
The Treasury Chest of Text Mining: Piling Available Resources for Powerful Biomedical Text Mining

 ,
,  , , , , , and
, , , , , and 
Abstract
1. Introduction
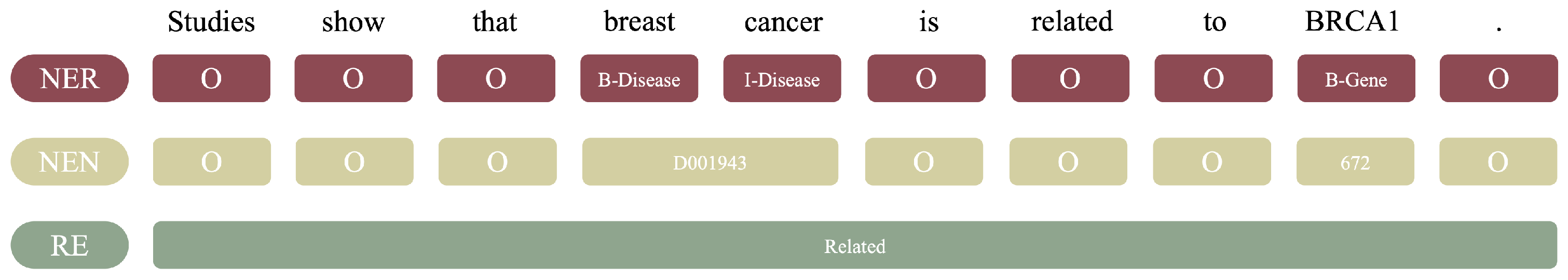
1.1. What Is Biomedical Text Mining
1.2. Text Mining Challenges—What Makes Text Mining Complex?
1.3. Traditional Versus Machine Learning Driven Text Mining
2. Resources for Text Mining
2.1. Biomedical Corpora
2.2. Text Mining Toolkits
2.3. Text Mining Tools for NER, NEN, and RE
2.4. Web-Based Applications
2.5. Public Databases That Incorporate Text Mining Models
3. Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Guo, J.W.; Radloff, C.L.; Wawrzynski, S.E.; Cloyes, K.G. Mining twitter to explore the emergence of COVID-19 symptoms. Public Health Nurs. 2020, 37, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Lazard, A.J.; Wilcox, G.B.; Tuttle, H.M.; Glowacki, E.M.; Pikowski, J. Public reactions to e-cigarette regulations on Twitter: A text mining analysis. Tobacco Control 2017, 26, e112–e116. [Google Scholar] [CrossRef] [PubMed]
- Nasralah, T.; El-Gayar, O.; Wang, Y. Social Media Text Mining Framework for Drug Abuse: Development and Validation Study With an Opioid Crisis Case Analysis. J. Med. Internet Res. 2020, 22, e18350. [Google Scholar] [CrossRef] [PubMed]
- Bach, M.P.; Krstić, Ž.; Seljan, S.; Turulja, L. Text Mining for Big Data Analysis in Financial Sector: A Literature Review. Sustainability 2019, 11, 1277. [Google Scholar] [CrossRef]
- Seljan, S.; Baretić, M.; Kučiš, V. Information retrieval and terminology extraction in online resources for patients with diabetes. Coll. Antropol. 2014, 38, 705–710. [Google Scholar]
- Seljan, S.; Dunđer, I.; Stančić, H. Extracting Terminology by Language Independent Methods. In Forum Translationswissenschaft: Translation Studies and Translation Practice 19; Peter Lang D: Bern, Switzerland, 2017; pp. 141–147. [Google Scholar]
- Fleuren, W.W.; Alkema, W. Application of text mining in the biomedical domain. Methods 2015, 74, 97–106. [Google Scholar] [CrossRef]
- Shorten, C.; Khoshgoftaar, T.M.; Furht, B. Deep Learning applications for COVID-19. J. Big Data 2021, 8. [Google Scholar] [CrossRef]
- Gachloo, M.; Wang, Y.; Xia, J. A review of drug knowledge discovery using BioNLP and tensor or matrix decomposition. Genom. Inform. 2019, 17, e18. [Google Scholar] [CrossRef]
- Zheng, S.; Dharssi, S.; Wu, M.; Li, J.; Lu, Z. Text Mining for Drug Discovery. In Methods in Molecular Biology; Springer: New York, NY, USA, 2019; pp. 231–252. [Google Scholar] [CrossRef]
- Gonzalez, G.H.; Tahsin, T.; Goodale, B.C.; Greene, A.C.; Greene, C.S. Recent Advances and Emerging Applications in Text and Data Mining for Biomedical Discovery. Briefings Bioinform. 2015, 17, 33–42. [Google Scholar] [CrossRef]
- Zhu, F.; Patumcharoenpol, P.; Zhang, C.; Yang, Y.; Chan, J.; Meechai, A.; Vongsangnak, W.; Shen, B. Biomedical text mining and its applications in cancer research. J. Biomed. Inform. 2013, 46, 200–211. [Google Scholar] [CrossRef]
- Perera, N.; Dehmer, M.; Emmert-Streib, F. Named Entity Recognition and Relation Detection for Biomedical Information Extraction. Front. Cell Dev. Biol. 2020, 8, 673. [Google Scholar] [CrossRef] [PubMed]
- Beheshti, S.M.R.; Venugopal, S.; Ryu, S.H.; Benatallah, B.; Wang, W. Big Data and Cross-Document Coreference Resolution: Current State and Future Opportunities. arXiv 2013, arXiv:1311.3987. [Google Scholar]
- Li, H.; Chen, Q.; Tang, B.; Wang, X.; Xu, H.; Wang, B.; Huang, D. CNN-based ranking for biomedical entity normalization. BMC Bioinform. 2017, 18. [Google Scholar] [CrossRef]
- Cho, H.; Choi, W.; Lee, H. A method for named entity normalization in biomedical articles: Application to diseases and plants. BMC Bioinform. 2017, 18. [Google Scholar] [CrossRef]
- Shirakawa, M.; Wang, H.; Song, Y.; Wang, Z.; Nakayama, K.; Hara, T. Entity Disambiguation based on a Probabilistic Taxonomy. Technical Report MSR-TR-2011-25. 2011. Available online: https://www.microsoft.com/en-us/research/publication/entity-disambiguation-based-on-a-probabilistic-taxonomy/ (accessed on 12 June 2021).
