Abstract
With increasing age, the chances of developing either MGUS or polyneuropathy increase as well. In some cases, there is a causative relationship between the IgM M-protein and polyneuropathy. In approximately half of these cases, IgM targets the myelin-associated glycoprotein (MAG). This results in chronic polyneuropathy with slowly progressive, predominantly sensory neurological deficits and distally demyelinating features in nerve conduction studies. Despite the disease being chronic and developing slowly, it can cause considerable impairment. We reviewed English medical publications between 1980 and May 2022 on IgM gammopathy-associated polyneuropathy, with special attention to studies addressing the pathophysiology or treatment of anti-MAG polyneuropathy. Treatment options have been limited to a temporizing effect of intravenous immunoglobulins in some patients and a more sustained effect of rituximab but in only 30 to 55 percent of patients. An increase in our knowledge concerning genetic mutations, particularly the MYD88L265P mutation, led to the development of novel targeted treatment options such as BTK inhibitors. Similarly, due to the increasing knowledge of the pathophysiology of anti-MAG polyneuropathy, new treatment options are emerging. Since anti-MAG polyneuropathy is a rare disease with diverse symptomatology, large trials with good outcome measures are a challenge.
Keywords:
IgM; monoclonal gammopathy; MGUS; Waldenström’s macroglobulinemia; anti-MAG; polyneuropathy; MYD88; CXCR4 1. Introduction
An M-protein develops in the context of a proliferative disorder of an antibody (Ig) secreting post-germinal center B-cells or plasma cells, which range from a premalignant disorder (i.e., monoclonal gammopathy of an undetermined significance, abbreviated as MGUS) to malignant disorders such as multiple myeloma (MM), Waldenström’s macroglobulinemia (WM) or chronic lymphocytic leukemia [1].
MGUS can be divided into three major groups: IgM gammopathies, non-IgM gammopathies and light chain gammopathies [2,3]. In the case of an IgM MGUS, there are no signs of anemia, constitutional symptoms, hyperviscosity, lymphadenopathy, hepatosplenomegaly, or other end-organ damage that can be attributed to the underlying lymphoproliferative disorder. In addition, the serum IgM monoclonal protein is <30 g/L and, if present, the clonal bone marrow plasma cells are <10% [2]. MGUS is a common disorder especially in the elderly, with prevalence in the general population ranging from approximately 3% [4,5], up to 5.1% [6] or even 6.9% [7] with more sensitive screening techniques of recent years. There is an increase in prevalence with age, with MGUS being diagnosed in 2.3%, 6.2% and 12.9% in age groups 40–59, 60–79 and 80–103 years, respectively [8]. Patients with IgM MGUS can experience clinical symptoms resulting from autoimmune or other properties of the IgM M-protein and are thus called monoclonal gammopathies with clinical significance [9,10]. When these M-protein-related mechanisms cause neurological diseases such as polyneuropathy (PNP), it is called monoclonal gammopathy of neurological significance [11].
Since MGUS is a precursor of malignant disorders, there is a risk of the development of a malignant disorder of approximately 1% each year [1]. IgM MGUS has a rate of progression to malignant disorders of 1.5% each year, markedly higher than the overall progression rate of non-IgM MGUS [12]. IgM MGUS usually develops into WM or lymphoma, rather than MM or (AL) amyloidosis [12].
Like MGUS, chronic PNP is also common, with an age-standardized prevalence of 4% in the general population [13], increasing to approximately 7–13% in the elderly [13,14]. Thus, MGUS and a chronic PNP could very well coincide in an elderly person. However, several studies show an especially elevated prevalence of PNP within MGUS cohorts, ranging from 7% to 42% [15,16,17,18]. Saemundur et al. found a risk ratio of 2.7 for PNP in an MGUS population compared to matched controls (comparable median age and stratified for diabetes mellitus), indicating a positive association between MGUS and PNP [18]. The percentage of an M-protein in patients with otherwise unexplained PNP is 10% [19], higher than the prevalence of gammopathies in the general population. A significantly larger number of PNP is caused by IgM compared to IgG or IgA gammopathies [15,16,17,20]. Baldini et al. described 45% of IgM MGUS patients with PNP [21]. Thus, there could be a causal relationship, especially for the IgM isotype. A causal relationship between IgM monoclonal gammopathy and PNP has become more robust, especially after the discovery of associated antigens such as the myelin-associated glycoprotein (MAG) [22,23,24,25,26,27,28].
The increase in our knowledge of pathophysiology and genetic alteration in IgM gammopathies itself and its related polyneuropathies, may lead to new therapy options. These therapies should be effective in treating both hematologic and neurologic diseases. The aim of this review is to give an up-to-date overview of the literature on IgM gammopathy-associated polyneuropathy, with particular attention to anti-MAG antibodies, as this is the most prevalent variant of IgM M-protein-associated PNP.
