
Inhibition of Lactate Dehydrogenase-A by Singlet Oxygen and Hypochlorous Acid via Cysteine Oxidation and Irreversible Conformational Changes



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. LDH-A Purification by Cibacron Blue (CB) Chromatography
2.3. Pyruvate Kinase Assay Using DNPH
2.4. Photo-Oxidation of LDH-A
2.5. Enhanced NADH Oxidation by LDH-A; Initiation by 1O2
2.6. LDH-A Activity Assay
2.7. Labeling of LDH-A Cysteines with M5F and IAF
2.8. Detection of LDH-A Oxidation Products by Western Blotting
2.9. Native Gel Electrophoresis of LDH-A with Activity Staining
2.10. Tryptophan Emission of HOCl-Treated LDH-A
2.11. Detection of Dityrosine by Fluorescence Emission
3. Results
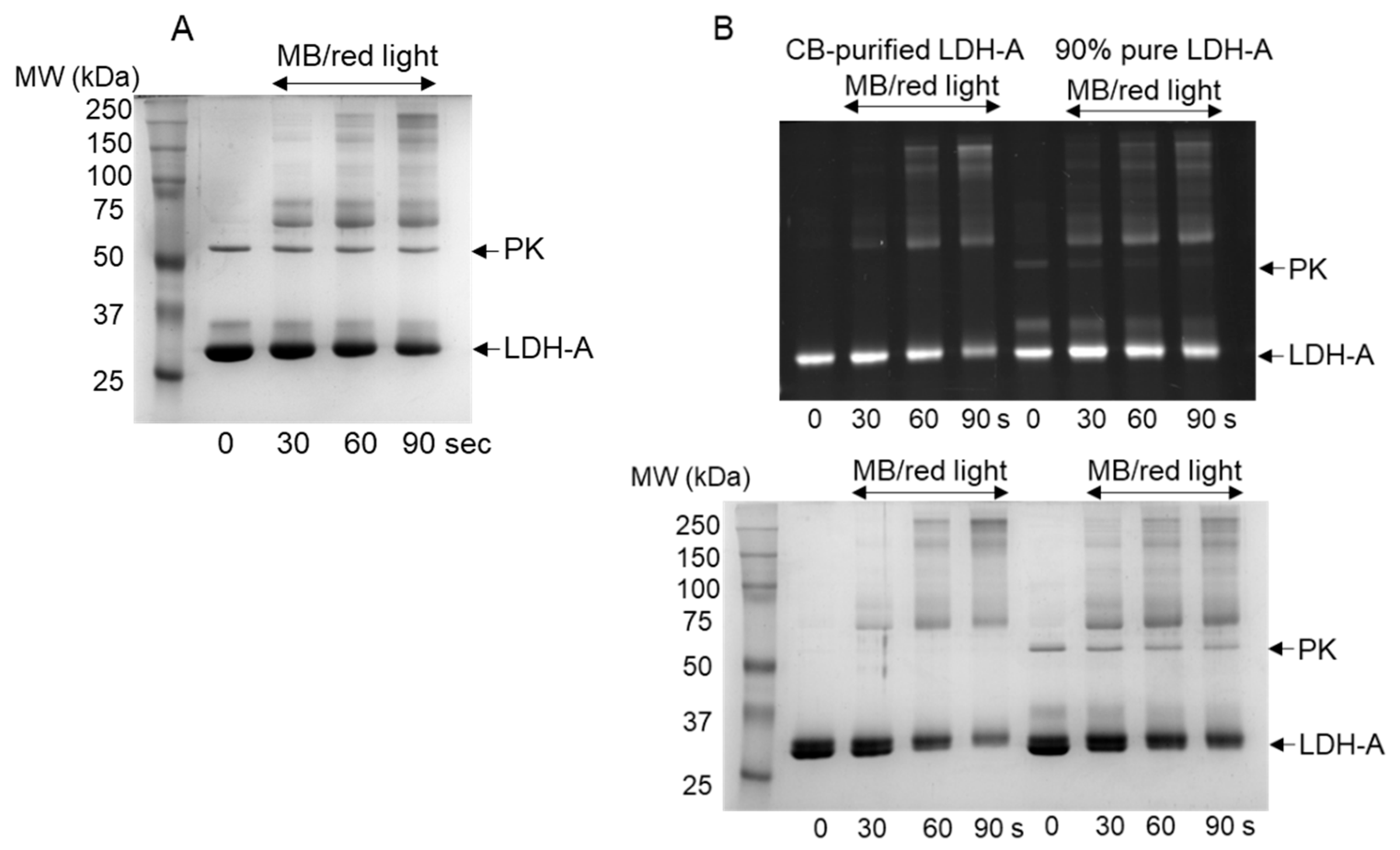
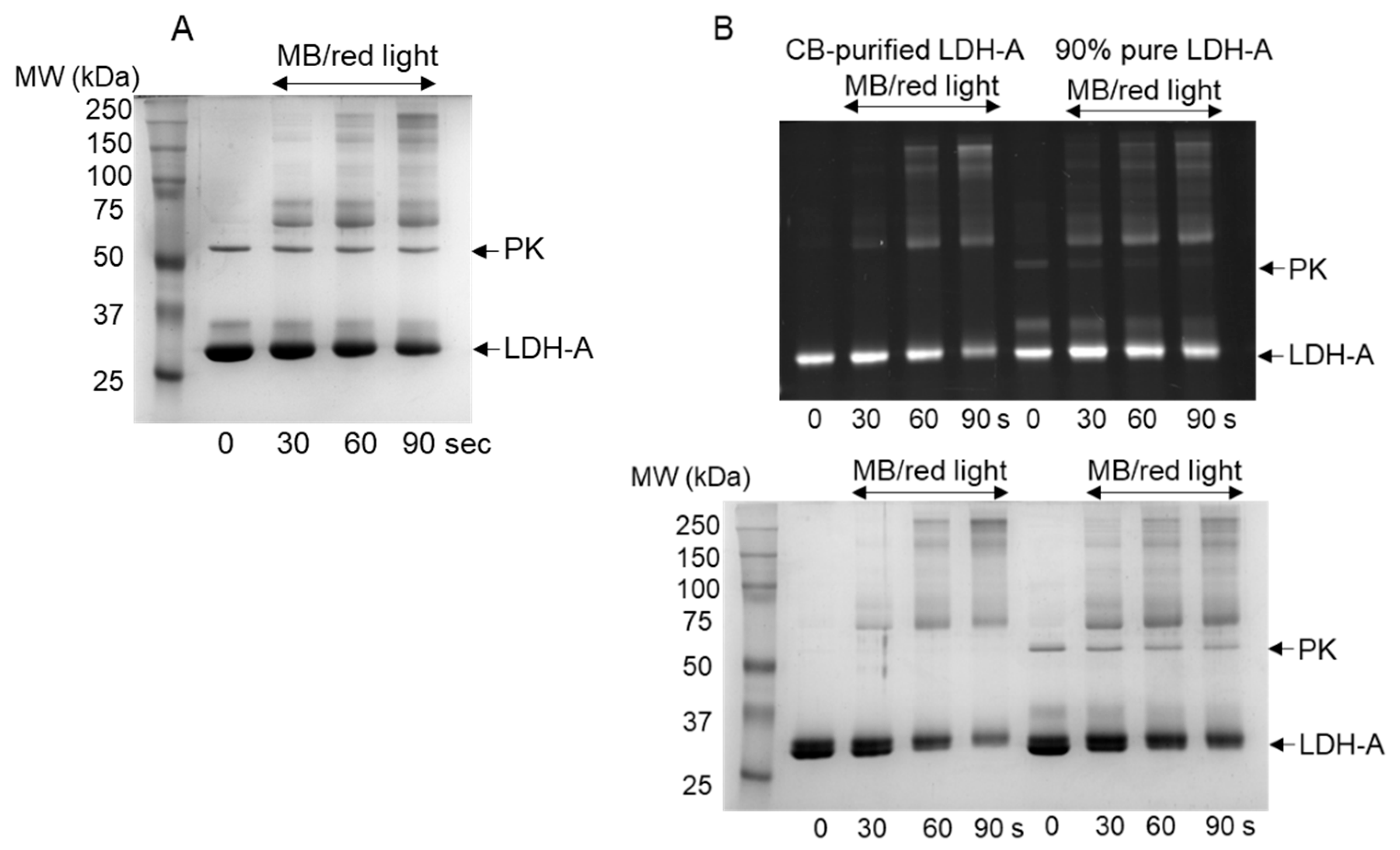
3.1. Oxidation of LDH-A by 1O2
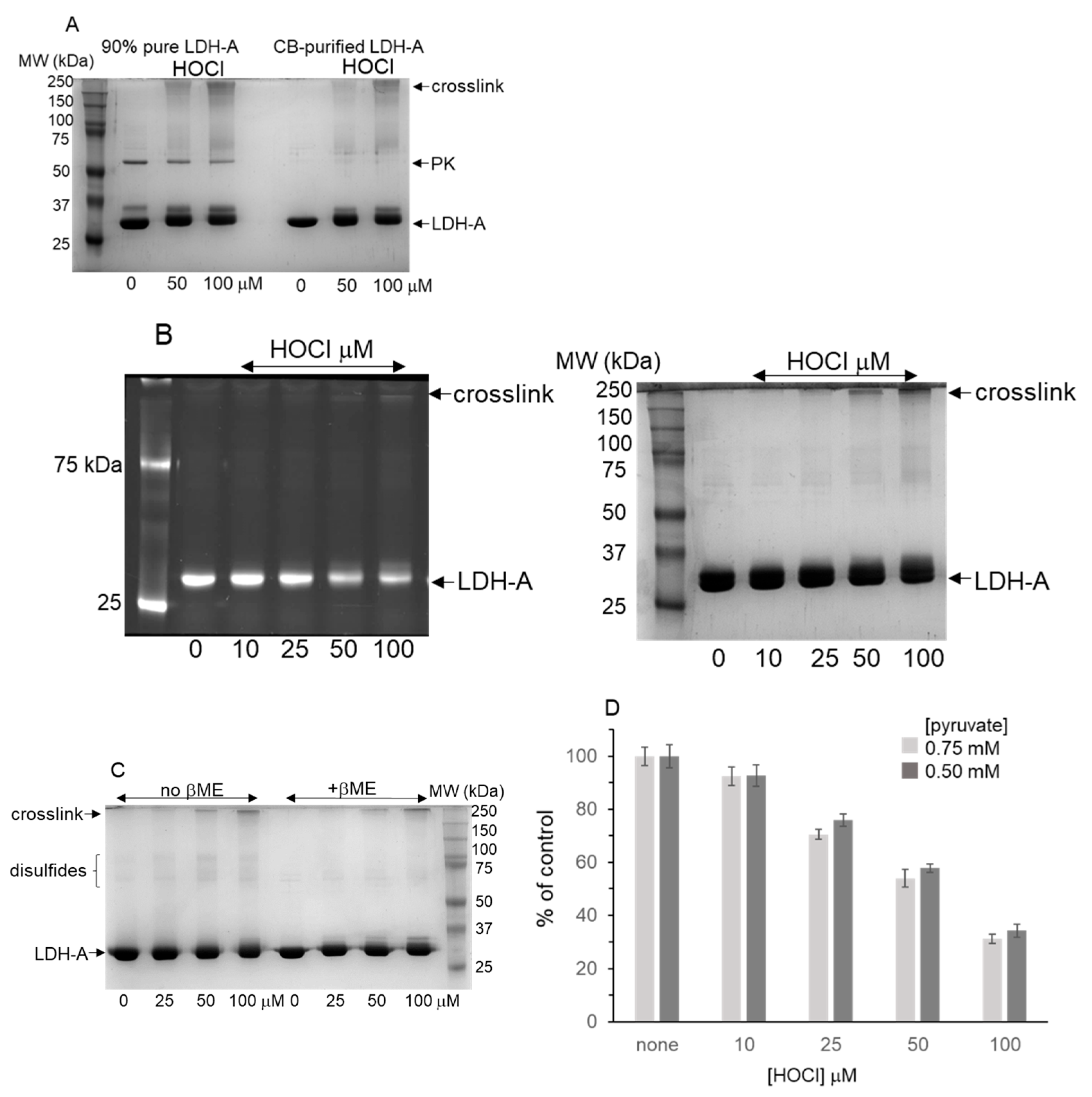
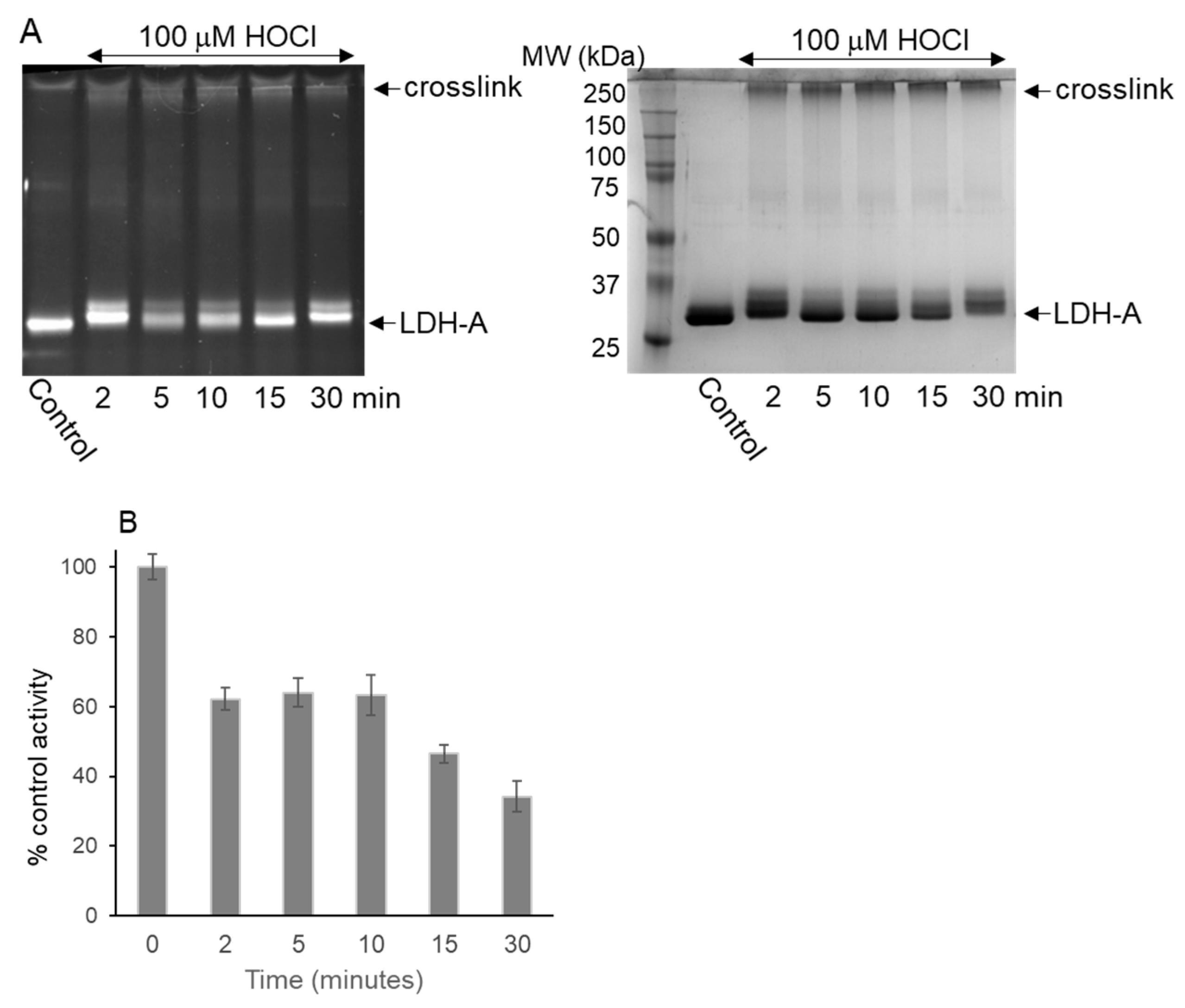
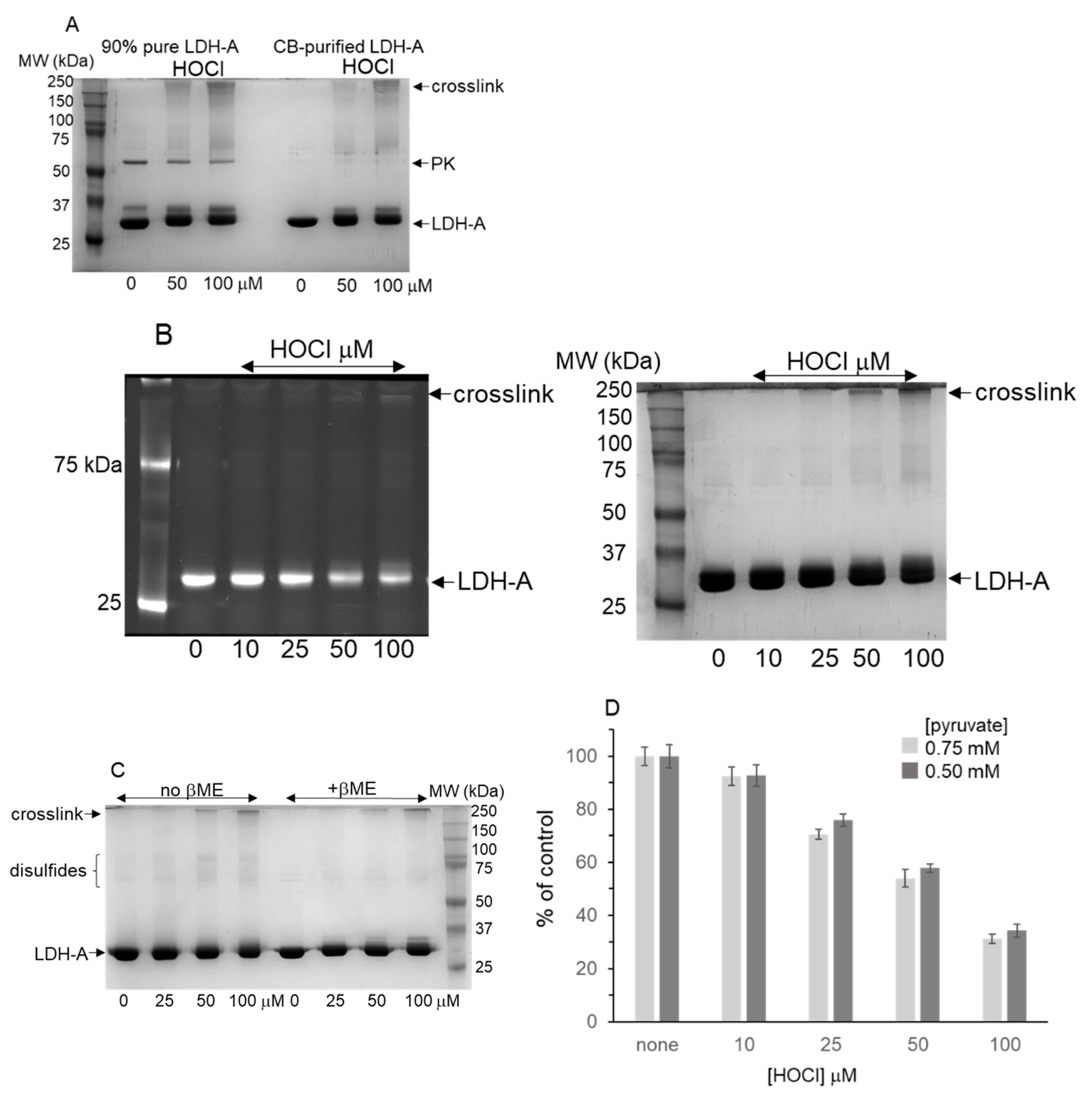
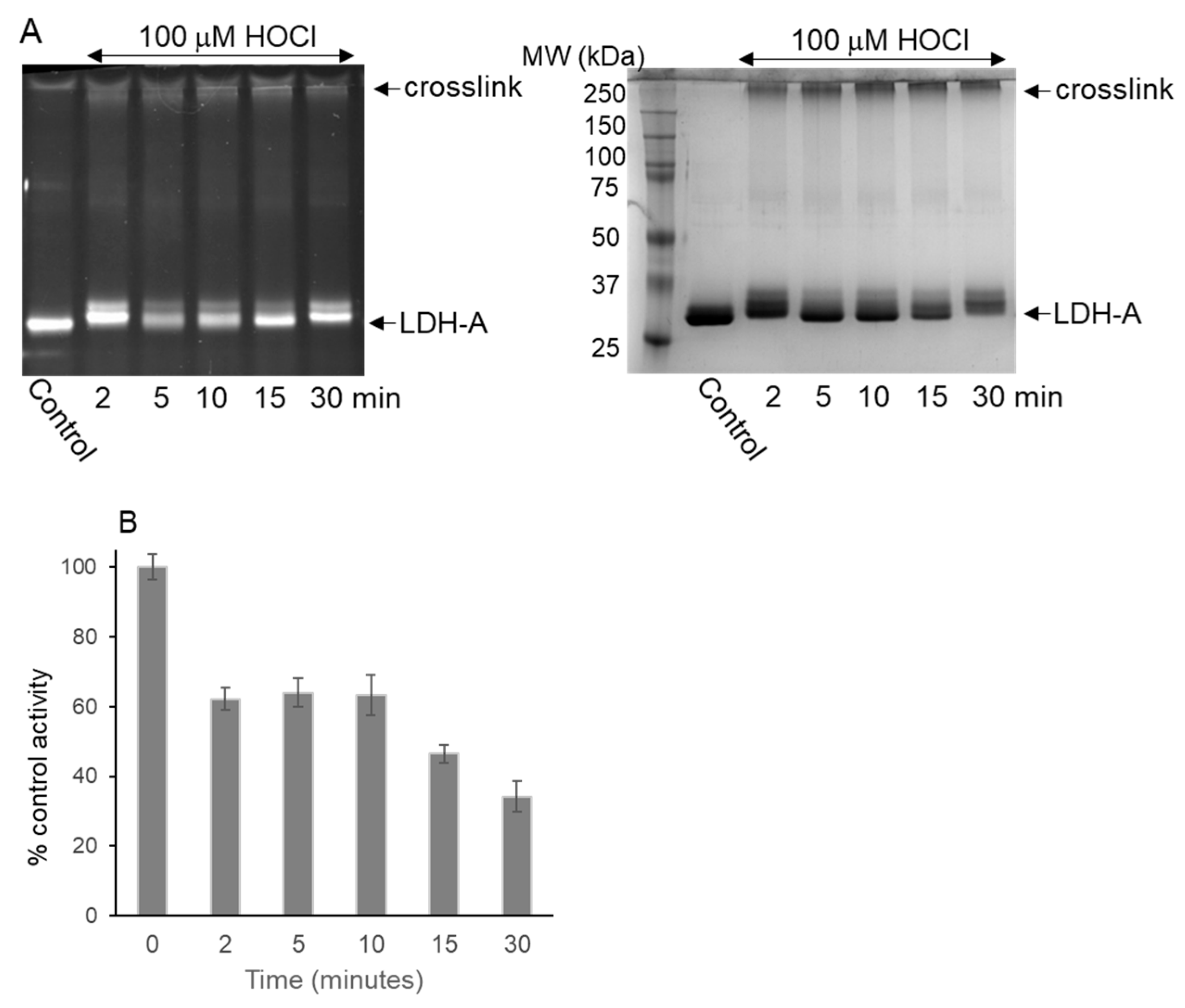
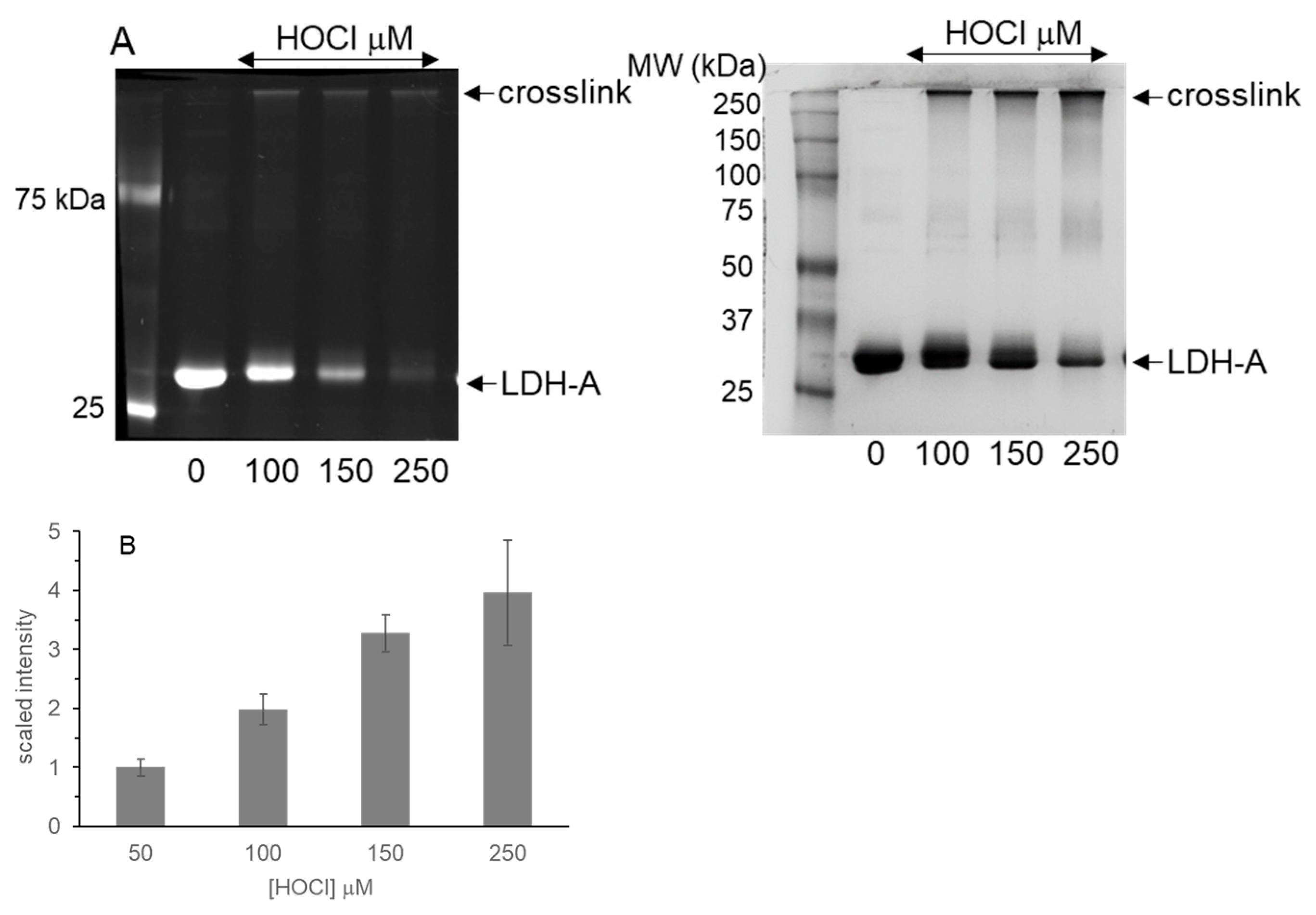
3.2. LDH-A Oxidation by HOCl
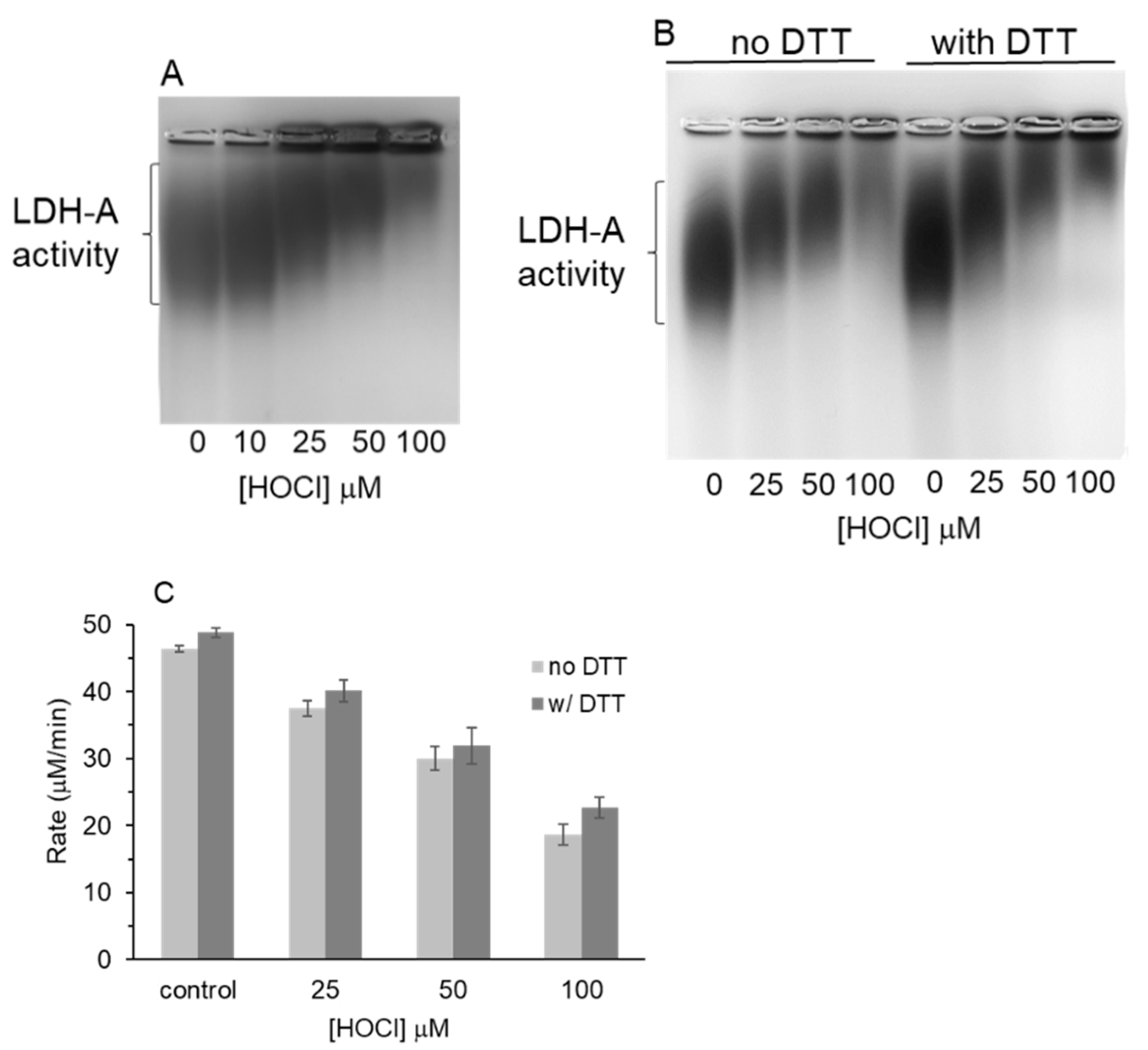
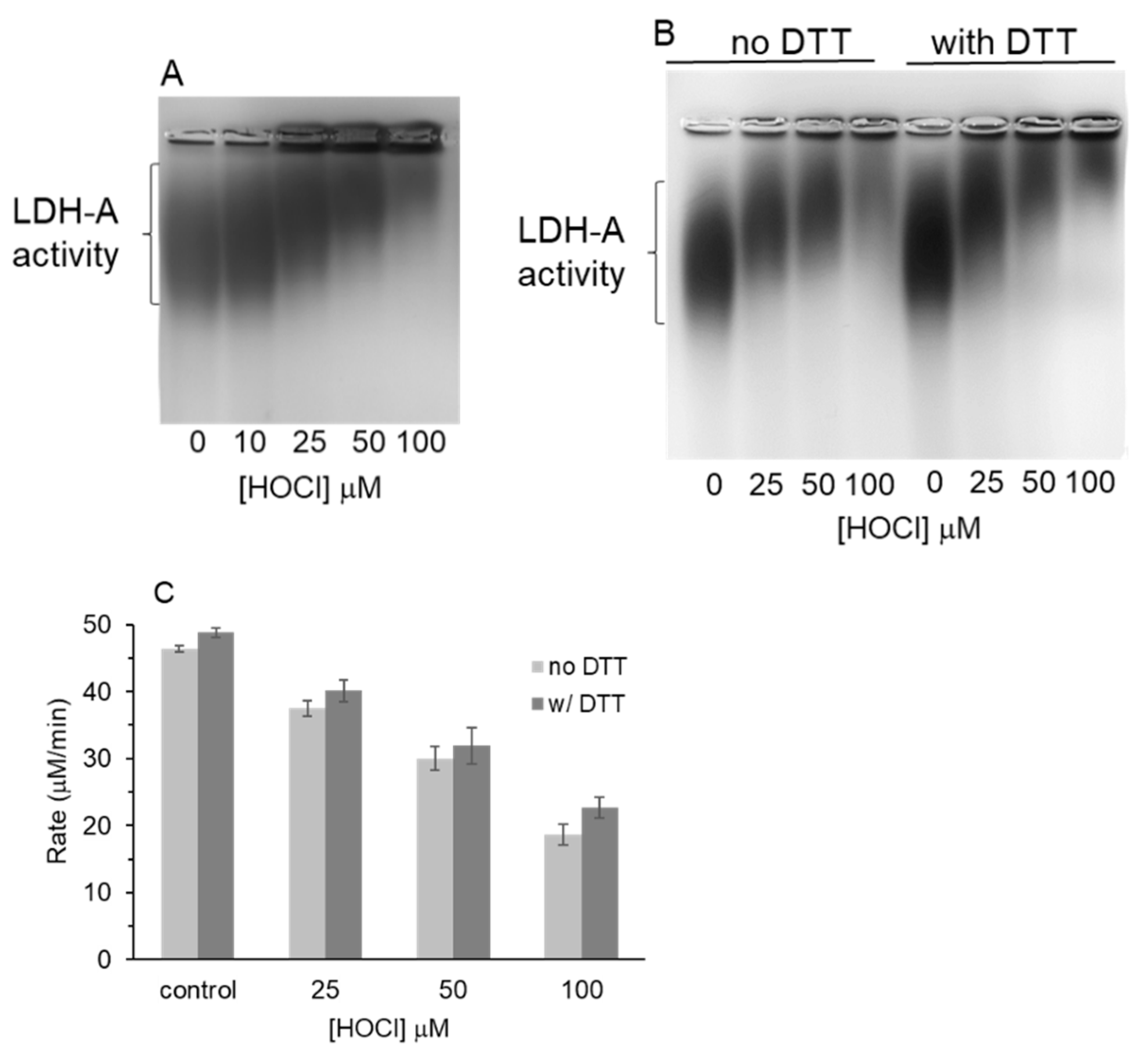
3.3. Native Gel Analysis of HOCl-Treated LDH-A; Activity Staining
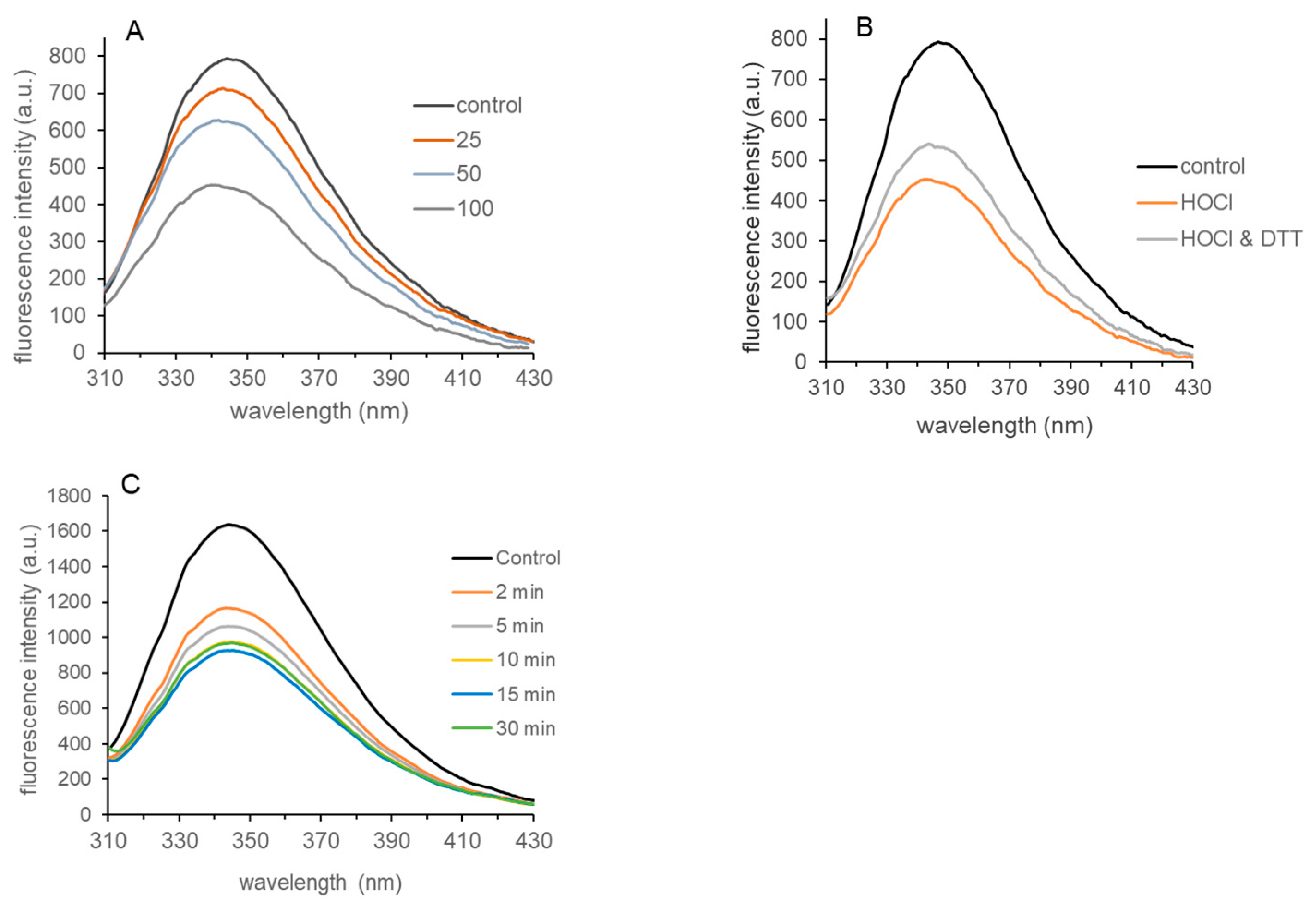
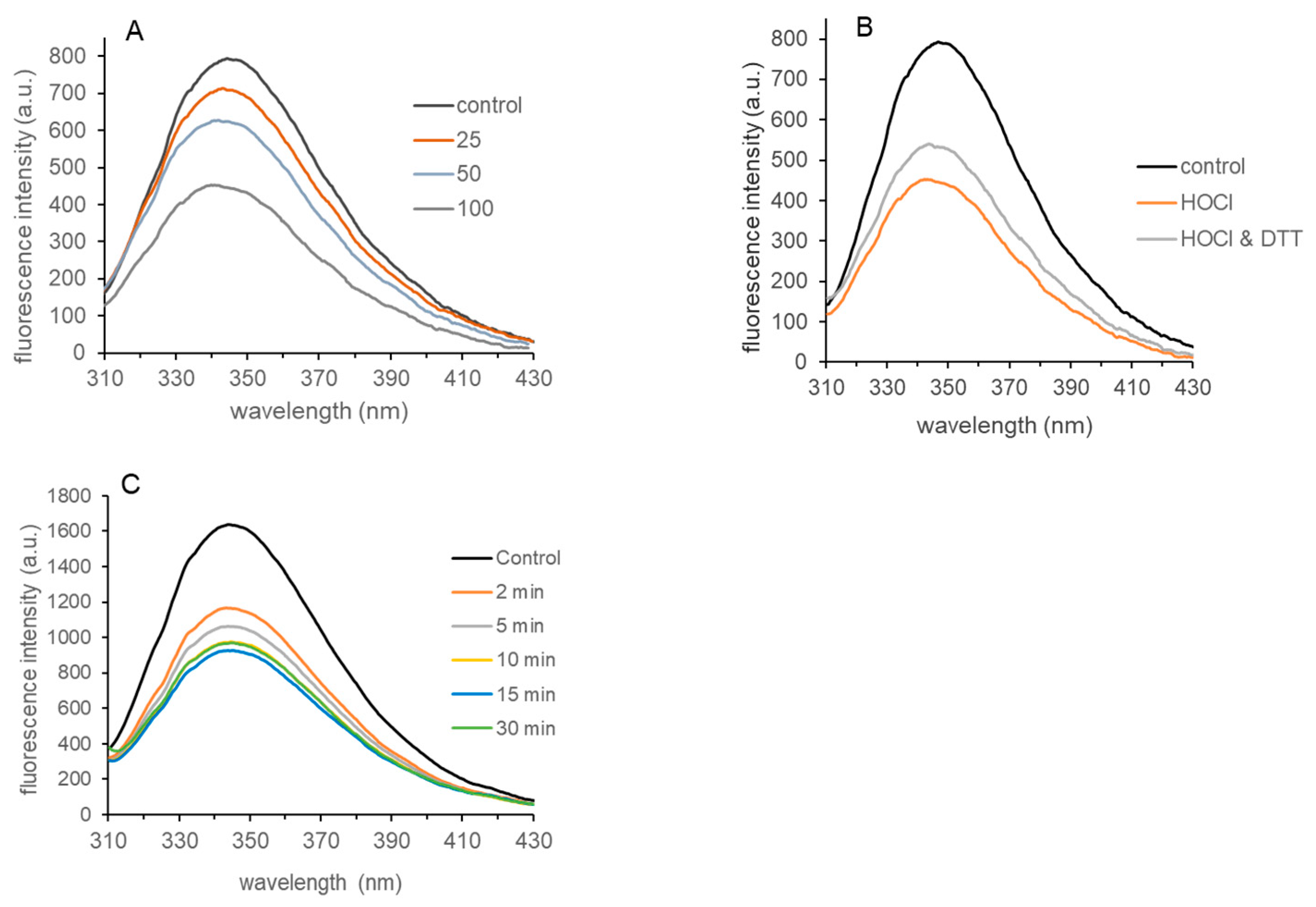
3.4. Tryptophan Fluorescence of HOCl-Treated LDH-A
3.5. Evidence for Dityrosine in HOCl-Treated LDH-A
3.6. Effects of HOCl Scavengers on LDH-A Inhibition
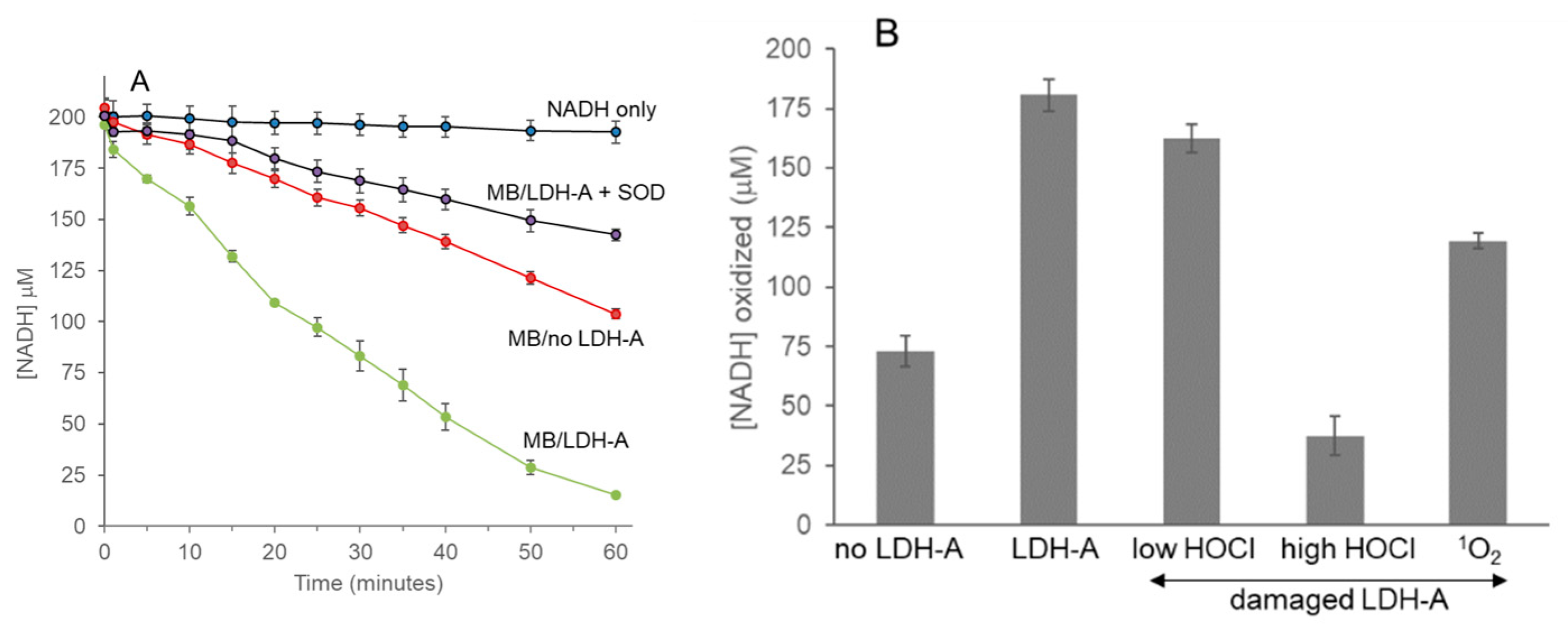
3.7. Enhanced NADH Oxidation by LDH-A; Activation by 1O2
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Holbrook, J.J.; Liljas, A.; Steindel, S.J.; Rossmann, M.J. Lactate Dehydrogenase. In The Enzymes; Boyer, P.D., Ed.; Academic Press: New York, NY, USA, 1975; Volume 11, pp. 191–292. [Google Scholar]
