Abstract
Background: The identification of body fluids collected from crime scenes is crucial for determining the type and nature of assaults and for advancing the resolution of crimes. Objectives: The primary aim of this study was to investigate tissue-specific DNA methylation markers that can effectively distinguish buccal samples from blood, semen, and vaginal epithelial tissue. Methods: We screened various markers and selected four genomic locations for further analysis. Genomic DNA was extracted from tissue samples, followed by bisulfite conversion, locus-specific polymerase chain reaction (PCR) amplification, and pyrosequencing. Results: Four loci—cg-9652652, cg-11536474, cg-3867465, and cg-10122865—along with several adjacent CpG sites, were found to be hypermethylated in buccal samples compared to other tissue types. The difference in DNA methylation of buccal samples was statistically significant (p < 0.0001) compared to other tissues, indicating the potential usefulness of these loci for forensic tissue identification. Two additional studies were conducted: (a) a species specificity study and (b) a mixture study involving two different tissue types. The species specificity study showed that the primers used in the assay were specific to primates and humans. They did not amplify five non-primate samples, while the two primate samples—chimpanzee and rhesus—provided usable methylation data. The mixture study involved DNA from two different tissues—buccal samples and semen—combined in varying proportions. The results showed a decrease in the overall percentage of DNA methylation at the locus cg-9652652 as well as five adjacent CpG sites when the amount of buccal cell DNA in the mixture was reduced. Conclusion: The specificity of the primers and the significant differences in percent DNA methylation between buccal cells and other tissues make these markers excellent candidates for forensic tissue identification.
1. Introduction
DNA evidence has become an essential tool in identifying the perpetrator of a crime in the justice system through forensic science. The development of restriction fragment length polymorphism (RFLP) probes by Alec Jefferys from 1985 to 1995 to detect “minisatellite” repeat polymorphisms in humans was a major success [1]. Current forensic DNA technology relies on the identification of short tandem repeat (STR) polymorphism using capillary electrophoresis (CE) and massively parallel sequencing (MPS) [2] and is constantly evolving with newer technologies. Identifying the biofluid present in forensic evidence is a routine practice in criminal investigations, serving two purposes: determining the nature of the assault and assisting in the reconstruction of events at a crime scene. It also helps analysts decide the right DNA extraction protocol for successful DNA typing. Specific body fluids, such as semen, may benefit from specialized DNA extraction protocols to maximize DNA recovery [3]. In everyday scenarios, the transfer of skin cells may occur through casual contact, while the transfer of blood or semen may suggest a violent event. Therefore, determining the tissue source of a DNA sample in criminal cases is important. Conventional serological tests, such as the Kastle–Mayer test for blood and the acid phosphatase test for seminal fluid, are routinely used to determine the presence of specific body fluids in evidence [4]. Other serological tests can be highly specific, such as immunodiffusion and immunochromatographic test strips for blood and semen (Independent Forensics, Lombard, IL), and the observation of spermatozoa via a microscope for the detection of semen in the evidence. However, these tests can lead to false negative or false positive results due to issues such as protein degradation and cross-reactivity with animal blood. Furthermore, many of these tests are destructive and may lead to a loss of evidence [5]. While blood and seminal fluid can be identified using serological analysis, there is currently no standardized serological test to identify skin cells and vaginal epithelial samples.
Several authors have proposed mRNA markers for identifying specific tissues [6,7,8]. These studies demonstrate the expression of tissue-specific mRNAs using quantitative reverse transcription PCR (RT-PCR) and real-time PCR to detect the presence of blood, semen, saliva, vaginal secretions, and menstrual blood. While these methods are reliable, there is a possibility of RNA degradation leading to inconclusive results. Vidaki et al. have pointed out that “mRNA instability” could be an issue with post-mortem samples, and RNase can degrade the gene transcripts [9]. Also, handling RNA samples and RT-PCR is not routine in forensic laboratories. To address the limitations associated with protein and RNA markers, it is ideal to develop and employ new and robust DNA technology for tissue identification, such as epigenetic/methylation analysis of DNA samples. The development of various DNA techniques has significantly improved our understanding of DNA methylation in different tissues and its use in forensic tissue identification. Genome-wide DNA methylation analysis studies have shown the presence of tissue-specific differentially methylated regions in the human genome [10,11,12]. In somatic tissues, 70–80% of all CpG sites in the genome are methylated, and approximately 18% of CpG islands in the human genome are subject to tissue-specific DNA methylation [13,14].
Recent advancements in forensic genetics include the introduction of massively parallel sequencing (MPS), which has significantly transformed our approach to DNA analysis. The primary advantages of MPS are its capability to simultaneously analyze thousands of markers. This technique is beneficial for several applications, including short tandem repeat (STR) marker analysis, single nucleotide polymorphism (SNP) analysis, differentiation between monozygotic twins, and determining the DNA methylation status of different genomic regions for forensic tissue identification [15,16]. Additionally, DNA methylation analysis using the SNaPshot multiplex assay has been employed for tissue identification and age prediction. This assay utilizes SNP genotyping technology, and the advantages of this technology include its ability to multiplex and its compatibility with capillary electrophoresis systems commonly used in forensic laboratories [17]. DNA methylation arrays utilize microarray technology to measure methylation levels at thousands to millions of CpG sites simultaneously. This is achieved by hybridizing bisulfite-converted DNA to probes on the array. The Infinium™ MethylationEPIC v2.0 (950 K) array can detect the methylation status of approximately 935,000 CpG sites across the human genome. These arrays are designed to offer high throughput, high precision, and sensitivity (Illumina.com; cd-genomics.com; URL accessed on 10 February 2025). Tissue-specific differential DNA methylation data using Illumina HumanMethylation BeadChips 27 and 450 K have also been reported [18].
