
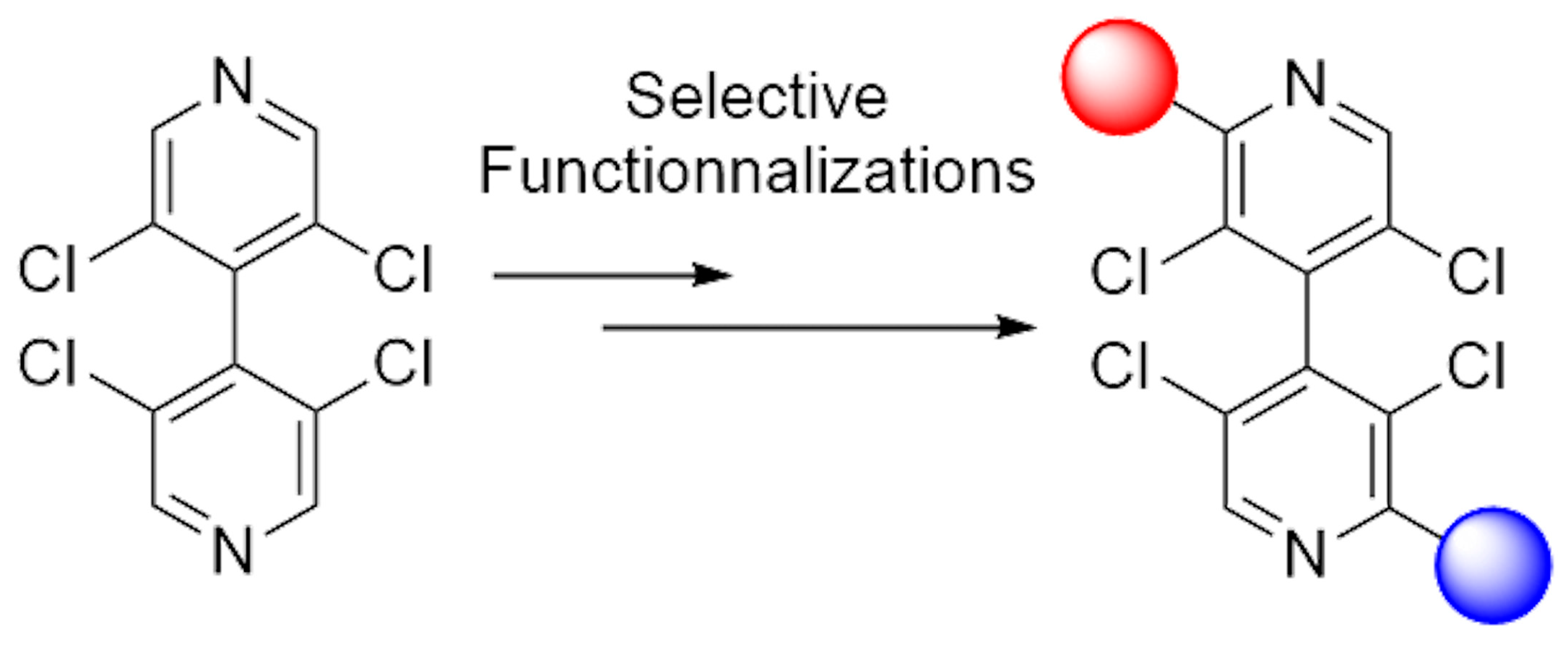
Convenient Access to Functionalized Non-Symmetrical Atropisomeric 4,4′-Bipyridines




Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. General Information
2.2. Syntheses
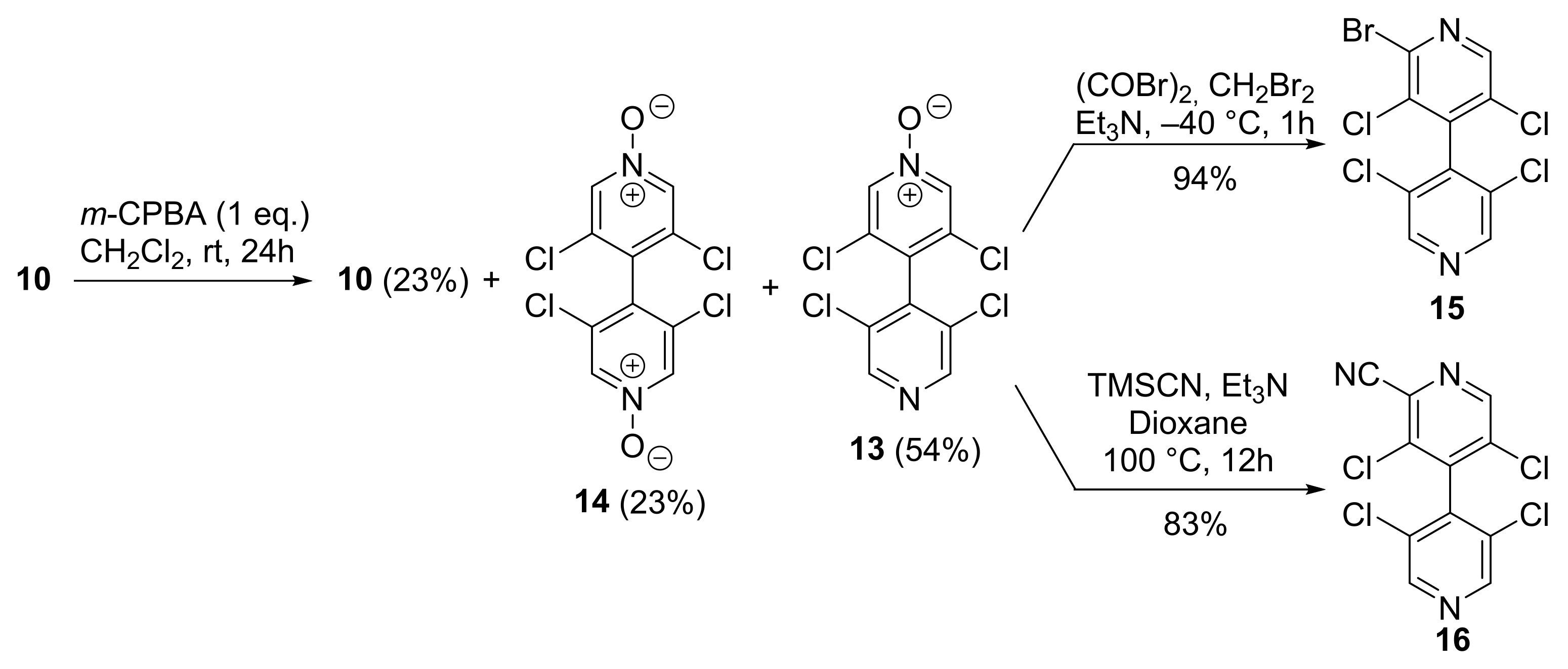
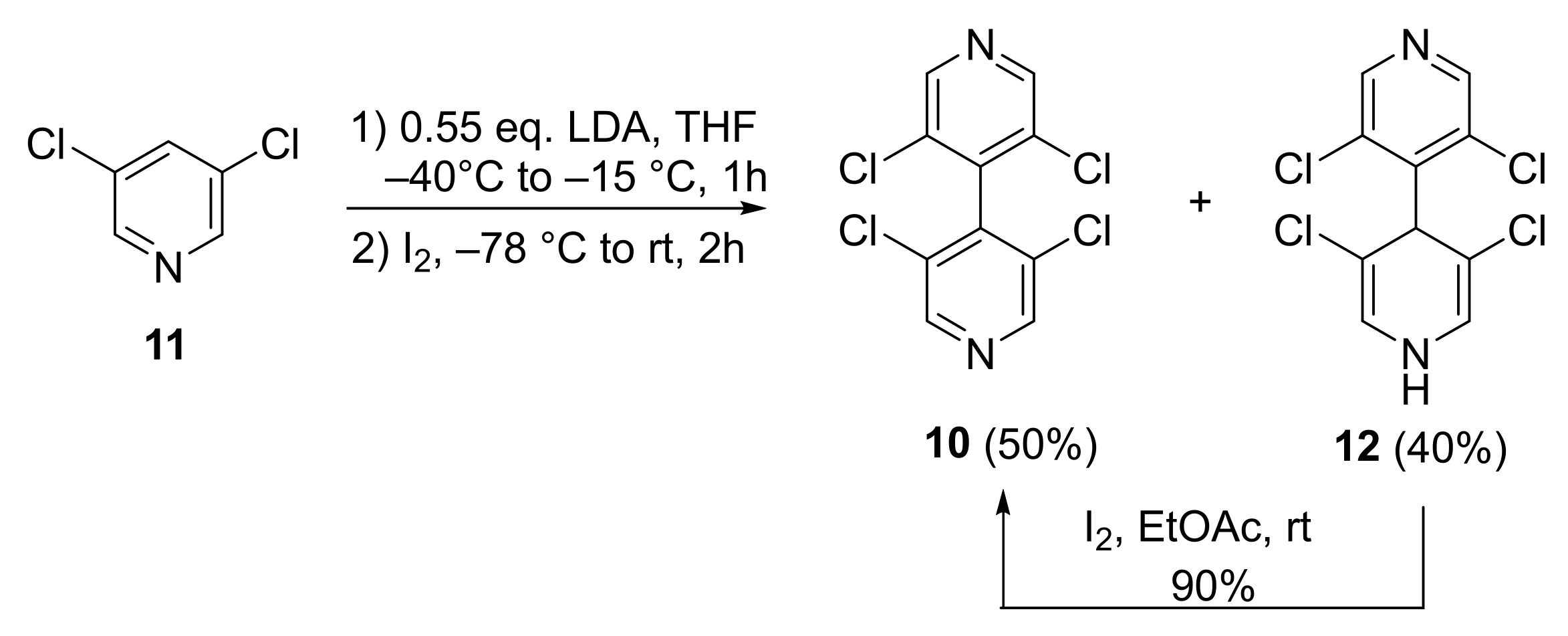
2.2.1. 3,3′,5,5′-Tetrachloro-4,4′-Bipyridine 10
2.2.2. 3,3′,5,5′-Tetrachloro-[4,4′-Bipyridine] 1-Oxide 13
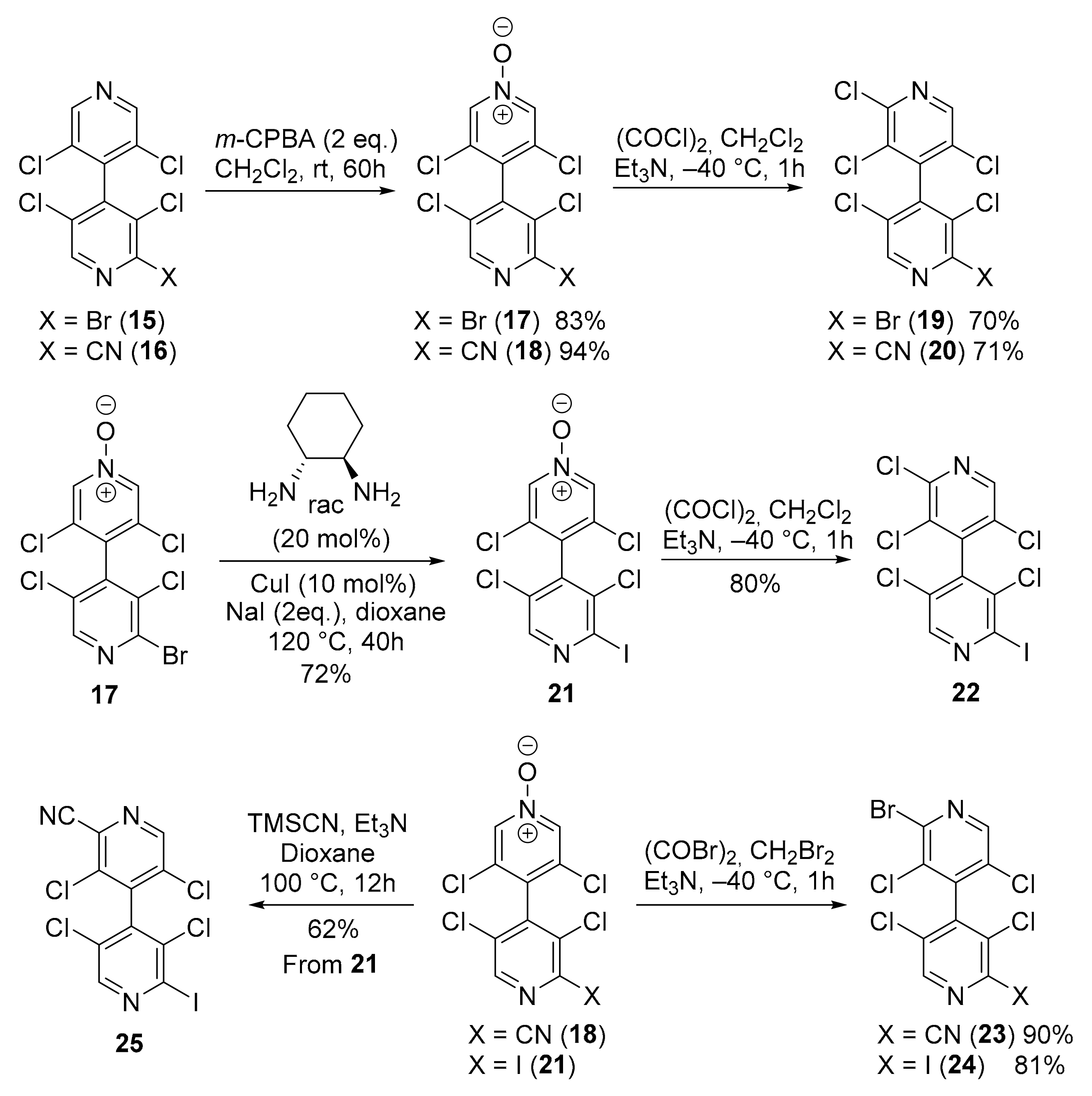
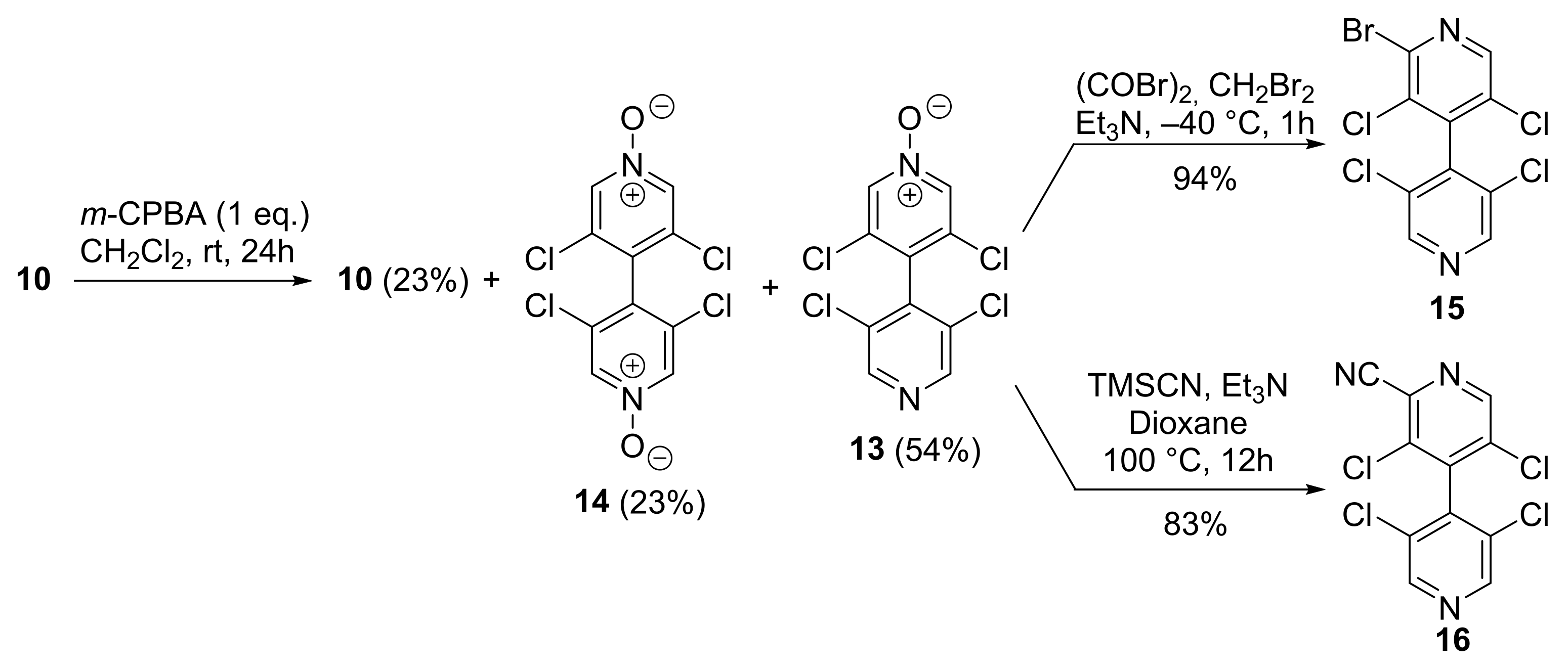
2.2.3. 2-Bromo-3,3′,5,5′-Tetrachloro-4,4′-Bipyridine 15
2.2.4. 3,3′,5,5′-Tetrachloro-[4,4′-Bipyridine]-2-Carbonitrile 16
2.2.5. 2-Bromo-3,3′,5,5′-Tetrachloro-[4,4′-Bipyridine] 1-Oxide 17
2.2.6. 3,3′,5,5′-Tetrachloro-2′-Cyano-[4,4′-Bipyridine] 1-Oxide 18
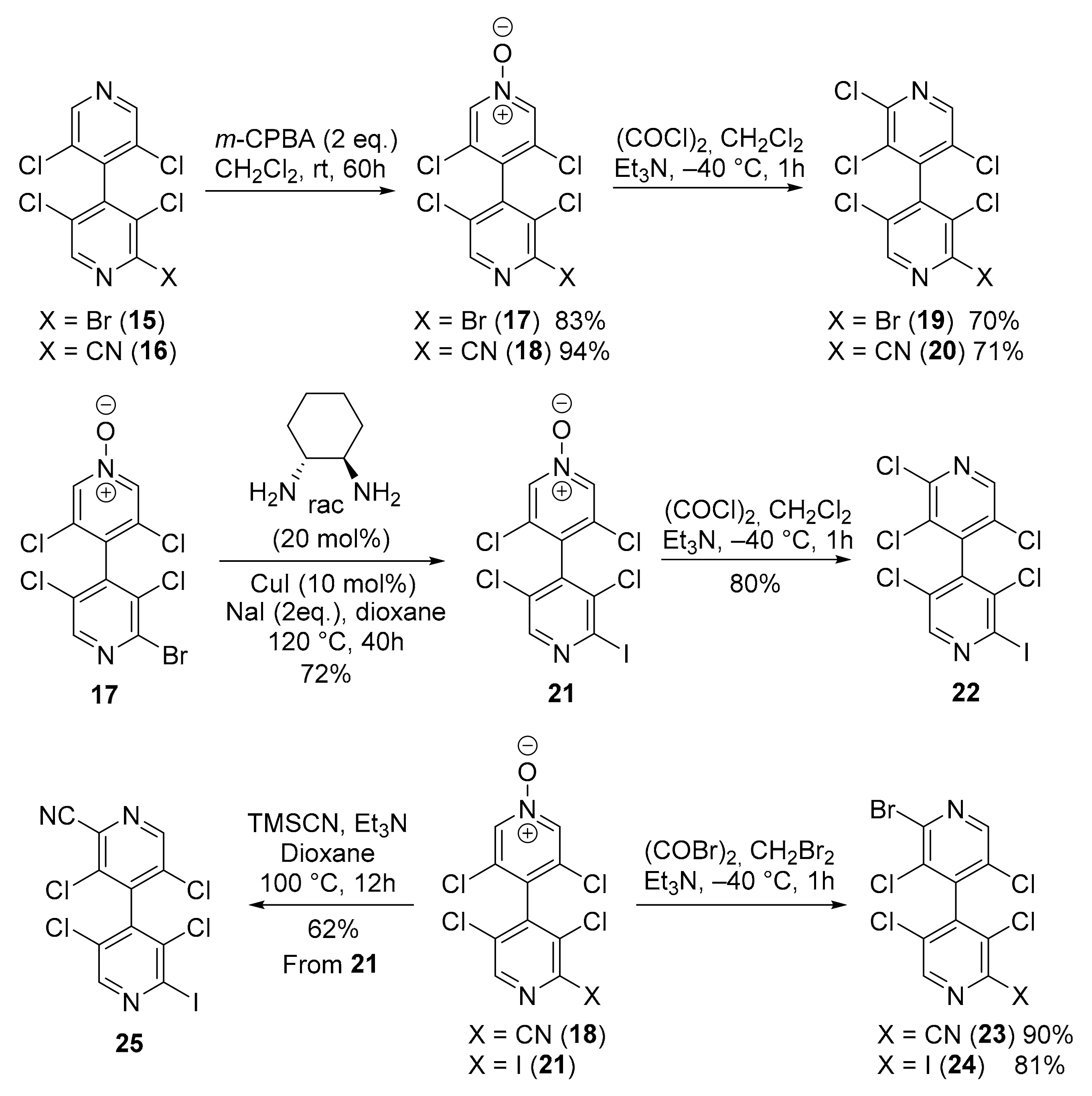
2.2.7. 2-Bromo-2′,3,3′,5,5′-Pentachloro-4,4′-Bipyridine 19
2.2.8. 2′,3,3′,5,5′-Pentachloro-[4,4′-Bipyridine]-2-Carbonitrile 20
2.2.9. 3,3′,5,5′-Tetrachloro-2′-Iodo-[4,4′-Bipyridine] 1-Oxide 21
2.2.10. 2,3,3′,5,5′-Pentachloro-2′-Iodo-4,4′-Bipyridine 22
2.2.11. 2′-Bromo-3,3′,5,5′-Tetrachloro-[4,4′-Bipyridine]-2-Carbonitrile 23
2.2.12. 2-Bromo-3,3′,5,5′-Tetrachloro-2′-Iodo-4,4′-Bipyridine 24
2.2.13. 3,3′,5,5′-Tetrachloro-2′-Iodo-[4,4′-Bipyridine]-2-Carbonitrile 25
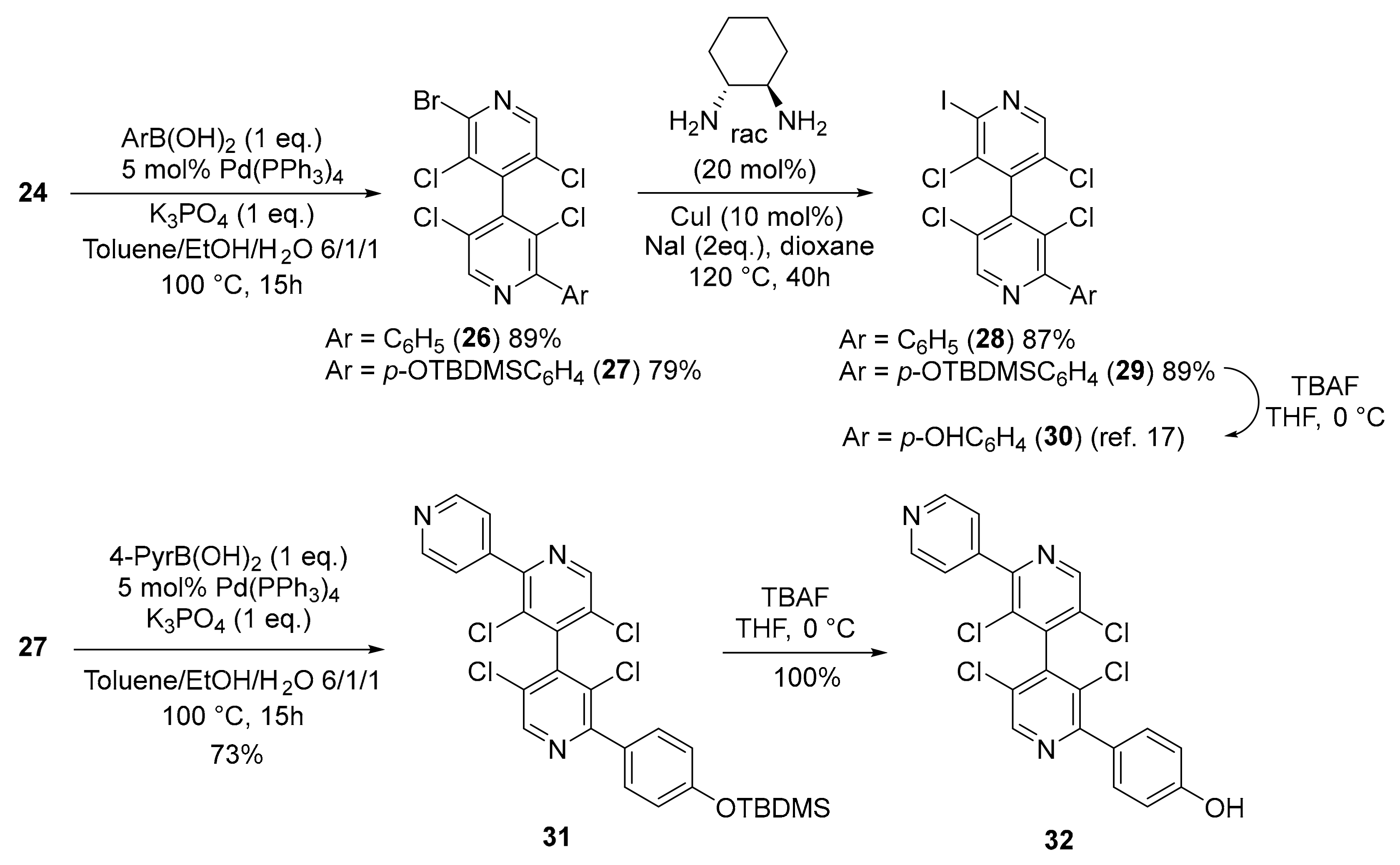
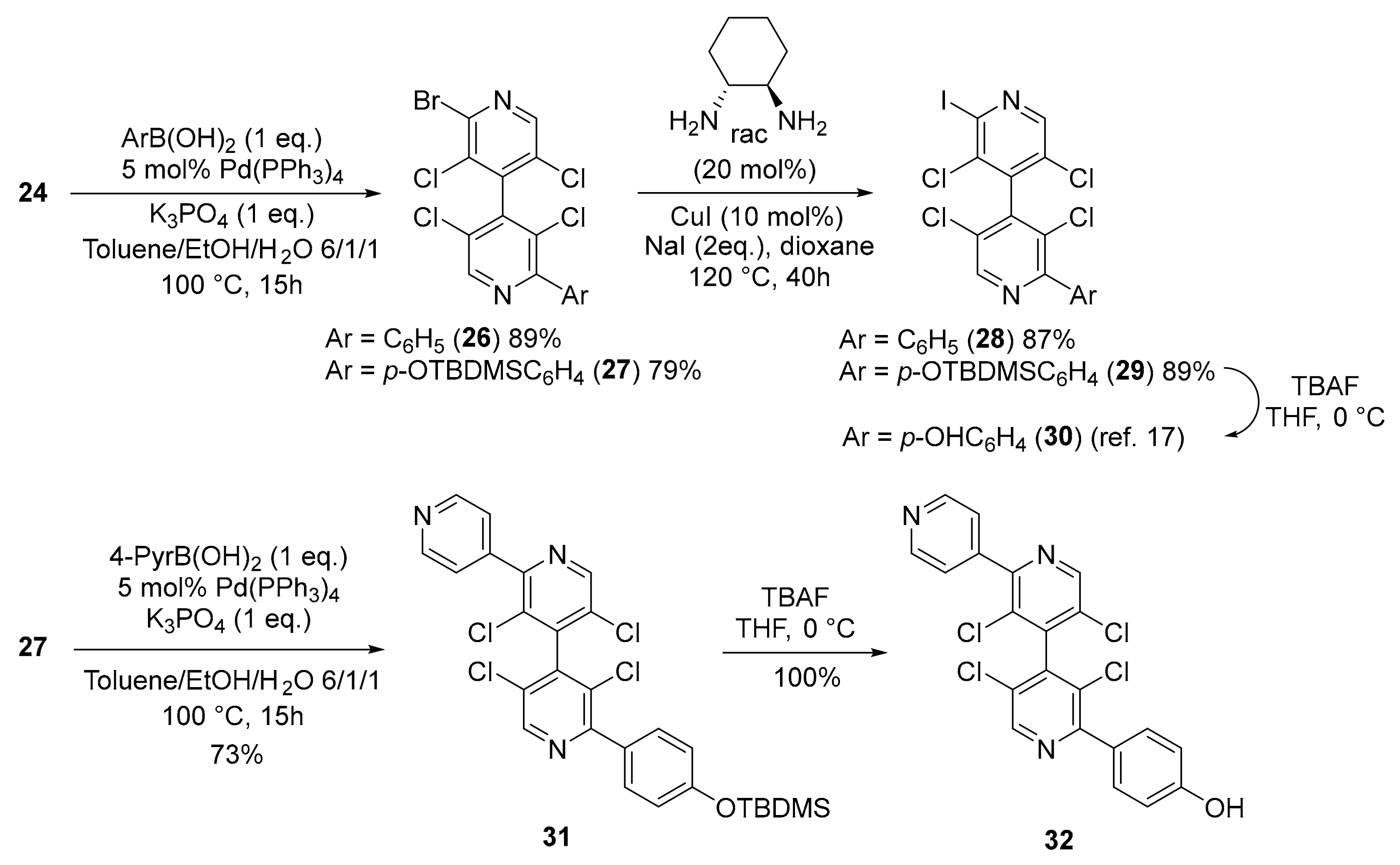
2.2.14. 2-Bromo-3,3′,5,5′-Tetrachloro-2′-Phenyl-4,4′-Bipyridine 26
