

Enhanced Assessment of Transition Metal Copper Sulfides via Classification of Density of States Spectra

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methodology
2.1. Dataset and Preprocessing
2.2. Quantifying Structural Relationships Through Cluster Centroids and Distances
2.3. The Convex Hull Analysis for Structural Classification and Outlier Identification
2.4. Deviations in Electronic Structure
3. Results and Discussion
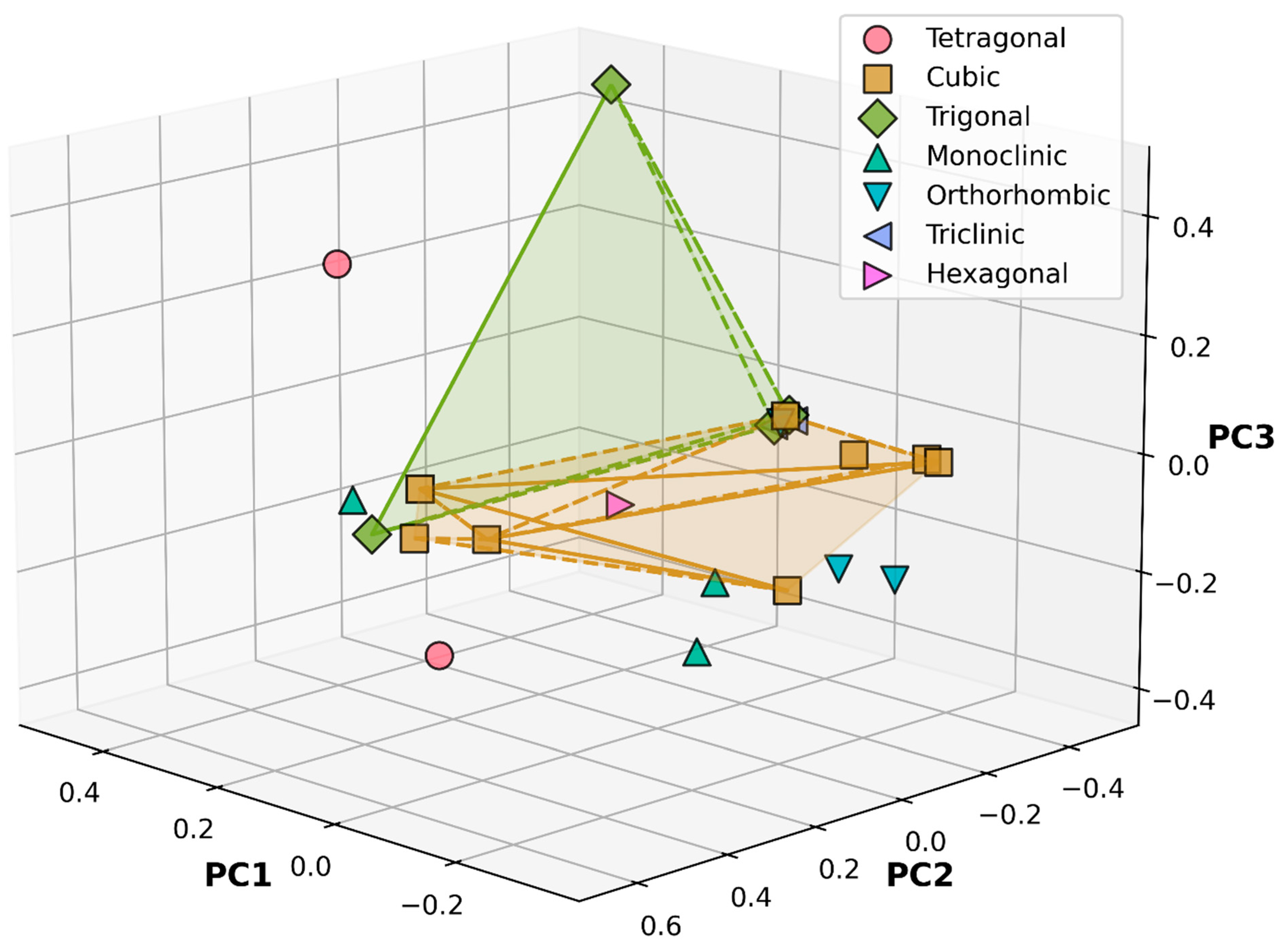
3.1. Crystal System Classification in Reduced-Dimensional KPCA Space
3.2. Orbital-Resolved PDOS of Outlier Compounds
3.3. Phase Stability and Transformation Pathways
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jaros, M. Electronic Properties of Semiconductor Alloy Systems. Rep. Prog. Phys. 1985, 48, 1091. [Google Scholar] [CrossRef]
- Keyes, R.W. Electronic Effects in the Elastic Properties of Semiconductors. In Solid State Physics; Seitz, F., Turnbull, D., Ehrenreich, H., Eds.; Academic Press: Cambridge, MA, USA, 1968; Volume 20, pp. 37–90. [Google Scholar]
- Poon, S.J. Electronic Properties of Quasicrystals an Experimental Review. Adv. Phys. 1992, 41, 303–363. [Google Scholar] [CrossRef]
- Tresca, C.; Giovannetti, G.; Capone, M.; Profeta, G. Electronic Properties of Superconducting FeS. Phys. Rev. B 2017, 95, 205117. [Google Scholar] [CrossRef]
- Lavanya, S.; Rajesh Kumar, T.; Prakash, B.; Rimal Isaac, R.S.; Ashraf, I.M.; Siddhartha; Shkir, M.; Kansal, L.; Payal, H.; Sehgal, S.S. Effect of Bi Doping on the Opto-Electronic Properties of ZnO Nanoparticles for Photodetector Applications. J. Photochem. Photobiol. A Chem. 2024, 446, 115119. [Google Scholar] [CrossRef]
- Chatzivasileiadi, A.; Ampatzi, E.; Knight, I. Characteristics of Electrical Energy Storage Technologies and Their Applications in Buildings. Renew. Sustain. Energy Rev. 2013, 25, 814–830. [Google Scholar] [CrossRef]
- Luo, B.; Wang, X.; Tian, E.; Yao, Y.; Cai, Z.; Xi, K.; Song, H.; Song, H.; Li, B.; Li, L. Interfacial Electronic Properties of Ferroelectric Nanocomposites for Energy Storage Application. Mater. Today Energy 2019, 12, 136–145. [Google Scholar] [CrossRef]
- Yeo, B.C.; Kim, D.; Kim, C.; Han, S.S. Pattern Learning Electronic Density of States. Sci. Rep. 2019, 9, 5879. [Google Scholar] [CrossRef]
- Rajshekar, K.; Kannadassan, D. A Comprehensive Density-of-States Model for Oxide Semiconductor Thin Film Transistors. J. Comput. Electron. 2021, 20, 2331–2341. [Google Scholar] [CrossRef]
- Huda, M.N.; Yan, Y.; Moon, C.-Y.; Wei, S.-H.; Al-Jassim, M.M. Density-Functional Theory Study of the Effects of Atomic Impurity on the Band Edges of Monoclinic WO3. Phys. Rev. B 2008, 77, 195102. [Google Scholar] [CrossRef]
