
Combined Therapies with Taxane-Based Chemotherapeutic Drugs in Prostate Cancer: Novel Insights and Future Directions



Abstract
:1. Introduction
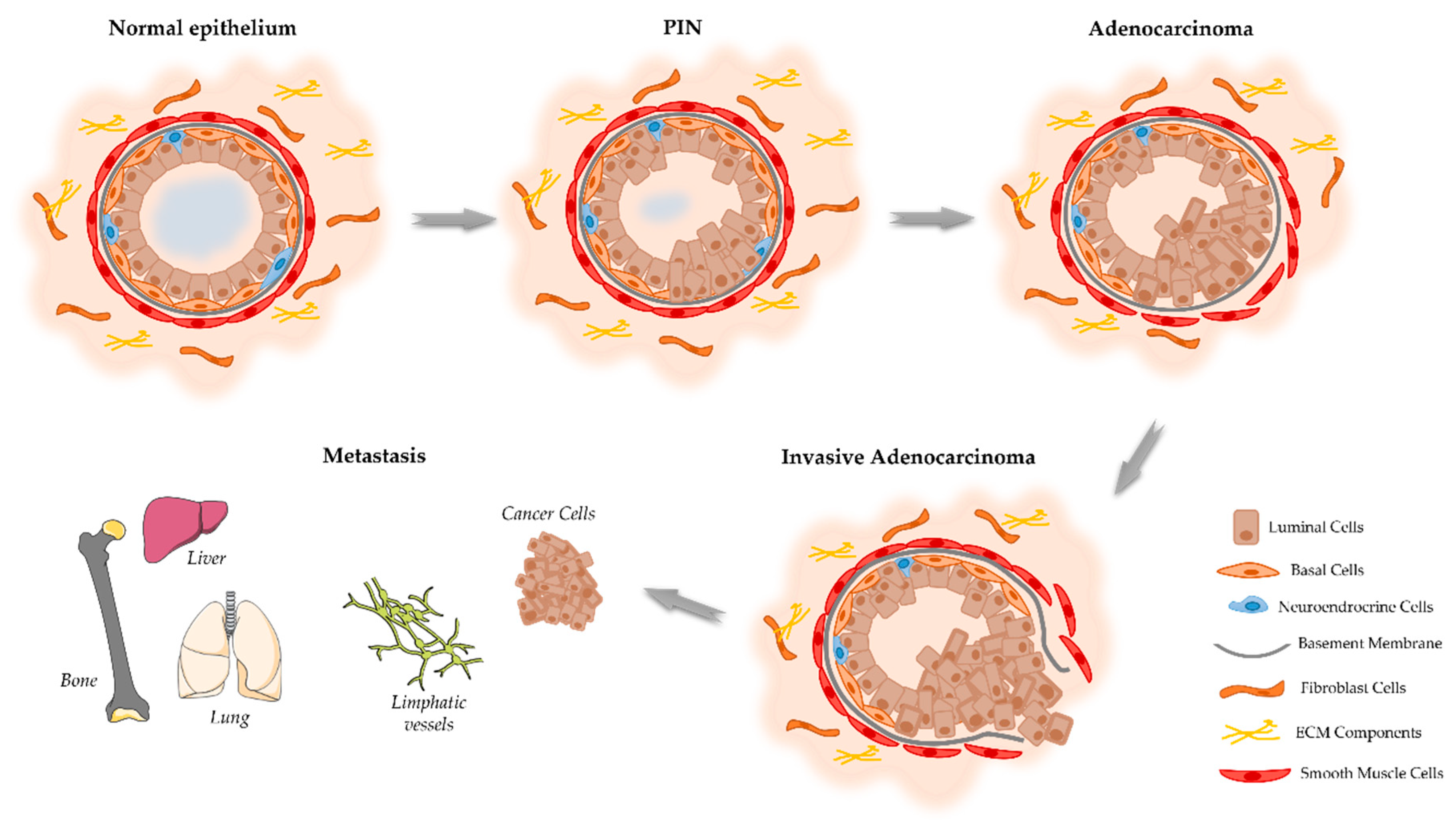
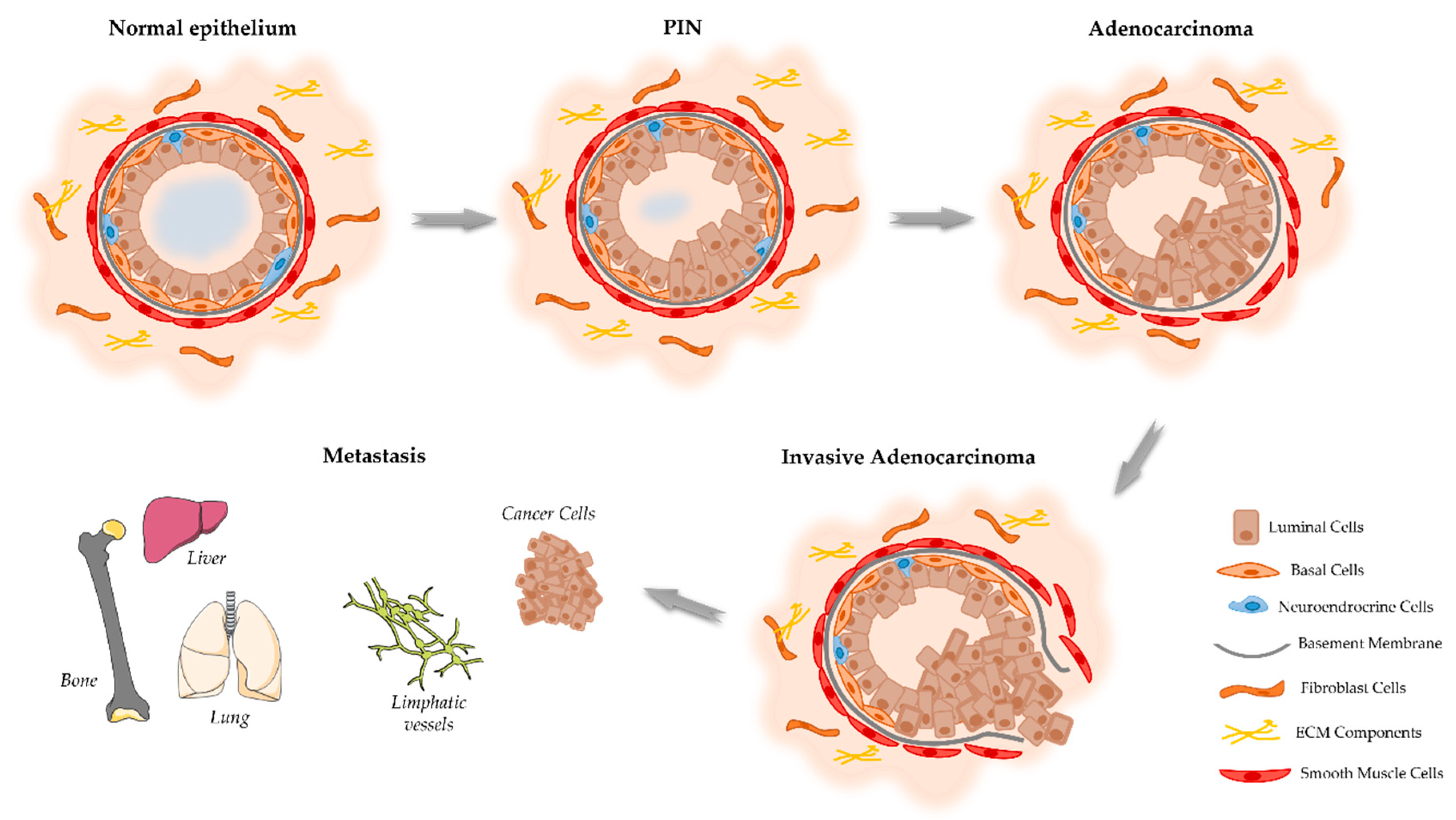
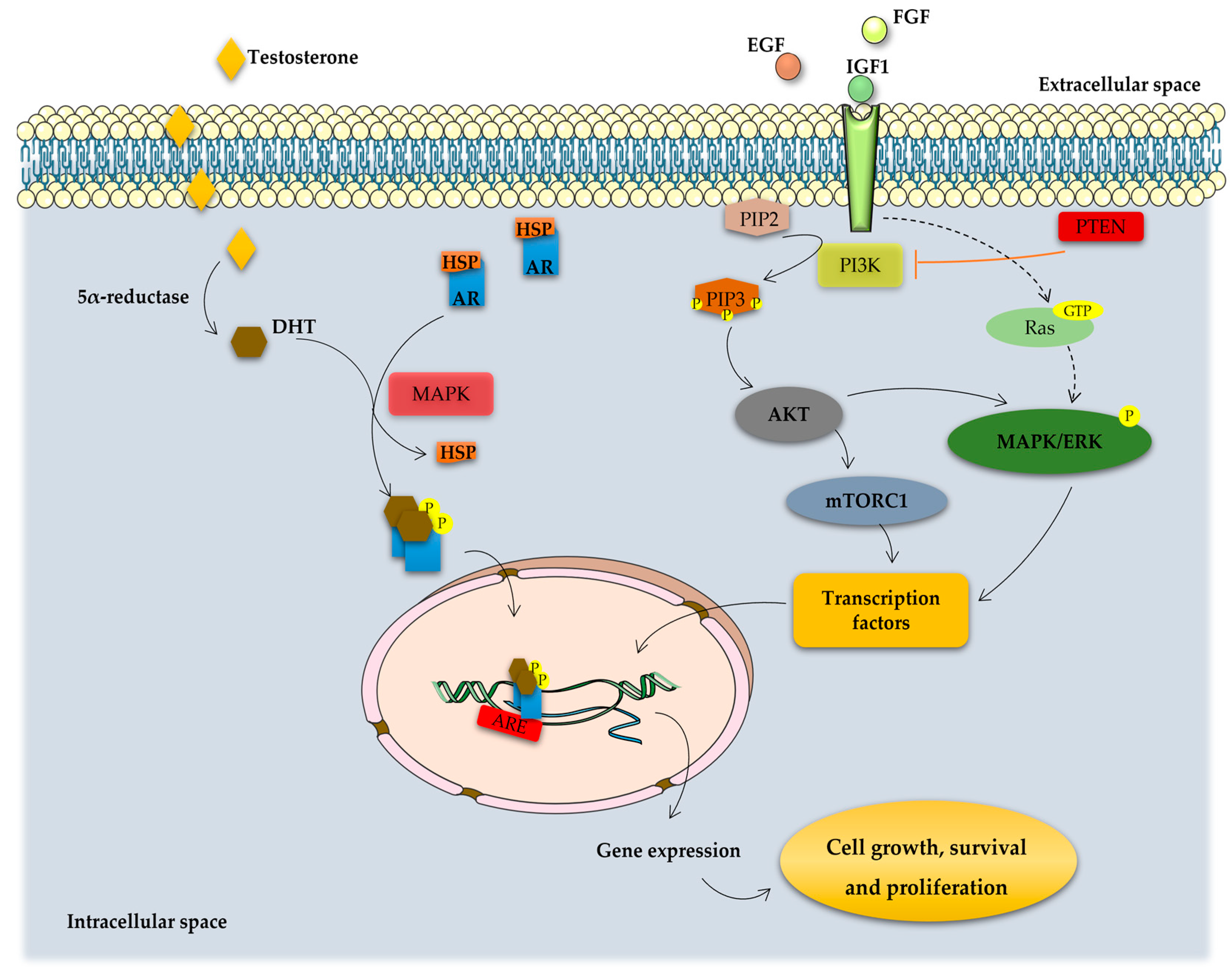
2. Onset and Development of PCa
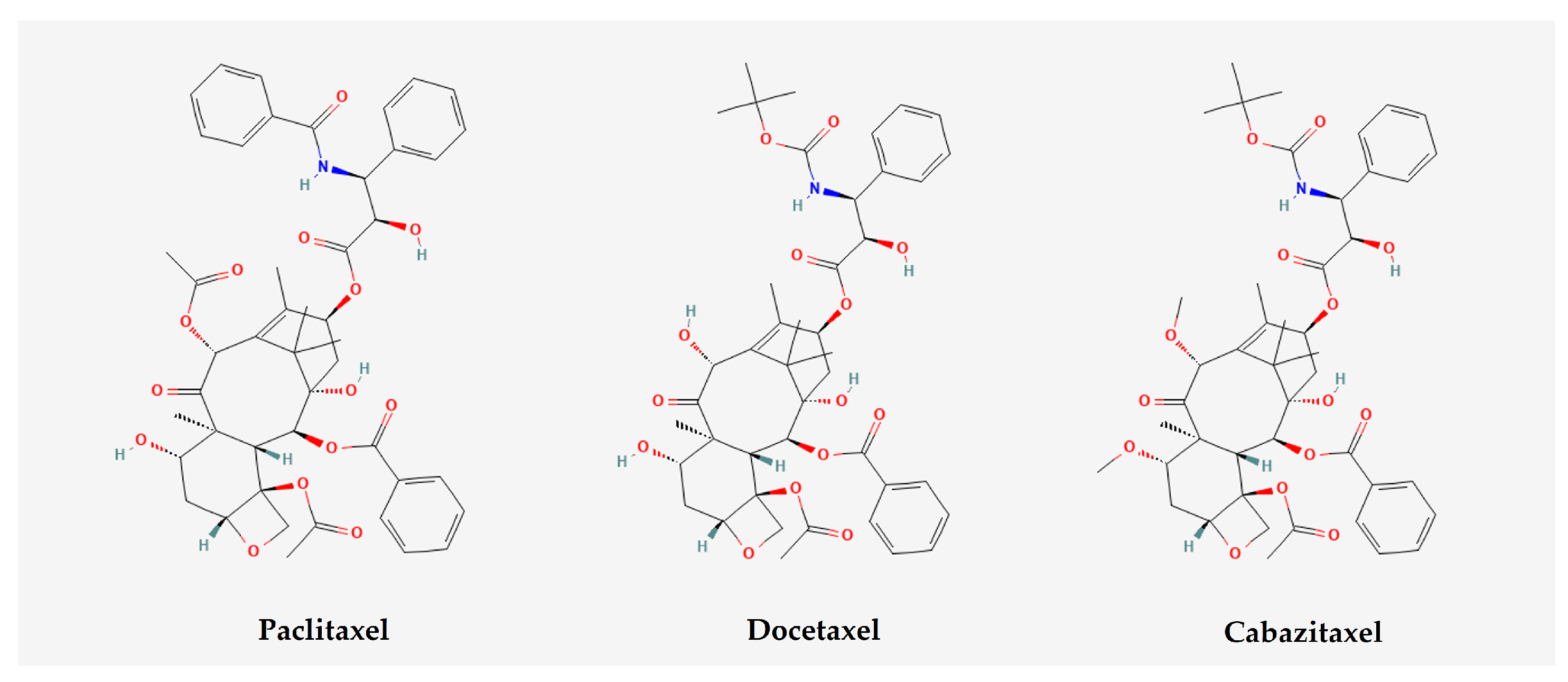
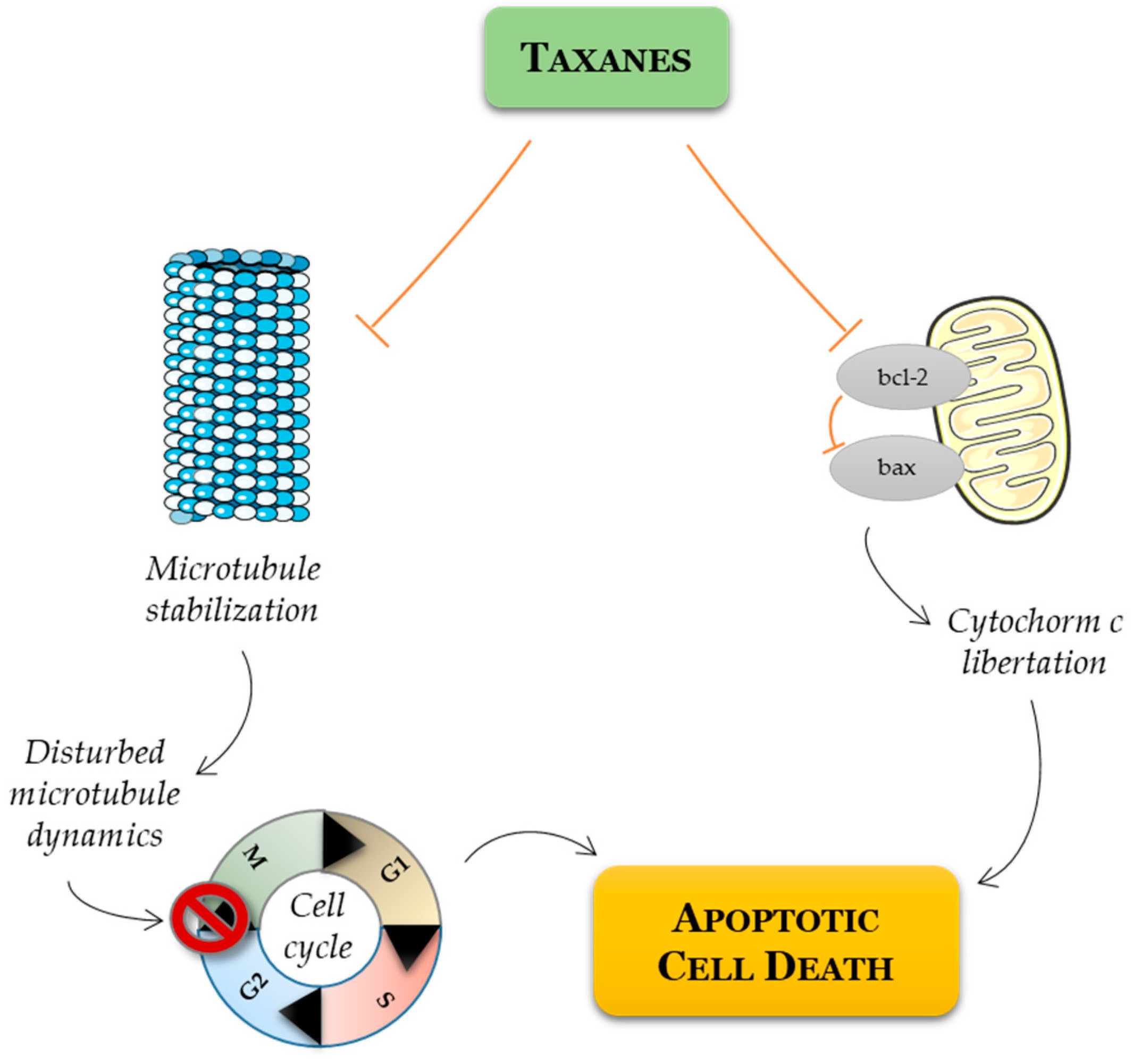
3. Current Use of Chemotherapy in PCa
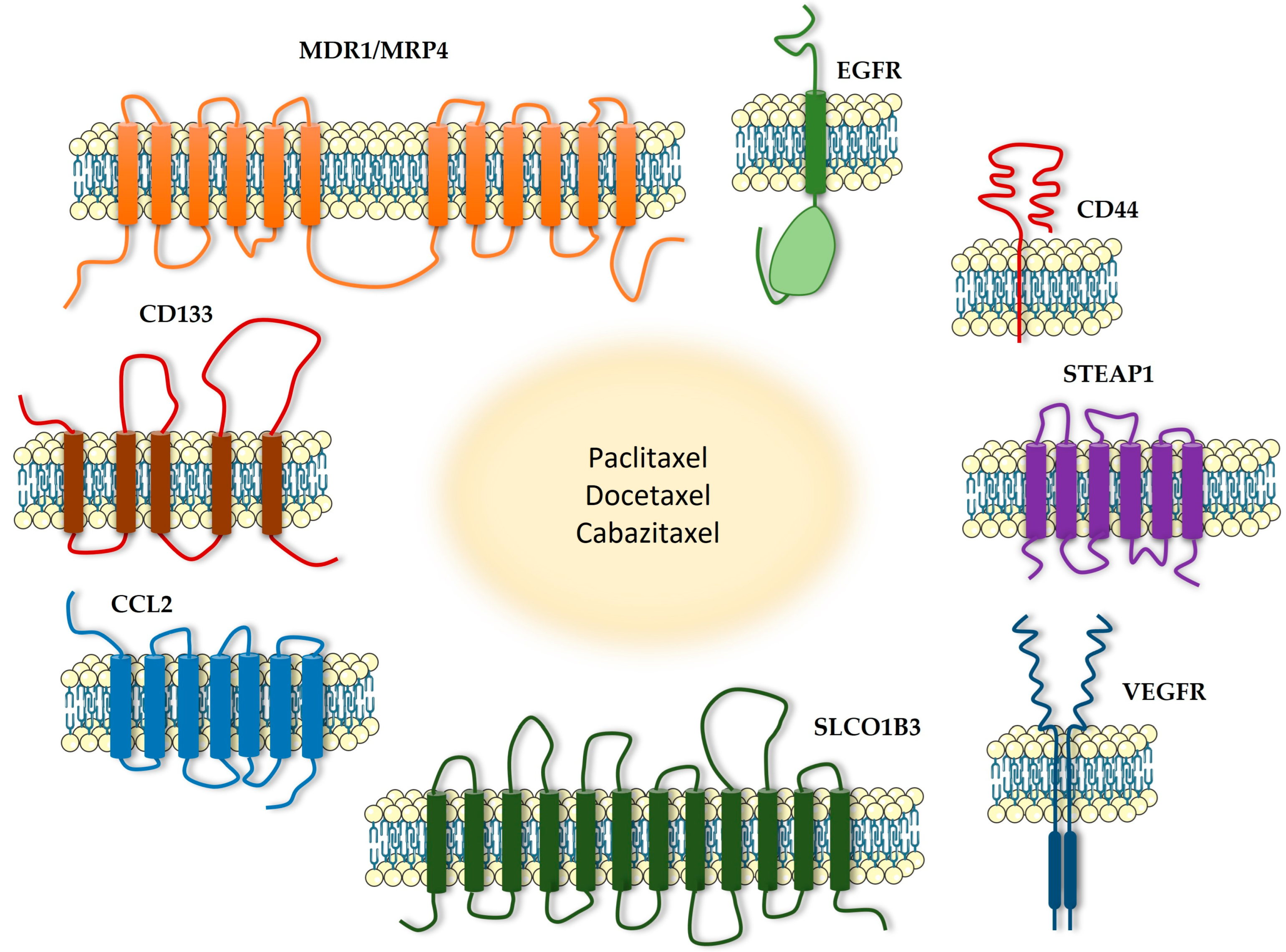
4. Transmembrane Proteins as a Potential Therapeutic Target in Combination with Taxanes
4.1. MDR1
4.2. MRP4
4.3. CD44
4.4. CD133
4.5. SLCO1B3
4.6. EGFR
4.7. STEAP1
4.8. CCL2/CCR2
4.9. VEGFR
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63. [Google Scholar] [CrossRef]
- Oar, A.; Moraes, F.Y.; Romero, Y.; Ilbawi, A.; Yap, M.L. Core elements of national cancer control plans: A tool to support plan development and review. Lancet Oncol. 2019, 20, e645–e652. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.L. The Etiology of Prostate Cancer. In Prostate Cancer; Exon Publications: Brisbane, Australia, 2021; pp. 17–28. [Google Scholar]
- Malik, S.S.; MPhil Batool, R.; Honors, B.S.; Masood, N.; Yasmin, A. Risk factors for prostate cancer: A multifactorial case-control study. Curr. Probl. Cancer 2018, 42, 337–343. [Google Scholar] [CrossRef]
- Leitzmann, M.F.; Rohrmann, S. Risk factors for the onset of prostatic cancer: Age, location, and behavioral correlates. Clin. Epidemiol. 2012, 4, 1–11. [Google Scholar] [CrossRef]
- Swami, U.; McFarland, T.R.; Nussenzveig, R.; Agarwal, N. Advanced Prostate Cancer: Treatment Advances and Future Directions. Trends Cancer 2020, 6, 702–715. [Google Scholar] [CrossRef]
- Corn, P.G.; Agarwal, N.; Araujo, J.C.; Sonpavde, G. Taxane-based Combination Therapies for Metastatic Prostate Cancer. Eur. Urol. Focus 2019, 5, 369–380. [Google Scholar] [CrossRef]
- Cournia, Z.; Allen, T.W.; Andricioaei, I.; Antonny, B.; Baum, D.; Brannigan, G.; Buchete, N.V.; Deckman, J.T.; Delemotte, L.; Del Val, C.; et al. Membrane Protein Structure, Function and Dynamics: A Perspective from Experiments and Theory. J. Membr. Biol. 2015, 248, 611–640. [Google Scholar] [CrossRef]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug. Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef]
- Lee, C.H.; Akin-Olugbade, O.; Kirschenbaum, A. Overview of prostate anatomy, histology, and pathology. Endocrinol. Metab. Clin. N. Am. 2011, 40, 565–575. [Google Scholar] [CrossRef]
- Ittmann, M. Anatomy and Histology of the Human and Murine Prostate. Cold Spring Harb. Perspect. Med. 2018, 8, a030346. [Google Scholar] [CrossRef] [PubMed]
- Leong, K.G.; Wang, B.-E.; Johnson, L.; Gao, W.-Q. Generation of a prostate from a single adult stem cell. Nature 2008, 456, 804–808. [Google Scholar] [CrossRef] [PubMed]
- Packer, J.R.; Maitland, N.J. The molecular and cellular origin of human prostate cancer. Biochim. Biophys. Acta 2016, 1863 Pt A, 1238–1260. [Google Scholar] [CrossRef]
- Corn, P.G. The tumor microenvironment in prostate cancer: Elucidating molecular pathways for therapy development. Cancer Manag. Res. 2012, 4, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Levesque, C.; Nelson, P.S. Cellular Constituents of the Prostate Stroma: Key Contributors to Prostate Cancer Progression and Therapy Resistance. Cold Spring Harb. Perspect. Med. 2018, 8, a030510. [Google Scholar] [CrossRef] [PubMed]
- Yadav, N.; Heemers, H.V. Androgen action in the prostate gland. Minerva Urol. Nefrol. 2012, 64, 35–49. [Google Scholar]
- Long, R.M.; Morrissey, C.; Fitzpatrick, J.M.; Watson, R.W. Prostate epithelial cell differentiation and its relevance to the under-standing of prostate cancer therapies. Clin. Sci. 2005, 108, 1–11. [Google Scholar] [CrossRef]
- Lawson, D.A.; Zong, Y.; Memarzadeh, S.; Xin, L.; Huang, J.; Witte, O.N. Basal epithelial stem cells are efficient targets for prostate cancer initiation. Proc. Natl. Acad. Sci. USA 2010, 107, 2610–2615. [Google Scholar] [CrossRef]
- Parimi, V.; Goyal, R.; Poropatich, K.; Yang, X.J. Neuroendocrine differentiation of prostate cancer: A review. Am. J. Clin. Exp. Urol. 2014, 2, 273–285. [Google Scholar]
- Wang, Z.A.; Mitrofanova, A.; Bergren, S.K.; Abate-Shen, C.; Cardiff, R.D.; Califano, A.; Shen, M.M. Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity. Nature 2013, 15, 274–283. [Google Scholar] [CrossRef]
- Wang, Z.A.; Toivanen, R.; Bergren, S.K.; Chambon, P.; Shen, M.M. Luminal Cells Are Favored as the Cell of Origin for Prostate Cancer. Cell Rep. 2014, 8, 1339–1346. [Google Scholar] [CrossRef]