- Newington, J.T.; Harris, R.A.; Cumming, R.C. Reevaluating Metabolism in Alzheimer’s Disease from the Perspective of the Astrocyte-Neuron Lactate Shuttle. J. Neurodegener. Dis. 2013, 2013, 1–13. [Google Scholar] [CrossRef]
- Ippolito, L.; Morandi, A.; Giannoni, E.; Chiarugi, P. Lactate: A Metabolic Driver in the Tumour Landscape. Trends Biochem. Sci. 2019, 44, 153–166. [Google Scholar] [CrossRef]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A Expression Uncovers a Link between Glycolysis, Mitochondrial Physiology, and Tumor Maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef]
- Miao, P.; Sheng, S.; Sun, X.; Liu, J.; Huang, G. Lactate Dehydrogenase a in Cancer: A Promising Target for Diagnosis and Therapy. Int. Union Biochem. Mol. Biol. 2013, 65, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C. V Inhibition of Lactate Dehydrogenase A Induces Oxidative Stress and Inhibits Tumor Progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Potter, M.; Newport, E.; Morten, K.J. The Warburg Effect: 80 Years On. Biochem. Soc. Trans. 2016, 44, 1499–1505. [Google Scholar] [CrossRef]
- Fan, J.; Hitosugi, T.; Chung, T.-W.; Xie, J.; Ge, Q.; Gu, T.-L.; Polakiewicz, R.D.; Chen, G.Z.; Boggon, T.J.; Lonial, S.; et al. Tyrosine Phosphorylation of Lactate Dehydrogenase A Is Important for NADH/NAD+ Redox Homeostasis in Cancer Cells. Mol. Cell. Biol. 2011, 31, 4938–4950. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Dang, C.V. Multifaceted Roles of Glycolytic Enzymes. Trends Biochem. Sci. 2005, 30, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Boukouris, A.E.; Zervopoulos, S.D.; Michelakis, E.D. Metabolic Enzymes Moonlighting in the Nucleus: Metabolic Regulation of Gene Transcription. Trends Biochem. Sci. 2016, 41, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Gallo, M.; Sapio, L.; Spina, A.; Naviglio, D.; Calogero, A.; Naviglio, S. Lactic Dehydrogenase and Cancer: An Overview. Front. Biosci. 2015, 20, 1234–1249. [Google Scholar]