2. Methods
This aim led to a search string that we used for the PubMed and Cochrane databases. This search string comprises four components: “polyneuropathy”, “IgM”, “gammopathy” and “antibodies”.
(polyneuropath*[Title/Abstract] OR “Polyneuropathies”[Mesh:NoExp] OR neuropath*[Title/Abstract]) AND (“Immunoglobulin M”[Mesh] OR IgM[Title/Abstract] OR immunoglobulin M[Title/Abstract] OR MGUS[Title/Abstract] OR monoclonal gammopathy of undetermined significance[Title/Abstract] OR anti-MAG[Title/Abstract])
We excluded case reports and only included articles with full-text availability that were written in English between 1980 and May 2022. This search generated 395 results on 27 May 2022. Thereafter, J.M. screened the abstracts and only included articles covering all components of the research question, which resulted in 259 articles (7 articles had no full-text availability). A subsequent selection was made using articles that address epidemiological and clinical features of IgM gammopathy-associated PNP in general, or pathophysiology and treatment of IgM anti-MAG PNP in particular. Regarding treatment, original trials and observational studies were included, along with recent (systematic) reviews. If available, we used randomized controlled trials and meta-analyses instead of other trial designs for treatment options. In the case of (updated versions of) similar reviews, the newest version was included to ensure up-to-date knowledge. This yielded 81 studies.
3. Discussion
3.1. Pathophysiology of IgM Gammopathies
The default immunoglobulin produced by B-cells is of the IgM isotype. Given that IgM MGUS develops more frequently into lymphoma or WM and rarely to (IgM) MM, it has been concluded that IgM MGUS typically arises from a CD20+ post-germinal lymphoplasmacytic cell that has not undergone switch recombination [2]. Specific immunoglobulin heavy chain (IGH) variable gene usage in IgM MGUS and WM patients suggests a cell of origin that may have appeared during a physiological immune response, with subsequent secondary (genetic) events leading to a (pre-)malignant transformation [29].
IgM MGUS and WM patients share clonal B-cells with similar phenotypic and molecular signatures, but specific genetic alterations commonly present in WM, i.e., +4, del(6q23.3–6q25.3), +12, and +18q11–18q23 (up to 73% in WM), are less frequently detected in IgM MGUS (approximately 20%). This suggests a multistep process with a primary genetic alteration allowing the B-cell to proliferate and avoid apoptosis [30]. A second hit in some cases could be the occurrence of del6q (4% in IgM MGUS compared to 30% in symptomatic WM) [31]. In addition, there might be a role for specific copy number variants as well, since the number of copy number alterations significantly increases from IgM MGUS (36%) to smoldering WM (73%) to symptomatic WM (82%) [30].
3.2. Genetic Mutations
In both IgM MGUS and WM, there is a recurring somatic mutation in the gene that encodes the myeloid differentiation factor 88 (MYD88). This MYD88L265P mutation (a switch of leucine to proline at amino acid position 265) was demonstrated in 90% of WM patients and in 10% of IgM MGUS patients [32]. Subsequent studies demonstrated an even higher frequency of 95% in WM patients and 44–87% in IgM MGUS patients [33,34,35,36,37,38]. The percentage differs according to the methods of MYD88 mutation detection, and although MYD88L265P is prevalent in the majority of IgM MGUS patients, the median mutation level was shown to be almost one logarithm lower in IgM MGUS compared to WM [36]. Rarely, the mutation is present in other (non-IgM) small B-cell lymphomas and large B-cell lymphomas. However, in MM, even of the IgM paraprotein subtype, the MYD88L265P mutation has not been found at all [39].
MYD88 is a Toll-interleukin receptor domain-containing adaptor protein that is part of the interleukin-1 receptor (IL-1R) and Toll-like receptor (TLR) pathways, except for TLR3, and plays an important role in the innate immune system [40,41]. MYD88 binds with IL-1R-associated kinase 4 (IRAK4), which auto-phosphorylates and trans-phosphorylates IRAK2/1 forming a large multimeric molecule: the “myddosome” [42,43]. This myddosome triggers further downstream signaling, eventually stimulating activation of the IκB kinase (IKK) complex. This facilitates phosphorylation and degradation of the inhibitor of NF-κB (IκBα), allowing canonical NF-κB signaling [44,45]. NF-κB promotes cell survival by initiating the transcription of genes encoding stress-response enzymes, cell-adhesion molecules, proinflammatory cytokines, and anti-apoptotic proteins [46]. This is important for the growth and survival of WM cells [47].
MYD88L265P stimulates IRAK1 phosphorylation, leading to continuous myddosome formation and increased NF-κB signaling [48]. Inhibition of MYD88 signaling decreases NF-κB activity and decreases survival of activated B-cell-type diffuse large cell lymphoma cells expressing MYD88L265P. Moreover, mutated MYD88 triggers HCK and IL-6 transcription, eliciting pro-survival signaling and inducing Bruton’s tyrosine kinase (BTK) signaling [49].