- Wang, X.; Kruithof-de Julio, M.; Economides, K.D.; Walker, D.; Yu, H.; Halili, M.V.; Hu, Y.-P.; Price, S.M.; Abate-Shen, C.; Shen, M.M. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature 2009, 461, 495–500. [Google Scholar] [CrossRef]
- Shen, M.M.; Abate-Shen, C. Molecular genetics of prostate cancer: New prospects for old challenges. Genes Dev. 2010, 24, 1967–2000. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.G.; Ro, J.Y. Prostatic intraepithelial neoplasia: Recent advances. Arch. Pathol. Lab. Med. 2007, 131, 1257–1266. [Google Scholar] [CrossRef]
- Joshua, A.; Evans, A.; Van der Kwast, T.; Zielenska, M.; Meeker, A.; Chinnaiyan, A.; Squire, J. Prostatic preneoplasia and beyond. Biochim. Biophys. Acta 2008, 1785, 156–181. [Google Scholar] [CrossRef] [PubMed]
- Murray, T.B.J. The Pathogenesis of Prostate Cancer. In Prostate Cancer; Exon Publications: Brisbane, Australia, 2021; pp. 29–42. [Google Scholar]
- Alexander, E.E.; Qian, J.; Wollan, P.C.; Myers, R.P.; Bostwick, D.G. Prostatic intraepithelial neoplasia does not appear to raise serum prostate-specific antigen concentration. Urology 1996, 47, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Bostwick, D.G.; Brawer, M.K. Prostatic Intra-Epithelial Neoplasia and Early Invasion in Prostate Cancer. Cancer 1987, 59, 788–794. [Google Scholar] [CrossRef]
- Schiebler, M.L.; E Tomaszewski, J.; Bezzi, M.; Pollack, H.M.; Kressel, H.Y.; Cohen, E.K.; Altman, H.G.; Gefter, W.B.; Wein, A.J.; Axel, L. Prostatic carcinoma and benign prostatic hyperplasia: Correlation of high-resolution MR and histopathologic findings. Radiology 1989, 172, 131–137. [Google Scholar] [CrossRef]
- Schiebler, M.L.; Schnall, M.D.; Pollack, H.M.; Lenkinski, R.E.; E Tomaszewski, J.; Wein, A.J.; Whittington, R.; Rauschning, W.; Kressel, H.Y. Current role of MR imaging in the staging of adenocarcinoma of the prostate. Radiology 1993, 189, 339–352. [Google Scholar] [CrossRef]
- Ulmert, D.; O’Brien, M.F.; Bjartell, A.S.; Lilja, H. Prostate kallikrein markers in diagnosis, risk stratification and prognosis. Nat. Rev. Urol. 2009, 6, 384–391. [Google Scholar] [CrossRef]
- Duffy, M.J. The role of proteolytic enzymes in cancer invasion and metastasis. Clin. Exp. Metastasis 1992, 10, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, D.; Spring, D.J.; DePinho, R.A. Genetics and biology of prostate cancer. Genes Dev. 2018, 32, 1105–1140. [Google Scholar] [CrossRef] [PubMed]
- Tuxhorn, J.A.; Ayala, G.E.; Smith, M.J.; Smith, V.C.; Dang, T.D.; Rowley, D.R. Reactive stroma in human prostate cancer: Induction of myofibroblast phenotype and extracellular matrix remodeling. Clin. Cancer Res. 2002, 8, 2912–2923. [Google Scholar]
- Shafi, A.A.; Yen, A.E.; Weigel, N.L. Androgen receptors in hormone-dependent and castration-resistant prostate cancer. Pharmacol. Ther. 2013, 140, 223–238. [Google Scholar] [CrossRef]
- Culig, Z.; Klocker, H.; Bartsch, G.; Hobisch, A. Androgen receptors in prostate cancer. Endocr. Relat. Cancer 2002, 9, 155–170. [Google Scholar] [CrossRef]
- Knuuttila, M.; Mehmood, A.; Mäki-Jouppila, J.; Ryberg, H.; Taimen, P.; Knaapila, J.; Ettala, O.; Boström, P.J.; Ohlsson, C.; Venäläinen, M.S.; et al. Intratumoral androgen levels are linked to TMPRSS2-ERG fusion in prostate cancer. Endocr.-Relat. Cancer 2018, 25, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, T.R.; Strittmatter, B.G.; Hollenhorst, P.C. Oncogenic ETS Factors in Prostate Cancer. Adv. Exp. Med. Biol. 2019, 1210, 409–436. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Lu, J.; Ma, T.; Huang, H.; Kocher, J.-P.; Wang, L. Re-Evaluate Fusion Genes in Prostate Cancer. Cancer Inform. 2021, 20, 11769351211027592. [Google Scholar] [CrossRef]
- Davey, R.A.; Grossmann, M. Androgen Receptor Structure, Function and Biology: From Bench to Bedside. Clin. Biochem. Rev. 2016, 37, 3–15. [Google Scholar]
- Bevan, C.; Parker, M. The Role of Coactivators in Steroid Hormone Action. Exp. Cell Res. 1999, 253, 349–356. [Google Scholar] [CrossRef]
- Tan, M.H.E.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.-L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Saraon, P.; Drabovich, A.P.; Jarvi, K.A.; Diamandis, E.P. Mechanisms of Androgen-Independent Prostate Cancer. EJIFCC 2014, 25, 42–54. [Google Scholar] [PubMed]
