
Structural and Energetic Aspects of Entacapone-Theophylline-Water Cocrystal

 ,
,  , ,
, ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Cocrystals and Physical Mixtures
2.3. X-ray Powder Diffraction
2.4. Attenuated Total Reflectance (ATR)-FTIR Spectroscopy
2.5. Thermal Analysis
2.5.1. Differential Scanning Calorimetry (DSC)
2.5.2. Thermogravimetric Analysis (TGA)
2.5.3. Hot Stage Polarized Light Microscopy (HSM)
2.6. Cocrystal Screening Methods
2.6.1. Construction of Binary Phase Diagram
2.6.2. Hansen Solubility Parameters (HSP)
2.6.3. Molecular Complementarity (MC)
2.6.4. Hydrogen Bond Propensity (HBP)
2.7. Molecular and Solid-State Modelling
2.7.1. Hirshfeld Surface Analysis
2.7.2. DFT Calculations and Topological Analysis
2.7.3. Lattice Energy Frameworks
2.7.4. Mechanical Properties
2.7.5. Crystal Morphology Modelling
2.8. Dehydration Kinetics
2.8.1. Mechanistic Kinetic Models
2.8.2. Vyazovkin’s Isoconversional Method
3. Results
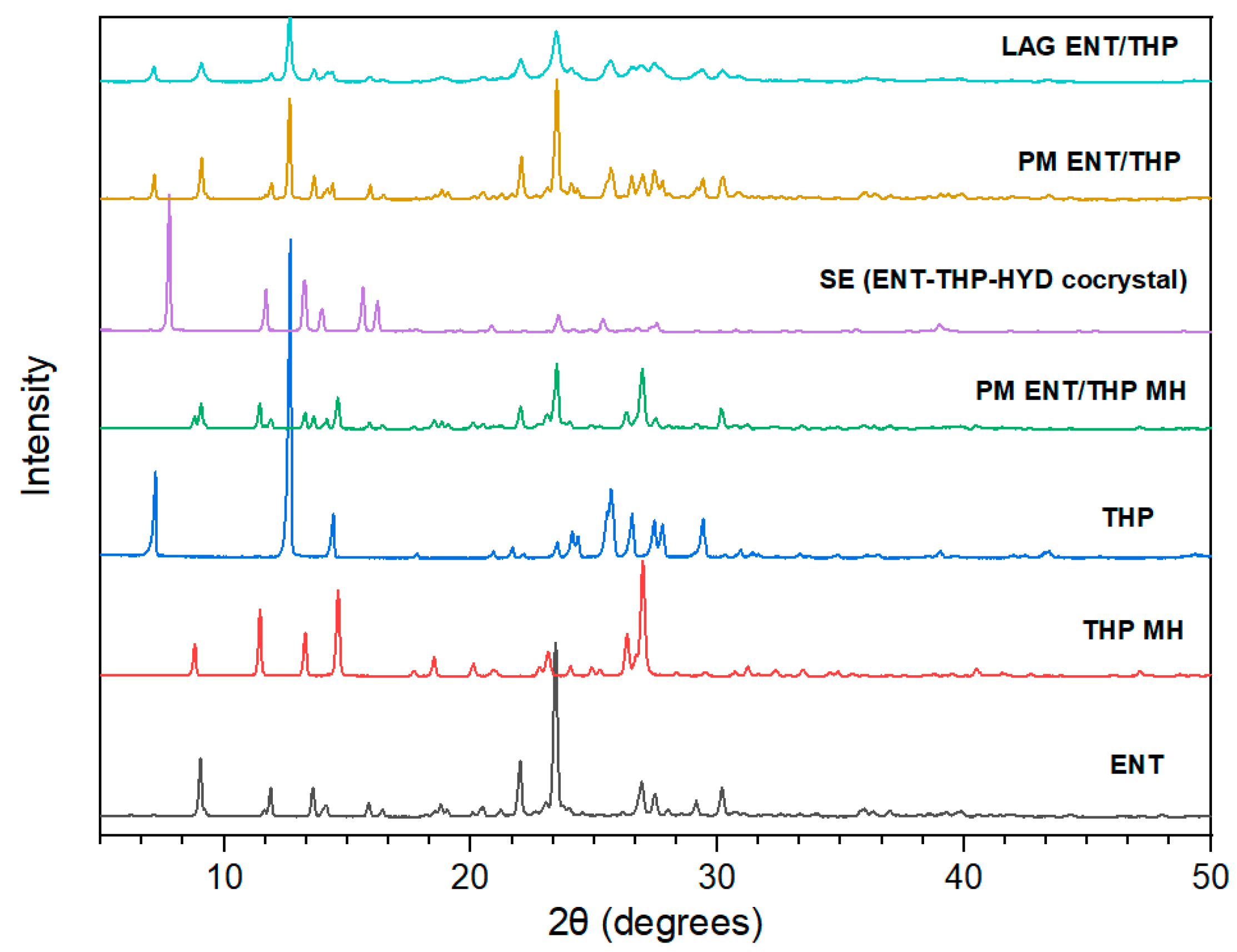
3.1. Preparation and Characterization of Cocrystals