BTK, an important adapter protein in B-cell receptor (BCR) signaling, also activates NF-κB. Triggering of the BCR activates NF-κB by IKK-dependent phosphorylation and proteasomal degradation of IκB-α. BCR triggering leads to phosphorylation of the B-cell linker (BLNK) adaptor, resulting in the recruitment of phospholipase C-isoform γ (PLC-γ) and BTK in the same complex, allowing the BTK-dependent phosphorylation and activation of PLC-γ. Activation of PLC-γ ultimately leads to NF-κB activation [50]. Intervening in this pathway by means of Bruton’s tyrosine kinase inhibitors such as ibrutinib and several second-generation drugs has been proven effective in treating WM [51,52].
In WM, the next most common somatic variant after MYD88L265P is in the C-terminal domain of the CXCR4 gene, which encodes for C-X-C chemokine receptor type 4. CXCR4 is a classical G-protein-coupled receptor. It is ubiquitously expressed in the human body and binds to CXCL12 (also known as stromal cell-derived factor-1 or SDF-1) [53]. CXCR4 is mutated in 24% [35] to 38% [54] or even 50% [55] of all WM patients, mostly along with MYD88L265P [35,54,55]. CXCR4 mutations have been reported in 4–20% of IgM MGUS patients [37].
CXCL12 binding induces conformational changes, leading to the activation of multiple signaling pathways involved in the activation and phosphorylation of cellular proteins and transcription factors to regulate the proliferation and migration of genes [56]. High expression of CXCR4 is observed in many (hematological) malignancies, with literature indicating that the binding of CXCL12 to CXCR4 on tumor cells of various types enhances proliferation [57]. CXCL12 can promote tumor growth and angiogenesis, participate in tumor metastasis and contribute to tumor immunosuppressive networks [58]. In contrast to MYD88 mutations, mutated CXCR4 is subclonal with the possibility to have several different CXCR4 mutations within an individual [59]. Most CXCR4 mutations lead to the loss of regulatory serines and promote sustained CXCL12-mediated activation [60].
3.3. Polyneuropathy in IgM Gammopathies
Myelin is used as an isolating layer in the majority of peripheral nerves to conduct a nerve impulse rapidly and efficiently, preventing the dissipation of the signal into the surrounding structures [61,62]. Myelin is formed by Schwann cells in the peripheral nervous system. Myelinated parts of the nerve are alternated by short unmyelinated parts, the nodes of Ranvier, to allow for fast, saltatory conduction. The nodes of Ranvier are flanked by paranodes and juxtaparanodes; the myelinated parts are called internodes. Myelin encircles the axons and consists primarily of lipids (70–80% [63]) and to a lesser degree of (glyco)proteins, of which P0 (MP0 or myelin protein zero), myelin basic protein (MBP) and periaxin are the most prevalent [64].
Myelin is organized in lamellae, compacted sheaths of myelin apposed to each other. This results in alternating phospholipid and protein layers, forming the major dense line (MDL) and intraperiod line (IPL), respectively. Compact myelin is interrupted by Schmidt–Lanterman incisures: characteristic funnel-shaped structures providing oblique cytosolic channels through compact myelin and thus forming a connection of cytoplasma between the myelin membranes juxtaposed to the axon (adaxonal membranes) and juxtaposed to the basal lamina of the nerve (abaxonal membranes). Each non-compacted membrane is called a mesaxonal membrane. The myelinated peripheral nerve configuration is summarized in Figure 1.
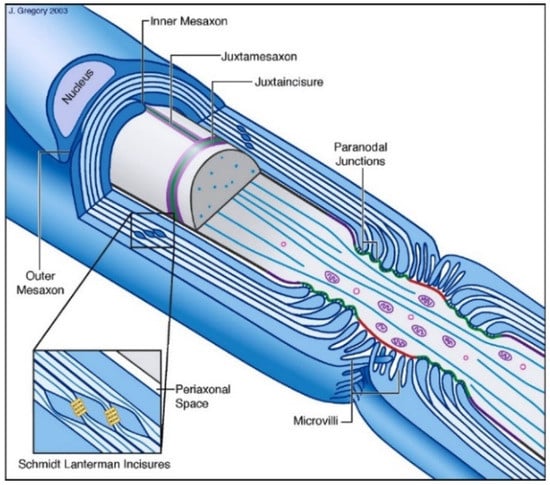
Figure 1.
Morphology of myelinated PNS nerve, borrowed from Salzer with permission from Elsevier [65].
PNP as a result of IgM gammopathy may be caused by different mechanisms that trigger a malfunctioning of the nerve [66]. The monoclonal IgM may act as an auto-reactive or toxic antibody. This antibody, in the majority of cases, has a target antigen [67,68,69,70,71,72,73] of which the prototype is anti-MAG but may also cause disease without an antigen, as seen in POEMS syndrome, which is related to cytokine release [74]. Myelin contains components that may act as antigens in several chronic inflammatory neuropathies and some of these antigens are targets for IgM antibodies [26].