- Gentile, A.L.; Zhang, Z.; Xia, L.; Iria, J. Semantic Relatedness Approach for Named Entity Disambiguation. In Communications in Computer and Information Science; Springer: Berlin/Heidelberg, Germany, 2010; pp. 137–148. [Google Scholar] [CrossRef]
- Zhu, G.; Iglesias, C.A. Exploiting semantic similarity for named entity disambiguation in knowledge graphs. Expert Syst. Appl. 2018, 101, 8–24. [Google Scholar] [CrossRef]
- Yadav, S.; Ramesh, S.; Saha, S.; Ekbal, A. Relation Extraction from Biomedical and Clinical Text: Unified Multitask Learning Framework. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, Z. Exploring semi-supervised variational autoencoders for biomedical relation extraction. Methods 2019, 166, 112–119. [Google Scholar] [CrossRef]
- Muzaffar, A.W.; Azam, F.; Qamar, U. A Relation Extraction Framework for Biomedical Text Using Hybrid Feature Set. Comput. Math. Methods Med. 2015, 2015, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Xing, R.; Luo, J.; Song, T. BioRel: Towards large-scale biomedical relation extraction. BMC Bioinform. 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Perez-Iratxeta, C.; Bork, P.; Andrade, M. Information extraction from full text scientific articles: Where are the keywords? BMC Bioinform. 2003, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Wu, C.Y.; Tzong, R.; Tsai, R.T.H.; Hsu, W.L. From Entity Recognition to Entity Linking: A Survey of Advanced Entity Linking Techniques. In Proceedings of the 26th Annual Conference of the Japanese Society for Artificial Intelligence, Tokyo, Japan, 12–15 June 2012; pp. 1–10. [Google Scholar]
- Collovini, S.; Bonamigo, T.; Vieira, R. A review on Relation Extraction with an eye on Portuguese. J. Braz. Comput. Soc. 2013, 19. [Google Scholar] [CrossRef]
- Sun, W.; Cai, Z.; Li, Y.; Liu, F.; Fang, S.; Wang, G. Data Processing and Text Mining Technologies on Electronic Medical Records: A Review. J. Healthc. Eng. 2018, 2018, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ghamami, F.; Keyvanpour, M. Why biomedical relation extraction is an open issue? ICIC Express Lett. Part B Appl. 2018. [Google Scholar] [CrossRef]
- Saffer, J.D.; Burnett, V.L. Introduction to Biomedical Literature Text Mining: Context and Objectives. In Methods in Molecular Biology; Springer: New York, NY, USA, 2014; pp. 1–7. [Google Scholar] [CrossRef]
- Nicholson, D.N.; Greene, C.S. Constructing knowledge graphs and their biomedical applications. Comput. Struct. Biotechnol. J. 2020, 18, 1414–1428. [Google Scholar] [CrossRef]
- Mikolov, T.; Chen, K.; Corrado, G.; Dean, J. Efficient estimation of word representations in vector space. arXiv 2013, arXiv:1301.3781. [Google Scholar]
- Sachan, D.S.; Xie, P.; Xing, E.P. Effective Use of Bidirectional Language Modeling for Medical Named Entity Recognition. arXiv 2017, arXiv:1711.07908. [Google Scholar]
- Devlin, J.; Chang, M.; Lee, K.; Toutanova, K. BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding. arXiv 2018, arXiv:1810.04805. [Google Scholar]
- Peters, M.E.; Neumann, M.; Iyyer, M.; Gardner, M.; Clark, C.; Lee, K.; Zettlemoyer, L. Deep contextualized word representations. arXiv 2018, arXiv:1802.05365. [Google Scholar]
- Lee, J.; Yoon, W.; Kim, S.; Kim, D.; Kim, S.; So, C.H.; Kang, J. BioBERT: A pre-trained biomedical language representation model for biomedical text mining. Bioinformatics 2019. [Google Scholar] [CrossRef]
- Dustin Wright, Y.K. NormCo: Deep Disease Normalization for Biomedical Knowledge Base Construction. 2019. Available online: https://openreview.net/forum?id=BJerQWcp6Q (accessed on 12 June 2021). [CrossRef]
- Ison, J.; Ménager, H.; Brancotte, B.; Jaaniso, E.; Salumets, A.; Raček, T.; Lamprecht, A.L.; Palmblad, M.; Kalaš, M.; Chmura, P.; et al. Community curation of bioinformatics software and data resources. Briefings Bioinform. 2020, 21, 1697–1705. [Google Scholar] [CrossRef]
- Sammartino, J.C.; Krallinger, M.; Valencia, A. Annotation Process, Guidelines and Text Corpus of Small Non-Coding RNA Molecules: The MiNCor for MicroRNA Annotations. In Proceedings of the Semantic Mining in Biomedicine (SMBM) 2016 CEUR Workshop Proceedings, Potsdam, Germany, 4–5 August 2016; pp. 56–63. [Google Scholar]
- Lamurias, A.; Couto, F.M. Text mining for bioinformatics using biomedical literature. Encycl. Bioinform. Comput. Biol. 2019, 1, 602–611. [Google Scholar]
- Campos, D.; Matos, S.; Oliveira, J.L. Biomedical named entity recognition: A survey of machine-learning tools. Theory Appl. Adv. Text Min. 2012, 11, 175–195. [Google Scholar]
- Li, F.; Zhang, M.; Fu, G.; Ji, D. A neural joint model for entity and relation extraction from biomedical text. BMC Bioinform. 2017, 18, 1–11. [Google Scholar] [CrossRef]
- Ananiadou, S.; Pyysalo, S.; Tsujii, J.; Kell, D.B. Event extraction for systems biology by text mining the literature. Trends Biotechnol. 2010, 28, 381–390. [Google Scholar] [CrossRef]