3.2. Cocrystal Screening Methods
3.3. Molecular and Solid-State Modelling
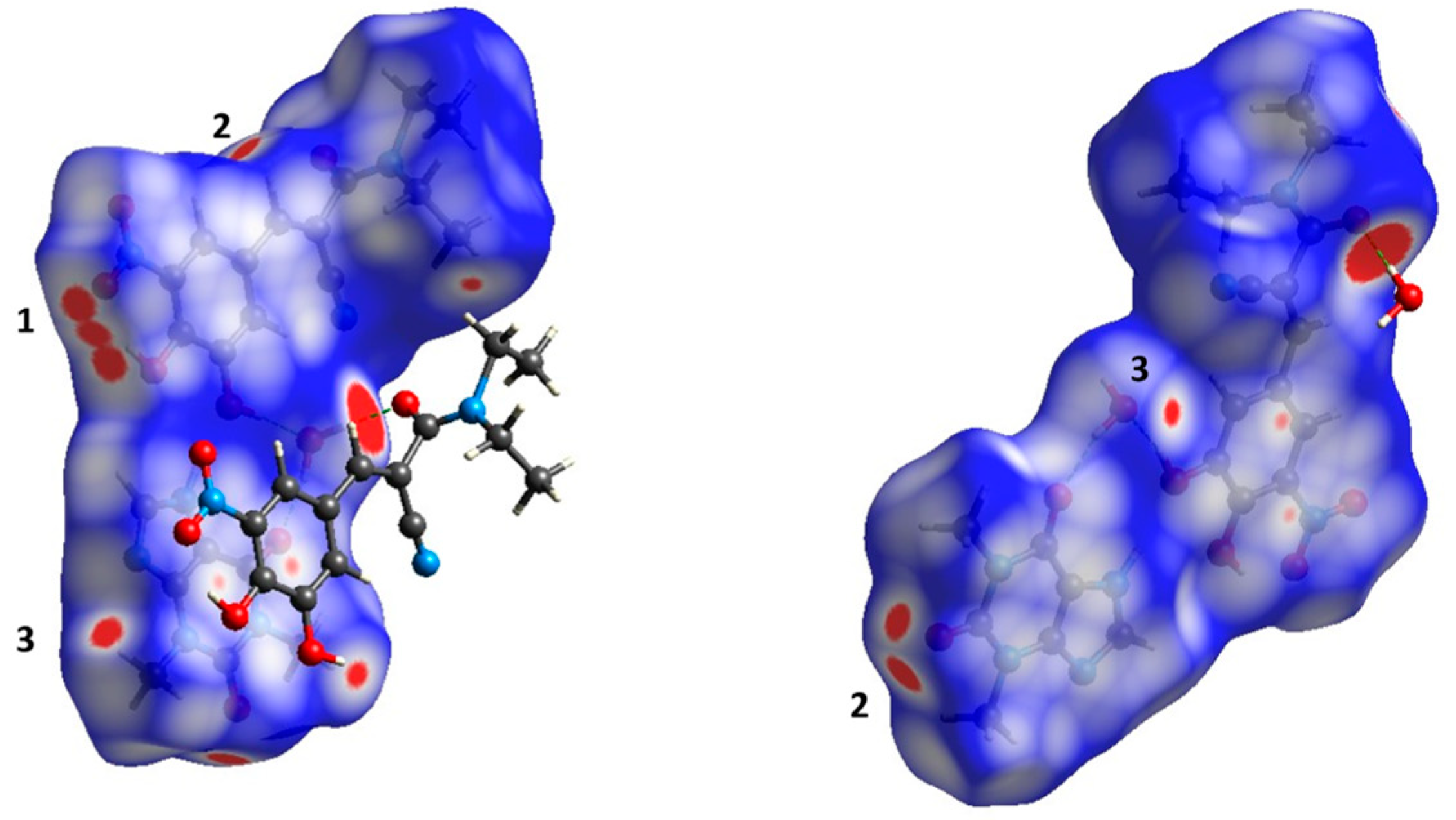
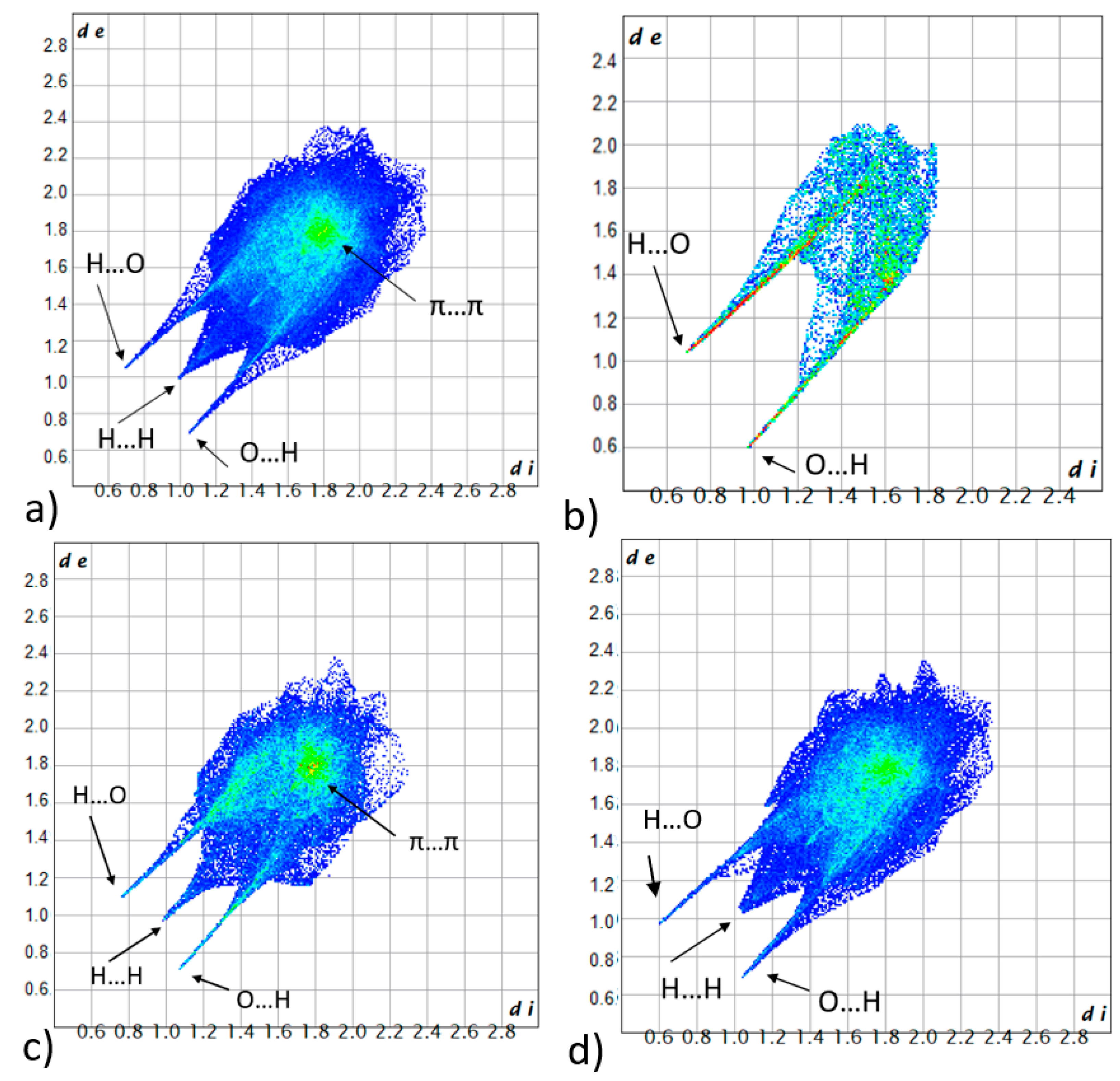
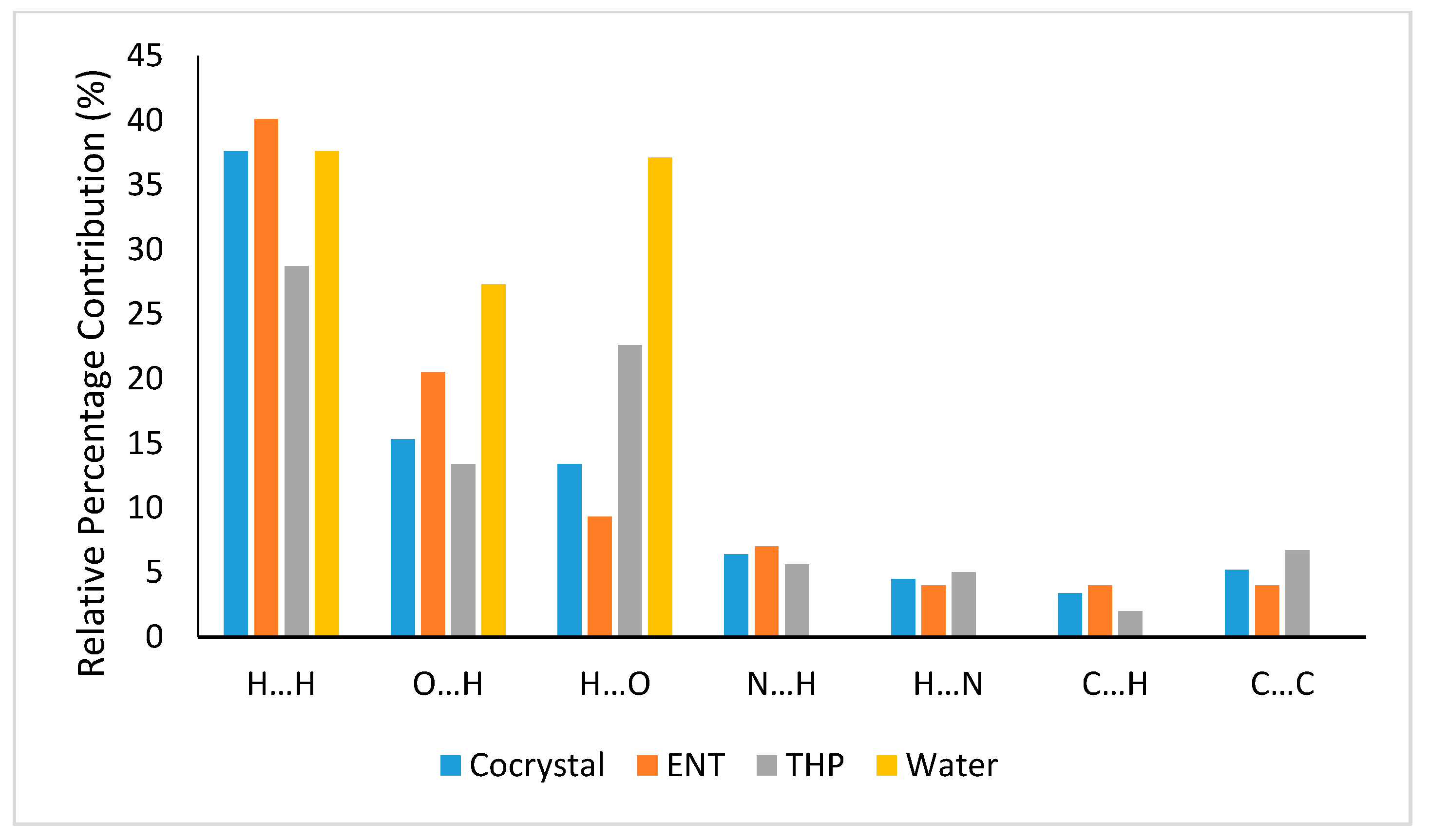
3.3.1. Hirshfeld Surface Analysis
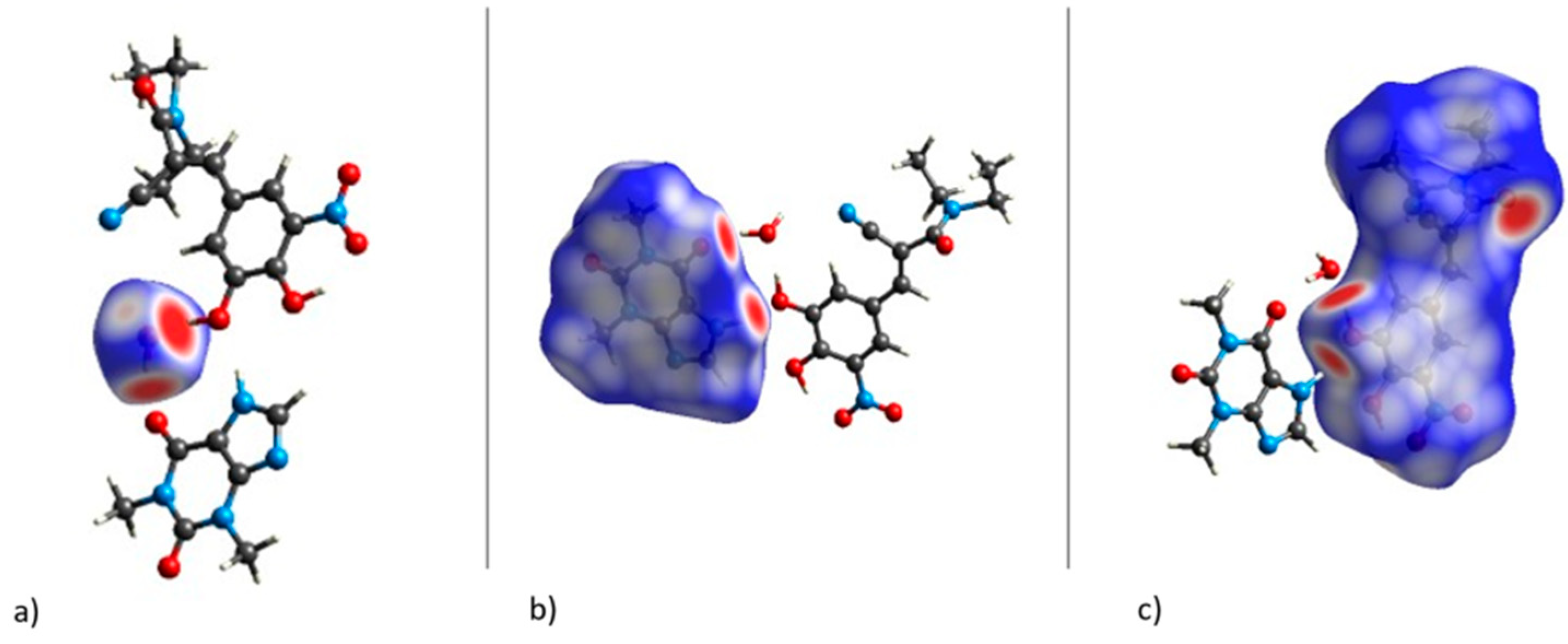
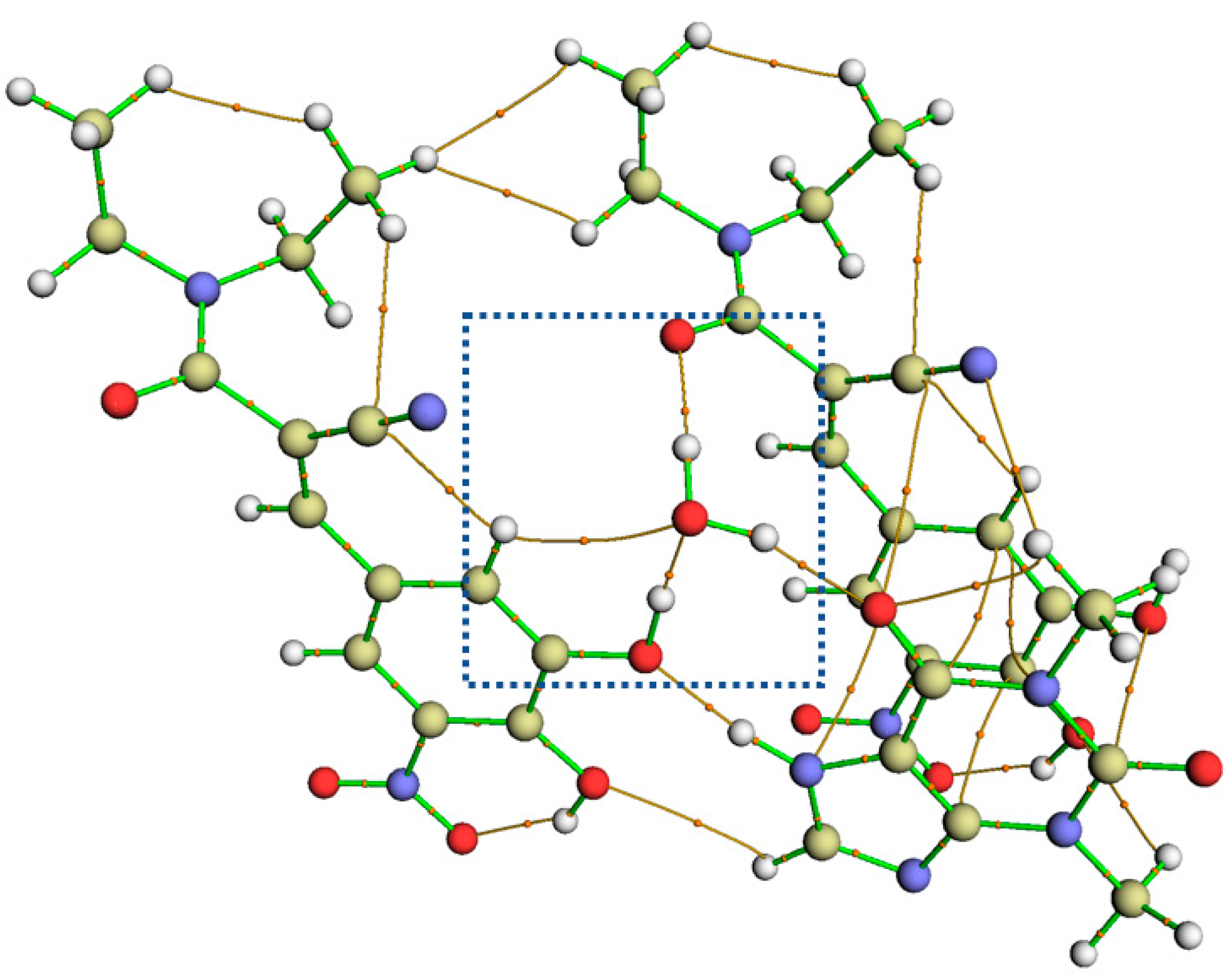
3.3.2. Non-Covalent Interaction Plots and Bond Critical Points via Quantum Theory of Atoms in Molecules (QTAIM) Analysis
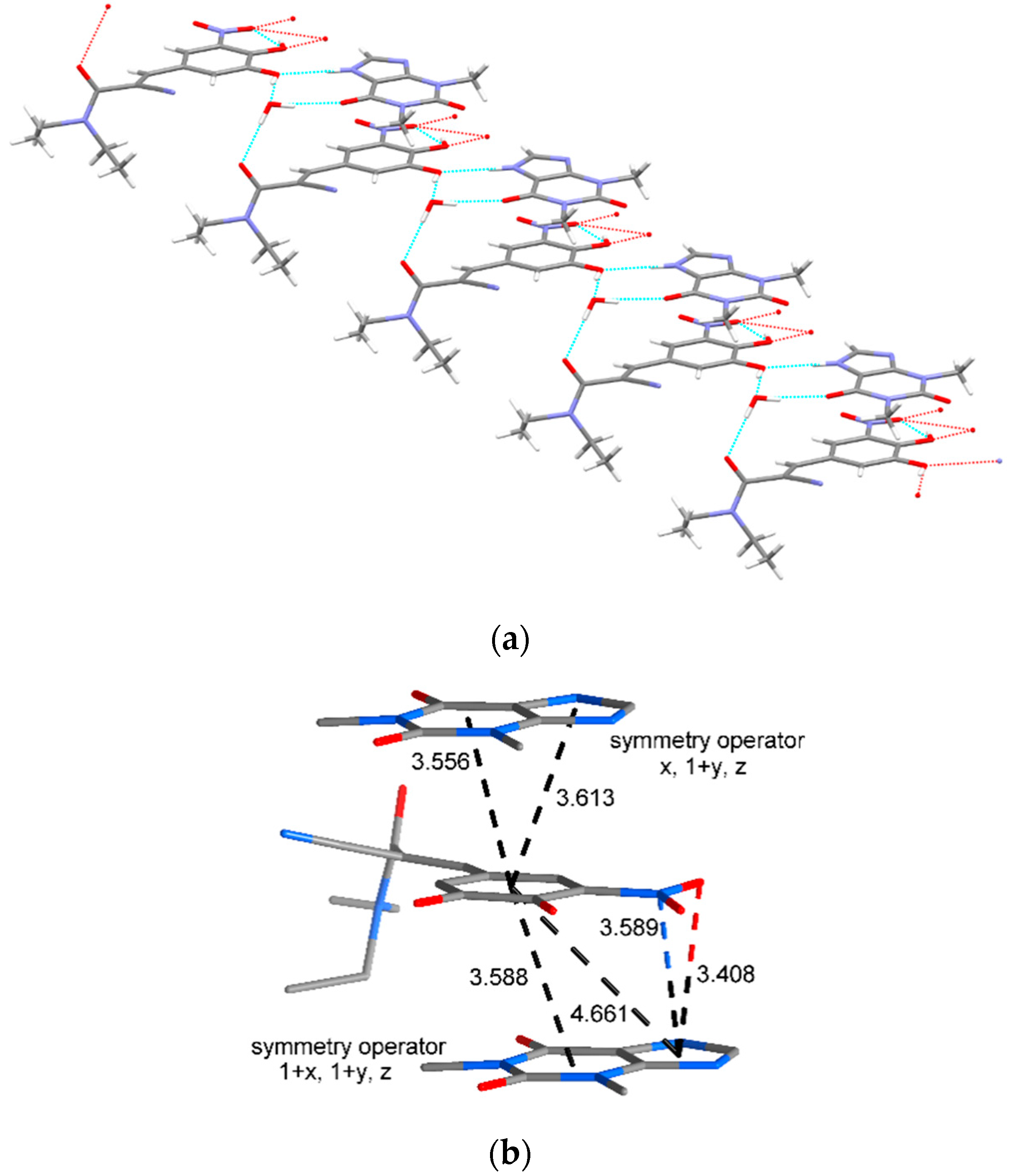
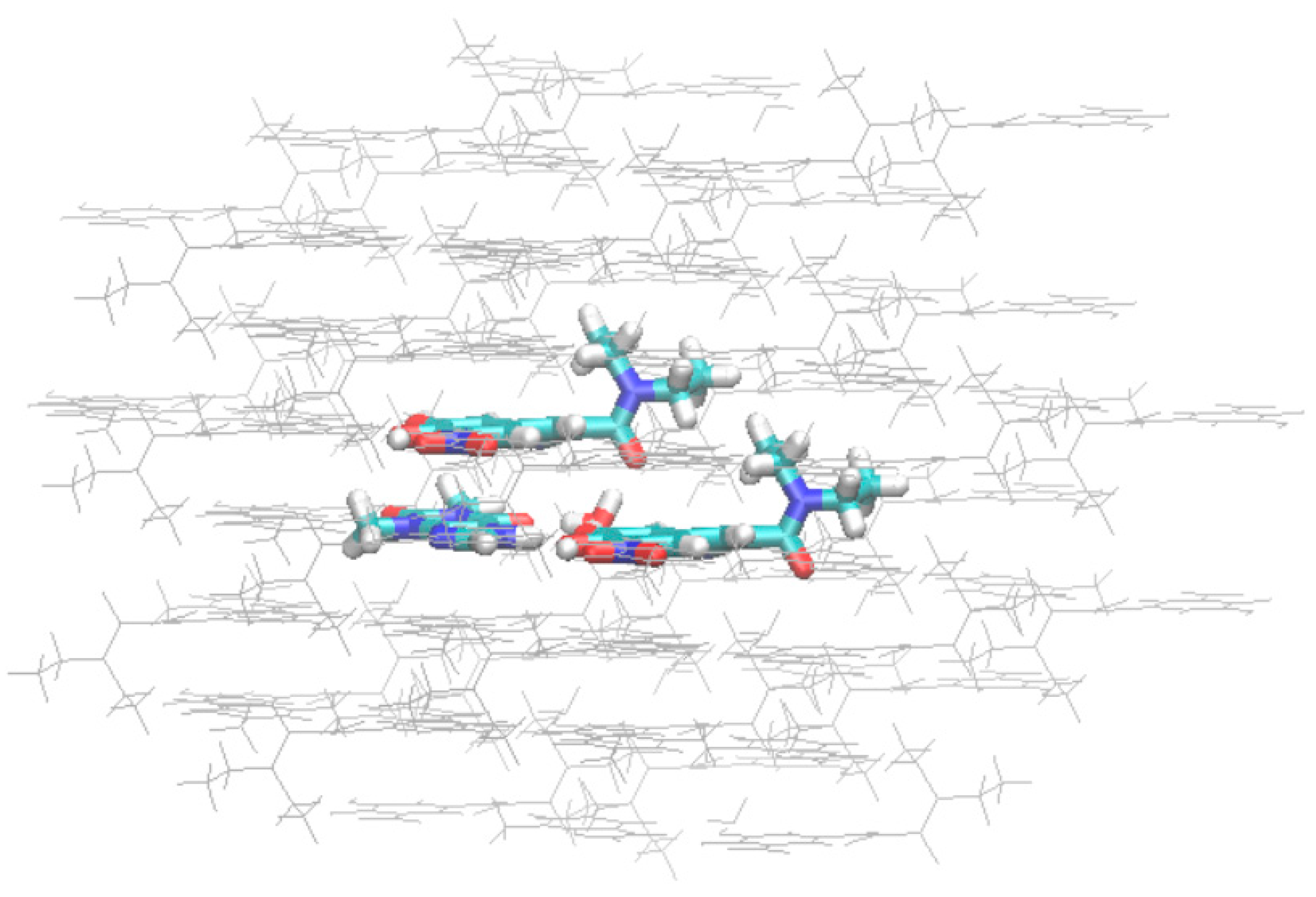