2.2.15. 2-Bromo-2′-(4-((Tert-Butyldimethylsilyl)oxy)phenyl)-3,3′,5,5′-Tetrachloro-4,4′-Bipyridine 27
2.2.16. 2″-(4-((Tert-Butyldimethylsilyl)oxy)phenyl)-3′,3″,5′,5″-Tetrachloro-4,2′:4′,4″-Terpyridine 31
2.2.17. 4-(3′,3″,5′,5″-Tetrachloro-[4,2′:4′,4″-Terpyridin]-2″-yl)phenol 32
2.3. Single Crystal X-ray Diffraction
2.3.1. Crystallizations and Analysis
2.3.2. Crystal Data
2.4. Isolated Molecule Calculations
3. Results
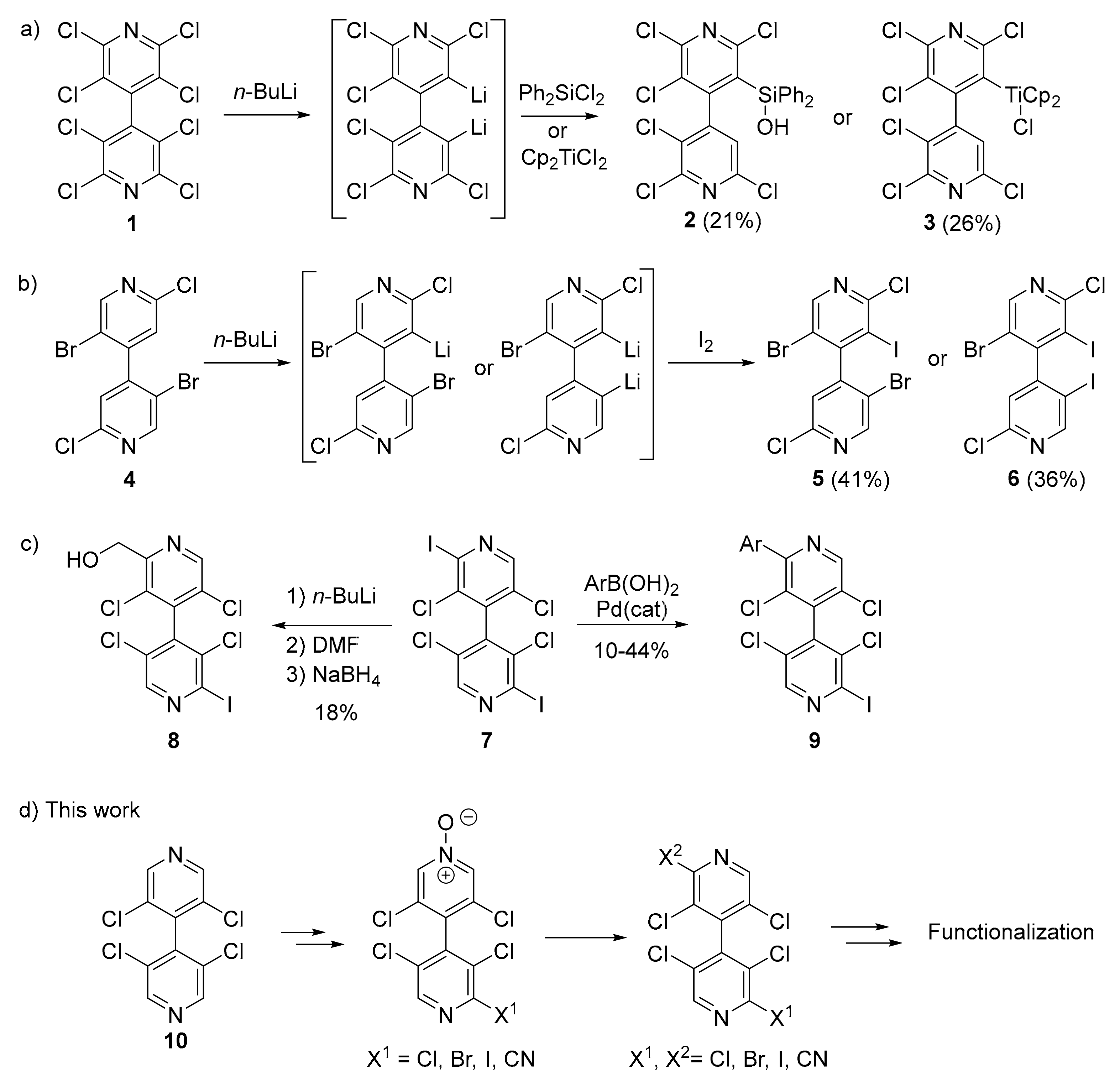
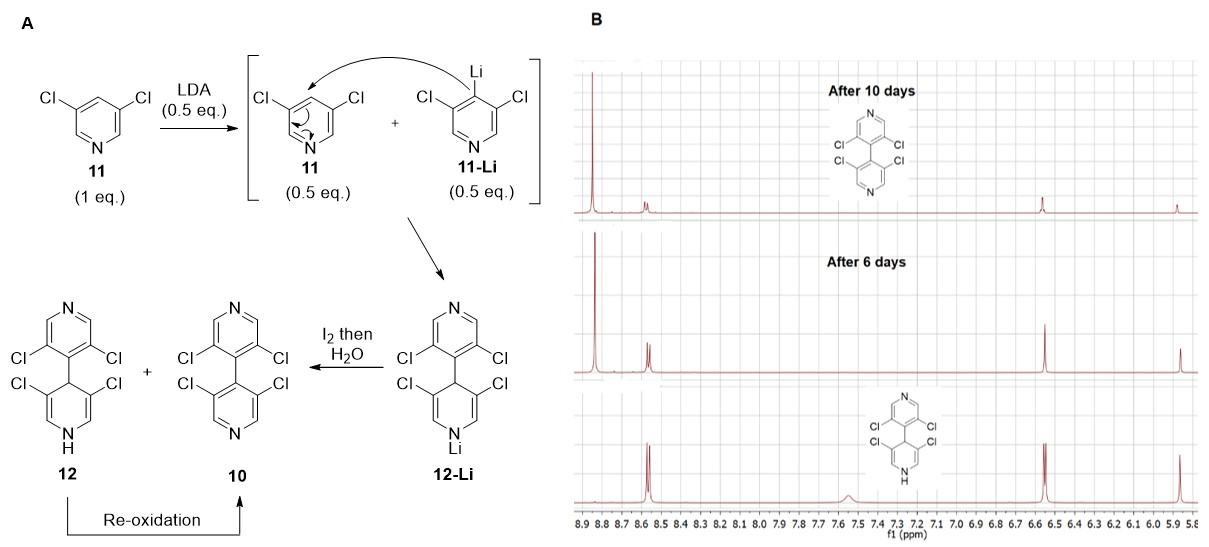
3.1. Improved Synthesis of 3,3′,5,5′-Tetrachloro-4,4′-Bipyridine 10
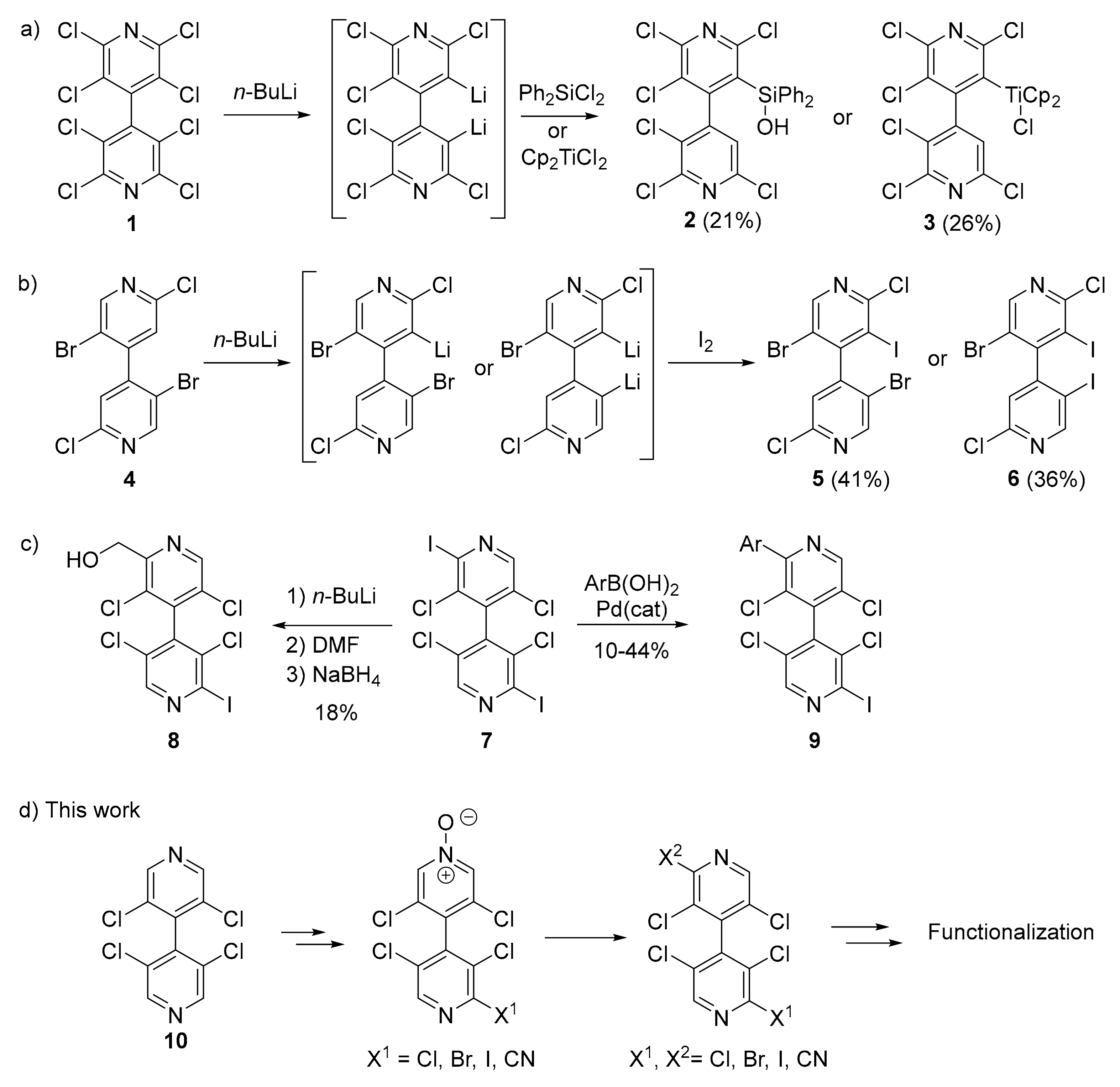
3.2. Desymmetrization of 3,3′,5,5′-Tetrachloro-4,4′-Bipyridine 10
3.3. New Chiral Non-Symmetrical 4,4′-Bipyridines Based on the 3,3′,5,5′-Tetrachloro-4,4′-Bipyridine Core
3.4. Cross-Coupling Reactions with 4,4′-Bipyridine 24
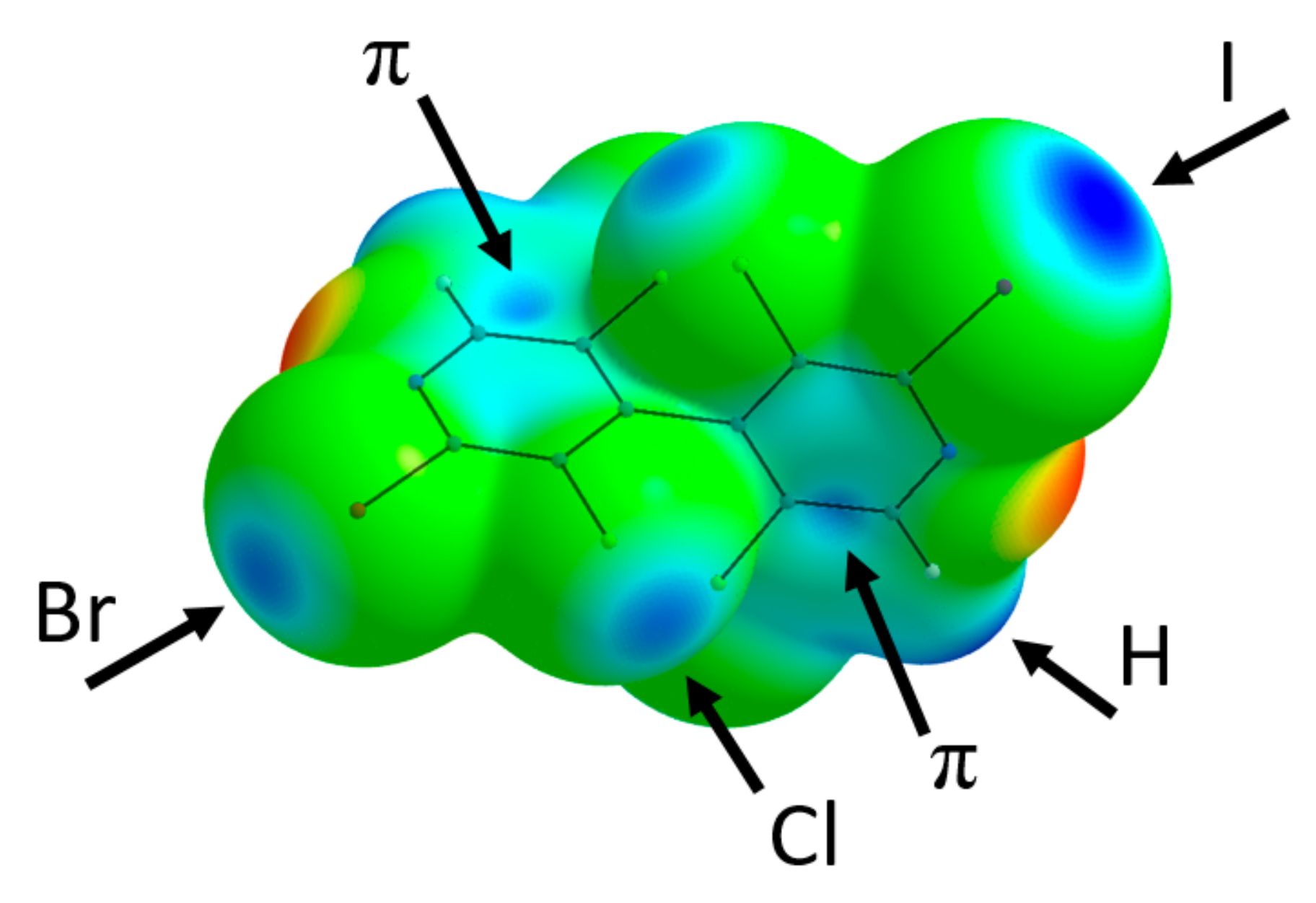
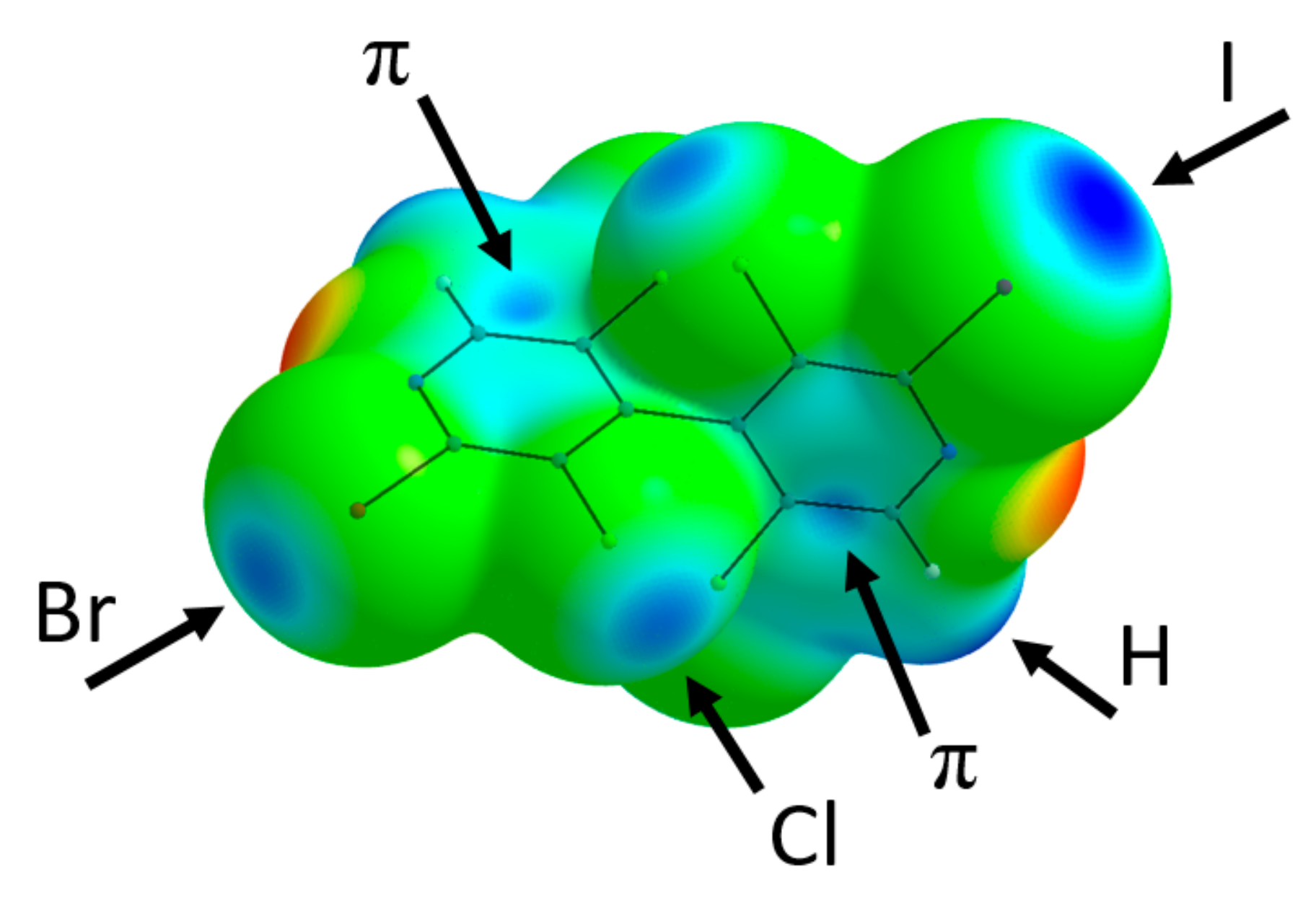
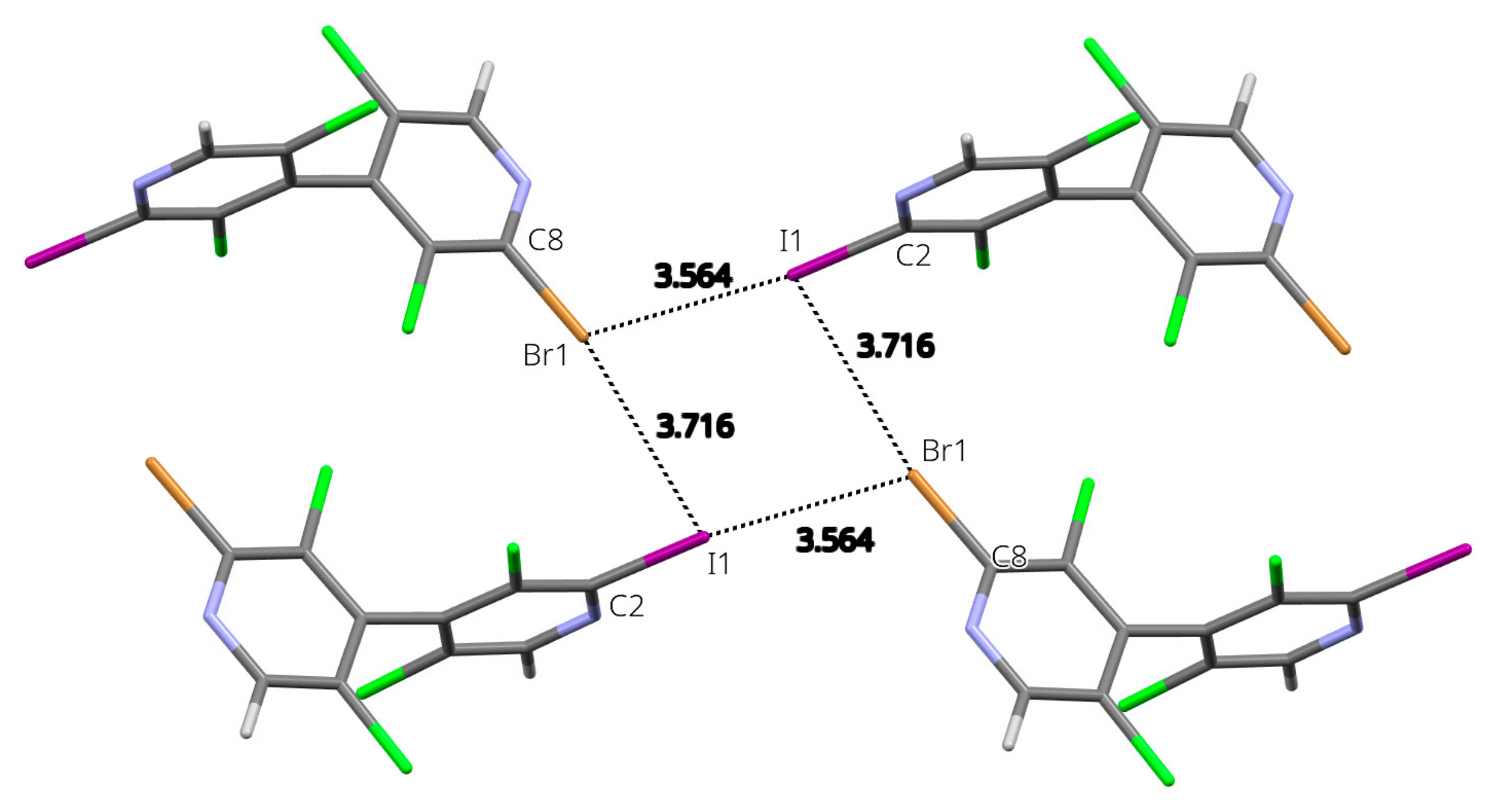
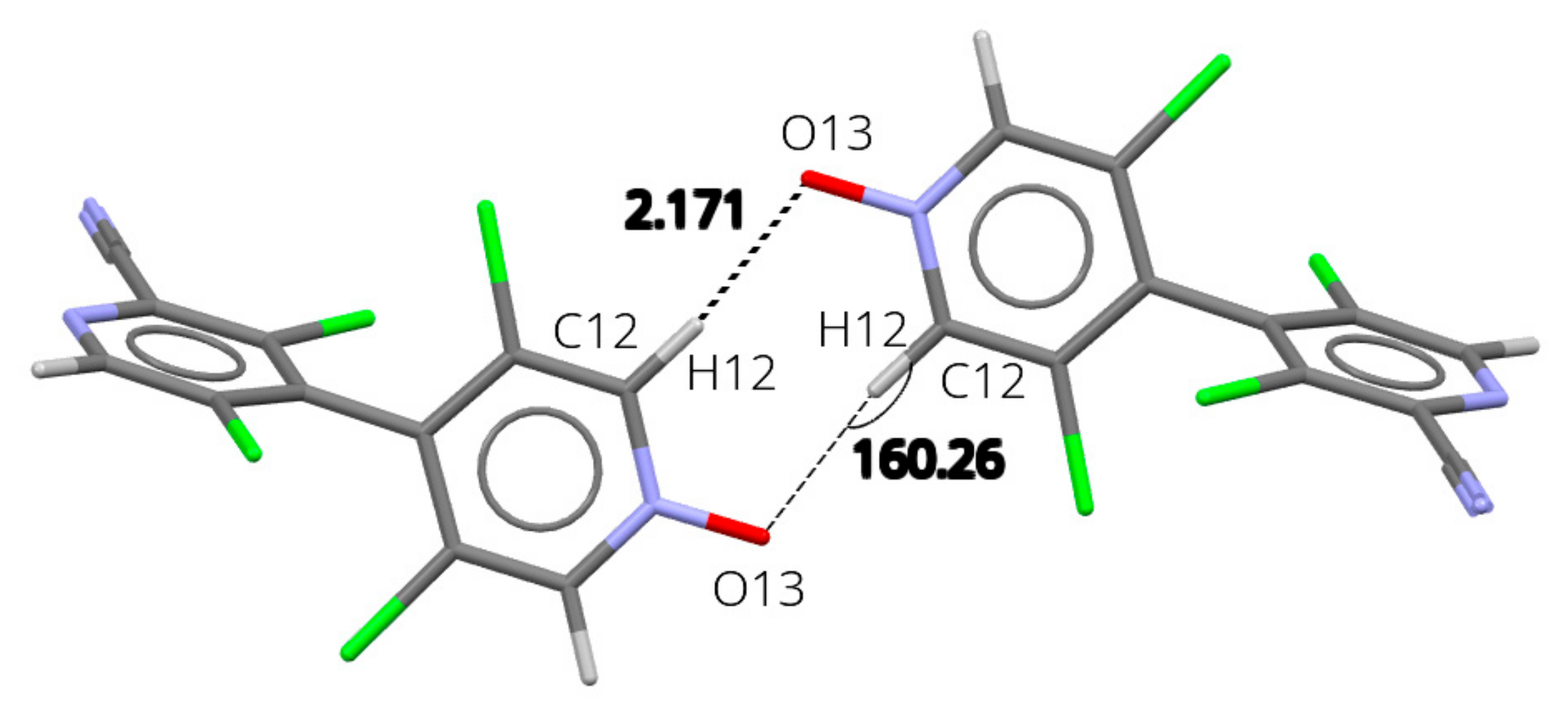
3.5. X-ray Diffraction Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Biradha, K.; Sarkar, M.; Rajput, L. Crystal engineering of coordination polymers using 4,4′-bipyridine as a bond between transition metal atoms. Chem. Commun. 2006, 40, 4169–4179. [Google Scholar] [CrossRef] [PubMed]
- Striepe, L.; Baumgartner, T. Viologens and their application as functional materials. Chem. Eur. J. 2017, 23, 16924–16940. [Google Scholar] [CrossRef] [PubMed]