Recently, there have been reports on the DNA methylation status of different genomic regions for various forensic applications such as tissue identification and age prediction by pyrosequencing of the bisulfite-converted genomic DNA. This bisulfite chemical modification converts unmethylated cytosine to uracil and subsequently to thymine following PCR. Methylated cytosine nucleotides remain as cytosine following bisulfite conversion and PCR. This method helps to quantitate the ratio of methylated versus unmethylated cytosine and provide direct quantitative DNA methylation data for the CpG sites analyzed.
The study by Madi et al. [19] describes the detection of blood, saliva, skin epithelial cells, and semen using bisulfite conversion and pyrosequencing technology. From the results of this study, it is evident that blood-, saliva-, and semen-containing samples can be clearly distinguished from each other based on the methylation levels of certain markers. The identification of spermatozoa using DNA methylation and pyrosequencing was reported using six genomic locations with multiple CpG sites [20]. In this study, the semen samples showed hypomethylation in five markers, while the sixth marker showed hypermethylation when compared to other tissues. Alghanim and colleagues conducted an epigenome-wide association study, identifying two markers, NMUR2 and UBE2U, that can effectively distinguish spermatozoa from other tissues using pyrosequencing and high-resolution melt (HRM) analysis. The spermatozoa were hypomethylated, while the other tissues were hypermethylated [21]. A DNA methylation profiling multiplex SNaPshot assay was developed by Lee et al. to confirm the presence of various body fluids. This resultant multiplex assay allowed for positive identification of blood, saliva, semen, vaginal fluid, and menstrual blood [22]. Recently, Ghai et al. reported two specific methylation markers, one for saliva and one for semen. In their study, the authors identified the CpG sites associated with the genes ZNF282 and HPCAL1 and showed that the CpG sites for ZNF282 exhibited hypomethylation specific to semen, whereas the CpG sites for HPCAL1 showed hypomethylation specific to saliva. [23]. Also, Konrad et. al. [24] developed a workflow for body fluid identification based on the methylation level of various CpG sites using bisulfite-modified pyrosequencing. Validation studies confirmed the identification of saliva, vaginal secretion, and semen in 100% of the samples.
In this study, we present the usefulness of DNA methylation data from four specific CpG sites and certain adjacent sites for forensic buccal tissue identification using bisulfite-modified pyrosequencing of DNA samples.
2. Materials and Methods
2.1. Sample Collection, DNA Extraction, and Bisulfite Conversion
Blood, buccal samples, semen, and vaginal epithelial cells were collected from normal volunteers aged 18–40. The collection was carried out following Institutional Review Board approved protocols at the University of Southern Mississippi, protocol #12010303. Blood samples were obtained through finger prick, collected with sterile cotton swabs, air-dried, and stored frozen. Buccal samples were collected by swabbing the inside of the cheek using sterile cotton swabs. Female participants collected vaginal epithelial cells in privacy using sterile cotton swabs, while male volunteers collected semen samples in privacy using sterile specimen cups, which were then transported to the laboratory. To maintain the privacy and anonymity of the donors, all samples were given a unique identifier number.
The genomic DNA was extracted using organic extraction techniques with the phenol/chloroform/isoamyl alcohol method [25]. DNA quantitation was performed using agarose gel electrophoresis and/or the Quantifiler® human DNA quantitation kit (Applied Biosystems, Foster City, CA, USA and the 7500 real-time PCR system (Applied Biosystems, Foster City, CA, USA). The extracted genomic DNA was modified using the Epitect® Bisulfite kit (Qiagen, Germantown, MD, USA), which can modify 1 ng–2 µg of DNA. Approximately 50–300 ng DNA was used for bisulfite conversion, following the manufacturer’s instructions (Qiagen).
2.2. Loci Selection and Assay Design
The markers chosen for this study were assessed from a group of 37 individual CpG sites identified by Park et al. [26] as having the potential to differentiate saliva from other bodily fluids. These 37 CpG sites were identified through a genome-wide study using the Illumina Human Methylation 450 K bead array, which contains over 450,000 CpG sites. The study by Park et al. provided the average beta values for the 37 candidate CpG sites across different tissues, where higher beta values indicate higher DNA methylation status and vice versa. From this group of 37 loci, we used pyrosequencing technology to screen multiple loci based on the beta value and selected four markers that had the highest percentage methylation difference between buccal samples and other body fluids to investigate in this study: cg-9652652, cg-11536474, cg-3867465, and cg-10122865, along with several adjacent CpG sites. The specific location of these CpG sites in the human genome was determined, and the information was entered into the University of California, Santa Cruz (UCSC) genome browser (Human GRCh37/hg19). We downloaded 200 base pairs of DNA sequence on both sides of the CpG site (400-base sequence in total) from the browser. This 400-base sequence was used to develop an assay for pyrosequencing using the Pyromark assay design software version 2.0.1 (Qiagen) that included the target CpG site and a few other adjacent CpG sites. The PCR and sequencing primers were obtained commercially from Qiagen, with one of the PCR primers labeled to produce biotinylated PCR products. The sequence of the PCR and sequencing primers as well as the target CpG loci involved are listed in Table 1.