Other mechanisms include deposits of the M-protein, or fragments of the M-protein, within various parts of the nerve (such as in IgM deposition disease or amyloid light chain amyloidosis, covered in the article by Sarosiek et al. in this issue). Moreover, the IgM M-protein can cause vasculitis (with or without cryoglobulinemia [75] resulting in ischemic damage to the nerve). This ischemic damage may also be caused by the direct blocking of blood vessels by cryoglobulins (covered in the article by Khwaja et al. in this issue) or the intrinsic increased viscosity of the blood because of the size of the IgM molecules [76]. Alternatively, the clonal B-cells may directly infiltrate the nerve, as is the case with neurolymphomatosis.
3.3.1. IgM M-Protein Related Polyneuropathies and Corresponding Phenotypes
Molecules in the peripheral nerve are capable of acting as antigens for the M-proteins being produced by IgM gammopathies (Table 1).

Table 1.
Possible antigens for M-proteins in the peripheral nerve. HNK1: human natural killer 1 carbohydrate epitope. SGPG: sulfated glucuronyl paragloboside. MAG: myelin-associated glycoprotein. MP0: myelin protein zero. Disialosyl epitope: specifically, the NeuAc(α2-8)NeuAc(α2-3)-Gal configuration. Sulfatide: galactosylceramide-3-O-sulfate.
This group of IgM gammopathy-associated PNPs, where the M-protein acts as an autoantibody, contains phenotypically, immunologically and electrophysiologically heterogeneous neuropathies [89]. Despite this heterogeneity, the majority of these neuropathies manifest as a chronic, slowly progressive demyelinating PNP [20,27,90,91,92].
Katz et al. showed that 60% of patients with an acquired demyelinating neuropathy and distal phenotype (DADS neuropathy) had an IgM M-protein, and 67% of these patients had anti-MAG antibodies [93]. Matà et al. showed in a cohort of 30 IgM gammopathy anti-MAG neuropathy patients that all of them cohered to the DADS phenotype [89]. Of all PNPs associated with IgM gammopathy, approximately 50% consist of PNP with anti-MAG antibodies [90,94,95], although this percentage might differ and might be up to 70% [96] or even 85% [86].
To detect the antibody activity of M-proteins (for instance with the HNK1 epitope in anti-MAG PNP), different techniques may be used [97,98,99]. While initially the Western blot method was widely used, approximately 15 years ago an enzyme-linked immunosorbent assay (ELISA) for MAG was developed that proved more sensitive compared to the Western blot [96], albeit less specific. The most used anti-MAG ELISA kit is from Bühlmann and measures the titer in Bühlmann titer units (BTU). While the initial lower limit for a positive test was >1000, studies suggest that a higher value of >7000 [100] gives a higher specificity and accuracy [26].
Although the entire group is heterogeneous, the IgM anti-MAG PNP patients are clinically and electrophysiologically more homogeneous, although differences can be seen [94,101] possibly related to lower anti-MAG titers [102]. Clinically, they classically manifest as a chronic, slowly progressive, predominantly sensory (atactic) PNP [73,89,90,91,92,94,101,103,104,105], regularly accompanied by a tremor [106,107,108]. Although the progression is usually slow, up to 24% and 50% of patients with anti-MAG IgM antibodies are disabled at 10 and 15 years after the onset of neuropathy, respectively [109]. At the most severe disease stage, with a mean follow-up of 8.4 years, 22.4% were found to be significantly disabled (Overall Neuropathy Limitations Scale > 3) [101].
Electrophysiologically, there is nearly always the presence of nerve conduction slowing indicating a demyelinating neuropathy, either with or without axonal degeneration [91,92,110,111,112] with distal conduction slowing increasing with nerve length [113,114]. The uncommon absence of demyelinating features in anti-MAG PNP is associated with a better prognosis regarding disability and progression, compared to demyelinating patterns in nerve conduction studies (NCS) [105]. Compared to controls, NCS of anti-MAG neuropathy showed statistically significant prolonged DML (distal motor latency) and reduced MCV (motor conduction velocity), SCV (sensory conduction velocity), CMAP (compound motor action potential) and SNAP (compound sensory nerve action potential) [115], indicative of distal demyelination. Anti-MAG neuropathy patients show a reduced terminal latency index (TLI) [114] and may have a reduced TLI of the median nerve more often compared to anti-MAG-negative patients [115]. This may be due to a higher susceptibility to nerve compression (in the wrist) in anti-MAG neuropathy since similar frequencies between anti-MAG-positive and negative patients of TLI < 0.25 were found in other nerves. The susceptibility to nerve compression has been suggested before [116]. Motor conduction slowing without conduction blocks remains stable over longer periods, whereas lower limb motor and sensory potentials frequently become unrecordable [117].
