
Laccase from Melanocarpus albomyces: Molecular Docking Analysis with First-Generation Tetracyclines Through a Mechanistic Approach

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Molecular Docking
2.2. Electronic Properties and Proton Transfer Mechanism
3. Results and Discussion
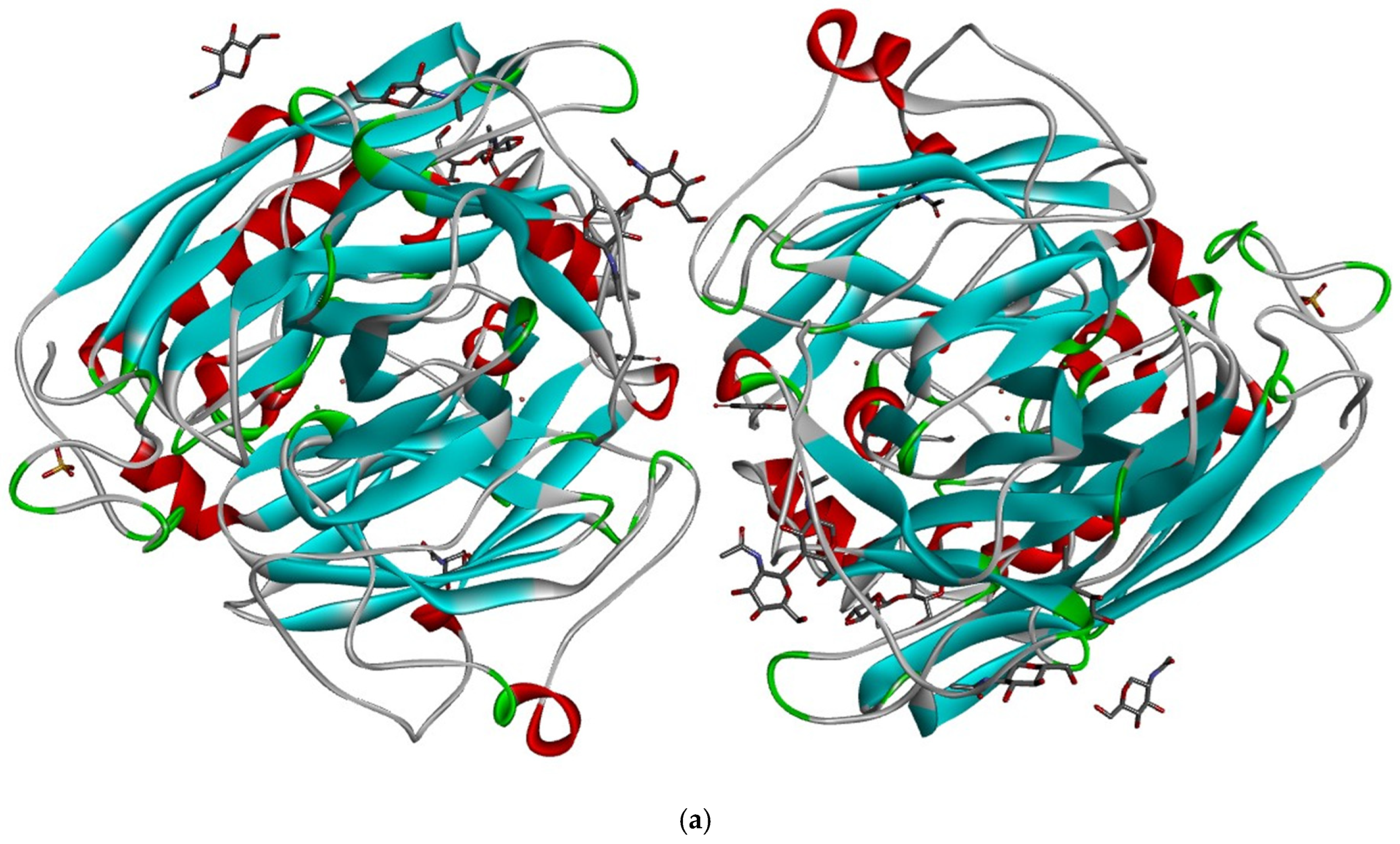
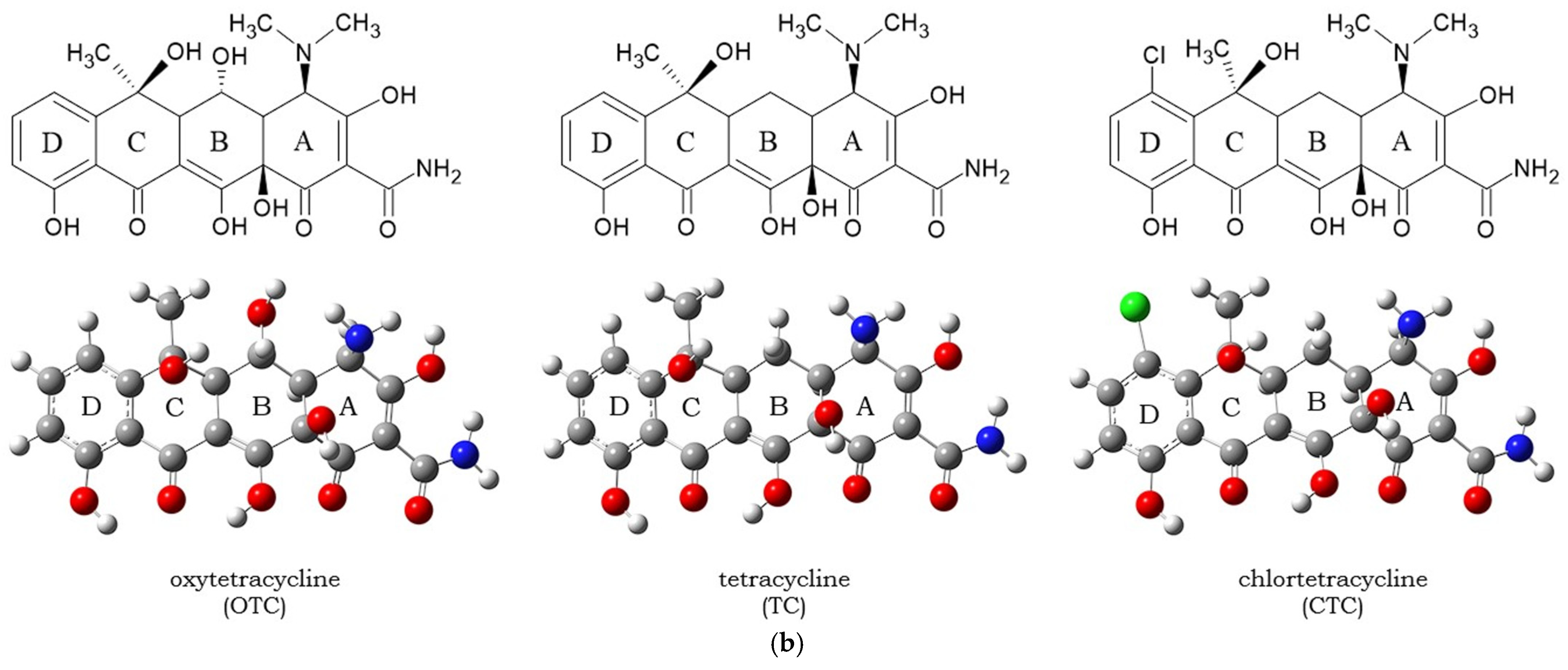
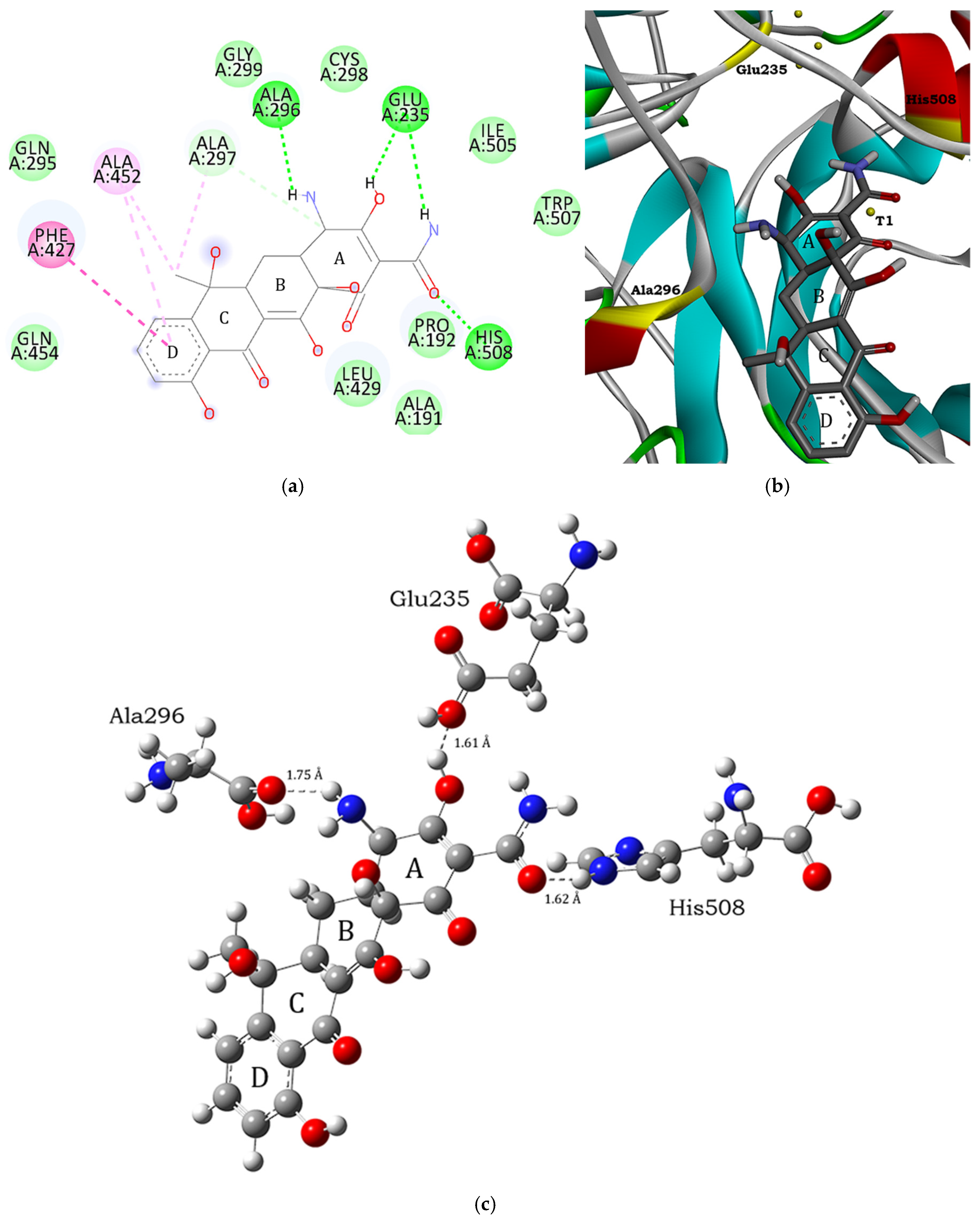
3.1. Molecular Docking and Intermolecular Interactions of Substrates–MaL Active Site
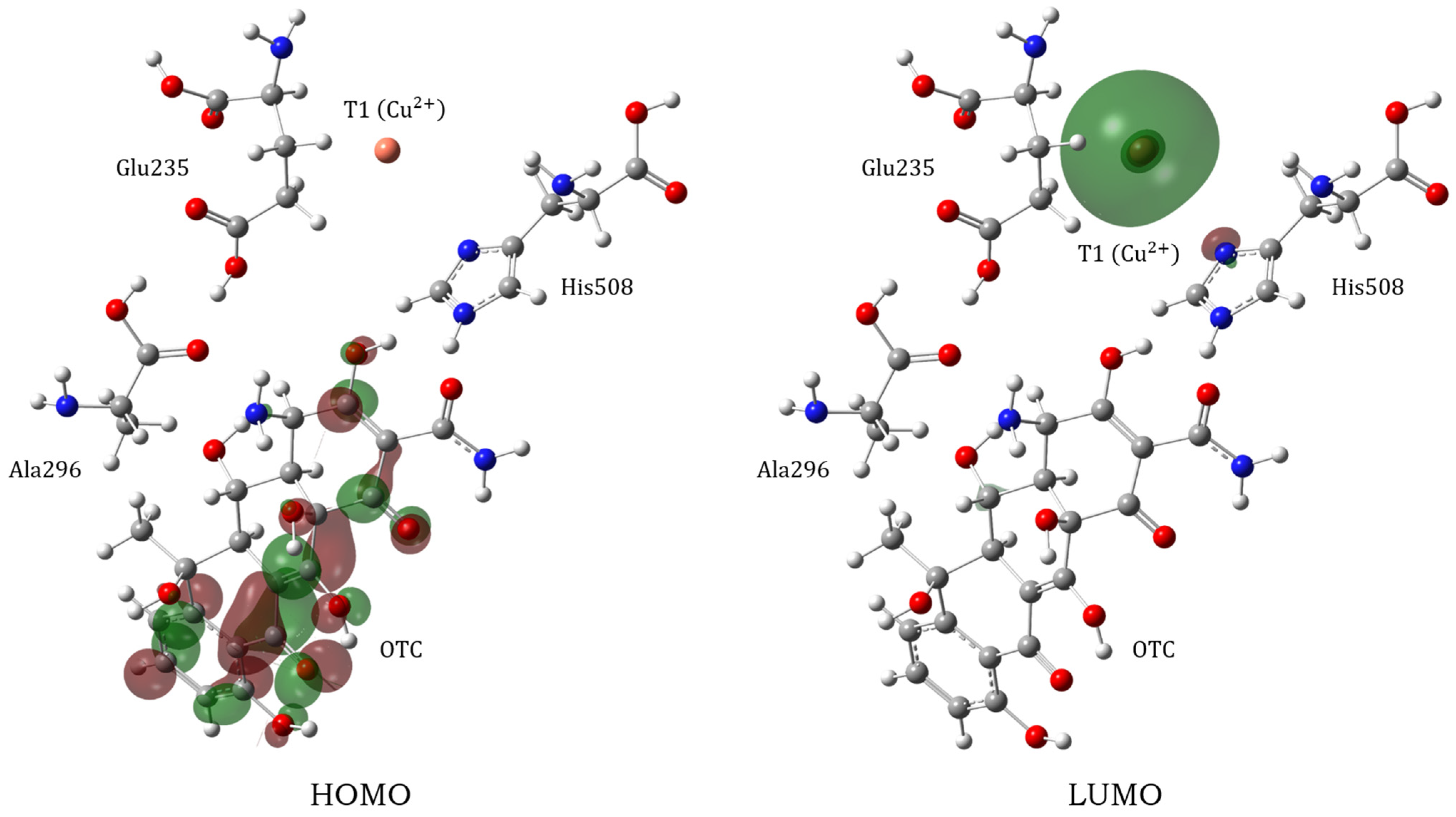
3.2. Electronic Properties and Proton Transfer Mechanism
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rivera-Hoyos, C.M.; Morales-Álvarez, E.D.; Poutou-Piñales, R.A.; Pedroza-Rodríguez, A.M.; Rodríguez-Vázquez, R.; Delgado-Boada, J.M. Fungal laccases. Fungal Biol. Rev. 2013, 27, 67–82. [Google Scholar] [CrossRef]
- Kudanga, T. Bacterial laccases: A general introduction. In Bacterial Laccases Engineering, Immobilization, Heterologous Production, and Industrial Applications; Academic Press: New York, NY, USA, 2024; Volume 1, pp. 1–9. [Google Scholar] [CrossRef]
- Tron, T. Laccases. In Encyclopedia of Metalloproteins; Kretsinger, R.H., Uversky, V.N., Permyakov, E.A., Eds.; Springer: New York, NY, USA, 2013. [Google Scholar]
- Jones, S.M.; Solomon, E.I. Electron transfer and reaction mechanism of laccases. Cell Mol. Life Sci. 2015, 72, 869–883. [Google Scholar] [CrossRef] [PubMed]