2.3. Template Preparation and Pyrosequencing
The bisulfite-converted DNA samples underwent PCR amplification for the four specified markers using custom-designed Pyromark CpG assays. These assays consist of a 10x PCR primer set, one of which is biotinylated, and a 10x sequencing primer. Individual PCR reactions for each marker were carried out following the protocols provided by the assay manufacturer (Qiagen). The thermal cycling program included Taq activation at 95 °C for 15 min, followed by 45 cycles of 94 °C for 30 s, Tm-5 °C for 30 s, and 72 °C for 30 s, with a final extension at 72 °C for 10 min [20]. The amplicons were evaluated on a 2% agarose gel to assess PCR efficiency and determine the quantity of amplicons for pyrosequencing. Approximately 200–400 ng of biotinylated PCR products were utilized for pyrosequencing, which was performed using a Pyromark Q24 pyrosequencer (Qiagen) following the manufacturer’s recommendations. A detailed pyrosequencing protocol can be found in Balamurugan et al. [20]. The Pyromark Q24 software was used to calculate the percent DNA methylation data, and the results are displayed as a program showing the DNA methylation data for each CpG site.
2.4. Mixture Study
In the mixture study, one DNA sample from semen and one DNA sample from buccal cells from a different donor were used. Each sample was diluted to a concentration of 10 ng/µL using TE buffer. Five different ratios of buccal cells vs. semen DNA were prepared: 90:10, 75:25, 50:50, 25:75, and 10:90, each containing 100 ng of DNA. In addition to the mixture, one neat buccal cell DNA sample and one neat semen DNA sample were used. The mixture was then bisulfite converted and used for PCR. The amplification targeted the cg-9652652 locus and five adjacent CpG sites. The average DNA methylation of all CpGs (1–6) for each mixture ratio was used for data analysis and histogram creation.
2.5. Species Specificity Study
To assess the specificity of the primers and determine whether any non-human species would cross-react with the human primers, we tested several non-human DNA samples along with one human control. The samples included five non-primate species: cat, dog, chicken, cow, and erythrobacter, as well as two primate species: chimpanzee and rhesus monkey. The DNA samples underwent bisulfite conversion and were then amplified using primers for the marker cg-9652652. We evaluated the amplification efficiency by checking the PCR amplicons on a 2% agarose gel. Regardless of the amplification success, all samples were sequenced using a pyrosequencer.
2.6. Data Analysis
The percent DNA methylation values for each CpG site for all samples were recorded in an Excel spreadsheet. Mean percent DNA methylation values were calculated for each CpG site across all tissue types studied for every marker. The mean DNA methylation data of different tissue types were compared using the SPSS statistical package with a one-way ANOVA and Tukey’s post hoc pairwise comparisons to determine whether there were any statistically significant differences in the DNA methylation data among the four tissues. DNA methylation differences were considered statistically significant when the p-values were less than 0.05 (p < 0.05). The statistical analysis of the data was performed using the SPSS software package (PASW Statistics 22).
3. Results
In this study, we identified four epigenetic markers that can distinguish buccal samples from three other body fluids: blood, semen, and vaginal epithelial cells. The DNA methylation data for the four CpG markers, along with some adjacent sites, are shown in Figure 1, Figure 2, Figure 3 and Figure 4. We found that all four markers and the adjacent CpG sites exhibited higher DNA methylation levels in buccal samples compared to the other three tissues.
3.1. Marker cg-9652652
The average DNA methylation range for the cg-9652652 marker was 49–61% for buccal samples, while it was 15% or lower for other tissues. The lowest DNA methylation value in buccal sample tissues differed by 46% from the highest value in other tissues, making this marker very useful for tissue differentiation. The standard deviation for this marker ranged from 8.9 to 15.9 for buccal samples, while for the other tissues it ranged from 1.6 to 15.1. Statistical analysis shows that the percent methylation of buccal sample DNA differed significantly from that of other body fluids (p < 0.0001). The graphical representation of the data for the six CpG sites is shown in Figure 1.
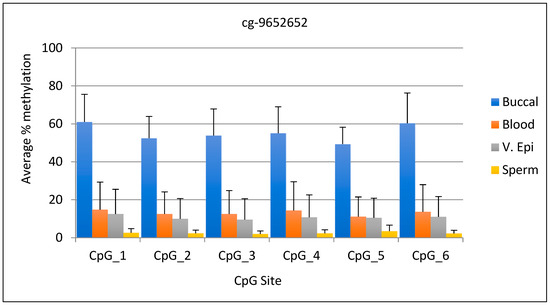
Figure 1.
Average DNA methylation data for marker cg-9652652 (CpG 5) and five adjacent sites. The fifth CpG site is listed in Park et al., 2014. N = 13 buccal, 10 blood, 11 v. epi, and 12 semen samples.