3.3.2. IgM Anti-MAG Antibody Polyneuropathy
In 1980, Latov et al. showed that in the setting of an IgM monoclonal gammopathy, in some cases a particular protein component on the peripheral nerve acts as an antigen and might be part of the pathophysiology of these PNPs [22,26]. Braun et al. [68] provided evidence that this protein is the myelin-associated glycoprotein (MAG).
MAG is a minor component of the peripheral nervous system myelin, accounting for less than 1% of the total protein weight [64]. It is a 100-kDa transmembrane adhesion protein of myelin-containing Schwann cell or glia cell membranes (in the CNS). MAG is located in the adaxonal membrane and on apposing myelin membranes in the non-compact myelin compartments, such as the Schmidt–Lanterman incisures and the paranodal loops [118,119].
Although the precise pathways are not known, there is evidence of bidirectional signaling between Schwann or glia cells and axon [120], concerning myelin maintenance and axon configuration. The fact that MAG knock-out mice develop a chronic axonopathy (with a decreased axonal diameter and progressive axonal degeneration [121,122]) and mild signs of demyelination on NCS [123] suggests that MAG promotes axonal stability, survival and maximal radial caliber and is thus needed for the maintenance of normal myelinated nerves [124].
MAG contains five extracellular immunoglobulin-like domains, a single transmembrane domain and a cytoplasmic domain [124]. MAG is a member of the immunoglobulin superfamily adhesion molecules [125], and because it binds sialic acid it also is a SIGLEC (sialic acid-binding immunoglobulin-like lectin) [126] and as such is named Siglec4a. SIGLECs recognize sialic acid groups on other molecules, and so does MAG. Sialic acid-containing glycosphingolipids are called gangliosides [127] and are abundant in the PNS and CNS. MAG recognizes the NeuAcα2→3Galβ1→3GalNAc (3-O) sialic acid [125], which is present on the GD1a and GT1b gangliosides, among others. By binding to axonal gangliosides, MAG maintains a defined spacing between the axon membrane and the adaxonal membrane of the Schwann cell [128].
In IgM anti-MAG PNP, IgM immunoglobulins cause disease by binding to the HNK1 carbohydrate epitope of MAG, which is shared with SGPG, MP0 and other proteins in peripheral nerves [79]. This has been shown by purifying MAG using deglycosylation, with MAG not causing reactivity afterward [77]. Biopsy studies show primary segmental demyelination, remyelination and IgM deposition on myelin sheaths [129], associated with the widening of myelin lamellae (WML) [130,131,132,133,134] and a decrease in neurofilament spacing [135], with axonal degeneration and tomaculous appearance (i.e., focal thickening [136]) of myelinated fibers [132]. There are multiple lines of evidence for the hypothesis that anti-MAG antibodies are the cause of these effects.
The deposition of IgM is associated with sites of MAG localization [131], mainly the Schmidt–Lanterman incisures and paranodal loops, but also additional HNK1-positive components of the basal lamina. Paranodal regions show an altered morphology, presumably because of IgM deposition on the terminal loops impairing paranodal function [137]. Dermal myelinated nerve fibers have diffuse deposits of anti-IgM antibodies, with increasing frequency distally compared to proximally in the limbs [138]. The IgM deposits colocalize with the aforementioned anti-MAG sites. Although MAG is also present in the adaxonal membrane, histological studies do not show IgM deposition at the membrane, most likely because the IgM immunoglobulins are unable to access these sites.
The fact that systemic transfusion of chickens with monoclonal IgM anti-MAG antibodies produced peripheral demyelination with a similar widening of the myelin lamellae, demyelination, remyelination and IgM deposition [139] strengthens the hypothesis that anti-MAG antibodies induce the demyelinating PNP. Similarly, intraneural injections of serum from patients with anti-MAG IgM PNP, supplemented with additional complements, produced an extensive inflammatory, macrophage-mediated demyelination of feline peripheral nerve, although pathological examination differed from human findings [140]. Moreover, immunization of cats with SGPG-induced anti-SGPG antibodies caused a sensory PNP clinically resembling the sensory ataxia of patients with monoclonal IgM anti-MAG/SGPG antibodies [141]. This has been shown in rats as well [142].
The widening of myelin lamellae may be one of the main mechanisms of demyelination in IgM anti-MAG PNP and is considered to be caused by the direct binding of IgM to the myelin membrane, with a positive correlation between the widening of the lamellae and demyelination [132], and the colocalization of IgM deposits and the finding of widening of the lamellae [132,143]. The fact that the frequency of these histological changes was correlated with anti-MAG titers suggests a causal role for IgM anti-MAG.
Histological studies suggest a role for the complement system in the pathogenesis of demyelination in IgM anti-MAG PNP, with the colocalization of sites with the widening of myelin lamellae and deposition of complement factors (C1q, C3d and C5) [132,134,144]. Deposits of both IgM and C3d were colocalized in the paranodal region, which suggests that the IgM antibody might injure myelin through activation of the complement pathway [132]. When classical and lectin pathways of complement activation are measured, there is no increase within anti-MAG patients [145]. Similarly, serum C3b and C4b were not elevated in MAG patients, nor were contactin-1 or neurofilament light chain (indicating axonal and paranodal damage) [146]. These findings might be the result of a sub-threshold effect due to extremely localized complement activation and particularly slow axonal damage. The exact influence of the complement system on IgM anti-MAG-induced demyelination is not known and needs further research, especially in light of the emerging potent complement inhibitors.
