry cofactor triggering hepatitis B virus internalization. Proc. Natl. Acad. Sci. USA 2019, 116, 8487–8492. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zhang, S.; Shen, Y.; Yang, Q. Epidermal growth factor receptor is a co-factor for transmissible gastroenteritis virus entry. Virology 2018, 521, 33–43. [Google Scholar] [CrossRef]
- Zheng, M.; Karki, R.; Williams, E.P.; Yang, D.; Fitzpatrick, E.; Vogel, P.; Jonsson, C.B.; Kanneganti, T.-D. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021, 22, 829–838. [Google Scholar] [CrossRef]
- DePaolo, R.W.; Lathan, R.; Rollins, B.J.; Karpus, W.J. The Chemokine CCL2 Is Required for Control of Murine Gastric Salmonella enterica Infection. Infect. Immun. 2005, 73, 6514–6522. [Google Scholar] [CrossRef]
- Gschwandtner, M.; Derler, R.; Midwood, K.S. More Than Just Attractive: How CCL2 Influences Myeloid Cell Behavior Beyond Chemotaxis. Front. Immunol. 2019, 10, 2759. [Google Scholar] [CrossRef]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef]
- Rath, P.C.; Aggarwal, B.B. TNF-induced signaling in apoptosis. J. Clin. Immunol. 1999, 19, 350–364. [Google Scholar] [CrossRef]
- Wu, B.; Peisley, A.; Richards, C.; Yao, H.; Zeng, X.; Lin, C.; Chu, F.; Walz, T.; Hur, S. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell 2013, 152, 276–289. [Google Scholar] [CrossRef]
- Alcami, A.; Koszinowski, U.H. Viral mechanisms of immune evasion. Trends Microbiol. 2000, 8, 410–418. [Google Scholar] [CrossRef]
- Sui, L.; Zhao, Y.; Wang, W.; Wu, P.; Wang, Z.; Yu, Y.; Hou, Z.; Tan, G.; Liu, Q. SARS-CoV-2 Membrane Protein Inhibits Type I Interferon Production Through Ubiquitin-Mediated Degradation of TBK1. Front. Immunol. 2021, 12, 662989. [Google Scholar] [CrossRef] [PubMed]
- Vaz de Paula, C.B.; Nagashima, S.; Liberalesso, V.; Collete, M.; da Silva, F.P.G.; Oricil, A.G.G.; Barbosa, G.S.; da Silva, G.V.C.; Wiedmer, D.B.; da Silva Dezidério, F.; et al. COVID-19: Immunohistochemical Analysis of TGF-β Signaling Pathways in Pulmonary Fibrosis. Int. J. Mol. Sci. 2021, 23, 168. [Google Scholar] [CrossRef]
- Ferner, R.E.; Aronson, J.K. Remdesivir in COVID-19. BMJ 2020, 369, m1610. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, J.; Hu, D.; Pan, P.; Liang, L.; Wu, W.; Tang, Y.; Huang, X.R.; Yu, X.; Wu, J.; et al. SARS-CoV-2 N Protein Induces Acute Kidney Injury via Smad3-Dependent G1 Cell Cycle Arrest Mechanism. Adv. Sci. 2022, 9, e2103248. [Google Scholar] [CrossRef]
- Kellici, T.F.; Pilka, E.S.; Bodkin, M.J. Therapeutic Potential of Targeting Plasminogen Activator Inhibitor-1 in COVID-19. Trends Pharmacol. Sci. 2021, 42, 431–433. [Google Scholar] [CrossRef]
- Angiolillo, A.L.; Sgadari, C.; Taub, D.D.; Liao, F.; Farber, J.M.; Maheshwari, S.; Kleinman, H.K.; Reaman, G.H.; Tosato, G. Human interferon-inducible protein 10 is a potent inhibitor of angiogenesis in vivo. J. Exp. Med. 1995, 182, 155–162. [Google Scholar] [CrossRef]
- Romagnani, P.; Annunziato, F.; Lazzeri, E.; Cosmi, L.; Beltrame, C.; Lasagni, L.; Galli, G.; Francalanci, M.; Manetti, R.; Marra, F.; et al. Interferon-inducible protein 10, monokine induced by interferon gamma, and interferon-inducible T-cell alpha chemoattractant are produced by thymic epithelial cells and attract T-cell receptor (TCR) alphabeta+ CD8+ single-positive T cells, TCRgammadelta+ T cells, and natural killer-type cells in human thymus. Blood 2001, 97, 601–607. [Google Scholar]
- Sidahmed, A.M.; León, A.J.; Bosinger, S.E.; Banner, D.; Danesh, A.; Cameron, M.J.; Kelvin, D.J. CXCL10 contributes to p38-mediated apoptosis in primary T lymphocytes in vitro. Cytokine 2012, 59, 433–441. [Google Scholar] [CrossRef]
- BAX BCL2 Associated X, Apoptosis Regulator [Homo sapiens (Human)]. Available online: https://www.ncbi.nlm.nih.gov/gene/581 (accessed on 18 July 2022).