- Orlando, C.; Rizzo, I.C.; Arrigoni, F.; Zampolli, J.; Mangiagalli, M.; Di Gennaro, P.; Lotti, M.; De Gioia, L.; Marino, T.; Greco, C.; et al. Mechanism of non-phenolic substrate oxidation by the fungal laccase Type 1 copper site from Trametes versicolor: The case of benzo[a]pyrene and anthracene. Dalton Trans. 2024, 53, 12152–12161. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xie, Q.; Luo, M.; Chen, X. Theoretical insights into the catalytic oxidation of phenols and arylamines by laccases via the proton-coupled electron transfer mechanism. J. Phys. Chem. B 2024, 128, 8915–8926. [Google Scholar] [CrossRef]
- Guan, Z.-B.; Luo, Q.; Wang, H.-R.; Chen, Y.; Liao, X.-R. Bacterial laccases: Promising biological green tools for industrial applications. Cell Mol. Life Sci. 2018, 75, 3569–3592. [Google Scholar] [CrossRef]
- Chopra, I.; Roberts, M. Tetracycline antibiotics: Mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol. Mol. Biol. Rev. 2001, 65, 232–260. [Google Scholar] [CrossRef]
- Dahl, E.L.; Shock, J.L.; Shenai, B.R.; Gut, J.; DeRisi, J.L.; Rosenthal, P.J. Tetracyclines specifically target the apicoplast of the malaria parasite Plasmodium falciparum. Antimicrob. Agents Chemother. 2006, 50, 3124–3131. [Google Scholar] [CrossRef]
- Amangelsin, Y.; Semenova, Y.; Dadar, M.; Aljofan, M.; Bjørklund, G. The impact of tetracycline pollution on the aquatic environment and removal strategies. Antibiotics 2023, 12, 440. [Google Scholar] [CrossRef]
- Suda, T.; Hata, T.; Kawai, S.; Okamura, H.; Nishida, T. Treatment of tetracycline antibiotics by laccase in the presence of 1-hydroxybenzotriazole. Bioresour. Technol. 2012, 103, 498–501. [Google Scholar] [CrossRef]
- Ding, H.; Wu, Y.; Zou, B.; Lou, Q.; Zhang, W.; Zhong, J.; Lu, L.; Dai, G. Simultaneous removal and degradation characteristics of sulfonamide, tetracycline, and quinolone antibiotics by laccase-mediated oxidation coupled with soil adsorption. J. Hazard. Mater. 2016, 307, 350–358. [Google Scholar] [CrossRef]