3.2. Marker cg-11536474
The average DNA methylation range for the cg-11536474 marker was 69–76% in buccal samples, while in other tissues it was 12% or lower. The difference between the lowest DNA methylation value in buccal samples and the highest value in other tissues was 57%, making this marker also useful for distinguishing between different tissues. The standard deviation for this marker ranged from 18.9 to 20.9 for buccal samples, while for the other tissues it ranged from 2.3 to 12.1. Statistical analysis shows that the percent methylation of buccal sample DNA differed significantly from that of other body fluids (p < 0.0001). The graphical representation of the data for the three CpG sites is shown in Figure 2. However, data for the cg-11536474 site (CpG 4) are not included in the figure due to a stretch of nine “C”s before the cg-11536474 site, which caused the data to skew and resulted in a “warning” outcome due to “peak height deviation” at the “C” stretch.
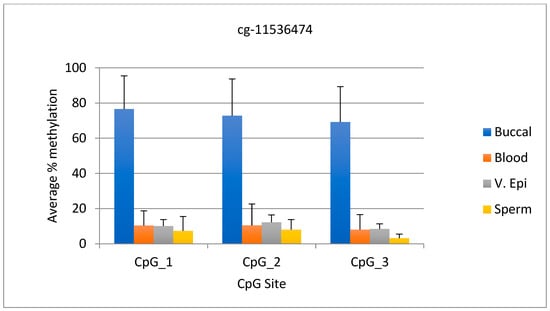
Figure 2.
The average DNA methylation data for three CpG sites upstream of cg-11536474 is presented. N = 12 buccal, 18 blood, 11 v. epi, and 12 semen samples.
3.3. Marker cg-3867465
The average DNA methylation range for the cg-3867465 marker was 47–74% in buccal samples, compared to 10% or lower in other tissues. The difference between the lowest DNA methylation value in buccal samples and the highest value in other tissues was 37%, making this marker useful for identifying buccal samples. The standard deviation for this marker ranged from 11 to 15.7 for buccal samples, while for the other tissues it ranged from 0.8–7.2. Statistical analysis shows the percent methylation of buccal sample DNA differ significantly from other body fluids (p < 0.0001). The data comparison for the five CpG sites is shown in Figure 3.
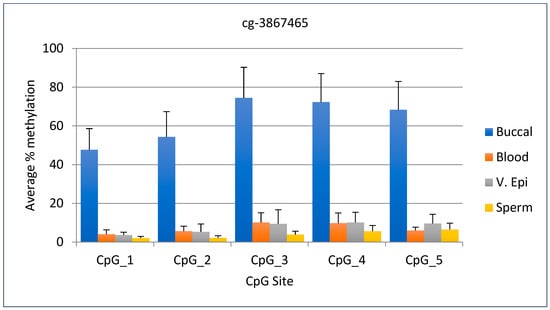
Figure 3.
Average DNA methylation data for cg-3867465 (CpG 4) and four other adjacent sites. The fourth CpG site is listed in Park et al. N = 11 buccal, 10 blood, 11 v. epi, and 11 semen samples.
3.4. Marker cg-10122865
The average DNA methylation range for the cg-10122865 marker was 59–85% in buccal samples, while it was 15% or lower in other tissues. The DNA methylation value for buccal samples was 44% higher than the highest value for other tissues, making this marker useful for identifying buccal samples. The standard deviation for this marker ranged from 17.2 to 21.4 for buccal samples, while for the other tissues it ranged from 1.6 to 9.0. Statistical analysis shows that the percent methylation of buccal sample DNA differed significantly from that of other body fluids (p < 0.0001). The graphical representation of the data for the five CpG sites is given in Figure 4.
The SPSS statistical analysis data for all the four markers are given in Tables S1–S4 (PASW Statistics v22). The p-values obtained for all the CpG sites and for all the markers tested are given in Table S5.
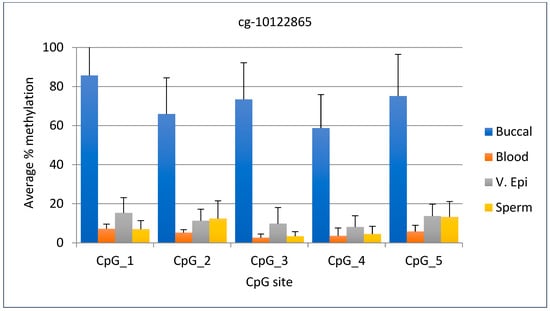
Figure 4.
Average DNA methylation data for marker cg-10122865 (CpG 1) and four adjacent sites. The first CpG site is listed in Park et al., 2014. N = 11 buccal, 11 blood, 11 v. epi, and 12 semen samples.
3.5. Mixture Analysis
The aim of this study was to analyze the methylation patterns of evidence samples when two different body fluids are mixed, and to compare these patterns with those from a single source of body fluid. We focused on two types of body fluids: buccal, which was hypermethylated, and semen, which was hypomethylated. In this study, we examined seven combinations of fluids: two neat samples, buccal and semen, along with five different mixtures of these two. The DNA methylation data obtained from these combinations revealed a clear trend: As the quantity of hypermethylated buccal sample DNA increased in the mixture, the overall percentage of DNA methylation also increased. Conversely, when the amount of buccal sample DNA decreased, the percentage of DNA methylation correspondingly decreased. This information is valuable for determining whether an analyzed biofluid came from a single source or is a mixed sample. In this study, we concentrated on only one marker because the DNA methylation pattern was consistent across all markers examined: Buccal sample DNA was hypermethylated, while all other tissues showed hypomethylation. The reduction in DNA methylation was apparent, as buccal sample DNA decreased in the mixture, as shown by the trendline in Figure 5.