- De Amézaga, C.S.P.; García-Suárez, V.M.; Fernández-Martínez, J.L. Classification and Prediction of Bulk Densities of States and Chemical Attributes with Machine Learning Techniques. Appl. Math. Comput. 2022, 412, 126587. [Google Scholar] [CrossRef]
- Jain, A.; Montoya, J.; Dwaraknath, S.; Zimmermann, N.E.R.; Dagdelen, J.; Horton, M.; Huck, P.; Winston, D.; Cholia, S.; Ong, S.P.; et al. The Materials Project: Accelerating Materials Design Through Theory-Driven Data and Tools. In Handbook of Materials Modeling: Methods: Theory and Modeling; Andreoni, W., Yip, S., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 1751–1784. ISBN 978-3-319-44677-6. [Google Scholar]
- Liu, H.; Cheng, J.; Dong, H.; Feng, J.; Pang, B.; Tian, Z.; Ma, S.; Xia, F.; Zhang, C.; Dong, L. Screening Stable and Metastable ABO3 Perovskites Using Machine Learning and the Materials Project. Comput. Mater. Sci. 2020, 177, 109614. [Google Scholar] [CrossRef]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. Commentary: The Materials Project: A Materials Genome Approach to Accelerating Materials Innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef]
- Lee, W.J.; Mendis, G.P.; Triebe, M.J.; Sutherland, J.W. Monitoring of a Machining Process Using Kernel Principal Component Analysis and Kernel Density Estimation. J. Intell. Manuf. 2020, 31, 1175–1189. [Google Scholar] [CrossRef]
- Rose, M.J. Semiconductor Band Structure, Symmetry, and Molecular Interface Hybridization for the Chemist. J. Am. Chem. Soc. 2024, 46, 5735–5748. [Google Scholar] [CrossRef]
- Imada, M.; Fujimori, A.; Tokura, Y. Metal-Insulator Transitions. Rev. Mod. Phys. 1998, 70, 1039–1263. [Google Scholar] [CrossRef]
- Filho, J.B.O.S.; Diniz, P.S.R. A Sparse Fixed-Point Online KPCA Extraction Algorithm. IEEE Trans. Signal Process. 2024, 72, 4604–4617. [Google Scholar] [CrossRef]
- Schlexer Lamoureux, P.; Winther, K.T.; Garrido Torres, J.A.; Streibel, V.; Zhao, M.; Bajdich, M.; Abild-Pedersen, F.; Bligaard, T. Machine Learning for Computational Heterogeneous Catalysis. ChemCatChem 2019, 11, 3581–3601. [Google Scholar] [CrossRef]
- Selvaratnam, B.; Oliynyk, A.O.; Mar, A. Interpretable Machine Learning in Solid-State Chemistry, with Applications to Perovskites, Spinels, and Rare-Earth Intermetallics: Finding Descriptors Using Decision Trees. Inorg. Chem. 2023, 62, 10865–10875. [Google Scholar] [CrossRef]
- Mangelis, P.; Ioannou, P.S.; So̷iland, A.-K.; Kyratsi, T. Mechanical Alloying: An Advantageous Method for the Development of Mg2 Si0.8 Sn0.2 and Mg2Si Thermoelectrics Using Commercial and Recyclable Silicon. ACS Appl. Energy Mater. 2025, 8, 1783–1795. [Google Scholar] [CrossRef]
- Almutairi, F.N. Synthesis and Investigation on the Structural and Complex Impedance Analysis in LISICON Compound, Li3Al2(PO4)3, for Solid Electrolyte Battery Applications. J. Mater. Sci: Mater. Electron. 2025, 36, 346. [Google Scholar] [CrossRef]