- de Fátima, N.G.; Barriga, A.; Cáceres, J.C.; Pinto, E.; Cabrera, R. Oxidation of chlortetracycline and its isomers by Botrytis aclada laccase in the absence of mediators: pH dependence and identification of transformation products by LC–MS. Biodegradation 2024, 35, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Meng, F. Construction of a continuous packed bed laccase reactor for the elimination of tetracyclines in seawater. J. Environ. Chem. Eng. 2024, 12, 111939. [Google Scholar] [CrossRef]
- Wang, X.; Meng, F.; Zhang, B.; Xia, Y. Elimination of tetracyclines in seawater by laccase-mediator system. Chemosphere 2023, 333, 138916. [Google Scholar] [CrossRef]
- Yang, J.; Lin, Y.; Yang, X.; Ng, T.B.; Ye, X.; Lin, J. Degradation of tetracycline by immobilized laccase and the proposed transformation pathway. J. Hazard. Mat. 2017, 322, 525–531. [Google Scholar] [CrossRef]
- Tian, Q.; Dou, X.; Huang, L.; Wang, L.; Meng, D.; Zhai, L.; Shen, Y.; You, C.; Guan, Z.; Liao, X. Characterization of a robust cold-adapted and thermostable laccase from Pycnoporus sp. SYBC-L10 with a strong ability for the degradation of tetracycline and oxytetracycline by laccase-mediated oxidation. J. Hazard. Mater. 2020, 382, 121084. [Google Scholar] [CrossRef] [PubMed]
- Kallio, J.P.; Auer, S.; Jänis, J.; Andberg, M.; Kruus, K.; Rouvinen, J.; Koivula, A.; Hakulinen, N. Structure–function studies of a Melanocarpus albomyces laccase suggest a pathway for oxidation of phenolic compounds. J. Mol. Biol. 2009, 392, 895–909. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Torres, P.H.M.; Sodero, A.C.R.; Jofily, P.; Silva, F.P., Jr. Key topics in molecular docking for drug design. Int. J. Mol. Sci. 2019, 20, 4574. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Szentpály, L.V.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Gazquez, J.L. Hardness and softness in density functional theory. In Chemical Hardness; Sen, K.D., Ed.; Springer: Berlin, Germany, 1993; Volume 80, p. 27. [Google Scholar]
- Parr, R.G.; Donnelly, R.A.; Levy, M.; Palke, W.E. Electronegativity: The density functional viewpoint. J. Chem. Phy. 1978, 68, 3801–3807. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Fox. Gaussian 09, Revision E. 01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Contreras, R.; Muñoz-Espinoza, J.; Sánchez, B. Theoretical models of solubility for organic solvents. A conceptual density functional theory approach. J. Mol. Liq. 2024, 403, 124736. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 1994; Chapters 4–5; pp. 70–104. [Google Scholar]
- Franco-Pérez, M.; Gázquez, J.L.; Ayers, P.W.; Vela, A. Revisiting the definition of the electronic chemical potential, chemical hardness, and softness at finite temperatures. J. Chem. Phys. 2015, 143, 154103. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Levy, M. Comment on “Significance of the highest occupied Kohn-Sham eigenvalue”. Phys. Rev. B Condens. Matter Mater. Phys. 1997, 56, 16021–16028. [Google Scholar] [CrossRef]