- Thompson, P.; Iqbal, S.A.; McNaught, J.; Ananiadou, S. Construction of an annotated corpus to support biomedical information extraction. BMC Bioinform. 2009, 10, 1–19. [Google Scholar] [CrossRef]
- Kim, J.D.; Ohta, T.; Tateisi, Y.; Tsujii, J. GENIA corpus—A semantically annotated corpus for bio-textmining. Bioinformatics 2003, 19, i180–i182. [Google Scholar] [CrossRef] [PubMed]
- Bada, M.; Eckert, M.; Evans, D.; Garcia, K.; Shipley, K.; Sitnikov, D.; Baumgartner, W.A.; Cohen, K.B.; Verspoor, K.; Blake, J.A.; et al. Concept annotation in the CRAFT corpus. BMC Bioinform. 2012, 13, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.; Tanabe, L.K.; nee Ando, R.J.; Kuo, C.J.; Chung, I.F.; Hsu, C.N.; Lin, Y.S.; Klinger, R.; Friedrich, C.M.; Ganchev, K.; et al. Overview of BioCreative II gene mention recognition. Genome Biol. 2008, 9, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Doğan, R.I.; Leaman, R.; Lu, Z. NCBI disease corpus: A resource for disease name recognition and concept normalization. J. Biomed. Inform. 2014, 47, 1–10. [Google Scholar] [CrossRef]
- Krallinger, M.; Rabal, O.; Leitner, F.; Vazquez, M.; Salgado, D.; Lu, Z.; Leaman, R.; Lu, Y.; Ji, D.; Lowe, D.M.; et al. The CHEMDNER corpus of chemicals and drugs and its annotation principles. J. Cheminformatics 2015, 7, 1–17. [Google Scholar] [CrossRef]
- Li, J.; Sun, Y.; Johnson, R.J.; Sciaky, D.; Wei, C.H.; Leaman, R.; Davis, A.P.; Mattingly, C.J.; Wiegers, T.C.; Lu, Z. BioCreative V CDR task corpus: A resource for chemical disease relation extraction. Database 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Lee, S.; Park, S.; Kim, S.; Kim, S.; Choi, K.; Tan, A.C.; Kang, J. BRONCO: Biomedical entity Relation ONcology COrpus for extracting gene-variant-disease-drug relations. Database 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Neves, M.; Damaschun, A.; Kurtz, A.; Leser, U. Annotating and evaluating text for stem cell research. In Proceedings of the Third Workshop on Building and Evaluation Resources for Biomedical Text Mining (BioTxtM 2012) at Language Resources and Evaluation (LREC), Manchester, UK, 26 May 2012; pp. 16–23. [Google Scholar]
- Krallinger, M.; Rabal, O.; Lourenço, A.; Perez, M.P.; Rodriguez, G.P.; Vazquez, M.; Leitner, F.; Oyarzabal, J.; Valencia, A. Overview of the CHEMDNER patents task. In Proceedings of the Fifth BioCreative Challenge Evaluation Workshop, 2015; pp. 63–75. Available online: https://www.jdb.uzh.ch/id/eprint/37857 (accessed on 12 June 2021).
- Lee, H.J.; Shim, S.H.; Song, M.R.; Lee, H.; Park, J.C. CoMAGC: A corpus with multi-faceted annotations of gene-cancer relations. BMC Bioinform. 2013, 14, 323. [Google Scholar] [CrossRef] [PubMed]
- Cohen, K.B.; Verspoor, K.; Fort, K.; Funk, C.; Bada, M.; Palmer, M.; Hunter, L.E. The Colorado Richly Annotated Full Text (CRAFT) Corpus: Multi-Model Annotation in the Biomedical Domain. In Handbook of Linguistic Annotation; Springer: Dordrecht, The Netherlands, 2017; pp. 1379–1394. [Google Scholar] [CrossRef]
- Herrero-Zazo, M.; Segura-Bedmar, I.; Martínez, P.; Declerck, T. The DDI corpus: An annotated corpus with pharmacological substances and drug–drug interactions. J. Biomed. Inform. 2013, 46, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Gerner, M.; Nenadic, G.; Bergman, C.M. An Exploration of Mining Gene Expression Mentions and Their Anatomical Locations from Biomedical Text. In Proceedings of the 2010 Workshop on Biomedical Natural Language Processing; Association for Computational Linguistics: Uppsala, Sweden, 2010; pp. 72–80. [Google Scholar]
- Oh, S.Y.; Kim, J.H.; Kim, S.J.; Nam, H.J.; Park, H.S. GNI Corpus version 1.0: Annotated full-text corpus of Genomics & Informatics to support biomedical information extraction. Genom. Inform. 2018, 16, 75. [Google Scholar]
- Smith, L.H.; Tanabe, L.; Rindflesch, T.C.; Wilbur, W.J. MedTag: A collection of biomedical annotations. In Proceedings of the ACL-ISMB Workshop on Linking Biological Literature, Ontologies and Databases: Mining Biological Semantics, Stroudsburg, PA, USA, 24 June 2005; pp. 32–37. [Google Scholar]
- Pyysalo, S.; Ohta, T.; Miwa, M.; Cho, H.C.; Tsujii, J.; Ananiadou, S. Event extraction across multiple levels of biological organization. Bioinformatics 2012, 28, i575–i581. [Google Scholar] [CrossRef] [PubMed]
- Shardlow, M.; Nguyen, N.; Owen, G.; O’Donovan, C.; Leach, A.; McNaught, J.; Turner, S.; Ananiadou, S. A new corpus to support text mining for the curation of metabolites in the Chebi database. In Proceedings of the Eleventh International Conference on Language Resources and Evaluation (LREC 2018), Miyazaki, Japan, 7–12 May 2018; pp. 280–285. [Google Scholar]