- Papadakis, R. Mono-and di-quaternized 4,4′-bipyridine derivatives as key building blocks for medium- and environment-responsive compounds and materials. Molecules 2019, 25, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chelucci, G.; Thummel, R.P. Chiral 2,2′-bipyridines, 1,10-phenanthrolines, and 2,2′:6′,2″-terpyridines: Syntheses and applications in asymmetric homogeneous catalysis. Chem. Rev. 2002, 102, 3129–3170. [Google Scholar] [CrossRef]
- Fletcher, N.C. Chiral 2,2′-bipyridines: Ligands for asymmetric induction. J. Chem. Soc. Perkin Trans. 1 2002, 16, 1831–1842. [Google Scholar] [CrossRef]
- Bednářová, E.; Malatinec, S.; Kotora, M. Applications of Bolm’s ligand in enantioselective synthesis. Molecules 2020, 25, 958. [Google Scholar] [CrossRef] [Green Version]
- Sbircea, L.; Sharma, N.D.; Clegg, W.; Harrington, R.W.; Horton, P.N.; Hursthouse, M.B.; Apperley, D.C.; Boyd, D.R.; James, S.L. Chemoenzymatic synthesis of chiral 4,4′-bipyridyls and their metal–organic frameworks. Chem. Commun. 2008, 43, 5538–5540. [Google Scholar] [CrossRef]
- Jouaiti, A.; Hosseini, M.W.; Kyritsakas, N. Non-centrosymmetric packing of 1-d coordination networks based on chirality. Chem. Commun. 2002, 17, 1898–1899. [Google Scholar] [CrossRef]
- Mamane, V.; Aubert, E.; Peluso, P.; Cossu, S. Synthesis, resolution, and absolute configuration of chiral 4,4′-bipyridines. J. Org. Chem. 2012, 77, 2579–2583. [Google Scholar] [CrossRef]