- Salvador, P.; Paizs, B.; Duran, M.; Suhai, S. On the effect of the BSSE on intermolecular potential energy surfaces. Comparison of a priori and a posteriori BSSE correction scheme. J. Comput. Chem. 2001, 22, 765–786. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Peng, C.; Bernhard Schlegel, H. Combining synchronous transit and Quasi-Newton methods to find transition states. Isr. J. Chem. 1993, 33, 449–454. [Google Scholar] [CrossRef]
- Agu, P.C.; Afiukwa, C.A.; Orji, O.U.; Ezeh, E.M.; Ofoke, I.H.; Ogbu, C.O.; Ugwaju, E.I.; Aja, P.M. Molecular docking as a tool for the discovery of molecular targets of nutraceuticals in diseases management. Sci. Rep. 2023, 13, 13398. [Google Scholar] [CrossRef]
- Pantsar, T.; Poso, A. Binding affinity via docking: Fact and fiction. Molecules 2018, 23, 1899. [Google Scholar] [CrossRef]
- Chen, T.; Shu, X.; Zhou, H.; Beckford, F.A.; Misir, M. Algorithm selection for protein-ligand docking strategies and analysis on ACE. Sci. Rep. 2023, 13, 8219. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Pritchard, B.P.; Altarawy, D.; Didier, B.T.; Gibson, T.D.; Windus, T.L. A new basis set exchange: An open, up-to-date resource for the molecular sciences community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef] [PubMed]
- Bender, M.L.; Kézdy, F.J. Proton-Transfer in Enzymatic Catalysis. In Proton-Transfer Reactions; Caldin, E., Gold, V., Eds.; Springer: Boston, MA, USA, 1975. [Google Scholar]
- Pusuluk, O.; Farrow, T.; Deliduman, C.; Burnett, K.; Vedral, V. Proton tunnelling in hydrogen bonds and its implications in an induced-fit model of enzyme catalysis. Proc. R. Soc. A 2018, 474, 20180037. [Google Scholar] [CrossRef]
- Galli, C.; Madzak, C.; Vadalà, R.; Jolivalt, C.; Gentili, P. Concerted electron/proton transfer mechanism in the oxidation of phenols by laccase. Chembiochem 2013, 14, 2500–2505. [Google Scholar] [CrossRef]