- Devlin, H.-L.; Mudryj, M. Progression of prostate cancer: Multiple pathways to androgen independence. Cancer Lett. 2009, 274, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, B.; Beitel, L.K.; Nadarajah, A.; Paliouras, M.; Trifiro, M. The androgen receptor gene mutations database: 2012 update. Hum. Mutat. 2012, 33, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Yelensky, R.; Frampton, G.M.; Park, K.; Downing, S.R.; MacDonald, T.Y.; Jarosz, M.; Lipson, D.; Tagawa, S.T.; Nanus, D.M.; et al. Targeted Next-generation Sequencing of Advanced Prostate Cancer Identifies Potential Therapeutic Targets and Disease Heterogeneity. Eur. Urol. 2013, 63, 920–926. [Google Scholar] [CrossRef]
- Chan, S.C.; Li, Y.; Dehm, S.M. Androgen Receptor Splice Variants Activate Androgen Receptor Target Genes and Support Aberrant Prostate Cancer Cell Growth Independent of Canonical Androgen Receptor Nuclear Localization Signal. J. Biol. Chem. 2012, 287, 19736–19749. [Google Scholar] [CrossRef]
- Sun, S.; Sprenger, C.C.; Vessella, R.L.; Haugk, K.; Soriano, K.; Mostaghel, E.A.; Page, S.T.; Coleman, I.M.; Nguyen, H.M.; Sun, H.; et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Investig. 2010, 120, 2715–2730. [Google Scholar] [CrossRef]
- Jenster, G. Ligand-independent activation of the androgen receptor in prostate cancer by growth factors and cytokines. J. Pathol. 2000, 191, 227–228. [Google Scholar] [CrossRef]
- Culig, Z.; Hobisch, A.; Cronauer, M.V.; Radmayr, C.; Trapman, J.; Hittmair, A.; Bartsch, G.; Klocker, H. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994, 54, 5474–5478. [Google Scholar] [CrossRef]
- Zhu, M.-L.; Kyprianou, N. Androgen receptor and growth factor signaling cross-talk in prostate cancer cells. Endocr.-Relat. Cancer 2008, 15, 841–849. [Google Scholar] [CrossRef]
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-mTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling. Int. J. Mol. Sci. 2020, 21, 4507. [Google Scholar] [CrossRef] [PubMed]
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. PI3K/AKT signaling pathway and cancer: An updated review. Ann. Med. 2014, 46, 372–383. [Google Scholar] [CrossRef]
- Labbé, D.P.; Brown, M. Transcriptional Regulation in Prostate Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a030437. [Google Scholar] [CrossRef] [PubMed]
- Schatten, H. Brief Overview of Prostate Cancer Statistics, Grading, Diagnosis and Treatment Strategies. Adv. Exp. Med. Biol. 2018, 1095, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Litwin, M.S.; Tan, H.J. The diagnosis and treatment of prostate cancer: A review. JAMA—J. Am. Med. Assoc. 2017, 317, 2532–2542. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.H.; Li, H.Y.; Chung, S.D.; Chiang, H.S.; Yu, H.J. Significant association between serum dihydrotestosterone level and prostate volume among Taiwanese men aged 40–79 years. Aging Male 2012, 15, 28–33. [Google Scholar] [CrossRef]
- Catt, S.; Matthews, L.; May, S.; Payne, H.; Mason, M.; Jenkins, V. Patients’ and partners’ views of care and treatment provided for met-astatic castrate-resistant prostate cancer in the UK. Eur. J. Cancer Care 2019, 28, e13140. [Google Scholar] [CrossRef]
- Tannock, I.F.; Osoba, D.; Stockler, M.R.; Ernst, D.S.; Neville, A.J.; Moore, M.J.; Armitage, G.R.; Wilson, J.J.; Venner, P.M.; Coppin, C.M.; et al. Chemotherapy with mitoxantrone plus prednisone or pred-nisone alone for symptomatic hormone-resistant prostate cancer: A Canadian randomized trial with palliative end points. J. Clin. Oncol. 1996, 14, 1756–1764. [Google Scholar] [CrossRef]
- Long, H.J. Paclitaxel (Taxol): A Novel Anticancer Chemotherapeutic Drug. Mayo Clin. Proc. 1994, 69, 341–345. [Google Scholar] [CrossRef]
- Kubník, J.; Pavlíčková, V.; Ruml, T.; Rimpelová, S. Current Perspectives on Taxanes: Focus on Their Bioactivity, Delivery and Combination Therapy. Plants 2021, 10, 569. [Google Scholar] [CrossRef]
- Pienta, K.J. Preclinical mechanisms of action of docetaxel and docetaxel combinations in prostate cancer. Semin. Oncol. 2001, 28 (Suppl. 15), 3–7. [Google Scholar] [CrossRef] [PubMed]
- Roumiguié, M.; Paoletti, X.; Neuzillet, Y.; Mathieu, R.; Vincendeau, S.; Kleinclauss, F.; Mejean, A.; Guy, L.; Timsit, M.O.; Lebret, T. Apalutamide, darolutamide and enzalutamide in nonmetastatic castration-resistant prostate cancer: A meta-analysis. Futur. Oncol. 2021, 17, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Rathkopf, D.E.; Morris, M.J.; Fox, J.J.; Danila, D.C.; Slovin, S.F.; Hager, J.H.; Rix, P.J.; Maneval, E.C.; Chen, I.; Gönen, M.; et al. Phase I study of ARN-509, a novel antiandrogen, in the treatment of castration-resistant prostate cancer. J. Clin. Oncol. 2013, 31, 3525–3530. [Google Scholar] [CrossRef]
- Madan, R.A.; Pal, S.K.; Sartor, O.; Dahut, W.L. Overcoming Chemotherapy Resistance in Prostate Cancer. Clin. Cancer Res. 2011, 17, 3892–3902. [Google Scholar] [CrossRef]