- Milhas, D.; Cuvillier, O.; Therville, N.; Clavé, P.; Thomsen, M.; Levade, T.; Benoist, H.; Ségui, B. Caspase-10 Triggers Bid Cleavage and Caspase Cascade Activation in FasL-induced Apoptosis. J. Biol. Chem. 2005, 280, 19836–19842. [Google Scholar] [CrossRef]
- Dzimianski, J.V.; Scholte, F.E.M.; Bergeron, É.; Pegan, S.D. ISG15: It’s Complicated. J. Mol. Biol. 2019, 431, 4203–4216. [Google Scholar] [CrossRef] [PubMed]
- Bizzotto, J.; Sanchis, P.; Abbate, M.; Lage-Vickers, S.; Lavignolle, R.; Toro, A.; Olszevicki, S.; Sabater, A.; Cascardo, F.; Vazquez, E.; et al. SARS-CoV-2 Infection Boosts MX1 Antiviral Effector in COVID-19 Patients. iScience 2020, 23, 101585. [Google Scholar] [CrossRef] [PubMed]
- Kausar, S.; Said Khan, F.; Ishaq Mujeeb Ur Rehman, M.; Akram, M.; Riaz, M.; Rasool, G.; Hamid Khan, A.; Saleem, I.; Shamim, S.; Malik, A. A review: Mechanism of action of antiviral drugs. Int. J. Immunopathol. Pharmacol. 2021, 35, 20587384211002621. [Google Scholar] [CrossRef] [PubMed]
- Piacentini, S.; La Frazia, S.; Riccio, A.; Pedersen, J.Z.; Topai, A.; Nicolotti, O.; Rossignol, J.F.; Santoro, M.G. Nitazoxanide inhibits paramyxovirus replication by targeting the Fusion protein folding: Role of glycoprotein-specific thiol oxidoreductase ERp57. Sci. Rep. 2018, 8, 10425. [Google Scholar] [CrossRef] [PubMed]
- Segrelles, C.; Contreras, D.; Navarro, E.M.; Gutiérrez-Muñoz, C.; García-Escudero, R.; Paramio, J.M.; Lorz, C. Bosutinib Inhibits EGFR Activation in Head and Neck Cancer. Int. J. Mol. Sci. 2018, 19, 1824. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Schmid-Bindert, G.; Zhou, C. Erlotinib in the treatment of advanced non-small cell lung cancer: An update for clinicians. Ther. Adv. Med. Oncol. 2012, 4, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Kim, J. Combination Effect of Cilengitide with Erlotinib on TGF-β1-Induced Epithelial-to-Mesenchymal Transition in Human Non-Small Cell Lung Cancer Cells. Int. J. Mol. Sci. 2022, 23, 3423. [Google Scholar] [CrossRef]
- Naik, R.R.; Shakya, A.K.; Aladwan, S.M.; El-Tanani, M. Kinase Inhibitors as Potential Therapeutic Agents in the Treatment of COVID-19. Front. Pharmacol. 2022, 13, 806568. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 5757, Estradiol. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/5757 (accessed on 25 July 2022).