- Siegbahn, P. How protons move in enzymes-The case of nitrogenase. J. Phys. Chem. B 2023, 127, 2156–2159. [Google Scholar] [CrossRef]
- Riccardi, D.; König, P.; Prat-Resina, X.; Yu, H.; Elstner, M.; Frauenheim, T.; Cui, Q. “Proton Holes” in Long-Range Proton Transfer Reactions in Solution and Enzymes: A Theoretical Analysis. J. Am. Chem. Soc. 2006, 128, 16302–16311. [Google Scholar] [CrossRef] [PubMed]
- Solomon, E.I.; Sundaram, U.M.; Machonkin, T.E. Multicopper Oxidases and Oxygenases. Chem. Rev. 1996, 96, 2563–2606. [Google Scholar] [CrossRef]
- Randall, D.W.; Gamelin, D.R.; LaCroix, L.B.; Solomon, E.I. Electronic Structure Contributions to Electron Transfer in Blue Cu and CuA. J. Biol. Inorg. Chem. 2000, 5, 16–29. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Conformers | RMSD | Conformers | RMSD | ||
|---|---|---|---|---|---|---|
| OTC | 1 | −5.80 | 0.00 | 11 | −5.33 | 0.22 |
| 2 | −5.52 | 0.00 | 12 | −5.32 | 0.16 | |
| 3 | −5.50 | 0.16 | 13 | −5.28 | 0.27 | |
| 4 | −5.50 | 0.21 | 14 | −5.29 | 0.00 | |
| 5 | −5.48 | 0.06 | 15 | −5.17 | 0.35 | |
| 6 | −5.48 | 0.11 | 16 | −5.16 | 0.36 | |
| 7 | −5.47 | 0.07 | 17 | −5.12 | 0.32 | |
| 8 | −5.46 | 0.15 | 18 | −4.85 | 0.00 | |
| 9 | −5.38 | 0.10 | 19 | −4.82 | 0.15 | |
| 10 | −5.37 | 0.15 | 20 | −4.76 | 0.10 | |
| TC | 1 | −6.20 | 0.00 | 11 | −5.79 | 0.08 |
| 2 | −5.95 | 0.00 | 12 | −5.73 | 0.23 | |
| 3 | −5.92 | 0.63 | 13 | −5.73 | 0.10 | |
| 4 | −5.03 | 1.04 | 14 | −5.73 | 0.23 | |
| 5 | −4.89 | 1.79 | 15 | −5.70 | 0.08 | |
| 6 | −5.86 | 0.00 | 16 | −5.66 | 0.22 | |
| 7 | −5.86 | 0.10 | 17 | −5.25 | 0.00 | |
| 8 | −5.83 | 0.11 | 18 | −5.22 | 0.06 | |