- Islamaj, R.; Leaman, R.; Kim, S.; Kwon, D.; Wei, C.H.; Comeau, D.C.; Peng, Y.; Cissel, D.; Coss, C.; Fisher, C.; et al. NLM-Chem, a new resource for chemical entity recognition in PubMed full text literature. Sci. Data 2021, 8, 1–12. [Google Scholar] [CrossRef]
- Islamaj, R.; Wei, C.H.; Cissel, D.; Miliaras, N.; Printseva, O.; Rodionov, O.; Sekiya, K.; Ward, J.; Lu, Z. NLM-Gene, a richly annotated gold standard dataset for gene entities that addresses ambiguity and multi-species gene recognition. J. Biomed. Informatics 2021, 118, 103779. [Google Scholar] [CrossRef]
- Sousa, D.; Lamúrias, A.; Couto, F.M. A silver standard corpus of human phenotype-gene relations. arXiv 2019, arXiv:1903.10728. [Google Scholar]
- Verspoor, K.; Jimeno Yepes, A.; Cavedon, L.; McIntosh, T.; Herten-Crabb, A.; Thomas, Z.; Plazzer, J.P. Annotating the biomedical literature for the human variome. Database 2013, 2013. [Google Scholar] [CrossRef]
- Cunningham, H.; Tablan, V.; Roberts, A.; Bontcheva, K. Getting More Out of Biomedical Documents with GATE’s Full Lifecycle Open Source Text Analytics. PLoS Comput. Biol. 2013, 9, e1002854. [Google Scholar] [CrossRef]
- Johansson, M.; Roberts, A.; Chen, D.; Li, Y.; Delahaye-Sourdeix, M.; Aswani, N.; Greenwood, M.A.; Benhamou, S.; Lagiou, P.; Holcátová, I.; et al. Using Prior Information from the Medical Literature in GWAS of Oral Cancer Identifies Novel Susceptibility Variant on Chromosome 4—The AdAPT Method. PLoS ONE 2012, 7, e36888. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, D.; Lally, A. UIMA: An architectural approach to unstructured information processing in the corporate research environment. Nat. Lang. Eng. 2004, 10, 327–348. [Google Scholar] [CrossRef]
- Ogren, P.V.; Wetzler, P.G.; Bethard, S. ClearTK: A UIMA toolkit for statistical natural language processing. In Proceedings of the Towards Enhanced Interoperability for Large HLT Systems: UIMA for NLP Workshop at Language Resources and Evaluation Conference (LREC), Marrakech, Morocco, 31 May 2008; Volume 32, pp. 32–38. [Google Scholar]
- Bethard, S.; Ogren, P.; Becker, L. ClearTK 2.0: Design patterns for machine learning in UIMA. In Proceedings of the 9th International Conference on Language Resources and Evaluation, LREC 2014; European Language Resources Association (ELRA): Reykjavik, Iceland, 2014; Volume 2014, pp. 3289–3293. [Google Scholar]
- Wang, Y.; Mehrabi, S.; Sohn, S.; Atkinson, E.J.; Amin, S.; Liu, H. Natural language processing of radiology reports for identification of skeletal site-specific fractures. BMC Med. Inform. Decis. Mak. 2019, 19. [Google Scholar] [CrossRef]
- Roeder, C.; Jonquet, C.; Shah, N.H.; Baumgartner, W.A.; Verspoor, K.; Hunter, L. A UIMA wrapper for the NCBO annotator. Bioinformatics 2010, 26, 1800–1801. [Google Scholar] [CrossRef]
- Comeau, D.C.; Dogan, R.I.; Ciccarese, P.; Cohen, K.B.; Krallinger, M.; Leitner, F.; Lu, Z.; Peng, Y.; Rinaldi, F.; Torii, M.; et al. BioC: A minimalist approach to interoperability for biomedical text processing. Database 2013, 2013, bat064. [Google Scholar] [CrossRef] [PubMed]
- Leaman, R.; Islamaj Doğan, R.; Lu, Z. DNorm: Disease name normalization with pairwise learning to rank. Bioinformatics 2013, 29, 2909–2917. [Google Scholar] [CrossRef]
- Wei, C.H.; Harris, B.R.; Kao, H.Y.; Lu, Z. tmVar: A text mining approach for extracting sequence variants in biomedical literature. Bioinformatics 2013, 29, 1433–1439. [Google Scholar] [CrossRef]
- Wei, C.H.; Kao, H.Y.; Lu, Z. SR4GN: A species recognition software tool for gene normalization. PLoS ONE 2012, 7, e38460. [Google Scholar] [CrossRef]
- Leaman, R.; Wei, C.H.; Lu, Z. tmChem: A high performance approach for chemical named entity recognition and normalization. J. Cheminformatics 2015, 7, 1–10. [Google Scholar] [CrossRef]
- Wei, C.H.; Kao, H.Y. Cross-species gene normalization by species inference. BMC Bioinform. 2011, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.H.; Kao, H.Y.; Lu, Z. PubTator: A web-based text mining tool for assisting biocuration. Nucleic Acids Res. 2013, 41, W518–W522. [Google Scholar] [CrossRef] [PubMed]
- Khare, R.; Wei, C.H.; Mao, Y.; Leaman, R.; Lu, Z. tmBioC: Improving interoperability of text-mining tools with BioC. Database 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, F.; Clematide, S.; Marques, H.; Ellendorff, T.; Romacker, M.; Rodriguez-Esteban, R. OntoGene web services for biomedical text mining. BMC Bioinform. 2014, 15. [Google Scholar] [CrossRef]
- Torii, M.; Li, G.; Li, Z.; Oughtred, R.; Diella, F.; Celen, I.; Arighi, C.N.; Huang, H.; Vijay-Shanker, K.; Wu, C.H. RLIMS-P: An online text-mining tool for literature-based extraction of protein phosphorylation information. Database 2014, 2014, bau081. [Google Scholar] [CrossRef] [PubMed]
- Casteleiro, M.A.; Demetriou, G.; Read, W.; Prieto, M.J.F.; Maroto, N.; Fernandez, D.M.; Nenadic, G.; Klein, J.; Keane, J.; Stevens, R. Deep learning meets ontologies: Experiments to anchor the cardiovascular disease ontology in the biomedical literature. J. Biomed. Semant. 2018, 9. [Google Scholar] [CrossRef]