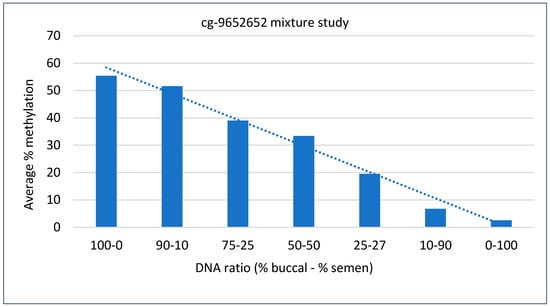
Figure 5.
DNA methylation percentage in the mixture study for the indicated locus. DNA methylation data from six adjacent CpG sites were averaged for each DNA ratio. The decreasing percentage of DNA methylation is evident by the trendline, as the quantity of buccal sample DNA in the mixture decreases.
3.6. Species Specificity Study
To evaluate the specificity of the primers and check for potential cross-reactivity with non-human species, we tested several non-human DNA samples, including those from cat, dog, chicken, cow, erythrobacter, chimpanzee, and rhesus monkey. The PCR amplicons from all samples were analyzed using a 2% agarose gel. The results indicate that both chimpanzee and rhesus monkey samples produced a visible band of the correct size, whereas the other samples did not yield any visible bands. Additionally, the pyrosequencing data confirmed that usable sequence data were generated from the chimpanzee and rhesus monkey samples, whereas the non-primate samples did not produce any sequencing data.
4. Discussion
Identifying body fluids in forensic casework is crucial. In addition to the current enzymatic and immunochromatographic assays used for detecting various body fluids, new identification methods have emerged. One innovative approach involves examining the methylation status of different genomic regions to determine tissue types. DNA methylation, an epigenetic modification, occurs naturally or in response to environmental factors such as age, smoking status, diet, and tissue type [9]. Recent studies have demonstrated that DNA methylation analysis is a viable option for forensic tissue identification, validating its effectiveness in casework samples. This includes the identification of blood, semen, epithelial tissues [19], and spermatozoa [20], as well as other tissues like saliva and vaginal secretions, through DNA methylation analysis [26]. High-resolution melt (HRM) analysis of DNA from different tissue sources is also a valuable tool for forensic tissue identification [21,27]. Beyond body fluid identification, methylation analysis has been utilized to determine an individual’s smoking status [28] and to estimate the age of a sample donor [29].
Even though several methods have been developed for DNA methylation analysis, bisulfite genomic sequencing is considered the gold standard [30]. Illumina Infinium Methylation BeadChip is widely used to measure individual CpG methylation on an epigenome-wide scale. Two methods have been proposed to measure the methylation level. The first one is called the beta-value, which is a continuous variable between 0 and 1. A value of “0” indicates the sample is unmethylated, while a value of “1” indicates it is fully methylated (www.illumina.com; URL accessed on 10 February 2025)). The second method is the log2 ratio of the intensities of a methylated probe versus an unmethylated probe, and this is referred to as the M-value method because it has been widely used in “mRNA expression microarray” analysis. A comparison of the M-value and beta-value methods has shown that the M-value approach yields better performance in terms of detection rate (DR) and true positive rate (TPR) for both highly methylated and unmethylated CpG sites [31].
In addition to methylation-based chip analysis, pyrosequencing of bisulfite-converted DNA provides quantitative DNA methylation values at one or more CpG sites with single-base resolution [19,20]. In bisulfite pyrosequencing, the CpG DNA methylation data are shown as percent DNA methylation for each CpG site studied in the target segment, providing a direct measure of DNA methylation. One limitation of bisulfite pyrosequencing is the incomplete bisulfite conversion at certain locations, which can result in a failed sequence due to unexpected peaks at the control site. Therefore, it is essential to incorporate a control to evaluate the quality of the sequence.
In this study, we present a set of four CpG sites, along with additional adjacent sites, that have the potential to differentiate buccal samples from three other body fluids: blood, vaginal epithelial cells, and semen. Identifying body fluids present at a crime scene is crucial in forensics, as it can help determine the sequence of events associated with the crime. In the human genome, some regions exhibit differential methylation for various reasons, including cell differentiation, gene expression, and X chromosome inactivation, and other factors such as age and smoking habits. We utilized these characteristics to identify specific segments of the genome that may differentiate one cell type from others based on their methylation status. A study by Park et al. [26] identified specific CpG sites presumed to be hypermethylated in saliva samples. We screened a selection of these sites using target-specific primers and identified four loci, along with certain adjacent sites. The target loci identified were cg-9652652, cg-11536474, cg-3867465, and cg-10122865. The results indicate that three of the four target CpG sites, as well as an additional 15 CpG sites, were hypermethylated in buccal samples compared to the other three body fluids: blood, vaginal epithelial cells, and semen. However, the CpG site cg-11536474 did not produce reliable results in the assay due to a stretch of nine “C” nucleotides preceding the site. This caused data skewing and triggered a “fail warning” flag due to “peak height deviation” at the “C” stretch. The DNA methylation data for the other three CpG sites located upstream of the target showed hypermethylation in buccal samples.