| 9 | −5.82 | 0.14 | 19 | −5.20 | 0.30 | |
| 10 | −5.81 | 0.07 | 20 | −4.71 | 0.00 | |
| CTC | 1 | −5.85 | 0.00 | 11 | −4.43 | 0.42 |
| 2 | −4.68 | 1.95 | 12 | −4.32 | 1.02 | |
| 3 | −4.49 | 0.89 | 13 | −4.29 | 1.07 | |
| 4 | −4.44 | 0.88 | 14 | −4.54 | 0.00 | |
| 5 | −5.47 | 0.00 | 15 | −4.49 | 0.13 | |
| 6 | −5.22 | 0.77 | 16 | −4.45 | 0.15 | |
| 7 | −4.84 | 0.00 | 17 | −4.38 | 0.32 | |
| 8 | −4.68 | 0.21 | 18 | −4.28 | 0.50 | |
| 9 | −4.58 | 0.00 | 19 | −4.21 | 0.00 | |
| 10 | −4.57 | 0.22 | 20 | −4.07 | 0.00 |
| Complex | |||
|---|---|---|---|
| OTC—MaL | −1780.22 | −5.30 | 5.95 |
| TC—MaL | −1780.51 | −5.59 | 5.95 |
| CTC—MaL | −1779.53 | −4.61 | 5.95 |
| Complex | H Bond | Distance | Donor NBO 1 | Acceptor NBO 1 | |||
|---|---|---|---|---|---|---|---|
| OTC (20)—MaL | −4.76 | −1.92 | 1.69 | BD(2)C82-O86 | BD*(1)N38-H39 | 14.90 | |
| BD(1)N38-H39 | BD*(2)C82-O86 | 0.46 | |||||
| TC (2)—MaL | −5.95 | −2.12 | 1.61 | LP(1)O10 | BD*(1)O77-H78 | 16.45 | |
| BD(1)O77-H78 | BD*(1)C8-010 | 0.78 | |||||
| 1.62 | LP(2)O83 | BD*(1)N38-H39 | 25.24 | ||||
| BD(1)C37-H44 | BD*(2)C79-O83 | 0.16 | |||||
| 1.75 | LP(1)O21 | BD*(1)N74-H75 | 10.24 | ||||
| BD(1)N74-H75 | BD*(1)C20-O21 | 0.53 | |||||
| CTC (1)—MaL | −5.85 | −2.21 | 1.64 | BD(1)O76-H77 | BD*(1)O96-H97 | 1.17 | |
| LP(1)O96 | BD*(1)O76-H77 | 0.67 | |||||
| 1.71 | BD(2)C78-O82 | BD*(1)N37-H38 | 5.66 | ||||
| BD(1)N37-H38 | BD*(2)C78-O82 | 0.69 | |||||
| 1.63 | BD(1)N73-H74 | BD*(1)C19-O20 | 0.81 | ||||
| BD(2)C19-O20 | BD*(1)N73-H74 | 1.33 |
| Isolated AS/Substrate | |||||||
|---|---|---|---|---|---|---|---|
| AS1–MaL a | −0.861 | 0.269 | −6.275 | 11.986 | 0.0834 | 1.642 | 0.282 |