- Doğan, R.I.; Kim, S.; Chatr-aryamontri, A.; Chang, C.S.; Oughtred, R.; Rust, J.; Wilbur, W.J.; Comeau, D.C.; Dolinski, K.; Tyers, M. The BioC-BioGRID corpus: Full text articles annotated for curation of protein–protein and genetic interactions. Database 2017, 2017, baw147. [Google Scholar] [CrossRef] [PubMed]
- Manning, C.; Surdeanu, M.; Bauer, J.; Finkel, J.; Bethard, S.; McClosky, D. The Stanford CoreNLP Natural Language Processing Toolkit. In Proceedings of 52nd Annual Meeting of the Association for Computational Linguistics: System Demonstrations; Association for Computational Linguistics: Baltimore, MD, USA, 2014; pp. 55–60. [Google Scholar] [CrossRef]
- Lu, H.; Kai, Z. How Do General-Purpose Sentiment Analyzers Perform when Applied to Health-Related Online Social Media Data? Stud. Health Technol. Inform. 2019, 264, 1208–1212. [Google Scholar] [CrossRef]
- Weber, L.; Münchmeyer, J.; Rocktäschel, T.; Habibi, M.; Leser, U. HUNER: Improving biomedical NER with pretraining. Bioinformatics 2019, 36, 295–302. [Google Scholar] [CrossRef]
- Weber, L.; Sänger, M.; Münchmeyer, J.; Habibi, M.; Leser, U.; Akbik, A. HunFlair: An easy-to-use tool for state-of-the-art biomedical named entity recognition. Bioinformatics 2021. [Google Scholar] [CrossRef]
- Cabot, C.; Darmoni, S.; Soualmia, L.F. Cimind: A phonetic-based tool for multilingual named entity recognition in biomedical texts. J. Biomed. Inform. 2019, 94, 103176. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; Rocktäschel, T.; Hakenberg, J.; Lichtblau, Y.; Leser, U. SETH detects and normalizes genetic variants in text. Bioinformatics 2016, 32, 2883–2885. [Google Scholar] [CrossRef]
- Lee, H.C.; Hsu, Y.Y.; Kao, H.Y. AuDis: An automatic CRF-enhanced disease normalization in biomedical text. Database 2016, 2016, baw091. [Google Scholar] [CrossRef][Green Version]
- Gupta, S.; Dingerdissen, H.; Ross, K.E.; Hu, Y.; Wu, C.H.; Mazumder, R.; Vijay-Shanker, K. DEXTER: Disease-Expression Relation Extraction from Text. Database 2018, 2018. [Google Scholar] [CrossRef]
- Dingerdissen, H.M.; Torcivia-Rodriguez, J.; Hu, Y.; Chang, T.C.; Mazumder, R.; Kahsay, R. BioMuta and BioXpress: Mutation and expression knowledgebases for cancer biomarker discovery. Nucleic Acids Res. 2017, 46, D1128–D1136. [Google Scholar] [CrossRef]
- Weber, L.; Thobe, K.; Lozano, O.A.M.; Wolf, J.; Leser, U. PEDL: Extracting protein–protein associations using deep language models and distant supervision. Bioinformatics 2020, 36, i490–i498. [Google Scholar] [CrossRef]
- Kim, D.; Lee, J.; So, C.H.; Jeon, H.; Jeong, M.; Choi, Y.; Yoon, W.; Sung, M.; Kang, J. A Neural Named Entity Recognition and Multi-Type Normalization Tool for Biomedical Text Mining. IEEE Access 2019, 7, 73729–73740. [Google Scholar] [CrossRef]
- Malarkodi, C.; Pattabhi, R.; Sobha, L.D. CLRG ChemNER: A Chemical Named Entity Recognizer@ ChEMU CLEF 2020. 2020. Available online: moz-extension://c64046de-9d28-4e46-a199-807c4d6ae096/pdf-viewer/web/viewer.html?file=http%3A%2F%2Fceur-ws.org%2FVol-2696%2Fpaper236.pdf (accessed on 12 June 2021).
- Yoon, W.; So, C.H.; Lee, J.; Kang, J. CollaboNet: Collaboration of deep neural networks for biomedical named entity recognition. BMC Bioinform. 2019, 20. [Google Scholar] [CrossRef]
- Dang, T.H.; Le, H.Q.; Nguyen, T.M.; Vu, S.T. D3NER: Biomedical named entity recognition using CRF-biLSTM improved with fine-tuned embeddings of various linguistic information. Bioinformatics 2018, 34, 3539–3546. [Google Scholar] [CrossRef]
- Wei, C.H.; Kao, H.Y.; Lu, Z. GNormPlus: An Integrative Approach for Tagging Genes, Gene Families, and Protein Domains. BioMed Res. Int. 2015, 2015, 1–7. [Google Scholar] [CrossRef]
- Giorgi, J.M.; Bader, G.D. Transfer learning for biomedical named entity recognition with neural networks. Bioinformatics 2018, 34, 4087–4094. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, G.; McDermott, M.; Szolovits, P. Reflex: Flexible framework for relation extraction in multiple domains. arXiv 2019, arXiv:1906.08318. [Google Scholar]
- Giorgi, J.M.; Bader, G.D. Towards reliable named entity recognition in the biomedical domain. Bioinformatics 2019, 36, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; King, D.; Beltagy, I.; Ammar, W. Scispacy: Fast and robust models for biomedical natural language processing. arXiv 2019, arXiv:1902.07669. [Google Scholar]
- Dao, M.H.; Nguyen, D.Q. VinAI at ChEMU 2020: An Accurate System for Named Entity Recognition in Chemical Reactions from Patents. 2020. Available online: https://www.vinai.io/publication-posts/vinai-at-chemu-2020-an-accurate-system-for-named-entity-recognition-in-chemical-reactions-from-patents (accessed on 12 June 2021).