- Hirschhaeuser, F.; Sattler, U.G.A.; Mueller-Klieser, W. Lactate: A Metabolic Key Player in Cancer. Cancer Res. 2011, 71, 6921–6925. [Google Scholar] [CrossRef]
- Latham, T.; Mackay, L.; Sproul, D.; Karim, M.; Culley, J.; Harrison, D.J.; Hayward, L.; Langridge-Smith, P.; Gilbert, N.; Ramsahoye, B.H. Lactate, a Product of Glycolytic Metabolism, Inhibits Histone Deacetylase Activity and Promotes Changes in Gene Expression. Nucleic Acids Res. 2012, 40, 4794–4803. [Google Scholar] [CrossRef]
- Cumming, R.C.; Andon, N.L.; Haynes, P.A.; Park, M.; Fischer, W.H.; Schubert, D. Protein Disulfide Bond Formation in the Cytoplasm during Oxidative Stress. J. Biol. Chem. 2004, 279, 21749–21758. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Perluigi, M.; Sultana, R. Oxidative Stress in Alzheimer’s Disease Brain: New Insights from Redox Proteomics. Eur. J. Pharmacol. 2006, 545, 39–50. [Google Scholar] [CrossRef]
- Green, P.S.; Mendez, A.J.; Jacob, J.S.; Crowley, J.R.; Growdon, W.; Hyman, B.T.; Heinecke, J.W. Neuronal Expression of Myeloperoxidase Is Increased in Alzheimer’s Disease. J. Neurochem. 2004, 90, 724–733. [Google Scholar] [CrossRef] [PubMed]
- Pullar, J.M.; Winterbourn, C.C.; Vissers, M.C.M. Loss of GSH and Thiol Enzymes in Endothelial Cells Exposed to Sublethal Concentrations of Hypochlorous Acid. Am. J. Physiol. 1999, 277, H1505–H1512. [Google Scholar] [CrossRef] [PubMed]
- McBean, G.J.; Aslan, M.; Griffiths, H.R.; Torrao, R.C. Thiol Redox Homeostasis in Neurodegenerative Disease. Redox Biol. 2015, 5, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Chida, A.S.; Rahman, I. Redox Modifications of Protein-Thiols: Emerging Roles in Cell Signaling. Biochem. Pharmacol. 2006, 71, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, J.J.; Gutfreund, H. Approaches to the Study of Enzyme Mechanisms Lactate Dehydrogenase. FEBS Lett. 1973, 31, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, J.J.; Stinson, R.A. Reactivity of the Essential Thiol Group of Lactate Dehydrogenase and Substrate Bindng. Biochem J 1970, 120, 289–297. [Google Scholar] [CrossRef]