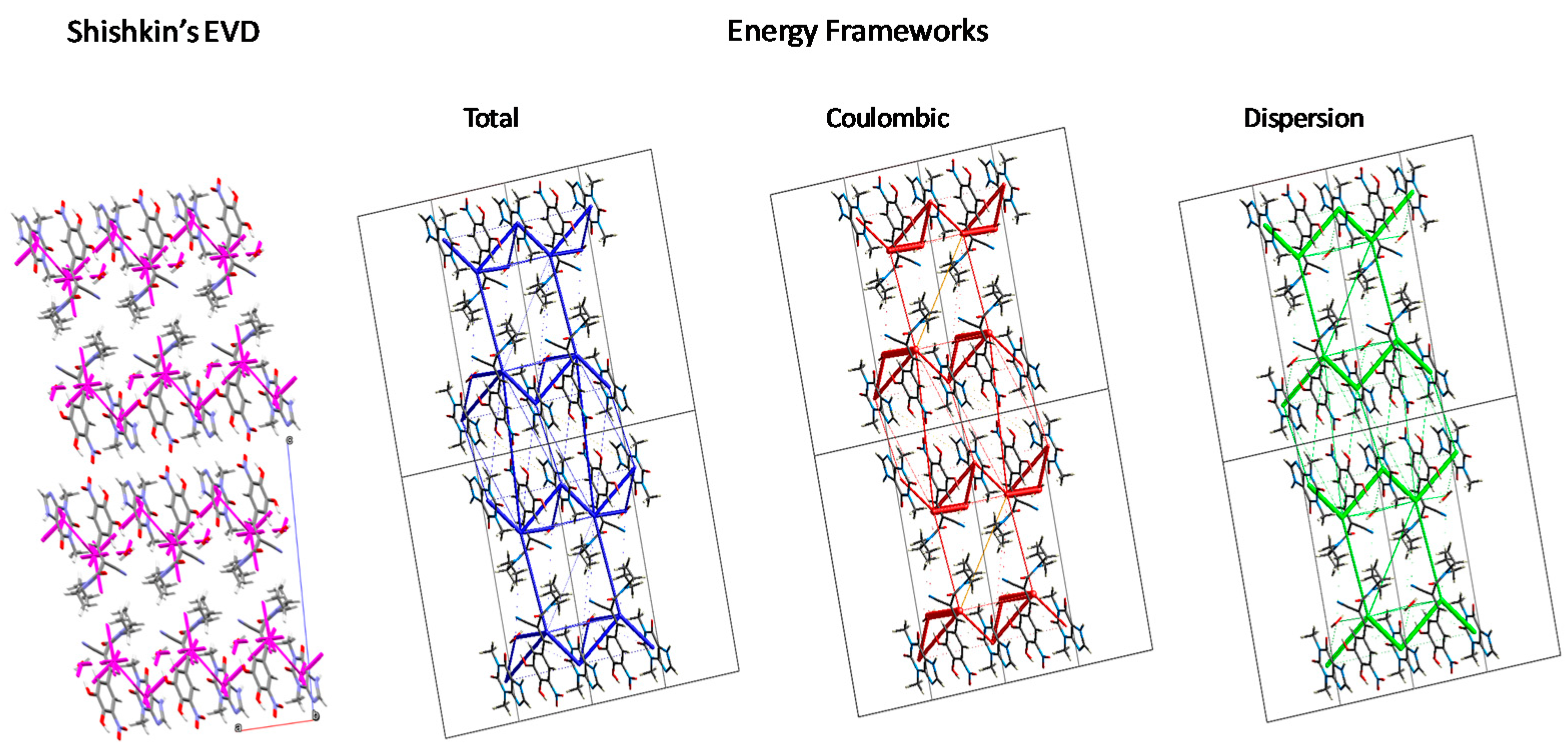
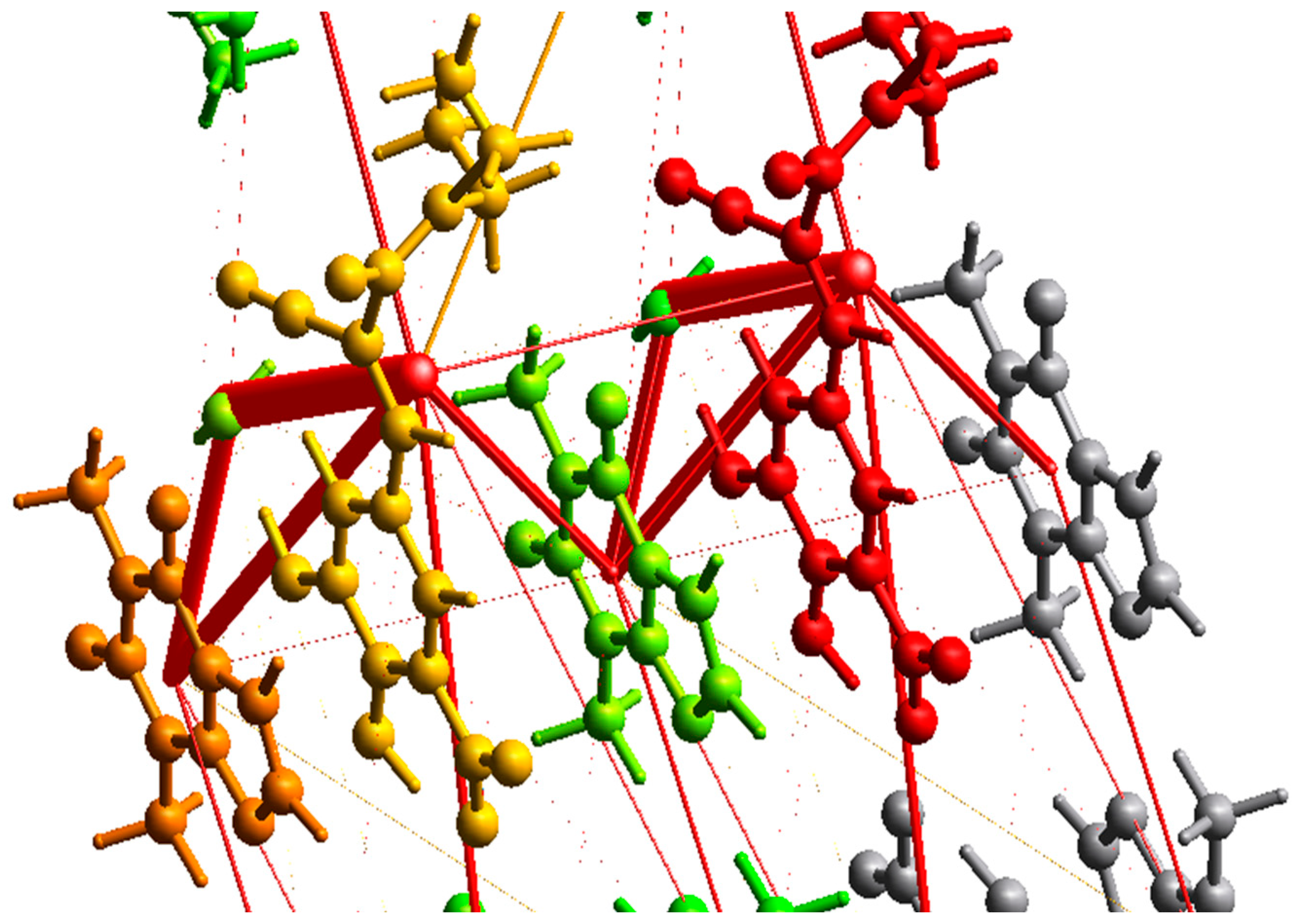
3.3.3. Intermolecular Interactions: Energy Vector Diagrams and Lattice Energy Frameworks
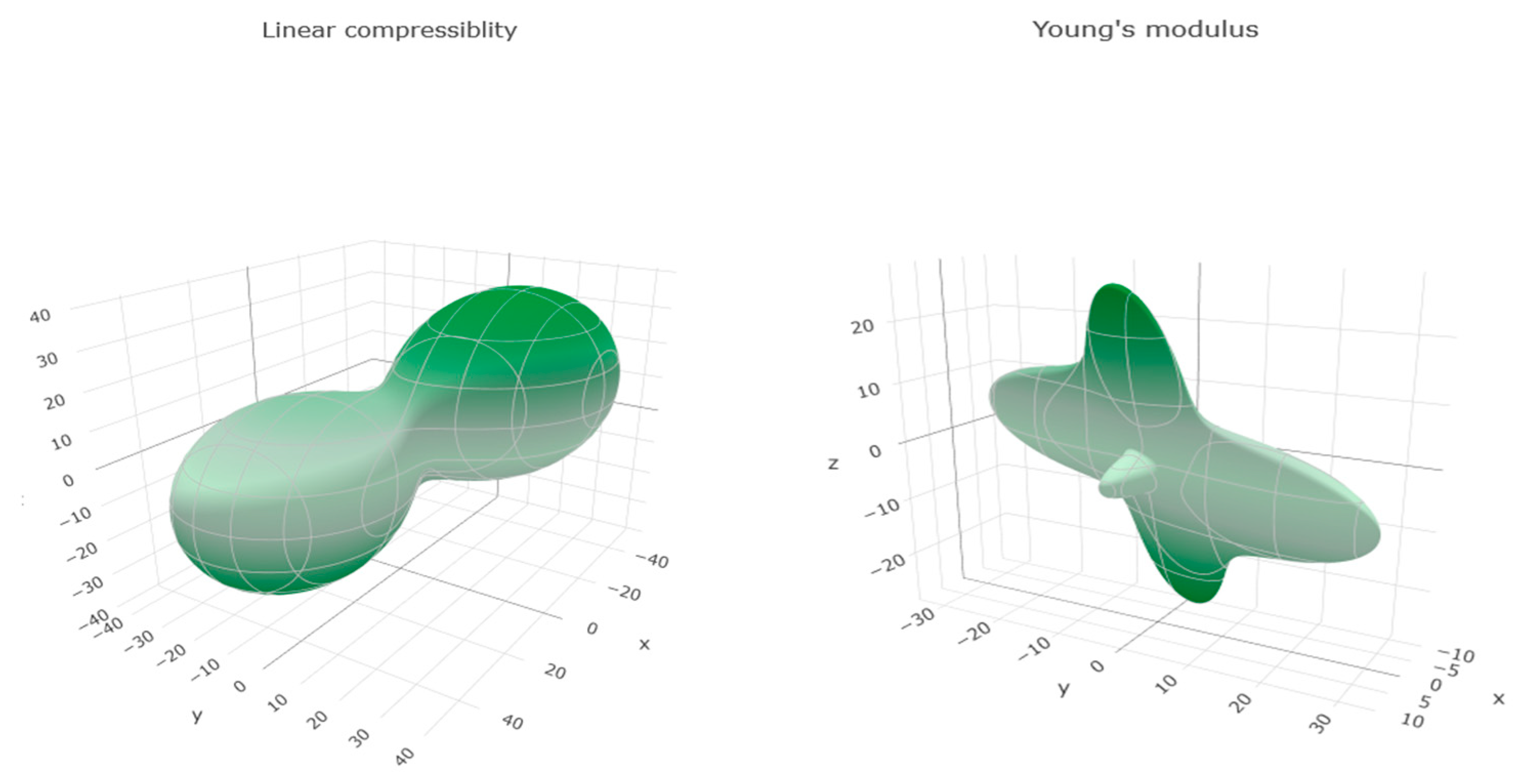
3.3.4. Mechanical Properties
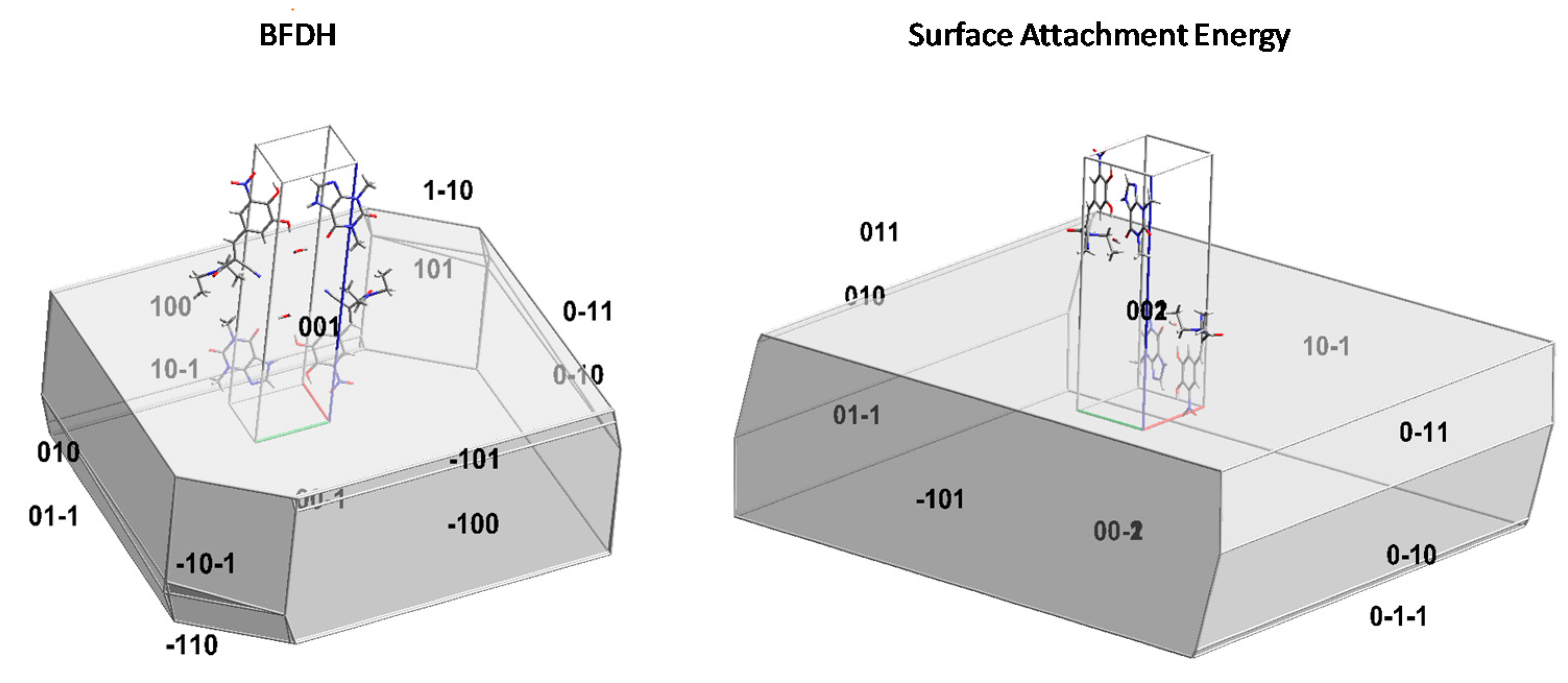
3.3.5. Crystal Morphology Modelling
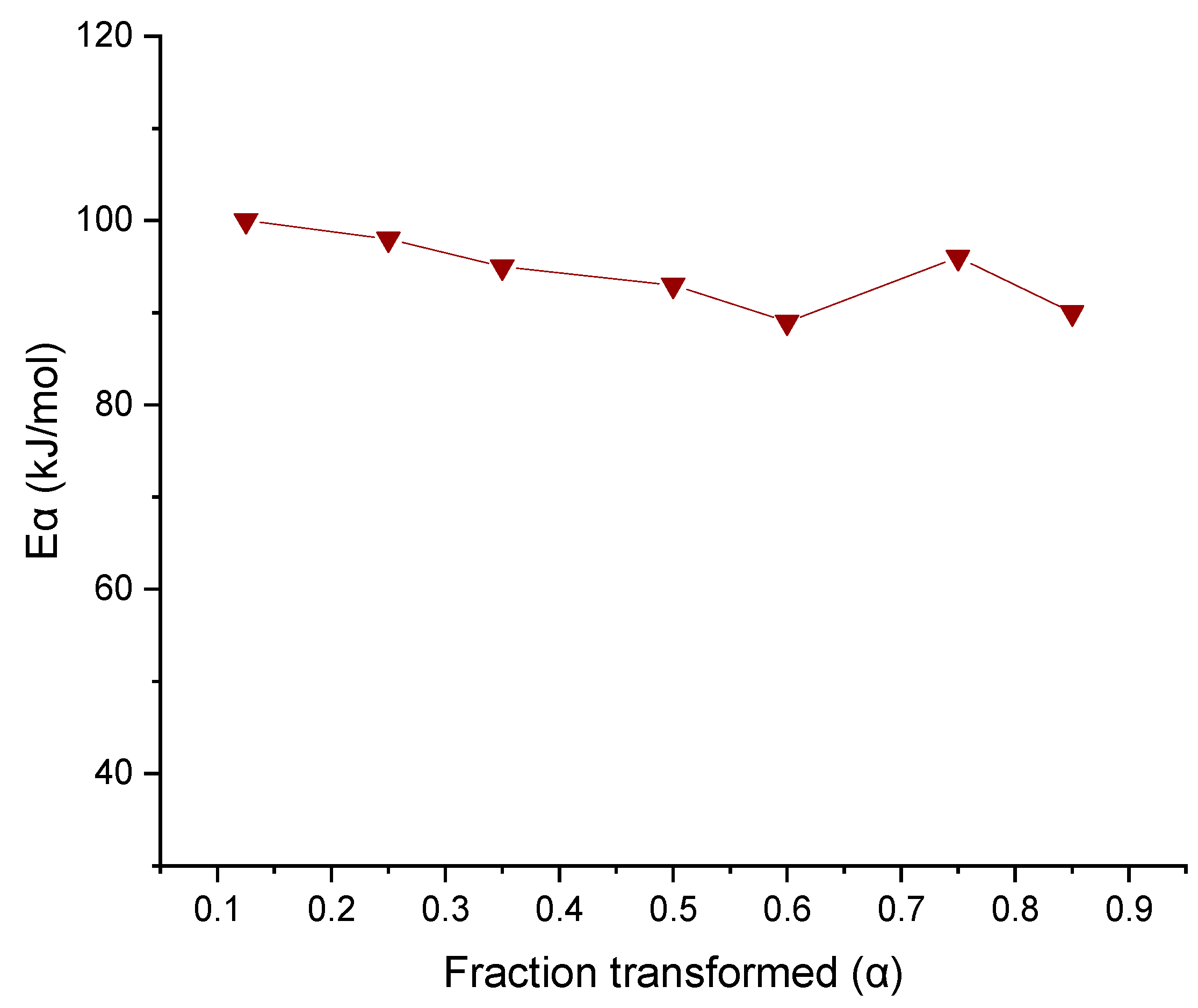
3.4. Dehydration Study of Cocrystal
3.4.1. Variable Temperature ATR-FTIR
3.4.2. Thermogravimetric Analysis (TGA) and Dehydration Kinetics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Aitipamula, S.; Banerjee, R.; Bansal, A.K.; Biradha, K.; Cheney, M.L.; Choudhury, A.R.; Desiraju, G.R.; Dikundwar, A.G.; Dubey, R.; Duggirala, N.; et al. Polymorphs, Salts, and Cocrystals: What’s in a Name? Cryst. Growth Des. 2012, 12, 2147–2152. [Google Scholar] [CrossRef]
- Regulatory Classification of Pharmaceutical Co-Crystals|FDA. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/regulatory-classification-pharmaceutical-co-crystals (accessed on 24 December 2021).