Interestingly, the MYD88L265P mutation is highly prevalent (approximately 60%) in patients with IgM M-protein anti-MAG-associated PNP [55,147]. The prevalence of CXCR4 mutations in this group is approximately 10%, although this might be an underestimate since only the CXCL4S338X mutation was analyzed [55]. The implication of the high frequency of these mutations is that the pathophysiology of anti-MAG PNP patients is close to WM and that patients therefore might be investigated and treated in this light [23].
3.4. Current Therapy for IgM Gammopathy Mediated Polyneuropathy
Evidence of pathological effects of IgM monoclonal proteins acting as antibodies (e.g., anti-MAG), led to the notion that lowering the M-protein titer would reduce the pathological and clinical effects. In the case of IgM anti-MAG PNP, clinical improvement is indeed associated with a relative decrease in anti-MAG titer, while a stable titer is not associated with improvement and a transient increase in anti-MAG titer is associated with acute worsening [148]. Since IgM M-protein-associated PNPs are slowly progressive and do not resolve without treatment [91,149], targeted treatment must be considered early to prevent axonal damage which might be irreversible. While the presence of an associated PNP is not a justification for treatment per se, steady deterioration of neurological findings with a disability may be a viable reason [66].
A decrease in IgM M-protein, or the amount of damage the M-protein does, can be achieved by different techniques. These include scavenging the M-protein, decreasing production by attacking the clonal B-cell population, preventing the M-protein from binding/attacking the target antigen or decreasing the damage done by the M-protein. In Table 2, we sum up these techniques with past and current treatment options for IgM monoclonal gammopathy anti-MAG PNP. In the next section, we will cover novel and possible future treatments. We used studies with the largest number of cases, and if possible we used RCTs. Treatment options in italic have not been proven effective.
Table 2.
Tried and current treatment options for IgM anti-MAG PNP.
Only a few studies on these treatment regimens were randomized controlled trials (RCTs) [169]. Many studies had a short follow-up period, had few participants and were different in design compared to each other. They often evaluate combinations of treatment with other combinations, instead of a placebo. Furthermore, there was no uniformity in selected outcome measures and outcome measures were mostly of the ordinal scale instead of the interval scale, being inadequate and insensitive as a measurement of response [170]. This might have contributed to trials with negative outcomes [171]. Rasch methodology-built outcome measures may offer better responsiveness in future trials [170].
Although two RCTs showed no significant improvement after plasma exchange [150,151], a temporal improvement cannot be ruled out [91,101,109,152,172], especially after a rapid clinical worsening [153]. A temporal improvement after IVIG in some patients was demonstrated in two RCTs [164,165], although a sustained effect was generally not shown in a large retro- and prospective study [101]. Prednisone is regarded as not effective, as are other immunomodulatory drugs except IVIG. Dexamethasone showed effects but caused a high degree of side effects such as severe mood disturbances classified as serious adverse events [167]. Immunomodulatory drugs may, however, improve the effects of other treatments [169]. Currently, the best technique seems to decrease M-protein production by directly targeting B-cells. While cyclophosphamide [158,159] and fludarabine [160,161] showed some effect, these therapies have considerable toxicity.
The most promising results regarding sustained treatment effects were found when using rituximab [169], shown through a meta-analysis of two RCTs [155,156]. Rituximab is a chimeric anti-CD20 monoclonal antibody that targets CD20-positive B-cells from pre-B-cells to memory B-cells [173]. Anti-CD20 activity is accomplished by several mechanisms, including complement-dependent cytotoxicity (CDC) and antibody-dependent cellular cytotoxicity (ADCC) [174]. ADCC is induced by NK cells and macrophages and is dependent on genetic polymorphisms in IgG Fcγ receptor II and III (FcγR II/III) [175]. Rituximab was first shown to be effective in IgM MGUS-associated PNP (anti-GM1 and anti-MAG) in 1999 [176]. An improvement is found in a portion of patients after approximately one year (circa 30–55% [101,157,177]), with an effect in sensory neuropathic tests, anti-MAG titers, total IgM and patient clinical impression of change, although some may not be clinically significant.