- Kröger, A.; Dallügge, A.; Kirchhoff, S.; Hauser, H. IRF-1 reverts the transformed phenotype of oncogenically transformed cells in vitro and in vivo. Oncogene 2003, 22, 1045–1056. [Google Scholar] [CrossRef][Green Version]
- Malek, D.; Gust, R.; Kleuser, B. 17-Beta-estradiol inhibits transforming-growth-factor-beta-induced MCF-7 cell migration by Smad3-repression. Eur. J. Pharmacol. 2006, 534, 39–47. [Google Scholar] [CrossRef]
- Penna, C.; Mercurio, V.; Tocchetti, C.G.; Pagliaro, P. Sex-related differences in COVID-19 lethality. Br. J. Pharmacol. 2020, 177, 4375–4385. [Google Scholar] [CrossRef] [PubMed]
- Bhopal, S.S.; Bhopal, R. Sex differential in COVID-19 mortality varies markedly by age. Lancet 2020, 396, 532–533. [Google Scholar] [CrossRef]
- Doerre, A.; Doblhammer, G. The influence of gender on COVID-19 infections and mortality in Germany: Insights from age-and gender-specific modeling of contact rates, infections, and deaths in the early phase of the pandemic. PLoS ONE 2022, 17, e0268119. [Google Scholar] [CrossRef] [PubMed]
- Zafari Zangeneh, F.; Shoushtari, M.S. Estradiol and COVID-19: Does 17-Estradiol Have an Immune-Protective Function in Women Against Coronavirus? J. Fam. Reprod. Health 2021, 15, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Suba, Z. Prevention and therapy of COVID-19 via exogenous estrogen treatment for both male and female patients: Prevention and therapy of COVID-19. J. Pharm. Pharm. Sci. 2020, 23, 75–85. [Google Scholar] [CrossRef]
- Gil-Ad, I.; Zolokov, A.; Lomnitski, L.; Taler, M.; Bar, M.; Luria, D.; Ram, E.; Weizman, A. Evaluation of the potential anti-cancer activity of the antidepressant sertraline in human colon cancer cell lines and in colorectal cancer-xenografted mice. Int. J. Oncol. 2008, 33, 277–286. [Google Scholar] [CrossRef]
- Chen, S.; Xuan, J.; Wan, L.; Lin, H.; Couch, L.; Mei, N.; Dobrovolsky, V.N.; Guo, L. Sertraline, an Antidepressant, Induces Apoptosis in Hepatic Cells Through the Mitogen-Activated Protein Kinase Pathway. Toxicol. Sci. 2014, 137, 404–415. [Google Scholar] [CrossRef]
- Xia, D.; Zhang, Y.T.; Xu, G.P.; Yan, W.W.; Pan, X.R.; Tong, J.H. Sertraline exerts its antitumor functions through both apoptosis and autophagy pathways in acute myeloid leukemia cells. Leuk. Lymphoma 2017, 58, 2208–2217. [Google Scholar] [CrossRef]
- Halperin, D.; Reber, G. Influence of antidepressants on hemostasis. Dialogues Clin. Neurosci. 2007, 9, 47–59. [Google Scholar] [CrossRef]
- Chen, B.S.; Wu, C.C. Systems Biology: An Integrated Platform for Bioinformatics, Systems Synthetic Biology and Systems Metabolic Engineering; Nova Publishers: Hauppauge, NY, USA, 2014. [Google Scholar]
- Puray-Chavez, M.; LaPak, K.M.; Schrank, T.P.; Elliott, J.L.; Bhatt, D.P.; Agajanian, M.J.; Jasuja, R.; Lawson, D.Q.; Davis, K.; Rothlauf, P.W.; et al. Systematic analysis of SARS-CoV-2 infection of an ACE2-negative human airway cell. Cell Rep. 2021, 36, 109364. [Google Scholar] [CrossRef]
- Dong, J.; Yao, Z.-J.; Zhang, L.; Luo, F.; Lin, Q.; Lu, A.-P.; Chen, A.F.; Cao, D.-S. PyBioMed: A python library for various molecular representations of chemicals, proteins and DNAs and their interactions. J. Cheminformatics 2018, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: A simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef] [PubMed]