- Rang, A.; Engeser, M.; Maier, N.M.; Nieger, M.; Lindner, W.; Schalley, C.A. Synthesis of axially chiral 4,4′-bipyridines and their remarkably selective self-assembly into chiral metallo-supramolecular squares. Chem. Eur. J. 2008, 14, 3855–3859. [Google Scholar] [CrossRef]
- Aubert, E.; Abboud, M.; Doudouh, A.; Durand, P.; Peluso, P.; Ligresti, A.; Vigolo, B.; Cossu, S.; Pale, P.; Mamane, V. Silver(I) coordination polymers with 3,3′,5,5′-tetrasubstituted 4,4′-bipyridine ligands: Towards new porous chiral materials. RSC Adv. 2017, 7, 7358–7367. [Google Scholar] [CrossRef] [Green Version]
- Peluso, P.; Mamane, V.; Aubert, E.; Dessì, A.; Dallocchio, R.; Dore, A.; Pale, P.; Cossu, S. Insights into halogen bond-driven enantioseparations. J. Chromatogr. A 2016, 1467, 228–238. [Google Scholar] [CrossRef]
- Peluso, P.; Mamane, V.; Dallocchio, R.; Dessì, A.; Villano, R.; Sanna, D.; Aubert, E.; Pale, P.; Cossu, S. Polysaccharide-based chiral stationary phases as halogen bond acceptors: A novel strategy for detection of stereoselective σ-hole bonds in solution. J. Sep. Sci. 2018, 41, 1247–1256. [Google Scholar] [CrossRef]
- Peluso, P.; Gatti, C.; Dessì, A.; Dallocchio, R.; Weiss, R.; Aubert, E.; Pale, P.; Cossu, S.; Mamane, V. Enantioseparation of fluorinated 3-arylthio-4,4′-bipyridines: Insights into chalcogen and π-hole bonds in high-performance liquid chromatography. J. Chromatogr. A 2018, 1567, 119–129. [Google Scholar] [CrossRef]