- Watanabe, H.; Kawakami, A.; Sato, R.; Watanabe, K.; Matsushita, Y.; Miyake, H. Molecular Mechanism Mediating Cytotoxic Activity of Cabazitaxel in Docetaxel-resistant Human Prostate Cancer Cells. Anticancer Res. 2021, 41, 3753–3758. [Google Scholar] [CrossRef] [PubMed]
- Cevik, O.; Acidereli, H.; Turut, F.A.; Yildirim, S.; Acilan, C. Cabazitaxel exhibits more favorable molecular changes compared to other taxanes in androgen-independent prostate cancer cells. J. Biochem. Mol. Toxicol. 2020, 34, e22542. [Google Scholar] [CrossRef] [PubMed]
- Takai, M.; Kato, S.; Nakano, M.; Fujimoto, S.; Iinuma, K.; Ishida, T.; Taniguchi, M.; Tamaki, M.; Uno, M.; Takahashi, Y.; et al. Efficacy of cabazitaxel and the influence of clinical factors on the overall survival of patients with castration-resistant prostate cancer: A local experience of a multicenter retrospective study. Asia-Pac. J. Clin. Oncol. 2021, 17, 238–244. [Google Scholar] [CrossRef]
- Miyake, H.; Sato, R.; Watanabe, K.; Matsushita, Y.; Watanabe, H.; Motoyama, D.; Ito, T.; Sugiyama, T.; Otsuka, A. Prognostic significance of third-line treatment for patients with metastatic castration-resistant prostate cancer: Comparative assessments between cabazitaxel and other agents. Int. J. Clin. Oncol. 2021, 26, 1745–1751. [Google Scholar] [CrossRef]
- Van Soest, R.J.; De Wit, R. Irrefutable evidence for the use of docetaxel in newly diagnosed metastatic prostate cancer: Results from the STAMPEDE and CHAARTED trials. BMC Med. 2015, 13, 304. [Google Scholar] [CrossRef]
- Damodaran, S.; Lang, J.M.; Jarrard, D.F. Targeting Metastatic Hormone Sensitive Prostate Cancer: Chemohormonal Therapy and New Combinatorial Approaches. J. Urol. 2019, 201, 876–885. [Google Scholar] [CrossRef]
- Huebner, N.A.; Shariat, S.F.; Resch, I.; Gust, K.; Kramer, G. The role of taxane-based chemotherapy in the treatment of prostate cancer. Curr. Opin. Urol. 2020, 30, 527–533. [Google Scholar] [CrossRef] [PubMed]
- James, N.D.; Sydes, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Spears, M.R.; Ritchie, A.W.S.; Parker, C.C.; Russell, J.M.; Attard, G.; et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): Survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet 2016, 387, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Clarke, N.W.; Ali, A.; Ingleby, F.C.; Hoyle, A.; Amos, C.L.; Attard, G.; Brawley, C.D.; Calvert, J.; Chowdhury, S.; Cook, A.; et al. Addition of docetaxel to hormonal therapy in low- and high-burden metastatic hormone sensitive prostate cancer: Long-term survival results from the STAMPEDE trial. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1992–2003. [Google Scholar] [CrossRef]
- Oudard, S.; Fizazi, K.; Sengeløv, L.; Daugaard, G.; Saad, F.; Hansen, S.; Hjälm-Eriksson, M.; Jassem, J.; Thiery-Vuillemin, A.; Caffo, O.; et al. Cabazitaxel Versus Docetaxel As First-Line Therapy for Patients With Metastatic Castration-Resistant Prostate Cancer: A Randomized Phase III Trial—FIRSTANA. J. Clin. Oncol. 2017, 35, 3189–3197. [Google Scholar] [CrossRef]
- Eisenberger, M.; Hardy-Bessard, A.-C.; Kim, C.S.; Géczi, L.; Ford, D.; Mourey, L.; Carles, J.; Parente, P.; Font, A.; Kacso, G.; et al. Phase III Study Comparing a Reduced Dose of Cabazitaxel (20 mg/m2) and the Currently Approved Dose (25 mg/m2) in Postdocetaxel Patients With Metastatic Castration-Resistant Prostate Cancer—PROSELICA. J. Clin. Oncol. 2017, 35, 3198–3206. [Google Scholar] [CrossRef] [PubMed]
- de Wit, R.; de Bono, J.; Sternberg, C.N.; Fizazi, K.; Tombal, B.; Wülfing, C.; Kramer, G.; Eymard, J.C.; Bamias, A.; Carles, J.; et al. Faculty Opinions recommendation of Cabazitaxel versus Abiraterone or Enzalutamide in Metastatic Prostate Cancer. N. Engl. J. Med. 2019, 381, 2506–2518. [Google Scholar] [CrossRef]
- Menges, D.; Piatti, M.C.; Cerny, T.; A Puhan, M. Patient Preference Studies for Advanced Prostate Cancer Treatment Along the Medical Product Life Cycle: Systematic Literature Review. Patient Prefer. Adherence 2022, 16, 1539–1557. [Google Scholar] [CrossRef]
- Baciarello, G.; Delva, R.; Gravis, G.; Tazi, Y.; Beuzeboc, P.; Gross-Goupil, M.; Bompas, E.; Joly, F.; Greilsamer, C.; Hon, T.N.T.; et al. Patient Preference Between Cabazitaxel and Docetaxel for First-line Chemotherapy in Metastatic Castration-resistant Prostate Cancer: The CABADOC Trial. Eur. Urol. 2022, 81, 234–240. [Google Scholar] [CrossRef]
- Schmit, K.; Michiels, C. TMEM Proteins in Cancer: A Review. Front. Pharmacol. 2018, 9, 1345. [Google Scholar] [CrossRef]