- Landino, L.M.; Shuckrow, Z.T.; Mooney, A.S.; Lauderback, C.O.; Lorenzi, K.E. Photo-Oxidation and Photoreduction of Catechols by Chlorophyll Metabolites and Methylene Blue. Chem. Res. Toxicol. 2022, 35, 1851–1862. [Google Scholar] [CrossRef] [PubMed]
- Dąbrowski, J.M. Chapter Nine—Reactive Oxygen Species in Photodynamic Therapy: Mechanisms of Their Genration and Potentiation. In Inorganic Reaction Mechanisms; van Eldik, R., Hubbard, C.D., Eds.; Academic Press: Cambridge, MA, USA, 2017; Volume 70, pp. 343–394. ISBN 0898-8838. [Google Scholar]
- Chan, P.C.; Bielski, H.J. Enzyme-Catalyzed Free Radical Reactions with Nicotinamide Adenine Nucleotides: II.Lactate Dehydrogenase-Catalyzed Oxidation of Reduced Nicotinamide Adenine Dinucleotide by Superoxide Radicals Denerated by Xanthine Oxidase. J. Biol. Chem. 1974, 249, 1317–1319. [Google Scholar] [CrossRef] [PubMed]
- Petrat, F.; Bramey, T.; Kirsch, M.; De Groot, H. Initiation of a Superoxide-Dependent Chain Oxidation of Lactate Dehydrogenase-Bound NADH by Oxidants of Low and High Reactivity. Free Radic. Res. 2005, 39, 1043–1057. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wang, Y.; Ying, M.; Jin, C.; Li, J.; Hu, X. Lactate Dehydrogenases Amplify Reactive Oxygen Species in Cancer Cells in Response to Oxidative Stimuli. Signal Transduct. Target. Ther. 2021, 6, 242. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Brennan, S.O. Characterization of the Oxidation Products of the Reaction between Reduced Glutathione and Hypochlorous Acid. Biochem. J. 1997, 326, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Landino, L.M.; Hagedorn, T.D.; Kim, S.B.; Hogan, K.M. Inhibition of Tubulin Polymerization by Hypochlorous Acid and Chloramines. Free Radic. Biol. Med. 2011, 50, 1000–1008. [Google Scholar] [CrossRef]