- Seçilmiş, D.; Hillerton, T.; Morgan, D.; Tjärnberg, A.; Nelander, S.; Nordling, T.E.M.; Sonnhammer, E.L.L. Uncovering cancer gene regulation by accurate regulatory network inference from uninformative data. NPJ Syst. Biol. Appl. 2020, 6, 37. [Google Scholar] [CrossRef]
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A.; et al. Discovering the anticancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 2020, 1, 235–248. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Node | Candidate HPI-GWGEN | Amplification Stage Real HPI-GWGEN | Saturation Stage Real HPI-GWGEN |
| Receptor | 2294 | 2294 | 2294 |
| Transcription factor | 1452 | 1449 | 1449 |
| Protein coding | 13,989 | 13,980 | 13,981 |
| miRNA | 154 | 153 | 152 |
| lncRNA | 8827 | 1023 | 664 |
| Virus | 11 | 11 | 11 |
| Total nodes | 26,727 | 18,910 | 18,551 |
| Edge | Candidate HPI-GWGEN | Amplification Stage Real HPI-GWGEN | Saturation Stage Real HPI-GWGEN |
| PPIs | 4,722,699 | 953,464 | 1,053,818 |
| TF -> Receptor | 14,633 | 9493 | 8697 |
| TF -> TF | 11,846 | 7168 | 6590 |
| TF -> Protein | 84,183 | 55,326 | 51,376 |
| TF -> miRNA | 178 | 102 | 88 |
| TF -> lncRNA | 301 | 290 | 291 |
| TF -> Virus | 15,972 | 131 | 59 |
| miRNA -> Receptor | 88,424 | 10,871 | 10,267 |
| miRNA -> TF | 71,046 | 9228 | 8256 |
| miRNA -> Protein | 570,830 | 72,563 | 67,652 |
| miRNA -> lncRNA | 5502 | 581 | 478 |
| miRNA -> Virus | 1694 | 23 | 12 |
| lncRNA -> Receptor | 436 | 313 | 306 |
| lncRNA -> TF | 472 | 270 | 287 |
| lncRNA -> Protein | 4274 | 2288 | 2337 |
| lncRNA -> miRNA | 7 | 5 | 4 |
| lncRNA -> lncRNA | 4 | 4 | 3 |
| lncRNA -> Virus | 97,097 | 753 | 244 |
| Virus -> Virus | 121 | 22 | 4 |
| Total edges | 5,689,719 | 1,122,895 | 1,210,769 |
| KEGG Pathway | Count | p-Value |
|---|---|---|
| Cell cycle | 86 | 6.32 × 10−17 |
| FoxO signaling pathway | 80 | 7.83 × 10−12 |
| Pathways in cancer | 236 | 1.37 × 10−10 |
| Hepatitis B | 90 | 4.19 × 10−12 |
| Hepatitis C | 84 | 1.81 × 10−8 |
| ErbB signaling pathway | 52 | 5.17 × 10−8 |
| Tight junction | 87 | 1.00 × 10−7 |
| MAPK signaling pathway | 133 | 6.95 × 10−7 |
| Endocytosis | 113 | 6.90 × 10−6 |
| KEGG Pathway | Count | p-Value |
|---|---|---|
| Pathways in cancer | 227 | 6.39 × 10−9 |
| Th17 cell differentiation | 59 | 8.13 × 10−7 |
| Cell cycle | 66 | 1.21 × 10−6 |
| Osteoclast differentiation | 66 | 2.46 × 10−6 |
| T cell receptor signaling pathway | 56 | 2.93 × 10−6 |
| Human T-cell leukemia virus 1 infection | 102 | 3.81 × 10−6 |
| Apoptosis | 68 | 6.66 × 10−6 |
| Hepatitis B | 77 | 1.59 × 10−5 |
| Hepatitis C | 75 | 1.7 × 10−5 |
| Validation Loss | Validation Accuracy | Test Loss | Test Accuracy | |
|---|---|---|---|---|
| 1 | 0.1656409 | 0.95065 | 0.2180897 | 0.9521126 |
| 2 | 0.1929858 | 0.9438001 | 0.1789017 | 0.9493726 |
| 3 | 0.1807019 | 0.9504943 | 0.1856036 | 0.9519569 |
| 4 | 0.1761861 | 0.9507278 | 0.2022759 | 0.951521 |
| 5 | 0.1868679 | 0.9527516 | 0.216308 | 0.9517078 |
| 6 | 0.1671205 | 0.9526701 | 0.1956254 | 0.9513031 |
| 7 | 0.1850127 | 0.9536821 | 0.1776747 | 0.9516767 |
| 8 | 0.1905898 | 0.9474545 | 0.1865388 | 0.9505246 |
| 9 | 0.1813395 | 0.9499455 | 0.1792253 | 0.951988 |
| 10 | 0.1789479 | 0.9497898 | 0.1836274 | 0.9529221 |
| Average | 0.1805393 | 0.9501966 | 0.192387 | 0.9515085 |