- Ryu, H.; Fuwad, A.; Yoon, S.; Jang, H.; Lee, J.C.; Kim, S.M.; Jeon, T.-J. Biomimetic Membranes with Transmembrane Proteins: State-of-the-Art in Transmembrane Protein Applications. Int. J. Mol. Sci. 2019, 20, 1437. [Google Scholar] [CrossRef]
- Marx, S.; Dal Maso, T.; Chen, J.W.; Bury, M.; Wouters, J.; Michiels, C.; Le Calvé, B. Transmembrane (TMEM) protein family members: Poorly charac-terized even if essential for the metastatic process. Semin. Cancer Biol. 2020, 60, 96–106. [Google Scholar] [PubMed]
- Bossennec, M.; Di Roio, A.; Caux, C.; Ménétrier-Caux, C. MDR1 in immunity: Friend or foe? Oncoimmunology 2018, 7, e1499388. [Google Scholar] [PubMed]
- Ganju, A.; Yallapu, M.M.; Khan, S.; Behrman, S.W.; Chauhan, S.C.; Jaggi, M. Nanoways to overcome docetaxel resistance in prostate cancer. Drug Resist. Updat. 2014, 17, 13–23. [Google Scholar] [CrossRef]
- Kawai, K.; Sakurai, M.; Sakai, T.; Misaki, M.; Kusano, I.; Shiraishi, T.; Yatani, R. Demonstration of MDR1 P-glycoprotein isoform expression in benign and malignant human prostate cells by isoform-specific monoclonal antibodies. Cancer Lett. 2000, 150, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Mizutani, K.; Kameyama, K.; Kawakami, K.; Fujita, Y.; Nakane, K.; Kanimoto, Y.; Ehara, H.; Ito, H.; Seishima, M.; et al. Serum exosomal P-glycoprotein is a potential marker to di-agnose docetaxel resistance and select a taxoid for patients with prostate cancer. Urol. Oncol. 2015, 33, e15–e385. [Google Scholar]
- Kato, T.; Fujita, Y.; Nakane, K.; Kojima, T.; Nozawa, Y.; Deguchi, T.; Ito, M. ETS1 promotes chemoresistance and invasion of paclitaxel-resistant, hormone-refractory PC3 prostate cancer cells by up-regulating MDR1 and MMP9 expression. Biochem. Biophys. Res. Commun. 2012, 417, 966–971. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, Z.; Chen, C.Z.; Liu, C.; Evans, C.P.; Gao, A.C.; Zhou, F.; Chen, H.-W. Therapeutic Targeting of MDR1 Expression by RORγ Antagonists Resensitizes Cross-Resistant CRPC to Taxane via Coordinated Induction of Cell Death Programs. Mol. Cancer Ther. 2020, 19, 364–374. [Google Scholar] [CrossRef]
- Hardy, D.; Bill, R.M.; Jawhari, A.; Rothnie, A.J. Functional Expression of Multidrug Resistance Protein 4 MRP4/ABCC4. SLAS Discov. 2019, 24, 1000–1008. [Google Scholar]
- Ravna, A.W.; Sager, G. Molecular model of the outward facing state of the human multidrug resistance protein 4 (MRP4/ABCC4). Bioorg. Med. Chem. Lett. 2008, 18, 3481–3483. [Google Scholar]
- Sauna, Z.E.; Nandigama, K.; Ambudkar, S.V. Multidrug resistance protein 4 (ABCC4)-mediated ATP hydrolysis: Effect of transport substrates and characterization of the post-hydrolysis transition state. J. Biol. Chem. 2004, 279, 48855–48864. [Google Scholar]
- Li, Y.-F.; Ji, H.-H.; Zhang, Z.-L.; Zhang, T.-T.; Gan, W.; Zhang, S.-F. Targeting MRP4 expression by anti-androgen treatment reverses MRP4-mediated docetaxel resistance in castration-resistant prostate cancer. Oncol. Lett. 2017, 14, 1748–1756. [Google Scholar] [CrossRef] [PubMed]
- Becerra, E.; Berumen, L.; Soto-Ontiveros, V.; García-Alcocer, G. Specific MRP4 Inhibitor Ceefourin-1 Enhances Apoptosis Induced by 6-Mercaptopurine in Jurkat Leukemic Cells, but Not in Normal Lymphoblast Cell Line CRL-1991. Medicina 2022, 58, 695. [Google Scholar] [CrossRef] [PubMed]
- Mesrati, M.H.; Syafruddin, S.E.; Mohtar, M.A.; Syahir, A. CD44: A Multifunctional Mediator of Cancer Progression. Biomolecules 2021, 11, 1850. [Google Scholar] [CrossRef]
- Li, W.; Qian, L.; Lin, J.; Huang, G.; Hao, N.; Wei, X.; Wang, W.; Liang, J. CD44 regulates prostate cancer proliferation, invasion and migration via PDK1 and PFKFB4. Oncotarget 2017, 8, 65143–65151. [Google Scholar] [CrossRef]
- Lai, C.-J.; Lin, C.-Y.; Liao, W.-Y.; Hour, T.-C.; Wang, H.-D.; Chuu, C.-P. CD44 Promotes Migration and Invasion of Docetaxel-Resistant Prostate Cancer Cells Likely via Induction of Hippo-Yap Signaling. Cells 2019, 8, 295. [Google Scholar] [CrossRef] [PubMed]
- Behrooz, A.B.; Syahir, A.; Ahmad, S. CD133: Beyond a cancer stem cell biomarker. J. Drug Target. 2019, 27, 257–269. [Google Scholar] [CrossRef]
- Glumac, P.M.; LeBeau, A.M. The role of CD133 in cancer: A concise review. Clin. Transl. Med. 2018, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Huang, X.; Zheng, X.; Wang, X.; Li, S.; Zhang, L.; Yang, Z.; Xia, Z. Enrichment of Prostate Cancer Stem-Like Cells from Human Prostate Cancer Cell Lines by Culture in Serum-Free Medium and Chemoradiotherapy. Int. J. Biol. Sci. 2013, 9, 472–479. [Google Scholar] [CrossRef]