- Makriyannis, T.; Clonis, Y.D. Simultaneous Separation and Purification of Pyruvate Kinase and Lactate Dehydrogenase by Dye-Ligand Chromatography. Process Biochem. 1993, 28, 179–185. [Google Scholar] [CrossRef]
- Rees, M.D.; Bottle, S.E.; Fairfull-Smith, K.E.; Malle, E.; Whitelock, J.M.; Davies, M.J. Inhibition of Myeloperoxidase-Mediated Hypochlorous Acid Production by Nitroxides. Biochem. J. 2009, 421, 79–86. [Google Scholar] [CrossRef]
- Pamp, K.; Bramey, T.; Kirsch, M.; de Groot, H.; Petrat, F. NAD(H) Enhances the Cu(II)-Mediated Inactivation of Lactate Dehydrogenase by Increasing the Accessibility of Sulfhydryl Groups. Free Radic. Res. 2005, 39, 31–40. [Google Scholar] [CrossRef]
- Storkey, C.; Davies, M.J.; Pattison, D.I. Reevaluation of the Rate Constants for the Reaction of Hypochlorous Acid (HOCl) with Cysteine, Methionine, and Peptide Derivatives Using a New Competition Kinetic Approach. Free Radic. Biol. Med. 2014, 73, 60–66. [Google Scholar] [CrossRef]
- Peskin, A.V.; Winterbourn, C.C. Kinetics of the Reactions of Hypochlorous Acid and Amino Acid Chloramines with Thiols, Methionine and Ascorbate. Free Radic. Biol. Med. 2001, 30, 572–579. [Google Scholar] [CrossRef]
- Landino, L.M.; Hasan, R.; McGaw, A.; Cooley, S.; Smith, A.W.; Masselam, K.; Kim, G. Peroxynitrite Oxidation of Tubulin Sulfhydryls Inhibits Microtubule Polymerization. Arch. Biochem. Biophys. 2002, 398, 213–220. [Google Scholar] [CrossRef]
- Sjoback, R.; Nygren, J.; Kubista, M. Absorption and Fluorescence Properties of Fluorescein. Spectrochim. Acta Part A 1995, 51, L7–L21. [Google Scholar] [CrossRef]