- Peluso, P.; Dessì, A.; Dallocchio, R.; Sechi, B.; Gatti, C.; Chankvetadze, B.; Mamane, V.; Weiss, R.; Pale, P.; Aubert, E.; et al. Enantioseparation of 5,5′-dibromo-2,2′-dichloro-3-selanyl-4,4′-bipyridines on polysaccharide-based chiral stationary phases: Exploring chalcogen bonds in liquid-phase chromatography. Molecules 2021, 26, 221. [Google Scholar] [CrossRef]
- Weiss, R.; Aubert, E.; Peluso, P.; Cossu, S.; Pale, P.; Mamane, V. Chiral chalcogen bond donors based on the 4,4′-bipyridine scaffold. Molecules 2019, 24, 4484. [Google Scholar] [CrossRef] [Green Version]
- Dessì, A.; Peluso, P.; Dallocchio, R.; Weiss, R.; Andreotti, G.; Allocca, M.; Aubert, E.; Pale, P.; Mamane, V.; Cossu, S. Rational design, synthesis, characterization and evaluation of iodinated 4,4′-bipyridines as new transthyretin fibrillogenesis inhibitors. Molecules 2020, 25, 2213. [Google Scholar] [CrossRef]
- Mamane, V.; Peluso, P.; Aubert, E.; Cossu, S.; Pale, P. Chiral hexahalogenated 4,4′-bipyridines. J. Org. Chem. 2016, 81, 4576–4587. [Google Scholar] [CrossRef]
- Foulger, N.J.; Wakefield, B.J. Polyhalogenoaromatic compounds. XXIX. Hexachloro-5,5′-dilithio-4,4′-bipyridine as an intermediate for organometallic and organic syntheses. J. Organomet. Chem. 1974, 69, 161–167. [Google Scholar] [CrossRef]