- Aghajani, M.; Mokhtarzadeh, A.; Aghebati-Maleki, L.; Mansoori, B.; Mohammadi, A.; Safaei, S.; Asadzadeh, Z.; Hajiasgharzadeh, K.; Khaze Shahgoli, V.; Baradaran, B. CD133 suppression increases the sen-sitivity of prostate cancer cells to paclitaxel. Mol. Biol. Rep. 2020, 47, 3691–3703. [Google Scholar]
- Sun, R.; Ying, Y.; Tang, Z.; Liu, T.; Shi, F.; Li, H.; Guo, T.; Huang, S.; Lai, R. The Emerging Role of the SLCO1B3 Protein in Cancer Resistance. Protein Pept. Lett. 2020, 27, 17–29. [Google Scholar] [CrossRef]
- Liu, S.; Peng, T.; Wang, Z.; Li, Y.; Zhang, H.; Gui, C. Effect of rare coding variants of charged amino acid residues on the function of human organic anion transporting polypeptide 1B3 (SLCO1B3). Biochem. Biophys. Res. Commun. 2021, 557, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Pressler, H.; Sissung, T.M.; Venzon, D.; Price, D.K.; Figg, W.D. Expression of OATP Family Members in Hormone-Related Cancers: Potential Markers of Progression. PLoS ONE 2011, 6, e20372. [Google Scholar] [CrossRef] [PubMed]
- König, J.; Cui, Y.; Nies, A.T.; Keppler, D. Localization and Genomic Organization of a New Hepatocellular Organic Anion Transporting Polypeptide. J. Biol. Chem. 2000, 275, 23161–23168. [Google Scholar] [CrossRef]
- Thakkar, N.; Kim, K.; Jang, E.R.; Han, S.; Kim, K.; Kim, D.; Merchant, N.; Lockhart, A.C.; Lee, W. A Cancer-Specific Variant of the SLCO1B3 Gene Encodes a Novel Human Organic Anion Transporting Polypeptide 1B3 (OATP1B3) Localized Mainly in the Cytoplasm of Colon and Pancreatic Cancer Cells. Mol. Pharm. 2013, 10, 406–416. [Google Scholar] [CrossRef]
- Wright, J.L.; Kwon, E.M.; Ostrander, E.A.; Montgomery, R.B.; Lin, D.W.; Vessella, R.; Stanford, J.L.; Mostaghel, E.A. Expression of SLCO transport genes in castration re-sistant prostate cancer and impact of genetic variation in SCLO1B3 and SLCO2B1 on prostate cancer outcomes. Cancer Epi-Demiol Biomark. Prev. 2011, 20, 619–627. [Google Scholar] [CrossRef]
- de Morrée, E.S.; Böttcher, R.; van Soest, R.J.; Aghai, A.; de Ridder, C.M.; A Gibson, A.; Mathijssen, R.H.; Burger, H.; Wiemer, E.A.; Sparreboom, A.; et al. Loss of SLCO1B3 drives taxane resistance in prostate cancer. Br. J. Cancer 2016, 115, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Malagnino, V.; Hussner, J.; Issa, A.; Midzic, A.; zu Schwabedissen, H.E.M. OATP1B3-1B7, a novel organic anion transporting polypeptide, is modulated by FXR ligands and transports bile acids. Am. J. Physiol. Liver Physiol. 2019, 317, G751–G762. [Google Scholar] [CrossRef]
- Fiorucci, S.; Distrutti, E. Chenodeoxycholic Acid: Update Its Ther. Applications. Handb. Exp. Pharmacol. 2019, 256, 265–282. [Google Scholar] [CrossRef]
- Bellezza, I.; Bracarda, S.; Caserta, C.; Minelli, A. Targeting of EGFR tyrosine kinase by ZD1839 (“Iressa”) in androgen-responsive prostate cancer in vitro. Mol. Genet. Metab. 2006, 88, 114–122. [Google Scholar] [CrossRef]
- Rude Voldborg, B.; Damstrup, L.; Spang-Thomsen, M.; Skovgaard Poulsen, H. Epidermal growth factor receptor (EGFR) and EGFR mutations, function and possible role in clinical trials. Ann. Oncol. 1997, 8, 1197–1206. [Google Scholar] [CrossRef]
- Jathal, M.K.; Steele, T.M.; Siddiqui, S.; Mooso, B.A.; D’abronzo, L.S.; Drake, C.M.; Whang, Y.E.; Ghosh, P.M. Dacomitinib, but not lapatinib, suppressed progression in castration-resistant prostate cancer models by preventing HER2 increase. Br. J. Cancer 2019, 121, 237–248. [Google Scholar] [CrossRef]
- Rossini, A.; Giussani, M.; Ripamonti, F.; Aiello, P.; Regondi, V.; Balsari, A.; Triulzi, T.; Tagliabue, E. Combined targeting of EGFR and HER2 against prostate cancer stem cells. Cancer Biol. Ther. 2020, 21, 463–475. [Google Scholar] [CrossRef]
- Day, K.C.; Hiles, G.L.; Kozminsky, M.; Dawsey, S.J.; Paul, A.; Broses, L.J.; Shah, R.; Kunja, L.P.; Hall, C.; Palanisamy, N.; et al. HER2 and EGFR Overexpression Support Metastatic Progression of Prostate Cancer to Bone. Cancer Res. 2017, 77, 74–85. [Google Scholar] [CrossRef]
- Vicentini, C.; Festuccia, C.; Gravina, G.L.; Angelucci, A.; Marronaro, A.; Bologna, M. Prostate cancer cell proliferation is strongly reduced by the epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 in vitro on human cell lines and primary cultures. J. Cancer Res. Clin. Oncol. 2003, 129, 165–174. [Google Scholar] [CrossRef]
- Monteverde, M.; Tonissi, F.; Fischel, J.-L.; Etienne-Grimaldi, M.-C.; Milano, G.; Merlano, M.; Nigro, C.L. Combination of docetaxel and vandetanib in docetaxel-sensitive or resistant PC3 cell line. Urol. Oncol. Semin. Orig. Investig. 2013, 31, 776–786. [Google Scholar] [CrossRef]