- Viskupová, K.; Fiantok, T.; Grančič, B.; Švec, P., Jr.; Roch, T.; Truchlý, M.; Šroba, V.; Satrapinskyy, L.; Kúš, P.; Mikula, M. Heterostructural Decomposition in V1-xWxB2-Δ Films Induced by B Deficiency. Materialia 2025, 39, 102351. [Google Scholar] [CrossRef]
- Ikeda, S.; Aono, N.; Iwase, A.; Kobayashi, H.; Kudo, A. Cu3MS4 (M=V, Nb, Ta) and Its Solid Solutions with Sulvanite Structure for Photocatalytic and Photoelectrochemical H2 Evolution under Visible-Light Irradiation. ChemSusChem 2019, 12, 1977–1983. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Zhang, Y.C.; Li, J.; Chen, Y.; Xu, B. Hydrothermal Synthesis of Cu2WS4 as a Visible-Light-Activated Photocatalyst in the Reduction of Aqueous Cr(VI). Mater. Lett. 2014, 117, 24–27. [Google Scholar] [CrossRef]
- Aslan, E.; Gonce, M.K.; Yigit, M.Z.; Sarilmaz, A.; Stathatos, E.; Ozel, F.; Can, M.; Patir, I.H. Photocatalytic H2 Evolution with a Cu2WS4 Catalyst on a Metal Free D-π-A Organic Dye-Sensitized TiO2. Appl. Catal. B Environ. 2017, 210, 320–327. [Google Scholar] [CrossRef]
- Calbo, J.; Golomb, M.J.; Walsh, A. Redox-Active Metal–Organic Frameworks for Energy Conversion and Storage. J. Mater. Chem. A 2019, 7, 16571–16597. [Google Scholar] [CrossRef]
- Gorai, P.; Toberer, E.S.; Stevanović, V. Thermoelectricity in Transition Metal Compounds: The Role of Spin Disorder. Phys. Chem. Chem. Phys. 2016, 18, 31777–31786. [Google Scholar] [CrossRef]
- Han, H.; Zhao, L.; Wu, X.; Zuo, B.; Bian, S.; Li, T.; Liu, X.; Jiang, Y.; Chen, C.; Bi, J.; et al. Advancements in Thermoelectric Materials: Optimization Strategies for Enhancing Energy Conversion. J. Mater. Chem. A 2024, 12, 24041–24083. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islam, M.T.; Ruiz, C.V.; Loyola, C.; Peralta, J.; Broderick, S.R. Enhanced Assessment of Transition Metal Copper Sulfides via Classification of Density of States Spectra. Solids 2025, 6, 32. https://doi.org/10.3390/solids6030032
Islam MT, Ruiz CV, Loyola C, Peralta J, Broderick SR. Enhanced Assessment of Transition Metal Copper Sulfides via Classification of Density of States Spectra. Solids. 2025; 6(3):32. https://doi.org/10.3390/solids6030032
Chicago/Turabian StyleIslam, Md Tohidul, Catalina Victoria Ruiz, Claudia Loyola, Joaquin Peralta, and Scott R. Broderick. 2025. "Enhanced Assessment of Transition Metal Copper Sulfides via Classification of Density of States Spectra" Solids 6, no. 3: 32. https://doi.org/10.3390/solids6030032
APA StyleIslam, M. T., Ruiz, C. V., Loyola, C., Peralta, J., & Broderick, S. R. (2025). Enhanced Assessment of Transition Metal Copper Sulfides via Classification of Density of States Spectra. Solids, 6(3), 32. https://doi.org/10.3390/solids6030032