- Zuo, M.; Zhang, Y. Dataset-aware multi-task learning approaches for biomedical named entity recognition. Bioinformatics 2020, 36, 4331–4338. [Google Scholar] [CrossRef]
- Habibi, M.; Weber, L.; Neves, M.; Wiegandt, D.L.; Leser, U. Deep learning with word embeddings improves biomedical named entity recognition. Bioinformatics 2017, 33, i37–i48. [Google Scholar] [CrossRef]
- Wei, C.H.; Allot, A.; Leaman, R.; Lu, Z. PubTator central: Automated concept annotation for biomedical full text articles. Nucleic Acids Res. 2019, 47, W587–W593. [Google Scholar] [CrossRef]
- Djekidel, M.N.; Rosikiewicz, W.; Peng, J.C.; Kanneganti, T.D.; Hui, Y.; Jin, H.; Hedges, D.; Schreiner, P.; Fan, Y.; Wu, G.; et al. CovidExpress: An Interactive Portal for Intuitive Investigation on SARS-CoV-2 Related Transcriptomes. 2021. Available online: https://www.biorxiv.org/content/10.1101/2021.05.14.444026v1 (accessed on 12 June 2021). [CrossRef]
- Wu, M.; Zhang, Y.; Grosser, M.; Tipper, S.; Venter, D.; Lin, H.; Lu, J. Profiling COVID-19 Genetic Research: A Data-Driven Study Utilizing Intelligent Bibliometrics. Front. Res. Metrics Anal. 2021, 6. [Google Scholar] [CrossRef]
- Desterke, C.; Turhan, A.G.; Bennaceur-Griscelli, A.; Griscelli, F. HLA-dependent heterogeneity and macrophage immunoproteasome activation during lung COVID-19 disease. J. Transl. Med. 2021, 19. [Google Scholar] [CrossRef]
- Venkatesan, A.; Kim, J.H.; Talo, F.; Ide-Smith, M.; Gobeill, J.; Carter, J.; Batista-Navarro, R.; Ananiadou, S.; Ruch, P.; McEntyre, J. SciLite: A platform for displaying text-mined annotations as a means to link research articles with biological data. Wellcome Open Res. 2016, 1, 25. [Google Scholar] [CrossRef]
- Palopoli, N.; Iserte, J.A.; Chemes, L.B.; Marino-Buslje, C.; Parisi, G.; Gibson, T.J.; Davey, N.E. The articles.ELM resource: Simplifying access to protein linear motif literature by annotation, text-mining and classification. Database 2020, 2020. [Google Scholar] [CrossRef]
- Firth, R.; Talo, F.; Venkatesan, A.; Mukhopadhyay, A.; McEntyre, J.; Velankar, S.; Morris, C. Automatic annotation of protein residues in published papers. Acta Crystallogr. Sect. Struct. Biol. Commun. 2019, 75, 665–672. [Google Scholar] [CrossRef]
- Müller, H.M.; Kenny, E.E.; Sternberg, P.W. Textpresso: An Ontology-Based Information Retrieval and Extraction System for Biological Literature. PLoS Biol. 2004, 2, e309. [Google Scholar] [CrossRef]
- Hu, Y.; Chung, V.; Comjean, A.; Rodiger, J.; Nipun, F.; Perrimon, N.; Mohr, S.E. BioLitMine: Advanced Mining of Biomedical and Biological Literature About Human Genes and Genes from Major Model Organisms. G3 Genes Genomes Genetics 2020, 10, 4531–4539. [Google Scholar] [CrossRef]
- Campos, D.; Lourenço, J.; Matos, S.; Oliveira, J.L. Egas: A collaborative and interactive document curation platform. Database 2014, 2014, bau048. [Google Scholar] [CrossRef]
- Nunes, T.; Campos, D.; Matos, S.; Oliveira, J.L. BeCAS: Biomedical concept recognition services and visualization. Bioinformatics 2013, 29, 1915–1916. [Google Scholar] [CrossRef]
- Liu, H.; Hu, Z.Z.; Zhang, J.; Wu, C. BioThesaurus: A web-based thesaurus of protein and gene names. Bioinformatics 2005, 22, 103–105. [Google Scholar] [CrossRef]
- Sernadela, P.; González-Castro, L.; Carta, C.; van der Horst, E.; Lopes, P.; Kaliyaperumal, R.; Thompson, M.; Thompson, R.; Queralt-Rosinach, N.; Lopez, E.; et al. Linked Registries: Connecting Rare Diseases Patient Registries through a Semantic Web Layer. BioMed Res. Int. 2017, 2017, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liang, Y.; Wishart, D. PolySearch2: A significantly improved text-mining system for discovering associations between human diseases, genes, drugs, metabolites, toxins and more. Nucleic Acids Res. 2015, 43, W535–W542. [Google Scholar] [CrossRef]
- Khan, F.; Radovanovic, A.; Gojobori, T.; Kaur, M. IBDDB: A manually curated and text-mining-enhanced database of genes involved in inflammatory bowel disease. Database 2021, 2021. [Google Scholar] [CrossRef]
- Liu, B.; Bai, C. Regulatory Mechanisms of Coicis Semen on Bionetwork of Liver Cancer Based on Network Pharmacology. BioMed Res. Int. 2020, 2020, 1–17. [Google Scholar] [CrossRef]
- Tsuruoka, Y.; Tsujii, J.; Ananiadou, S. FACTA: A text search engine for finding associated biomedical concepts. Bioinformatics 2008, 24, 2559–2560. [Google Scholar] [CrossRef] [PubMed]
- Tsuruoka, Y.; Miwa, M.; Hamamoto, K.; Tsujii, J.; Ananiadou, S. Discovering and visualizing indirect associations between biomedical concepts. Bioinformatics 2011, 27, i111–i119. [Google Scholar] [CrossRef]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef]
- Humphreys, B.L.; Lindberg, D.A.B.; Schoolman, H.M.; Barnett, G.O. The Unified Medical Language System: An Informatics Research Collaboration. J. Am. Med. Inform. Assoc. 1998, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The Human Metabolome Database. Nucleic Acids Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2007, 36, D901–D906. [Google Scholar] [CrossRef]
- Le, N.; Ho, T.; Ho, B.; Tran, D. A nucleosomal approach to inferring causal relationships of histone modifications. BMC Genom. 2014, 15, S7. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2018, 47, D607–D613. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Santos, A.; von Mering, C.; Jensen, L.J.; Bork, P.; Kuhn, M. STITCH 5: Augmenting protein–chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2015, 44, D380–D384. [Google Scholar] [CrossRef]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; Veij, M.D.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2018, 47, D930–D940. [Google Scholar] [CrossRef]