Since rituximab only has an effect on a portion of patients, it is interesting and helpful to know if there are correlations between patient characteristics and response to treatment. There is a positive correlation between an anti-MAG ELISA titer of ≥10 000 BTU and response to treatment [101]. Rituximab seems to have less effect on functional outcomes in patients with a longer disease course [157,178,179]. Clinically, proximal weakness of the lower limbs seems to predict significant treatment response, although the number of patients was small [179]. Patients with more profound axonal damage have a tendency to have an inferior response to rituximab treatment [180], which can be explained by a longer disease course. Furthermore, a certain FcγR polymorphism (FcγRIIIA-V/V158) was associated with (better) improvement in outcomes in a study with IgM PNP patients, of which 67% had anti-MAG antibodies [181]. This suggests different receptor binding properties of patients with this specific genotype, with subsequent better recruitment of effector cells. The effect of rituximab is similar in IgM MGUS anti-MAG and WM anti-MAG patients, with 36% (9/25) and 37.5% (3/8) of patients responding on the used neurological scales, respectively [157].
Sporadically, treatment of IgM anti-MAG PNP with rituximab can cause a paradoxical acute and severe worsening of clinical features [182]. The reason for this flare-up phenomenon is not known. A rapid rise in total IgM has been reported before in the light of WM treatment with rituximab [183,184], and this could include IgM anti-MAG antibodies, thus causing the worsening of clinical features. This rise in total IgM was, however, only seen in a portion of patients with an acute worsening after rituximab treatment and can therefore not explain all cases [182]. Other mechanisms that may be of influence on the transient worsening include an increase in pro-inflammatory cytokines and a higher permeability of the blood–brain or blood–nerve barrier to antibodies (such as anti-MAG), complement or cytokines [185].
Similar to current practice in WM [186], where rituximab combined with chemotherapy yields superior responses compared to rituximab monotherapy, rituximab might be more effective in treating IgM M-protein anti-MAG PNP if combined with chemotherapies such as cyclophosphamide [187], fludarabine [188] and bendamustine [189].
The addition of these agents may elicit an earlier response compared to rituximab monotherapy. Moreover, patients that do not respond to rituximab monotherapy might respond to rituximab combined with chemotherapy [190]. This study by Hospital et al. [190] was reproduced by Nivet et al. [191] and showed a larger neurological response rate (modified Rankin scale) with rituximab combined with chemotherapy at different measuring moments (at 1 year; 46% vs. 18%), compared to rituximab monotherapy. Both therapies were initiated at similar moments after disease onset. The study being retrospective, however, likely caused selection bias, with rituximab monotherapy patients being less disabled and thus less able to have a large improvement. Moreover, these regimens give an increased risk of prolonged immunosuppression and myeloid neoplasms [186]. However, in some cases, such as rapidly deteriorating patients with IgM anti-MAG M-protein autoantibodies, rituximab combined with chemotherapy can be considered.
Current guidelines for WM give suggestions for the treatment of WM-associated PNP which can be used in clinical practice [186,192,193]. Considering the proposed shared etiology of disorders, these therapies seem viable for IgM MGUS anti-MAG PNP patients as well. Moreover, BTK inhibitors can be considered, such as ibrutinib. Since these are not yet investigated in large trials, much less in RCTs, we will cover these therapies in the next section.
3.5. Novel Therapy Options
Advances in attacking the M-protein-producing B-cells have been made through specific characteristics of B-cells. These techniques are increasingly used in WM patients (covered by Kapoor and Abeykoon in this issue). The MYD88L265P mutation makes the B-cells susceptible to BTK inhibition. This has been shown with ibrutinib, a first-generation BTK inhibitor, in WM and also in IgM gammopathy anti-MAG PNP [51,52]. This response was positively influenced by the MYD88L265P mutation and was negatively influenced by CXCR4 mutations. Treon et al. showed a high and durable response rate in WM and furthermore showed that IgM-related peripheral polyneuropathy improved in five of nine patients and stayed stable in the remaining four [51]. Castellani et al. showed both a subjective and objective neurological improvement (by means of sensory, motor and ataxia scales) in three anti-MAG patients with the MYD88L265P mutation with a concomitant decrease in M-protein and IgM levels, but not of MAG-titers [52]. Larger studies are needed to confirm a significant improvement in neurological parameters in IgM gammopathy-associated PNP.
In recent years, second- and third-generation BTK inhibitors have been developed, aiming to minimize the side effects of ibrutinib and reduce resistance to ibrutinib by more selectively targeting BTK. The second-generation BTK inhibitors bind to BTK covalently and irreversibly, similar to ibrutinib, and include acalabrutinib, zanabrutinib and tirabrutinib [194]. Third-generation BTK inhibitors bind BTK reversibly and are versatile in the binding site, aiming to overcome resistance. These agents are currently still being tested in clinical trials, including pirtobrutinib [195]. In the near future, we will conduct a multicenter, open-label, phase II study in patients with IgM MGUS and anti-MAG antibodies to analyze the treatment effects of zanubrutinib in combination with rituximab on neurological and hematological outcomes (MAGNAZ trial) [196].