| Standard Deviation | 0.0086033 | 0.0027209 | 0.0143861 | 0.0009184 |
| Candidate Drugs | Regulation Ability (L1000) | Sensitivity (PRISM) | Toxicity (LC50, mol/kg) |
|---|---|---|---|
| Downregulation of EGFR | |||
| Fursultiamine | −0.932 | −0.035 | 2.928 |
| fasudil | −0.791 | 0.367 | 3.083 |
| * Bosutinib | −0.585 | −0.017 | 6.273 |
| cefaclor | −0.383 | −0.099 | 3.666 |
| * Erlotinib | −0.229 | −0.332 | 5.73 |
| Downregulation of AKT1 | |||
| Iproniazid | −0.802 | −0.337 | 2.82 |
| gabexate | −0.733 | −0.134 | 4.487 |
| diazoxide | −0.544 | 0.393 | 3.058 |
| * Bosutinib | −0.434 | −0.017 | 6.273 |
| Apoptosis-activator-II | −0.302 | 0.037 | 5.695 |
| Upregulation of IFNB1 | |||
| topiramate | 0.848 | 0.161 | 2.289 |
| * 17-beta-estradiol | 0.72 | −0.27 | 5.215 |
| nitrofural | 0.691 | −0.404 | 3.88 |
| raclopride | 0.514 | 0.078 | 3.851 |
| Acyclovir | 0.363 | 0.3078 | 2.452 |
| Downregulation of SMAD3 | |||
| niridazole | −0.772 | 0.264 | 2.746 |
| * Erlotinib | −0.537 | −0.332 | 5.730 |
| * 17-beta-estradiol | −0.503 | −0.27 | 5.215 |
| Azacitidine | −0.412 | −0.393 | 2.049 |
| Nobiletin | −0.312 | −0.448 | 5.214 |
| Upregulation of JUN | |||
| oleoylethanolamide | 0.878 | −0.15 | 3.54 |
| carmoxirole | 0.776 | −0.006 | 4.477 |
| zibotentan | 0.611 | 0.209 | 3.013 |
| * Sertraline | 0.557 | 0.097 | 7.434 |
| Limonin | 0.367 | −0.36 | 6.726 |
| Target | EGFR | AKT1 | IFNB1 | SMAD3 | |
|---|---|---|---|---|---|
| Drug | |||||
| Bosutinib | V | V | |||
| Erlotinib | V | V | |||
| 17-beta-estradiol | V | V | |||
| Structure of multiple-molecule drug | |||||
| Bosutinib | Erlotinib | 17-beta-estradiol | |||
 |  |  | |||
| Target | SMAD3 | IFNB1 | JUN | |
|---|---|---|---|---|
| Drug | ||||
| Erlotinib | V | |||
| 17-beta-estradiol | V | V | ||
| Sertraline | V | |||
| Structure of multiple-molecule drug | ||||
| Erlotinib | 17-beta-estradiol | Sertraline | ||
 |  |  | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.-G.; Chen, B.-S. Multiple-Molecule Drug Repositioning for Disrupting Progression of SARS-CoV-2 Infection by Utilizing the Systems Biology Method through Host-Pathogen-Interactive Time Profile Data and DNN-Based DTI Model with Drug Design Specifications. Stresses 2022, 2, 405-436. https://doi.org/10.3390/stresses2040029
Wang C-G, Chen B-S. Multiple-Molecule Drug Repositioning for Disrupting Progression of SARS-CoV-2 Infection by Utilizing the Systems Biology Method through Host-Pathogen-Interactive Time Profile Data and DNN-Based DTI Model with Drug Design Specifications. Stresses. 2022; 2(4):405-436. https://doi.org/10.3390/stresses2040029
Chicago/Turabian StyleWang, Cheng-Gang, and Bor-Sen Chen. 2022. "Multiple-Molecule Drug Repositioning for Disrupting Progression of SARS-CoV-2 Infection by Utilizing the Systems Biology Method through Host-Pathogen-Interactive Time Profile Data and DNN-Based DTI Model with Drug Design Specifications" Stresses 2, no. 4: 405-436. https://doi.org/10.3390/stresses2040029
APA StyleWang, C.-G., & Chen, B.-S. (2022). Multiple-Molecule Drug Repositioning for Disrupting Progression of SARS-CoV-2 Infection by Utilizing the Systems Biology Method through Host-Pathogen-Interactive Time Profile Data and DNN-Based DTI Model with Drug Design Specifications. Stresses, 2(4), 405-436. https://doi.org/10.3390/stresses2040029