- Roth, B.L.; Lopez, E.; Patel, S.; Kroeze, W.K. The Multiplicity of Serotonin Receptors: Uselessly Diverse Molecules or an Embarrassment of Riches? Neuroscientist 2000, 6, 252–262. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Costanzo, L.D.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2020, 49, D437–D451. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2017, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Okuno, Y.; Tamon, A.; Yabuuchi, H.; Niijima, S.; Minowa, Y.; Tonomura, K.; Kunimoto, R.; Feng, C. GLIDA: GPCR ligand database for chemical genomics drug discovery database and tools update. Nucleic Acids Res. 2007, 36, D907–D912. [Google Scholar] [CrossRef]
- Gunther, S.; Kuhn, M.; Dunkel, M.; Campillos, M.; Senger, C.; Petsalaki, E.; Ahmed, J.; Urdiales, E.G.; Gewiess, A.; Jensen, L.J.; et al. SuperTarget and Matador: Resources for exploring drug-target relationships. Nucleic Acids Res. 2007, 36, D919–D922. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, S.; Li, F.; Zhou, Y.; Zhang, Y.; Wang, Z.; Zhang, R.; Zhu, J.; Ren, Y.; Tan, Y.; et al. Therapeutic target database 2020: Enriched resource for facilitating research and early development of targeted therapeutics. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef]
- Davis, A.P.; Wiegers, T.C.; Wiegers, J.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; Mattingly, C.J. CTD anatomy: Analyzing chemical-induced phenotypes and exposures from an anatomical perspective, with implications for environmental health studies. Curr. Res. Toxicol. 2021, 2, 128–139. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2020, 49, D545–D551. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Karp, P.D.; Billington, R.; Caspi, R.; Fulcher, C.A.; Latendresse, M.; Kothari, A.; Keseler, I.M.; Krummenacker, M.; Midford, P.E.; Ong, Q.; et al. The BioCyc collection of microbial genomes and metabolic pathways. Briefings Bioinform. 2017, 20, 1085–1093. [Google Scholar] [CrossRef]
- Huang, H.Y.; Lin, Y.C.D.; Li, J.; Huang, K.Y.; Shrestha, S.; Hong, H.C.; Tang, Y.; Chen, Y.G.; Jin, C.N.; Yu, Y.; et al. miRTarBase 2020: Updates to the experimentally validated microRNA–target interaction database. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef]
- Oughtred, R.; Stark, C.; Breitkreutz, B.J.; Rust, J.; Boucher, L.; Chang, C.; Kolas, N.; O’Donnell, L.; Leung, G.; McAdam, R.; et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 2018, 47, D529–D541. [Google Scholar] [CrossRef] [PubMed]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2020, 48, D845–D855. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Rodríguez, G.; Pérez-Pérez, M.; Fdez-Riverola, F.; Lourenço, A. Online visibility of software-related web sites: The case of biomedical text mining tools. Inf. Process. Manag. 2019, 56, 565–583. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Corpus | Description | Number of Documents | Annotations | References |
|---|---|---|---|---|
| BC5CDR | BioCreative V Chemical Disease Relation—disease and chemicals annotations for chemicals-disease interactions retrieval | 1500 PubMed abstracts | Diseases, chemicals, chemical-disease interactions | [49] |
| BRONCO | Biomedical entity Relation ONcology COrpus—focused on cancer research and anti-tumor drug screening containing 400 genomic variants and their relation to genes, diseases, drugs, and cell lines from 108 full-text articles | 108 full-length articles | Variants, genes, diseases, cell lines, drugs | [50] |
| CellFinder | A corpus based on stem cells | 10 full-length articles | Anatomical parts, cell components, cell lines, cell types, genes/proteins, species, several binary relationships, biological processes | [51] |
| ChemDNER | CHEMicals Disease Named Entity Recognition—focused on chemical substances and their characteristics | 10,000 PubMed abstracts | Chemicals | [48] |
| ChemDNER patents | CHEMicals Disease Named Entity Recognition patents—focused on detecting mentions in running patent text. Manual annotation led to 2 gold standard corpora Chemical Entity Mention in Patents (CEMP) and Gene and Protein Related Object (GPRO) | 21,000 medicinal chemistry-related patents abstracts | Chemicals, gene and gene products | [52] |
| CoMAGC | Corpus with Multi-faceted Annotations of Gene-Cancer relations—focused on gene-cancer relations (namely regarding prostate, breast, and ovarian cancers) | 821 sentences from 408 documents | Change in gene expression, change in cell state, proposition type, and initial gene expression level | [53] |
| CRAFT | Colorado Richly Annotated Full-Text Corpus | 97 full-length articles | Chemicals, cell types, biological processes, cellular and extracellular components and regions, molecular function, chemical reactions, biological taxa, proteins, biomacromolecular entities and sequences, anatomical entities | [54] |
| DDI corpus | Drug-Drug Interactions—focused on pharmacological substances and their relationships | 792 texts from DrugBank database and 233 MEDLINE abstracts | Pharmacological substances; DDIs | [55] |
| GENIA | GENome Information Acquisition—focused on biological reactions base on transcription factors in human blood cells | 2000 MEDLINE abstracts | 47 biologically relevant nominal categories | [44] |
| GETM | GeneExpression Text Miner corpus—gold standard corpus focused on gene expression events and their anatomical locations | 150 MEDLINE abstracts | Genes, anatomical locations | [56] |
| GNI corpus | GeNomics And Informatics - Originally developed to identify trends from publications from the Genomics and Informatics Journal | 499 full texts from Genomics and Informatics Journal | Proteins; DNA; RNA; cell lines; cell types | [57] |