- Mamane, V.; Aubert, E.; Peluso, P.; Cossu, S. Lithiation of prochiral 2,2′-dichloro-5,5′-dibromo-4,4′-bipyridine as a tool for the synthesis of chiral polyhalogenated 4,4′-bipyridines. J. Org. Chem. 2013, 78, 7683–7689. [Google Scholar] [CrossRef]
- Kutasevich, A.V.; Perevalov, V.P.; Mityanov, V.S. Recent progress in non-catalytic C–Hunctionalization of heterocyclic N-oxides. Eur. J. Org. Chem. 2021, 2021, 357–373. [Google Scholar] [CrossRef]
- Abboud, M.; Mamane, V.; Aubert, E.; Lecomte, C.; Fort, Y. Synthesis of polyhalogenated 4,4′-bipyridines via a simple dimerization procedure. J. Org. Chem. 2010, 75, 3224–3231. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, J.; Hwang, T.L.; Chen, M.J.; Tedrow, J.S.; Farrell, R.P.; Bio, M.M.; Cui, S. Highly regioselective halogenation of pyridine N-oxide: Practical access to 2-halo-substituted pyridines. Org. Lett. 2015, 17, 2948–2951. [Google Scholar] [CrossRef] [PubMed]
- Carson, M.W.; Giese, M.W.; Coghlan, M.J. An intra/intermolecular Suzuki sequence to benzopyridyloxepines containing geometrically pure exocyclic tetrasubstituted alkenes. Org. Lett. 2008, 10, 2701–2704. [Google Scholar] [CrossRef] [PubMed]
- Klapars, A.; Buchwald, S.L. Copper-catalyzed halogen exchange in aryl halides: An aromatic Finkelstein reaction. J. Am. Chem. Soc. 2002, 124, 14844–14845. [Google Scholar] [CrossRef] [PubMed]