- Jiao, Q.; Bi, L.; Ren, Y.; Song, S.; Wang, Q.; Wang, Y.-S. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol. Cancer 2018, 17, 36. [Google Scholar] [CrossRef]
- Xu, M.; Evans, L.; Bizzaro, C.L.; Quaglia, F.; Verrillo, C.E.; Li, L.; Stieglmaier, J.; Schiewer, M.J.; Languino, L.R.; Kelly, W.K. STEAP1–4 (Six-Transmembrane Epithelial Antigen of the Prostate 1–4) and Their Clinical Implications for Prostate Cancer. Cancers 2022, 14, 4034. [Google Scholar] [CrossRef]
- Gomes, I.M.; Maia, C.J.; Santos, C.R. STEAP proteins: From structure to applications in cancer therapy. Mol. Cancer Res. 2012, 10, 573–587. [Google Scholar] [CrossRef]
- Zhao, C.; Xiong, K.; Ji, Z.; Liu, F.; Li, X. The Prognostic Value and Immunological Role of STEAP1 in Pan-Cancer: A Result of Da-ta-Based Analysis. Oxid. Med. Cell Longev. 2022, 2022, 8297011. [Google Scholar] [CrossRef]
- Rocha, S.M.; Nascimento, D.; Coelho, R.S.; Cardoso, A.M.; Passarinha, L.A.; Socorro, S.; Maia, C.J. STEAP1 Knockdown Decreases the Sensitivity of Prostate Cancer Cells to Paclitaxel, Docetaxel and Cabazitaxel. Int. J. Mol. Sci. 2023, 24, 6643. [Google Scholar] [CrossRef]
- Gomes, I.M.; Rocha, S.M.; Gaspar, C.; Alvelos, M.I.; Santos, C.R.; Socorro, S.; Maia, C.J. Knockdown of STEAP1 inhibits cell growth and induces apoptosis in LNCaP prostate cancer cells counteracting the effect of androgens. Med. Oncol. 2018, 35, 40. [Google Scholar] [CrossRef] [PubMed]
- Loberg, R.D.; Ying, C.; Craig, M.; Day, L.L.; Sargent, E.; Neeley, C.; Wojno, K.; Snyder, L.A.; Yan, L.; Pienta, K.J. Targeting CCL2 with systemic delivery of neutralizing an-tibodies induces prostate cancer tumor regression in vivo. Cancer Res. 2007, 67, 9417–9424. [Google Scholar] [CrossRef] [PubMed]
- Kadomoto, S.; Izumi, K.; Mizokami, A. Roles of CCL2-CCR2 Axis in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 8530. [Google Scholar] [CrossRef]
- Loberg, R.D.; Day, L.L.; Harwood, J.; Ying, C.; John, L.N.S.; Giles, R.; Neeley, C.K.; Pienta, K.J. CCL2 is a Potent Regulator of Prostate Cancer Cell Migration and Proliferation. Neoplasia 2006, 8, 578–586. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, Q.; Corey, E.; Xie, W.; Fan, J.; Mizokami, A.; Zhang, J. Activation of the MCP-1/CCR2 axis promotes prostate cancer growth in bone. Clin. Exp. Metastasis 2009, 26, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Mary, L.G.-S.; Oh, W.K.; Freedman, M.L.; Pomerantz, M.; Pienta, K.J.; Kantoff, P.W. Inherited Variants in the Chemokine CCL2 Gene and Prostate Cancer Aggressiveness in a Caucasian Cohort. Clin. Cancer Res. 2015, 17, 1546–1552. [Google Scholar] [CrossRef]
- Natsagdorj, A.; Izumi, K.; Hiratsuka, K.; Machioka, K.; Iwamoto, H.; Naito, R.; Makino, T.; Kadomoto, S.; Shigehara, K.; Kadono, Y.; et al. CCL2 induces resistance to the antiproliferative effect of cabazitaxel in prostate cancer cells. Cancer Sci. 2019, 110, 279–288. [Google Scholar] [CrossRef]
- Qian, D.Z.; Rademacher, B.L.; Pittsenbarger, J.; Huang, C.-Y.; Myrthue, A.; Higano, C.S.; Garzotto, M.; Nelson, P.S.; Beer, T.M. CCL2 is induced by chemotherapy and protects prostate cancer cells from docetaxel-induced cytotoxicity. Prostate 2010, 70, 433–442. [Google Scholar] [CrossRef]
- Kirk, P.S.; Koreckij, T.; Nguyen, H.M.; Brown, L.G.; Snyder, L.A.; Vessella, R.L.; Corey, E. Inhibition of CCL2 Signaling in Combination with Docetaxel Treatment Has Profound Inhibitory Effects on Prostate Cancer Growth in Bone. Int. J. Mol. Sci. 2013, 14, 10483–10496. [Google Scholar] [CrossRef]
- Rozel, S.; Galbán, C.J.; Nicolay, K.; Lee, K.C.; Sud, S.; Neeley, C.; Snyder, L.A.; Chenevert, T.L.; Rehemtulla, A.; Ross, B.D.; et al. Synergy between anti-CCL2 and docetaxel as determined by DW-MRI in a metastatic bone cancer model. J. Cell. Biochem. 2009, 107, 58–64. [Google Scholar] [CrossRef]
- Pienta, K.J.; Machiels, J.-P.; Schrijvers, D.; Alekseev, B.; Shkolnik, M.; Crabb, S.J.; Li, S.; Seetharam, S.; Puchalski, T.A.; Takimoto, C.; et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Investig. New Drugs 2013, 31, 760–768. [Google Scholar] [CrossRef]
- Secker, G.A.; Harvey, N.L. Regulation of VEGFR Signalling in Lymphatic Vascular Development and Disease: An Update. Int. J. Mol. Sci. 2021, 22, 7760. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Hong, S.K.; Oh, M.M.; Yoon, C.Y.; Jeong, S.J.; Byun, S.S.; Cheon, J.; Lee, S.E.; Moon, D.G. Synergistic antitumor effect of NVP-BEZ235 and sunitinib on docetaxel-resistant human castration-resistant prostate cancer cells. Anticancer. Res. 2014, 34, 3457–3468. [Google Scholar] [PubMed]