| GREC | Gene Regulation Event Corpus—developed to train text mining systems to extract biologically meaningful events | 240 MEDLINE abstracts | Biological events (13 semantic roles) and biological concepts (10 categories for E. coli and 10 categories for Human) | [43] |
| MedTag | A biomedical corpus that combines 3 corpora: MedPost, ABGene and GENETAG | MedPost: 6700 sentences; ABGene: 4265 sentences; GENETAG: 15,000 sentences | Genes, proteins, clinical medicine semantics | [58] |
| MLEE | Multi-Level Event Extraction—focused on event extraction | 262 PubMed abstracts on angiogenesis | 3 entity categories: organism, anatomy (11 subcategories), and molecule (2 subcategories); and 4 event types: anatomical (7 subcategories), molecular (6 subcategories), general (5 subcategories), and planned | [59] |
| NCBI disease corpus | National Center for Biotechnology Information disease corpus—A corpus to disease recognition | 793 PubMed abstracts | Diseases | [47] |
| new corpus | Unnamed corpus meant to automatize curation of the ChEBI database | 200 abstracts and 100 full-text articles | 6 entities (metabolites, chemicals, proteins, species, biological activities, spectral data) and 4 relations (isolated from, associated with, binds with, metabolite of) | [60] |
| NLM-Chem | National Library of Medicine—Chemical—gold standard dataset focused on chemical NER | 150 Pubmed full-text articles | Chemicals | [61] |
| NLM-Gene | National Library of Medicine—Gene—gold standard dataset focused on gene NER | 550 Pubmed full-text articles | Gene | [62] |
| PGR | Phenotype-Gene Relations—a silver standard corpus based on human genes and phenotype, as well as their relations | 1712 abstracts | Genes, human phenotypes and phenotype-gene relations | [63] |
| Variome | A corpus focused on the relationship between the inherited colorectal cancer and human genetic variation | 10 articles | 11 entities and 13 relations | [64] |
| Tool/Model | NER | NEN | RE | Class | Architecture | Reference |
|---|---|---|---|---|---|---|
| AuDis | X | X | — | Diseases | CRF | [90] |
| BERN | X | X | — | Chemical, Disease, Specie, Gene/Protein, and Mutation | NER—Transformers (BioBERT based) | [94] |
| BioBERT | X | — | X | Fine-tuning is available for desired classes when criteria are met | Transformers | [35] |
| BioRel | X | X | X | Entities: Clinical drugs, pharmacologic substance, organic chemical, disease or syndrome, biologically active substance, molecular function, food, organ or tissue function and neoplastic process; Relations: 124 classes + NA | DNN-based approaches and distance supervised learning | [23] |
| Cimind | X | — | — | Diseases | Double metaphone phonetic algorithm and weighted distance scale algorithm | [88] |
| CLRG | X | — | — | Chemical compounds and their specific types | CRF and Artificial Neural Networks (ANN) | [95] |
| CollaboNet | X | — | — | Chemical, gene/protein, and disease | BiLSTM-CRF | [96] |
| D3NER | X | — | — | Any type of entity as long as criteria are met | CRF-biLSTM | [97] |
| DEXTER | X | — | X | Gene/microRNA, disease, and expression information | Standard dependency graph | [91] |
| Dnorm | X | X | — | Diseases | [73] | |
| HunFlair | X | — | — | Cell line, chemical, disease, gene and species | BiLSTM- CRF | [87] |
| GNormPlus | X | X | — | Gene, gene family, protein domain | CRF | [98] |
| NeuroNER | X | — | — | Chemicals, disease, species and gene/protein | LSTM-CRF | [99] |
| PEDL | X | X | X | Protein-protein association | Multi-instance learning (NER and NEN are BERT-based) | [93] |
| REflex | X | — | X | CNN | [100] | |
| Saber | X | X | — | Chemical, disorder, organism, gene/gene product | BiLSTM-CRF | [101] |
| SciSpacy | X | X | — | Depends on the chosen model from the repository | spaCy based | [102] |
| SETH | X | X | — | Genetic variants | [89] | |
| tmChem | X | X | — | Chemicals, drugs | CRF | [76] |
| tmVar | X | X | — | Variants at protein and gene level | CRF | [74] |
| VinAI | X | — | — | Chemicals | BiLSTM-CNN-CRF | [103] |
| NA | X | — | — | 29 entities types from 15 datasets | Multi-Task Learning (MTL)-BC and MTL-LBC | [104] |
| NA | X | — | — | Chemicals, diseases, species, genes/proteins and cell lines | BiLSTM-CRF | [105] |
| NA | — | X | — | Diseases | CNN | [15] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosário-Ferreira, N.; Marques-Pereira, C.; Pires, M.; Ramalhão, D.; Pereira, N.; Guimarães, V.; Santos Costa, V.; Moreira, I.S. The Treasury Chest of Text Mining: Piling Available Resources for Powerful Biomedical Text Mining. BioChem 2021, 1, 60-80. https://doi.org/10.3390/biochem1020007
Rosário-Ferreira N, Marques-Pereira C, Pires M, Ramalhão D, Pereira N, Guimarães V, Santos Costa V, Moreira IS. The Treasury Chest of Text Mining: Piling Available Resources for Powerful Biomedical Text Mining. BioChem. 2021; 1(2):60-80. https://doi.org/10.3390/biochem1020007
Chicago/Turabian StyleRosário-Ferreira, Nícia, Catarina Marques-Pereira, Manuel Pires, Daniel Ramalhão, Nádia Pereira, Victor Guimarães, Vítor Santos Costa, and Irina Sousa Moreira. 2021. "The Treasury Chest of Text Mining: Piling Available Resources for Powerful Biomedical Text Mining" BioChem 1, no. 2: 60-80. https://doi.org/10.3390/biochem1020007
APA StyleRosário-Ferreira, N., Marques-Pereira, C., Pires, M., Ramalhão, D., Pereira, N., Guimarães, V., Santos Costa, V., & Moreira, I. S. (2021). The Treasury Chest of Text Mining: Piling Available Resources for Powerful Biomedical Text Mining. BioChem, 1(2), 60-80. https://doi.org/10.3390/biochem1020007