- Bloxham, D.P.; Sharma, R.P.; Wilton, D.C. A Detailed Investigation of the Properties of Lactate Dehydrogenase in Which the Essential Cysteine-165 Is Modified by Thioalkylation. Biochem. J. 1979, 177, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Braz, V.A.; Howard, K.J. Separation of Protein Oligomers by Blue Native Gel Electrophoresis. Anal. Biochem. 2009, 388, 170–172. [Google Scholar] [CrossRef] [PubMed]
- Malencik, D.A.; Sprouse, J.F.; Swanson, C.A.; Anderson, S.R. Dityrosine: Preparation, Isolation and Analysis. Anal. Biochem. 1996, 242, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Milzani, A.; Colombo, R. Fluorometric Detection of Dityrosine Coupled with HPLC Separation for Determining Actin Oxidation. Am. Biotechnol. Lab. 2001, 19, 34–36. [Google Scholar]
- Eiserich, J.P.; Cross, C.E.; Jones, A.D.; Halliwell, B.; Van der Vliet, A. Formation of Nitrating and Chlorinating Species by Reaction of Nitrite with Hypochlorous Acid. J. Biol. Chem. 1996, 271, 19199–19208. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Rose, P.; Siau, J.L.; Halliwell, B. Nitrite-Mediated Protection against Hypochlorous Acid-Induced Chondrocyte Toxicity. Arthritis Rheum. 2003, 48, 3140–3150. [Google Scholar] [CrossRef] [PubMed]
- Peters, G.; Rodgers, M.A.J. Single-Electron Transfer from NADH Analogues to Singlet Oxygen. Biochim. Biophys. Acta-Bioenerg. 1981, 637, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Petrat, F.; Pindiur, S.; Kirsch, M.; de Groot, H. NAD(P)H, a Primary Target of 1O2 in Mitochondria of Intact Cells. J. Biol. Chem. 2003, 278, 3298–3307. [Google Scholar] [CrossRef]
- Prütz, W.A.; Kissner, R.; Koppenol, W.H.; Rüegger, H. On the Irreversible Destruction of Reduced NicotinamideNucleotides by Hypohalous Acids. Arch. Biochem. Biophys. 2000, 380, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Ma, K. Determination of Hydrogen Peroxide Generated by Reduced Nicotinamide Adenine Dinucleotide Oxidase. Anal. Biochem. 