- Ortholan, C.; Durivault, J.; Hannoun-Levi, J.-M.; Guyot, M.; Bourcier, C.; Ambrosetti, D.; Safe, S.; Pagès, G. Bevacizumab/docetaxel association is more efficient than docetaxel alone in reducing breast and prostate cancer cell growth: A new paradigm for understanding the therapeutic effect of combined treatment. Eur. J. Cancer 2010, 46, 3022–3036. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Chiu, C.; Liu, I.; Chang, Y.; Liu, Y.; Wu, H. Novel human Ab against vascular endothelial growth factor receptor 2 shows therapeutic potential for leukemia and prostate cancer. Cancer Sci. 2019, 110, 3773–3787. [Google Scholar] [CrossRef]
- Adesunloye, B.A.; Karzai, F.H.; Dahut, W.L. Angiogenesis Inhibitors in the Treatment of Prostate Cancer. Chem. Immunol. Allergy 2014, 99, 197–215. [Google Scholar] [CrossRef]
- Michaelson, M.D.; Regan, M.M.; Oh, W.K.; Kaufman, D.S.; Olivier, K.; Michaelson, S.Z.; Spicer, B.; Gurski, C.; Kantoff, P.W.; Smith, M.R. Phase II study of sunitinib in men with advanced prostate cancer. Ann. Oncol. 2009, 20, 913–920. [Google Scholar] [CrossRef]
- Momeny, M.; Sankanian, G.; Hamzehlou, S.; Yousefi, H.; Esmaeili, F.; Alishahi, Z.; Karimi, B.; Zandi, Z.; Shamsaiegahkani, S.; Sabourinejad, Z.; et al. Cediranib, an inhibitor of vascular endothelial growth factor receptor kinases, inhibits proliferation and invasion of prostate adenocarcinoma cells. Eur. J. Pharmacol. 2020, 882, 173298. [Google Scholar] [CrossRef]
- Najy, A.J.; Jung, Y.S.; Won, J.J.; Conley-LaComb, M.K.; Saliganan, A.; Kim, C.J.; Heath, E.; Cher, M.L.; Bonfil, R.D.; Kim, H.-R.C. Cediranib inhibits both the intraosseous growth of PDGF D-Positive prostate cancer cells and the associated bone reaction. Prostate 2012, 72, 1328–1338. [Google Scholar] [CrossRef]
- Yin, J.J.; Zhang, L.; Munasinghe, J.; Linnoila, R.I.; Kelly, K. Cediranib/AZD2171 inhibits bone and brain metastasis in a preclinical model of advanced prostate cancer. Cancer Res. 2010, 70, 8662–8673. [Google Scholar] [CrossRef]
- Ryan, C.J.; Stadler, W.M.; Roth, B.; Hutcheon, D.; Conry, S.; Puchalski, T.; Morris, C.; Small, E.J. Phase I dose escalation and pharmacokinetic study of AZD2171, an inhibitor of the vascular endothelial growth factor receptor tyrosine kinase, in patients with hormone refractory prostate cancer (HRPC). Investig. New Drugs 2007, 25, 445–451. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Function | Effect of Knockdown Alone | Effect of Knockdown + Taxane Treatment |
|---|---|---|---|
| MDR1 | Efflux pump | - | Improvement in docetaxel sensitivity |
| MRP4 | Efflux pump | - | Resensitization to docetaxel treatment |
| CD44 | Hyaluronate receptor | Reduced cell migration | Decrease viability of PC3 cells |
| CD133 | Membrane organization | No alteration in cell proliferation and viability | Decrease in survival rate of cell, reduced metastatic potential, sensibilization to paclitaxel |
| SLCO1B3 | Sodium-independent transporter | Reduction in cellular uptake of docetaxel | - |
| EGFR | Membrane receptor | Reduce cell proliferation | Tumor regression |
| STEAP1 | Metalloreductase | Reduce cell viability and proliferation | Increase cell viability |
| CCL2/CCR2 | Immune cell recruitment | Decreased cell viability in culture and decreased tumor burden in animals | Increased survival period of animals and tumor growth inhibition |
| VEGFR | Development of blood and lymphatic vascular networks | Decreased tumor growth | Enhanced sensitivity to docetaxel |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coelho, R.S.; Rocha, S.M.; Maia, C.J. Combined Therapies with Taxane-Based Chemotherapeutic Drugs in Prostate Cancer: Novel Insights and Future Directions. BioChem 2023, 3, 118-141. https://doi.org/10.3390/biochem3030009
Coelho RS, Rocha SM, Maia CJ. Combined Therapies with Taxane-Based Chemotherapeutic Drugs in Prostate Cancer: Novel Insights and Future Directions. BioChem. 2023; 3(3):118-141. https://doi.org/10.3390/biochem3030009
Chicago/Turabian StyleCoelho, Rafaella S., Sandra M. Rocha, and Cláudio J. Maia. 2023. "Combined Therapies with Taxane-Based Chemotherapeutic Drugs in Prostate Cancer: Novel Insights and Future Directions" BioChem 3, no. 3: 118-141. https://doi.org/10.3390/biochem3030009
APA StyleCoelho, R. S., Rocha, S. M., & Maia, C. J. (2023). Combined Therapies with Taxane-Based Chemotherapeutic Drugs in Prostate Cancer: Novel Insights and Future Directions. BioChem, 3(3), 118-141. https://doi.org/10.3390/biochem3030009