| AS2–MaL b | −0.816 | 0.264 | −5.860 | 11.453 | 0.0873 | 1.499 | 0.134 |
| AS3–MaL c | −0.812 | 0.269 | −5.765 | 11.461 | 0.0872 | 1.450 | 0.034 |
| OTC | −0.302 | 0.005 | −3.158 | 3.255 | 0.3072 | 1.532 | |
| TC | −0.202 | 0.004 | −2.095 | 2.179 | 0.4590 | 1.008 | |
| CTC | −0.309 | 0.003 | −3.248 | 3.310 | 0.3010 | 1.593 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muñoz-Espinoza, J.; Barriga-González, G.; Corsini, G.; Lagos, S.; Barriga González, A.; de Fátima, N.G. Laccase from Melanocarpus albomyces: Molecular Docking Analysis with First-Generation Tetracyclines Through a Mechanistic Approach. Compounds 2025, 5, 17. https://doi.org/10.3390/compounds5020017
Muñoz-Espinoza J, Barriga-González G, Corsini G, Lagos S, Barriga González A, de Fátima NG. Laccase from Melanocarpus albomyces: Molecular Docking Analysis with First-Generation Tetracyclines Through a Mechanistic Approach. Compounds. 2025; 5(2):17. https://doi.org/10.3390/compounds5020017
Chicago/Turabian StyleMuñoz-Espinoza, José, Germán Barriga-González, Gino Corsini, Sebastián Lagos, Andrés Barriga González, and Nadia Gavilán de Fátima. 2025. "Laccase from Melanocarpus albomyces: Molecular Docking Analysis with First-Generation Tetracyclines Through a Mechanistic Approach" Compounds 5, no. 2: 17. https://doi.org/10.3390/compounds5020017
APA StyleMuñoz-Espinoza, J., Barriga-González, G., Corsini, G., Lagos, S., Barriga González, A., & de Fátima, N. G. (2025). Laccase from Melanocarpus albomyces: Molecular Docking Analysis with First-Generation Tetracyclines Through a Mechanistic Approach. Compounds, 5(2), 17. https://doi.org/10.3390/compounds5020017