- Lara-Ochoa, F.; Espinosa-Pérez, G. Cocrystals Definitions. Supramol. Chem. 2007, 19, 553–557. [Google Scholar] [CrossRef]
- Karimi-Jafari, M.; Padrela, L.; Walker, G.M.; Croker, D.M. Creating cocrystals: A review of pharmaceutical cocrystal preparation routes and applications. Cryst. Growth Des. 2018, 18, 6370–6387. [Google Scholar] [CrossRef]
- Wong, S.N.; Chen, Y.C.S.; Xuan, B.; Sun, C.C.; Chow, S.F. Cocrystal engineering of pharmaceutical solids: Therapeutic potential and challenges. CrystEngComm 2021, 23, 7005–7038. [Google Scholar] [CrossRef]
- Karagianni, A.; Malamatari, M.; Kachrimanis, K. Pharmaceutical cocrystals: New solid phase modification approaches for the formulation of APIs. Pharmaceutics 2018, 10, 18. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Cabeza, A.J. Acid-base crystalline complexes and the pKa rule. CrystEngComm 2012, 14, 6362–6365. [Google Scholar] [CrossRef]
- Mohammad, M.A.; Alhalaweh, A.; Velaga, S.P. Hansen solubility parameter as a tool to predict cocrystal formation. Int. J. Pharm. 2011, 407, 63–71. [Google Scholar] [CrossRef]
- Galek, P.T.A.; Pidcock, E.; Wood, P.A.; Bruno, I.J.; Groom, C.R. One in half a million: A solid form informatics study of a pharmaceutical crystal structure. CrystEngComm 2012, 14, 2391–2403. [Google Scholar] [CrossRef]
- Fábián, L. Cambridge structural database analysis of molecular complementarity in cocrystals. Cryst. Growth Des. 2009, 9, 1436–1443. [Google Scholar] [CrossRef]
- Perlovich, G.L. Two-component molecular crystals: Relationship between the entropy term and the molecular volume of co-crystal formation. CrystEngComm 2018, 20, 3634–3637. [Google Scholar] [CrossRef]
- Perlovich, G.L. Formation Thermodynamics of Two-Component Molecular Crystals: Polymorphism, Stoichiometry, and Impact of Enantiomers. Cryst. Growth Des. 2020, 20, 5526–5537. [Google Scholar] [CrossRef]
- Heng, T.; Yang, D.; Wang, R.; Zhang, L.; Lu, Y.; Du, G. Progress in Research on Artificial Intelligence Applied to Polymorphism and Cocrystal Prediction. ACS Omega 2021, 6, 15543–15550. [Google Scholar] [CrossRef]
- Devogelaer, J.J.; Meekes, H.; Tinnemans, P.; Vlieg, E.; de Gelder, R. Co-crystal Prediction by Artificial Neural Networks. Angew. Chem. Int. Ed. 2020, 59, 21711–21718. [Google Scholar] [CrossRef]
- Mswahili, M.E.; Lee, M.J.; Martin, G.L.; Kim, J.; Kim, P.; Choi, G.J.; Jeong, Y.S. Cocrystal Prediction Using Machine Learning Models and Descriptors. Appl. Sci. 2021, 11, 1323. [Google Scholar] [CrossRef]
- Wicker, J.G.P.; Crowley, L.M.; Robshaw, O.; Little, E.J.; Stokes, S.P.; Cooper, R.I.; Lawrence, S.E. Will they co-crystallize? CrystEngComm 2017, 19, 5336–5340. [Google Scholar] [CrossRef] [Green Version]
- Friščić, T.; Fábián, L.; Burley, J.C.; Jones, W.; Motherwell, W.D.S. Exploring cocrystal–cocrystal reactivity via liquid-assisted grinding: The assembling of racemic and dismantling of enantiomeric cocrystals. Chem. Commun. 2006, 48, 5009–5011. [Google Scholar] [CrossRef]
- Haskins, M.M.; Zaworotko, M.J. Screening and Preparation of Cocrystals: A Comparative Study of Mechanochemistry vs Slurry Methods. Cryst. Growth Des. 2021, 21, 4141–4150. [Google Scholar] [CrossRef]
- Saganowska, P.; Wesolowski, M. DSC as a screening tool for rapid co-crystal detection in binary mixtures of benzodiazepines with co-formers. J. Therm. Anal. Calorim. 2018, 133, 785–795. [Google Scholar] [CrossRef] [Green Version]
- Berry, D.J.; Seaton, C.C.; Clegg, W.; Harrington, R.W.; Coles, S.J.; Horton, P.N.; Hursthouse, M.B.; Storey, R.; Jones, W.; Friščić, T.; et al. Applying hot-stage microscopy to co-crystal screening: A study of nicotinamide with seven active pharmaceutical ingredients. Cryst. Growth Des. 2008, 8, 1697–1712. [Google Scholar] [CrossRef]
- Malamatari, M.; Ross, S.A.; Douroumis, D.; Velaga, S.P. Experimental cocrystal screening and solution based scale-up cocrystallization methods. Adv. Drug Deliv. Rev. 2017, 117, 162–177. [Google Scholar] [CrossRef]
- Wang, X.; Du, S.; Zhang, R.; Jia, X.; Yang, T.; Zhang, X. Drug-drug cocrystals: Opportunities and challenges. Asian J. Pharm. Sci. 2021, 16, 307–317. [Google Scholar] [CrossRef]
- Taylor, C.R.; Day, G.M. Evaluating the Energetic Driving Force for Cocrystal Formation. Cryst. Growth Des. 2018, 18, 892–904. [Google Scholar] [CrossRef] [Green Version]
- Chethan, B.S.; Lokanath, N.K. Study of the crystal structure, H-bonding and noncovalent interactions of novel cocrystal by systematic computational search approach. J. Mol. Struct. 2021, 1251, 131936. [Google Scholar] [CrossRef]
- Drozd, K.V.; Manin, A.N.; Voronin, A.P.; Boycov, D.E.; Churakov, A.V.; Perlovich, G.L. A combined experimental and theoretical study of miconazole salts and cocrystals: Crystal structures, DFT computations, formation thermodynamics and solubility improvement. Phys. Chem. Chem. Phys. 2021, 23, 12456–12470. [Google Scholar] [CrossRef]
- Ross, S.A.; Lamprou, D.A.; Douroumis, D. Engineering and manufacturing of pharmaceutical co-crystals: A review on solvent-free manufacturing technologies. Chem. Commun. 2016, 52, 8772–8786. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.W.; Kendrick, J.; Leusen, F.J.J.; Yu, L. Co-crystallization with nicotinamide in two conformations lowers energy but expands volume. J. Pharm. Sci. 2014, 103, 2896–2903. [Google Scholar] [CrossRef]
- Li, F.; Zheng, Z.; Xia, S.; Yu, L. Synthesis, co-crystal structure, and DFT calculations of a multicomponent co-crystal constructed from 1H-benzotriazole and tetrafluoroterephthalic acid. J. Mol. Struct. 2020, 1219, 128480. [Google Scholar] [CrossRef]
- Kiely, E.; Zwane, R.; Fox, R.; Reilly, A.M.; Guerin, S. Density functional theory predictions of the mechanical properties of crystalline materials. CrystEngComm 2021, 23, 5697–5710. [Google Scholar] [CrossRef]
- Surov, A.O.; Voronin, A.P.; Vasilev, N.A.; Churakov, A.V.; Perlovich, G.L. Cocrystals of Fluconazole with Aromatic Carboxylic Acids: Competition between Anhydrous and Hydrated Solid Forms. Cryst. Growth Des. 2020, 20, 1218–1228. [Google Scholar] [CrossRef]
- Bommaka, M.K.; Chaitanya Mannava, M.K.; Suresh, K.; Gunnam, A.; Nangia, A. Entacapone: Improving aqueous solubility, diffusion permeability, and cocrystal stability with theophylline. Cryst. Growth Des. 2018, 18, 6061–6069. [Google Scholar] [CrossRef]
- Vebber, G.C.; Pranke, P.; Pereira, C.N. Calculating hansen solubility parameters of polymers with genetic algorithms. J. Appl. Polym. Sci. 2014, 131, 1–12. [Google Scholar] [CrossRef]
- Hansen, C.M. Methods of characterization-surfaces. In Hansen Solubility Parameters A Users Handbook, 2nd ed.; Taylor and Francis Group: Abingdon, UK, 2007; pp. 113–123. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; Van De Streek, J.; Wood, P.A. Mercury CSD 2.0-new features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Spackman, M.A.; Mitchell, A.S. Novel Tools for Visualizing and Exploring Intermolecular Interactions in Molecular Crystals. Acta Crystallogr. Sect. B Struct. Sci. 2004, 60, 627–668. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Turner, M.J.; Thomas, S.P.; Shi, M.W.; Jayatilaka, D.; Spackman, M.A. Energy frameworks: Insights into interaction anisotropy and the mechanical properties of molecular crystals. Chem. Commun. 2015, 51, 3735–3738. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. Quantum espresso: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision C.01.; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Weingart, O.; Nenov, A.; Altoè, P.; Rivalta, I.; Segarra-Martí, J.; Dokukina, I.; Garavelli, M. COBRAMM 2.0—A software interface for tailoring molecular electronic structure calculations and running nanoscale (QM/MM) simulations. J. Mol. Model. 2018, 24, 1–30. [Google Scholar] [CrossRef] [Green Version]
- Otero-de-la-Roza, A.; Johnson, E.R.; Contreras-García, J. Revealing non-covalent interactions in solids: NCI plots revisited. Phys. Chem. Chem. Phys. 2012, 14, 12165. [Google Scholar] [CrossRef]
- Bader, R.F.W. A Quantum Theory of Molecular Structure and Its Applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Shishkin, O.V.; Zubatyuk, R.I.; Shishkina, S.V.; Dyakonenko, V.V.; Medviediev, V.V. Role of supramolecular synthons in the formation of the supramolecular architecture of molecular crystals revisited from an energetic viewpoint. Phys. Chem. Chem. Phys. 2014, 16, 6773. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; Van De Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Spackman, P.; Turner, M.; McKinnon, J.; Wolff, S.; Grimwood, D.; Jayatilaka, D.; Spackman, M. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- Gale, J.D.; Rohl, A.L. The General Utility Lattice Program (GULP). Mol. Simul. 2003, 29, 291–341. [Google Scholar] [CrossRef]