2005, 344, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-J.; Tsoi, S.C.-M. Phylogenetic Analysis of Vertebrate Lactate Dehydrogenase (LDH) Multigene Families. J. Mol. Evol. 2002, 54, 614–624. [Google Scholar] [CrossRef]
- Landino, L.M.; Moynihan, K.L.; Todd, J.V.; Kennett, K.L. Modulation of the Redox State of Tubulin by the Glutathione/Glutaredoxin Reductase System. Biochem. Biophys. Res. Commun. 2004, 314, 555–560. [Google Scholar] [CrossRef]
- Heinecke, J.W.; Li, W.; Daehnke, H.L.; Goldstein, J.A. Dityrosine, a Specific Marker of Oxidation, Is Synthesized by the Myeloperoxidase-Hydrogen Peroxide System of Human Neutrophils and Macrophages. J. Biol. Chem. 1993, 268, 4069–4077. [Google Scholar] [CrossRef]
- Vivian, J.T.; Callis, P.R. Mechanisms of Tryptophan Fluorescence Shifts in Proteins. Biophys. J. 2001, 80, 2093–2109. [Google Scholar] [CrossRef]
- Freund, E.; Miebach, L.; Stope, M.B.; Bekeschus, S. Hypochlorous Acid Selectively Promotes Toxicity and the Expression of Danger Signals in Human Abdominal Cancer Cells. Oncol. Rep. 2021, 45, 71. [Google Scholar] [CrossRef]
- Chiang, C.L.-L.; Ledermann, J.A.; Aitkens, E.; Benjamin, E.; Katz, D.R.; Chain, B.M. Oxidation of Ovarian Epithelial Cancer Cells by Hypochlorous Acid Enhances Immunogenicity and Stimulates T Cells That Recognize Autologous Primary Tumor. Clin. Cancer Res. 2008, 14, 4898–4907. [Google Scholar] [CrossRef]
- Landino, L.M.; Hagedorn, T.D.; Kennett, K.L. Evidence for Thiol/Disulfide Exchange Reactions Between Tubulin and Glyceraldehyde-3-Phosphate Dehydrogenase. Cytoskeleton 2014, 71, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Di Mascio, P.; Martinez, G.R.; Miyamoto, S.; Ronsein, G.E.; Medeiros, M.H.G.; Cadet, J. Singlet Molecular Oxy gen Reactions with Nucleic Acids, Lipids, and Proteins. Chem. Rev. 2019, 119, 2043–2086. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [HOCl] μM | Mol Cys/Mol LDH-A | % Control Labeling (Integration of Gel Bands) | |