- Peluso, P.; Sechi, B.; Lai, G.; Dessì, A.; Dallocchio, R.; Cossu, S.; Aubert, E.; Weiss, R.; Pale, P.; Mamane, V.; et al. Comparative enantioseparation of chiral 4,4′-bipyridine derivatives on coated and immobilized amylose-based chiral stationary phases. J. Chromatogr. A 2020, 1625, 461303. [Google Scholar] [CrossRef] [PubMed]
- Dallocchio, R.; Sechi, B.; Dessì, A.; Chankvetadze, B.; Cossu, S.; Mamane, V.; Weiss, R.; Pale, P.; Peluso, P. Enantioseparations of polyhalogenated 4,4′-bipyridines on polysaccharide-based chiral stationary phases and molecular dynamics simulations of selector–selectand interactions. Electrophoresis 2021. [Google Scholar] [CrossRef] [PubMed]
- Perdomo Rivera, R.; Ehlers, P.; Ohlendorf, L.; Torres Rodríguez, E.; Villinger, A.; Langer, P. Chemoselective synthesis of arylpyridines through Suzuki–Miyaura cross-coupling reactions. Eur. J. Org. Chem. 2018, 8, 990–1003. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17 (2017). University of Western Australia. Available online: https://hirshfeldsurface.net (accessed on 26 February 2021).
- Desiraju, G.R.; Parthasarathy, R. The nature of halogen⋯halogen interactions: Are short halogen contacts due to specific attractive forces or due to close packing of nonspherical atoms? J. Am. Chem. Soc. 1989, 111, 8725–8726. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G. Type II halogen⋯halogen contacts are halogen bonds. IUCrJ 2014, 1, 5–7. [Google Scholar] [CrossRef]