- Mayo, S.L.; Olafson, B.D.; Goddard, W.A. Dreiding: A generic force field for molecular simulations. J. Phys. Chem. 2002, 94, 8897–8909. [Google Scholar] [CrossRef]
- Breneman, C.M.; Wiberg, K.B. Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J. Comput. Chem. 1990, 11, 361–373. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Gaillac, R.; Pullumbi, P.; Coudert, F.X. Elate: An open-source online application for analysis and visualization of elastic tensors. J. Phys. Condens. Matter 2016, 28, 275201. [Google Scholar] [CrossRef] [PubMed]
- Mazhnik, E.; Oganov, A.R. A model of hardness and fracture toughness of solids. J. Appl. Phys. 2019, 126, 125109. [Google Scholar] [CrossRef]
- McArdle, P. Oscail, a program package for small-molecule single-crystal crystallography with crystal morphology prediction and molecular modelling. J. Appl. Crystallogr. 2017, 50, 320–326. [Google Scholar] [CrossRef]
- Lifson, S.; Hagler, A.T.; Dauber, P. Consistent Force Field Studies of Intermolecular Forces in Hydrogen-Bonded Crystals. 1. Carboxylic Acids, Amides and the C=O...H-Hydrogen Bonds. J. Am. Chem. Soc. 1979, 101, 5111–5121. [Google Scholar] [CrossRef]
- Kachrimanis, K.; Griesser, U.J. Dehydration kinetics and crystal water dynamics of carbamazepine dihydrate. J. Chem. Inf. Model. 2012, 29, 1143–1157. [Google Scholar] [CrossRef]
- Khawam, A.; Flanagan, D.R. Solid-state kinetic models: Basics and mathematical fundamentals. J. Phys. Chem. B 2006, 110, 17315–17328. [Google Scholar] [CrossRef]
- Vyazovkin, S. Evaluation of Activation Energy of Thermally Stimulated Solid-State Reactions under Arbitrary Variation of Temperature. J. Comput. Chem. 1997, 18, 393–402. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Wight, C.A. Model-free and model-fitting approaches to kinetic analysis of isothermal and nonisothermal data. Thermochim. Acta 1999, 341, 53–68. [Google Scholar] [CrossRef]
- Joseph, A.; Bernardes, C.E.S.; Druzhinina, A.I.; Varushchenko, R.M.; Nguyen, T.Y.; Emmerling, F.; Yuan, L.; Dupray, V.; Coquerel, G.; Da Piedade, M.E.M. Polymorphic Phase Transition in 4′-Hydroxyacetophenone: Equilibrium Temperature, Kinetic Barrier, and the Relative Stability of Z′ = 1 and Z′ = 2 Forms. Cryst. Growth Des. 2017, 17, 1918–1932. [Google Scholar] [CrossRef]
- Jayasankar, A.; Good, D.J.; Rodríguez-Hornedo, N. Mechanisms by Which Moisture Generates Cocrystals. Mol. Pharm. 2007, 4, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Rager, T.; Hilfiker, R. Application of phase diagrams. In Co-Crystal Search and Preparation; Royal Society of Chemistry: Cambridge, UK, 2011; pp. 280–299, Chapter 12. [Google Scholar] [CrossRef]
- Abdelkader, H.; Abdallah, O.Y.; Salem, H.; Alani, A.W.G.; Alany, R.G. Eutectic, monotectic and immiscibility systems of nimesulide with water-soluble carriers: Phase equilibria, solid-state characterisation and in-vivo/pharmacodynamic evaluation. J. Pharm. Pharmacol. 2014, 66, 1439–1450. [Google Scholar] [CrossRef] [PubMed]
- Salem, A.; Nagy, S.; Pál, S.; Széchenyi, A. Reliability of the Hansen solubility parameters as co-crystal formation prediction tool. Int. J. Pharm. 2019, 558, 319–327. [Google Scholar] [CrossRef]
- Khalaji, M.; Potrzebowski, M.J.; Dudek, M.K. Virtual Cocrystal Screening Methods as Tools to Understand the Formation of Pharmaceutical Cocrystals-A Case Study of Linezolid, a Wide-Range Antibacterial Drug. Cryst. Growth Des. 2021, 21, 2301–2314. [Google Scholar] [CrossRef]
- Sarkar, N.; Gonnella, N.C.; Krawiec, M.; Xin, D.; Aakeröy, C.B. Evaluating the Predictive Abilities of Protocols Based on Hydrogen-Bond Propensity, Molecular Complementarity, and Hydrogen-Bond Energy for Cocrystal Screening. Cryst. Growth Des. 2020, 20, 7320–7327. [Google Scholar] [CrossRef]
- Galek, P.T.A.; Fábián, L.; Motherwell, W.D.S.; Allen, F.H.; Feeder, N. Knowledge-based model of hydrogen-bonding propensity in organic crystals. Acta Crystallogr. B. 2007, 63, 768–782. [Google Scholar] [CrossRef]
- Wood, P.A.; Feeder, N.; Furlow, M.; Galek, P.T.A.; Groom, C.R.; Pidcock, E. Knowledge-based approaches to co-crystal design. CrystEngComm 2014, 16, 5839–5848. [Google Scholar] [CrossRef]
- Sarkar, N.; Sinha, A.S.; Aakeroÿ, C.B. Systematic investigation of hydrogen-bond propensities for informing co-crystal design and assembly. CrystEngComm 2019, 21, 6048–6055. [Google Scholar] [CrossRef]
- Clausen, H.F.; Chevallier, M.S.; Spackman, M.A.; Iversen, B.B. Three new co-crystals of hydroquinone: Crystal structures and Hirshfeld surface analysis of intermolecular interactions. New J. Chem. 2010, 34, 193–199. [Google Scholar] [CrossRef]
- Álvarez-Vidaurre, R.; Castiñeiras, A.; Frontera, A.; García-Santos, I.; Gil, D.M.; González-Pérez, J.M.; Niclós-Gutiérrez, J.; Torres-Iglesias, R. Weak Interactions in Cocrystals of Isoniazid with Glycolic and Mandelic Acids. Crystals 2021, 11, 328. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Janiak, C. A critical account on π–π stacking in metal complexes with aromatic nitrogen-containing ligands. J. Chem. Soc. Dalt. Trans. 2000, 21, 3885–3896. [Google Scholar] [CrossRef]
- Bjornsson, R.; Bühl, M. Modeling molecular crystals by QM/MM: Self-consistent electrostatic embedding for geometry optimizations and molecular property calculations in the solid. J. Chem. Theory Comput. 2012, 8, 498–508. [Google Scholar] [CrossRef]
- Hernández-Paredes, J.; Carrillo-Torres, R.C.; López-Zavala, A.A.; Sotelo-Mundo, R.R.; Hernández-Negrete, O.; Ramírez, J.Z.; Alvarez-Ramos, M.E. Molecular structure, hydrogen-bonding patterns and topological analysis (QTAIM and NCI) of 5-methoxy-2-nitroaniline and 5-methoxy-2-nitroaniline with 2-amino-5-nitropyridine (1:1) co-crystal. J. Mol. Struct. 2016, 1119, 505–516. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C.; Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. CPL 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Mata, I.; Alkorta, I.; Espinosa, E.; Molins, E. Relationships between interaction energy, intermolecular distance and electron density properties in hydrogen bonded complexes under external electric fields. Chem. Phys. Lett. 2011, 507, 185–189. [Google Scholar] [CrossRef]
- Lin, H.; Zhu, S.G.; Li, H.Z.; Peng, X.H. Synthesis, characterization, AIM and NBO analysis of HMX/DMI cocrystal explosive. J. Mol. Struct. 2013, 1048, 339–348. [Google Scholar] [CrossRef]
- Boraei, A.; Haukka, M.; Sarhan, A.; Soliman, S.; A, B. Intramolecular Hydrogen Bond, Hirshfeld Analysis, AIM.; DFT Studies of Pyran-2,4-dione Derivatives. Crystals 2021, 11, 896. [Google Scholar] [CrossRef]
- Rozas, I.; Alkorta, I.; Elguero, J. Behavior of ylides containing N, O, and C atoms as hydrogen bond acceptors. J. Am. Chem. Soc. 2000, 122, 11154–11161. [Google Scholar] [CrossRef]
- Vener, M.V.; Egorova, A.N.; Churakov, A.V.; Tsirelson, V.G. Intermolecular hydrogen bond energies in crystals evaluated using electron density properties: DFT computations with periodic boundary conditions. J. Comput. Chem. 2012, 33, 2303–2309. [Google Scholar] [CrossRef] [PubMed]
- Filarowski, A.; Majerz, I. AIM Analysis of Intramolecular Hydrogen Bonding in O-Hydroxy Aryl Schiff Bases. J. Phys. Chem. A 2008, 112, 3119–3126. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. What Is the Covalency of Hydrogen Bonding? Chem. Rev. 2011, 111, 2597–2625. [Google Scholar] [CrossRef] [PubMed]
- Shishkin, O.V.; Dyakonenko, V.V.; Maleev, A.V. Supramolecular architecture of crystals of fused hydrocarbons based on topology of intermolecular interactions. CrystEngComm 2012, 14, 1795–1804. [Google Scholar] [CrossRef]
- Shishkin, O.V.; Medvediev, V.V.; Zubatyuk, R.I.; Shyshkina, O.O.; Kovalenko, N.V.; Volovenko, J.M. Role of different molecular fragments in formation of the supramolecular architecture of the crystal of 1,1-dioxo-tetrahydro-1λ6-thiopyran-3-one. CrystEngComm 2012, 14, 8698–8707. [Google Scholar] [CrossRef]
- Luan, X.; Qin, H.; Liu, F.; Dai, Z.; Yi, Y.; Li, Q. The Mechanical Properties and Elastic Anisotropies of Cubic Ni3Al from First Principles Calculations. Crystals 2018, 8, 307. [Google Scholar] [CrossRef] [Green Version]