|---|---|---|---|
| 0 | 2.7 ± 0.2 | M5F | IAF |
| 10 | 2.6 ± 0.2 | 85 ± 3 | 86 ± 4 |
| 25 | 2.3 ± 0.1 | 67 ± 4 | 66 ± 3 |
| 50 | 2.0 ± 0.1 | 56 ± 4 | 48 ± 5 |
| 100 | 1.4 ± 0.2 | 46 ± 4 | 36 ± 3 |
| HOCl Scavenger | LDH-A Activity (% of Control) | Rate Constant (M−1 s−1) |
|---|---|---|
| none | 100 | |
| +HOCl | 62 ± 4 | |
| +HOCl + 50 μM GSH | 88 ± 2 | |
| +HOCl + 100 μM GSH | 97 ± 3 | 1.2 × 108 [33] |
| +HOCl + 500 μM NO2− | 70 ± 5 | |
| +HOCl + 1 mM NO2− | 92 ± 4 | 7.3 × 103 [42] |
| +HOCl + 50 μM S-methyl-cys | 77 ± 5 | |
| +HOCl + 100 μM S-methyl-cys | 88 ± 4 | |
| +HOCl + 200 μM S-methyl-cys | 95 ± 3 | 3.1 × 107 [33] * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Landino, L.; Boike, L.; Lain, T. Inhibition of Lactate Dehydrogenase-A by Singlet Oxygen and Hypochlorous Acid via Cysteine Oxidation and Irreversible Conformational Changes. BioChem 2024, 4, 18-37. https://doi.org/10.3390/biochem4010002
Landino L, Boike L, Lain T. Inhibition of Lactate Dehydrogenase-A by Singlet Oxygen and Hypochlorous Acid via Cysteine Oxidation and Irreversible Conformational Changes. BioChem. 2024; 4(1):18-37. https://doi.org/10.3390/biochem4010002
Chicago/Turabian StyleLandino, Lisa, Lydia Boike, and Taylor Lain. 2024. "Inhibition of Lactate Dehydrogenase-A by Singlet Oxygen and Hypochlorous Acid via Cysteine Oxidation and Irreversible Conformational Changes" BioChem 4, no. 1: 18-37. https://doi.org/10.3390/biochem4010002
APA StyleLandino, L., Boike, L., & Lain, T. (2024). Inhibition of Lactate Dehydrogenase-A by Singlet Oxygen and Hypochlorous Acid via Cysteine Oxidation and Irreversible Conformational Changes. BioChem, 4(1), 18-37. https://doi.org/10.3390/biochem4010002