Although the average methylation data for all CpG sites across the markers showed that the buccal samples were hypermethylated compared to the other three body fluids, the standard deviations for these markers were modestly elevated, particularly in the buccal samples. This increase in standard deviation may have been due to several factors, including run-to-run variations, differences in the performance of various lots of the kits used in the study, the efficiency of bisulfite conversion, instrument service and calibration, individual variations among the sample population or different relative concentration of buccal versus spit cells. Despite these factors and the increased standard deviation, the percent methylation of the buccal samples was statistically significant (p < 0.0001) compared to the other three body fluids across all CpG sites studied for all markers. This significant difference in methylation highlights the four target CpG sites and adjacent sites as ideal candidates for differentiating buccal samples from other tissues. The two markers, cg-10122865 and cg-11536474, exhibited superior discriminatory power compared to the other markers due to their higher methylation percentages in the buccal samples, despite a modest increase in standard deviation. Specifically, the methylation percentage for these two markers in buccal samples was 59% for cg-10122865 and 69% or higher for cg-11536474 across all the studied CpG sites. In contrast, the methylation percentages for the other body fluids at these two markers were 15% or lower for all the CpG sites examined.
While the study of buccal samples may have limited applications in forensic casework compared to saliva, collecting one sample without the other can be challenging, and co-extraction of saliva with leukocytes cannot be excluded. Buccal swabs contain a higher proportion of epithelial cells than saliva. Saliva, on the other hand, contains a mix of epithelial cells and various types of leukocytes. A study by Theda et al. [32] found that the mean proportion of buccal epithelial cells in cheek swabs was 83%, compared to 47% in saliva. This indicates that nearly half of the cell types in the saliva were epithelial cells. In addition to examining the epithelial cell composition in saliva and buccal swabs, the study also analyzed the leukocyte composition in both sample types. It is noteworthy that the proportions of different types of leukocytes in the two samples were quite similar, although the overall quantities may have differed. Two types of leukocytes were identified: mature granulocytes and lymphocytes. In buccal swabs, approximately 60% of leukocytes were mature granulocytes and 25% were lymphocytes, while in saliva, these figures were about 55% and 40%, respectively. The authors concluded that the saliva and buccal swab samples almost always contain a mix of leukocytes and epithelial cells in a wide range of proportions, especially in saliva. To differentiate the methylation levels between buccal cells and saliva, it is essential to study both sample types for the specific marker of interest and determine whether there are any statistically significant differences between them before arriving at a conclusion.
The results of the mixture study for the cg-9652652 marker involving buccal sample and semen show that as the quantity of buccal cell DNA increased, the percentage of DNA methylation also increased, and as the quantity of buccal cell DNA decreased, the percentage of DNA methylation decreased (Figure 5). This information can be valuable when dealing with suspected mixed stains. For example, observing half of the expected DNA methylation data in a suspected saliva sample may indicate a stain of mixed body fluid origin [24,33]. The species specificity study demonstrated that the primers used are specific to primates and humans, as non-primate samples did not show amplification for the locus studied. Although the primer specificity study and the mixture study were conducted on a small scale, they provide a foundation for the reliability and human specificity of this marker. Further studies evaluating the primers for additional loci will help assess the effectiveness of the current primers. One technical limitation of this study is the method’s sensitivity, and a detailed sensitivity analysis will help determine the lowest amplification threshold for the sample.
In conclusion, these epigenetic markers hold great promise as candidates for forensic tissue identification. They offer several advantages, including the availability of extracted DNA from casework, primer specificity, and significant differences in DNA methylation levels between buccal samples and other bodily fluids. The specificity of the primers for primate and human samples ensures there is no interference from non-primate DNA, making them ideal for this purpose. Although the primer specificity study is still in its exploratory phase, it lays a foundation for larger-scale studies involving other markers of interest. This could enhance our understanding of these markers for forensic purposes. Additionally, these markers could serve as valuable tools for detecting the presence of mixed stains, particularly when other bodily fluids are present alongside buccal and/or saliva samples.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/forensicsci5020026/s1, Table S1. Statistical analysis data for marker cg-9652652. The DNA methylation levels of significance for Buccal/saliva samples, compared to other body fluids are highlighted in yellow for all CpG sites studied. Table S2. Statistical analysis data for marker cg-11536474. The DNA methylation levels of significance for Buccal/saliva samples, compared to other body fluids are highlighted in yellow for all CpG sites studied. Table S3. Statistical analysis data for marker cg-3867465. The DNA methylation levels of significance for Buccal/saliva samples, compared to other body fluids are highlighted in yellow for all CpG sites studied. Table S4. Statistical analysis data for marker cg-10122865. The DNA methylation levels of significance for Buccal/saliva samples, compared to other body fluids are highlighted in yellow for all CpG sites studied. Table S5. p values obtained for sample comparisons. The buccal/saliva DNA methylation was compared to the DNA methylation of other tissues and the p value results are given.