- Tumanov, I.A.; Achkasov, A.F.; Myz, S.A.; Boldyreva, E.V.; Boldyrev, V.V. Different effect of impact and shear mechanical treatment on mechanochemical cocrystallization of piroxicam and succinic acid. Dokl. Chem. 2014, 457, 154–159. [Google Scholar] [CrossRef]
- Liu, X.; Wang, H.; Wang, W.; Fu, Z. A simple bulk modulus model for crystal materials based on the bond valence model. Phys. Chem. Chem. Phys. 2017, 19, 22177–22189. [Google Scholar] [CrossRef]
- Toziopoulou, F.; Malamatari, M.; Nikolakakis, I.; Kachrimanis, K. Production of aprepitant nanocrystals by wet media milling and subsequent solidification. Int. J. Pharm. 2017, 533, 324–334. [Google Scholar] [CrossRef]
- Hadjittofis, E.; Isbell, M.A.; Karde, V.; Varghese, S.; Ghoroi, C.; Heng, J.Y.Y. Influences of Crystal Anisotropy in Pharmaceutical Process Development. Pharm. Res. 2018, 35, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Pugh, S.F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dublin Philos. Mag. J. Sci. 2009, 45, 823–843. [Google Scholar] [CrossRef]
- Liao, X.; Zhou, N. Dehydration Study of Piracetam Co-Crystal Hydrates. J. Pharm. Sci. 2018, 107, 2804–2809. [Google Scholar] [CrossRef] [PubMed]
- Clarke, H.D.; Arora, K.K.; Bass, H.; Kavuru, P.; Ong, T.T.; Pujari, T.; Wojtas, L.; Zaworotko, M.J. Structure-stability relationships in cocrystal hydrates: Does the promiscuity of water make crystalline hydrates the nemesis of crystal engineering? Cryst. Growth Des. 2010, 10, 2152–2167. [Google Scholar] [CrossRef]
- Matsuo, K.; Matsuoka, M. Solid-State Polymorphic Transition of Theophylline Anhydrate and Humidity Effect. Cryst. Growth Des. 2007, 7, 411–415. [Google Scholar] [CrossRef]
- Paiva, E.M.; Li, Q.; Zaczek, A.J.; Pereira, C.F.; Rohwedder, J.J.R.; Zeitler, J.A. Understanding the Metastability of Theophylline FIII by Means of Low-Frequency Vibrational Spectroscopy. Mol. Pharm. 2021, 18, 3578–3587. [Google Scholar] [CrossRef]
- Nunes, C.; Mahendrasingam, A.; Suryanarayanan, R. Investigation of the multi-step dehydration reaction of theophylline monohydrate using 2-dimensional powder X-ray diffractometry. Pharm. Res. 2006, 23, 2393–2404. [Google Scholar] [CrossRef]
- Sovizi, M.R.; Fakhrpour, G.; Bagheri, S.; Rezanejade Bardajee, G. Non-isothermal dehydration kinetic study of a new swollen biopolymer silver nanocomposite hydrogel. J. Therm. Anal. Calorim. 2015, 121, 1383–1391. [Google Scholar] [CrossRef]
- Zhang, Q.; Ye, G. Dehydration kinetics of Portland cement paste at high temperature. J. Therm. Anal. Calorim. 2012, 110, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Galwey, A.K. What is meant by the term ‘variable activation energy’ when applied in the kinetic analyses of solid state decompositions (crystolysis reactions)? Thermochim. Acta 2003, 397, 249–268. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Linert, W. Kinetic analysis of reversible thermal decomposition of solids. Int. J. Chem. Kinet. 1995, 27, 73–84. [Google Scholar] [CrossRef]
- Sanii, R.; Patyk-Kaźmierczak, E.; Hua, C.; Darwish, S.; Pham, T.; Forrest, K.A.; Space, B.; Zaworotko, M.J. Toward an Understanding of the Propensity for Crystalline Hydrate Formation by Molecular Compounds. Part 2. Cryst. Growth Des. 2021, 21, 4927–4939. [Google Scholar] [CrossRef] [PubMed]
- Shishkina, S.V.; Ukrainets, I.V.; Petrushova, L.A. Competition between intermolecular hydrogen bonding & stacking in the crystals of 4-Hydroxy- N-(pyridin-2-yl)-2,2-dioxo-1H-2λ6,1-benzothiazine- 3-carboxamides. Z. Fur Krist. Cryst. Mater. 2017, 232, 307–316. [Google Scholar] [CrossRef]
- Ireta, J.; 1rg Neugebauer, J.; Scheffler, M. On the Accuracy of DFT for Describing Hydrogen Bonds: Dependence on the Bond Directionality. J. Phys. Chem. A 2004, 108, 5692–5698. [Google Scholar] [CrossRef] [Green Version]
- Vener, M.V.; Levina, E.O.; Koloskov, O.A.; Rykounov, A.A.; Voronin, A.P.; Tsirelson, V.G. Evaluation of the lattice energy of the two-component molecular crystals using solid-state density functional theory. Cryst. Growth Des. 2014, 14, 4997–5003. [Google Scholar] [CrossRef]
- Shishkina, A.V.; Zhurov, V.V.; Stash, A.I.; Vener, M.V.; Pinkerton, A.A.; Tsirelson, V.G. Noncovalent interactions in crystalline picolinic acid N-oxide: Insights from experimental and theoretical charge density analysis. Cryst. Growth Des. 2013, 13, 816–828. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kinetic Models | Differential form f(α) | Integral form g(α) |
|---|---|---|
| Diffusion models | ||
| D1 (1D-diffusion) | 1/2α | α2 |
| D2 (Valensi-Carter 2D-diffusion) | [−ln(1 − α)] – 1 | (1 − α)ln(1 − α)+α |
| D3 (Jander) | (3/2)(1 − α)2/3/[1 − (1 − α)1/3 ] | [1 − (1 − α)1/3]2 |
| D4 (Ginstling-Brounshtein) | (3/2)/[(1 − α)−1/3 − 1] | 1 − (2α/3) − (1 − α)2/3 |
| D5 (Zhuravlev, lesokin, Tempelman) | (3/2)(1 − α)4/3/[(1 − α)−1/3 − 1] | [(1 − α)−1/3 − 1]2 |
| D6 (Anti-Jander) | (3/2)(1 + α)2/3/[(1 + α)1/3 − 1] | [(1 + α)1/3 − 1]2 |
| Nucleation and growth models | ||
| n = 1 (Prout-Tomkins Au) | α(1 − α) | ln[α/(1 − α)] |
| n = 3/2 (Avarami-Erofeyev) | (3/2)(1 − α)[−ln(1 − α)]1/3 | [−ln(1 − α)]2/3 |
| n = 2 (Avarami-Erofeyev) | 2(1 − α)[−ln(1 − α)]1/2 | [−ln(1 − α)]1/2 |
| n = 3 (Avarami-Erofeyev) | 3(1 − α)[−ln(1 − α)]2/3 | [−ln(1 − α)]1/3 |
| n = 4 (Avarami-Erofeyev) | 4(1 − α)[−ln(1 − α)]3/4 | [−ln(1 − α)]1/4 |
| P2 (Power law) | 2α ½ | α 1/2 |
| P3 (Power law) | 3α 2/3 | α 1/3 |
| P4 (Power law) | 4α ¾ | α 1/4 |
| P3/2 (Power law) | (2/3)α−1/2 | α 2/3 |
| Geometrical contraction models | ||
| R2 (Area contraction) | 2(1 − α)1/2 | 1 − (1 − α)1/2 |
| R3 (Volume contraction) | 3(1 − α)2/3 | 1 − (1 − α)1/3 |
| Reaction order models | ||
| F1 (1st order) | 1 – α | −ln(1 − α) |
| F2 (2nd order) | (1 − α)2 | (1 − α)−1 – 1 |
| F3 (3rd order) | (1 − α)3 | [(1 − α)−2 − 1]/2 |
| Hydrogen Bonds | Molecules | ρ(r) (a.u.) | ∇2ρ(r) (a.u.) | G(r) (a.u.) | H(r) (a.u.) | Eint (kcal/mol) |
|---|---|---|---|---|---|---|
| O5···H24-O8 | ENT-water | 0.0456 | 0.1343 | 0.0342 | −0.0656 | −9.22 |
| O6···H25-O8 | THP-water | 0.0349 | 0.1223 | 0.0291 | 0.0016 | −7.84 |
| O2-H1···O8 | ENT-water | 0.0838 | 0.1502 | 0.0616 | −0.0240 | −16.57 |
| O1···H23-N4 | ENT-THP | 0.0395 | 0.123 | 0.0302 | 0.0007 | −8.14 |
| C8-H4···O8 | ENT-water | 0.0083 | 0.0333 | 0.007 | 0.0014 | −1.88 |
| Molecule 1 | Molecule 2 | Symmetry Operator | Interaction Energy (kcal/mol) | Contact |
|---|---|---|---|---|
| ENT | THP | x, y, z | −9.15 | O1···H23-N4 |
| ENT | water | x, y, z | −6.78 | O2-H1···O8 |
| THP | water | x, y, z | −6.05 | O6···H25-O8 |
| ENT | THP | x, 1 + y, z | −10.47 | π-π stacking interactions a |
| ENT | water | x, 1 + y, z | −5.96 | O5···H24-O8 |
| ΕΝΤ | ΤHP | 1 + x, 1 + y, z | −11.87 | π-π stacking interactions a |
| ENT | THP | 1 + x, 2 + y, z | −8.61 | C-H···O |
| ENT | ENT | 1 − x, 3 − y, 1 − z | −8.13 | C-H···C-H C14-H12···N3 |
| Mechanical Property (Units) | Value |
|---|---|
| Bulk modulus (K) (GPa) | 11.86 |
| Shear modulus (G) (GPa) | 4.49 |
| Young modulus (E) (GPa) | 11.97 |
| Ea (GPa) | 6.91 |
| Eb (GPa) | 16.48 |
| Ec (GPa) | 14.61 |
| Compressibility (GPa−1) | 0.09 |
| Elastic anisotropy | 8.13 |
| Vickers Hardness (GPa) | 0.66 |
| Fracture Τoughness (MPa m1/2) | 0.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karagianni, A.; Quodbach, J.; Weingart, O.; Tsiaxerli, A.; Katsanou, V.; Vasylyeva, V.; Janiak, C.; Kachrimanis, K. Structural and Energetic Aspects of Entacapone-Theophylline-Water Cocrystal. Solids 2022, 3, 66-92. https://doi.org/10.3390/solids3010006
Karagianni A, Quodbach J, Weingart O, Tsiaxerli A, Katsanou V, Vasylyeva V, Janiak C, Kachrimanis K. Structural and Energetic Aspects of Entacapone-Theophylline-Water Cocrystal. Solids. 2022; 3(1):66-92. https://doi.org/10.3390/solids3010006
Chicago/Turabian StyleKaragianni, Anna, Julian Quodbach, Oliver Weingart, Anastasia Tsiaxerli, Vasiliki Katsanou, Vera Vasylyeva, Christoph Janiak, and Kyriakos Kachrimanis. 2022. "Structural and Energetic Aspects of Entacapone-Theophylline-Water Cocrystal" Solids 3, no. 1: 66-92. https://doi.org/10.3390/solids3010006
APA StyleKaragianni, A., Quodbach, J., Weingart, O., Tsiaxerli, A., Katsanou, V., Vasylyeva, V., Janiak, C., & Kachrimanis, K. (2022). Structural and Energetic Aspects of Entacapone-Theophylline-Water Cocrystal. Solids, 3(1), 66-92. https://doi.org/10.3390/solids3010006