- Zhu, X.; Kreutter, K.D.; Hu, H.; Player, M.R.; Gaul, M.D. A novel reagent combination for the oxidation of highly electron deficient pyridines to N-oxides: Trifluoromethanesulfonic anhydride/sodium percarbonate. Tetrahedron Lett. 2008, 49, 832–834. [Google Scholar] [CrossRef]
- Caron, S.; Do, N.D.; Sieser, J.E. A practical, efficient, and rapid method for the oxidation of electron deficient pyridines using trifluoroacetic anhydride and hydrogen peroxide–urea complex. Tetrahedron Lett. 2000, 41, 2299–2302. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aubert, E.; Wenger, E.; Peluso, P.; Mamane, V. Convenient Access to Functionalized Non-Symmetrical Atropisomeric 4,4′-Bipyridines. Compounds 2021, 1, 58-74. https://doi.org/10.3390/compounds1020006
Aubert E, Wenger E, Peluso P, Mamane V. Convenient Access to Functionalized Non-Symmetrical Atropisomeric 4,4′-Bipyridines. Compounds. 2021; 1(2):58-74. https://doi.org/10.3390/compounds1020006
Chicago/Turabian StyleAubert, Emmanuel, Emmanuel Wenger, Paola Peluso, and Victor Mamane. 2021. "Convenient Access to Functionalized Non-Symmetrical Atropisomeric 4,4′-Bipyridines" Compounds 1, no. 2: 58-74. https://doi.org/10.3390/compounds1020006
APA StyleAubert, E., Wenger, E., Peluso, P., & Mamane, V. (2021). Convenient Access to Functionalized Non-Symmetrical Atropisomeric 4,4′-Bipyridines. Compounds, 1(2), 58-74. https://doi.org/10.3390/compounds1020006