Author Contributions
Conceptualization, K.B. and B.M.; methodology, K.B. and E.S.; data analysis, E.S. and K.B. writing—original daft preparation, K.B.; writing—review and editing, G.D. and H.A.; funding acquisition, B.M. and K.B. All authors have read and agreed to the published version of the manuscript.
Funding
This work received partial support from a research grant from the National Institute of Justice (NIJ), Department of Justice, USA, under project number 2017-NE-BX-0001. All opinions expressed in this document are those of the authors and do not necessarily represent the official view of the NIJ.
Institutional Review Board Statement
This study was conducted after obtaining approval from the Institutional Review Board at the University of Southern Mississippi, protocol #12010303 dated 20 January 2018 with additional yearly renewals.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
All data are available in the results section and in the Supplementary Tables.
Acknowledgments
The authors want to thank the volunteers who provided samples for this research. This work received partial support from a research grant from the National Institute of Justice (NIJ), Department of Justice, USA, under project number 2017-NE-BX-0001. All opinions expressed in this document are those of the authors and do not necessarily represent the official view of the NIJ.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Jeffreys, A.; Wilson, V.; Thein, S.L. Hypervariable ‘minisatellite’ regions in human DNA. Nature 1985, 314, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Sawaguchi, T.; Sawaguchi, A. Forensic Application of STR Polymorphic Markers. In Proceedings of the 16th Congress of the International Society for Forensic Haemogenetics (Internationale Gesellschaft für Forensische Hämogenetik e.V.), Santiago de Compostela, Spain, 12–16 September 1995; Advances in Forensic Haemogenetics. Carracedo, A., Brinkmann, B., Bär, W., Eds.; Springer: Berlin/Heidelberg, Germany, 1996; Volume 6. [Google Scholar]
- Fernandez-Tejero, N.; Ghemrawi, M.; McCord, B.R.; Lee, S.B. Deoxyribonucleic Acid Extraction Methods in Forensic Analysis. Encycl. Anal. Chem. Appl. Theory Instrum. 2006, 1–21. [Google Scholar] [CrossRef]
- Li, R. Forensic Biology, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2015. [Google Scholar]
- Frumkin, D.; Wasserstrom, A.; Budowle, B.; Davidson, A. DNA methylation-based forensic tissue identification. Forensic Sci. Int. Genet. 2011, 5, 517–524. [Google Scholar] [CrossRef]
- Juusola, J.; Ballantyne, J. mRNA profiling for body fluid identification by multiplex quantitative RT-PCR. J. Forensic Sci. 2007, 52, 1252–1262. [Google Scholar] [CrossRef]
- Haas, C.; Klesser, B.; Maake, C.; Bär, W.; Kratzer, A. mRNA profiling for body fluid identification by reverse transcription endpoint PCR and realtime PCR. Forensic Sci. Int. Genet. 2009, 3, 80–88. [Google Scholar] [CrossRef]
- Haas, C.; Hänggi, N.; Hanson, E.; Ballantyne, J. Body Fluid Identification by mRNA and MicroRNA. In Encyclopedia of Forensic Sciences, 3rd ed.; Houck, M.M., Ed.; Springer: Berlin/Heidelberg, Germany, 2023; Volume 1, pp. 390–401. [Google Scholar]
- Vidaki, A.; Daniel, B.; Court, D.S. Forensic DNA methylation profiling—Potential opportunities and challenges. Forensic Sci. Int. Genet. 2013, 7, 499–507. [Google Scholar] [CrossRef]
- Song, F.; Smith, J.F.; Kimura, M.T.; Morrow, A.D.; Matsuyama, T.; Nagase, H.; Held, W.A. Association of tissue-specific differentially methylated regions (TDMs) with differential gene expression. Proc. Natl. Acad. Sci. USA 2005, 102, 3336–3341. [Google Scholar] [CrossRef]
- Lokk, K.; Modhukur, V.; Rajashekar, B.; Märtens, K.; Mägi, R.; Kolde, R.; Koltšina, M.; Nilsson, T.K.; Vilo, J.; Salumets, A.; et al. DNA methylome profiling of human tissues identifies global and tissue-specific methylation patterns. Genome Biol. 2014, 15, r54. [Google Scholar] [CrossRef]
- Boers, R.; Boers, J.; Hoon, B.; Kockx, C.; Ozgur, Z.; Molijn, A.; IJcken, W.; Laven, J.; Gribnau, J. Genome-wide DNA methylation profiling using the methylation-dependent restriction enzyme LpnPI. Genome Res. 2018, 28, 88–99. [Google Scholar] [CrossRef]
- Li, E.; Zhang, Y. DNA methylation in mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a019133. [Google Scholar] [CrossRef]
- Rakyan, V.K.; Down, T.A.; Thorne, N.P.; Flicek, P.; Kulesha, E.; Gräf, S.; Tomazou, E.M.; Bäckdahl, L.; Johnson, N.; Herberth, M.; et al. An integrated resource for genome-wide identification and analysis of human tissue-specific differentially methylated regions (tDMRs). Genome Res. 2008, 18, 1518–1529. [Google Scholar] [CrossRef] [PubMed]
- Bruijns, B.; Tiggelaar, R.; Gardeniers, H. Massively parallel sequencing techniques for forensics: A review. Electrophoresis 2018, 39, 2642–2654. [Google Scholar] [CrossRef] [PubMed]
- Carratto, T.M.T.; Moraes, V.M.S.; Recalde, T.S.F.; Oliveira, M.L.G.; Teixeira Mendes-Junior, C. Applications of massively parallel sequencing in forensic genetics. Genet. Mol. Biol. 2022, 45 (Suppl. S1), e20220077. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Lee, J.M.; Naue, J.; Fleckhaus, J.; Freire-Aradas, A.; Neubauer, J.; Pośpiech, E.; McCord, B.; Kalamara, V.; Gauthier, Q.; et al. A collaborative exercise on DNA methylation-based age prediction and body fluid typing. Forensic Sci. Int. Genet. 2022, 57, 102656. [Google Scholar] [CrossRef]
- Forat, S.; Huettel, B.; Reinhardt, R.; Fimmers, R.; Haidl, G.; Denschlag, D.; Olek, K. Methylation Markers for the Identification of Body Fluids and Tissues from Forensic Trace Evidence. PLoS ONE 2016, 11, e0147973. [Google Scholar] [CrossRef]
- Madi, T.; Balamurugan, K.; Bombardi, R.; Duncan, G.; McCord, B. The determination of tissue specific DNA methylation patterns in forensic biofluids using bisulfite modification and pyrosequencing. Electrophoresis 2012, 33, 1736–1745. [Google Scholar] [CrossRef]
- Balamurugan, K.; Bombardi, R.; Duncan, G.; McCord, B. Identification of spermatozoa by tissue-specific differential DNA methylation using bisulfite modification and pyrosequencing. Electrophoresis 2014, 35, 3079–3086. [Google Scholar] [CrossRef]
- Alghanim, H.; Balamurugan, K.; McCord, B. Development of DNA methylation markers for sperm, saliva and blood identification using pyrosequencing and qPCR/HRM. Anal. Biochem. 2020, 611, 113933. [Google Scholar] [CrossRef]
- Lee, H.Y.; Jung, S.E.; Lee, E.H.; Yang, W.I.; Shin, K.J. DNA methylation profiling for a confirmatory test for blood, saliva, semen, vaginal fluid and menstrual blood. Forensic Sci. Int. Genet. 2016, 24, 75–82. [Google Scholar] [CrossRef]
- Ghai, M.; Naidoo, N.; Evans, D.L.; Kader, F. Identification of novel semen and saliva specific methylation markers and its potential application in forensic analysis. Forensic Sci. Int. Genet. 2020, 49, 102392. [Google Scholar] [CrossRef]
- Konrad, H.; Jürgens, L.; Hartung, B.; Poetsch, M. More than just blood, saliva, or sperm-setup of a workflow for body fluid identification by DNA methylation analysis. Int. J. Legal Med. 2023, 137, 1683–1692. [Google Scholar] [CrossRef] [PubMed]
- Budowle, B.; Smith, J.; Moretti, T.; DiZinno, J. DNA Typing Protocols: Molecular Biology and Forensic Analysis; Eaton Publishing: Natick, MA, USA, 2000. [Google Scholar]
- Park, J.L.; Kwon, O.H.; Kim, J.H.; Yoo, H.S.; Lee, H.C.; Woo, K.M.; Kim, S.Y.; Lee, S.H.; Kim, Y.S. Identification of body fluid-specific DNA methylation markers for use in forensic science. Forensic Sci. Int. Genet. 2014, 13, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Antunes, J.; Silva, D.; Balamurugan, K.; Duncan, G.; Alho, C.; McCord, B. High Resolution Melt analysis of DNA methylation to discriminate semen in biological stains. Anal. Biochem. 2016, 494, 40–45. [Google Scholar] [CrossRef]
- Alghanim, H.; Wu, W.; McCord, B. DNA methylation assay based on pyrosequencing for determination of smoking status. Electrophoresis 2018, 39, 2806–2814. [Google Scholar] [CrossRef]
- Poussard, D.A.; Curci, J.; Siatka, C.; Hermitte, F.; Pussiau, A.; Singla-Sanchez, H.; Hubac, S. Evaluation of DNA Methylation-Based Age-Prediction Models from Saliva and Buccal Swab Samples Using Pyrosequencing. Forensic Sci. 2023, 3, 192–204. [Google Scholar] [CrossRef]
- Singer, B.D. A Practical Guide to the Measurement and Analysis of DNA Methylation. Am. J. Respir. Cell Mol. Biol. 2019, 61, 417–428. [Google Scholar] [CrossRef]
- Du, P.; Zhang, X.; Huang, C.C.; Jafari, N.; Kibbe, W.A.; Hou, L.; Lin, S.M. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef]
- Theda, C.; Hwang, S.H.; Czajko, A.; Loke, Y.J.; Leong, P.; Craig, J.M. Quantitation of the cellular content of saliva and buccal swab samples. Sci. Rep. 2018, 8, 6944. [Google Scholar] [CrossRef]
- Wang, H.X.; Liu, X.Z.; He, X.M.; Xiao, C.; Huang, D.X.; Yi, S.H. Identification of Mixtures of Two Types of Body Fluids Using the Multiplex Methylation System and Random Forest Models. Curr. Med. Sci. 2023, 43, 908–918. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).