In the field of WM treatment, many novel options are being developed. These include BCL2 inhibitors such as venetoclax [197], anti-CXCR4 monoclonal antibodies (ulocuplumab) [198], next-generation proteasome inhibitors [199] and next-generation anti-CD20 agents, such as ofatumumab and obinutuzumab [200]. Obinutuzumab causes enhanced B-cell-depleting activity compared to rituximab [201] and may be effective in treating (rituximab non-responding) IgM gammopathy anti-MAG polyneuropathy [202,203].
Briani et al. published a case of CLL with IgM-kappa monoclonal protein with anti-MAG antibody activity without MYD88 or CXCL4 mutations. There was a sensorimotor demyelinating polyneuropathy present. The patient had only a minor improvement with rituximab with cyclophosphamide. Since BTK inhibitor response might be low due to the lack of an MYD88 mutation, venetoclax was given in a dose-increasing regimen, combined with rituximab. Venetoclax may improve BTK inhibition and, as such, can improve response rates of BTK inhibitors used to treat anti-MAG PNP [197]. There was a decrease in total IgM, anti-MAG titer, and importantly, the neurological symptomatology. Being only one case report, additional observations and controlled studies are needed to form conclusions [204].
Considering the notion that IgM anti-MAG PNP needs complement activation to elicit damage to the nerve and myelin sheath, complement activation inhibiting therapy could be an interesting way to prevent IgM anti-MAG antibodies from causing damage. Complement activation plays a role in the pathophysiology of several neurological disorders, including myasthenia gravis [205], Guillain–Barré syndrome [206], CIDP [207] and MMN [208].
Since the complement activation cascade consists of many steps and pathways [209], there are several points in this cascade at which therapies can intervene. Therapies acting on C1 (e.g., Cinryze [210]), C2 (e.g., ARGX-117 [211]), C3 (the compstatin family of C3 inhibitors [212]), C5 (e.g., eculizumab [213]) and other components of the complement system are being investigated for a variety of (neurological) disorders wherein complement activation plays a role [214]. To our knowledge, up to now there have been no clinical trials conducted that evaluated direct complement-inhibiting therapies in gammopathy-associated polyneuropathy.
Analogous to plasma exchange and plasmapheresis, the circulating antibodies could be removed by novel techniques as well. Since the M-protein acts as a de facto autoantibody, an autoantibody competitor decoy may be used to scavenge the harmful IgM M-proteins by acting as the actual antigen [215]. In this fashion, glycopolymers mimicking the HNK1 epitope have been designed to selectively decoy the circulating HNK1 epitope targeting antibody-acting M-protein [216]. This poly(phenyl disodium 3-O-sulfo-β-d-glucopyranuronate)-(1→3)-β-d-galactopyranoside (PPSGG, also called PN-1007) glycopolymer quickly removed the anti-HNK1 IgM immunoglobulin ex vivo [217]. A clinical trial was set up to evaluate this effect and its safety in a clinical setting (NCT04568174) but was terminated early. Glycopolymers acting as a decoy can elicit a potentially dangerous complement-mediated reaction called CARPA (complement activation-related pseudoallergy), which may be a reason not to try this technique [218].
In conclusion, in recent years there have been many developments in treating IgM gammopathies and, subsequently, the associated (anti-MAG) PNP as well. Similarly, in other polyneuropathies and chronic neurological disorders, new treatment options may be effective in treating IgM (anti-MAG) PNP too. While there have not been controlled trials yet to confirm the efficacy and clinical significance of treatment effects, small case reports and case series show promising results and warrant larger studies. In Table 3, we sum up the novel therapy options.
Table 3.
Novel treatment options for IgM anti-MAG PNP.
4. Conclusions
With increasing age, the chances of developing either MGUS or polyneuropathy increase as well. In some cases, this is no coincidence. Particularly in IgM gammopathies, it is important to identify when polyneuropathy is caused by the IgM M-protein. In approximately half of these cases, IgM targets MAG. This results in chronic polyneuropathy with slowly progressive, predominantly sensory neurological deficits and distally demyelinating features in nerve conduction studies. Despite the disease being chronic and developing slowly, it can cause considerable impairment. Treatment options have been limited for a long time, with a temporizing effect of intravenous immunoglobulins in some patients and a more sustained effect of rituximab, but in only 30 to 55 percent of patients. An increase in our knowledge concerning genetic mutations in WM, IgM MGUS and IgM anti-MAG polyneuropathy patients, particularly the MYD88L265P mutation, has led to the development of novel targeted treatment options such as BTK inhibitors in these settings. Similarly, due to the increasing knowledge about the pathophysiology of anti-MAG polyneuropathy, new treatment options are emerging. Since anti-MAG polyneuropathy is a rare disease with diverse symptomatology, large trials with good outcome measures are a challenge. However, promising results of novel therapy options in recent case series warrant larger studies to confirm potential good treatment options in this difficult-to-treat disease.
Author Contributions
Writing—original draft preparation, J.P.M.v.d.M.; writing—review and editing, S.D., A.F.J.E.V., N.C.N., J.M.I.V. and M.C.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kyle, R.A.; Therneau, T.M.; Rajkumar, S.V.; Offord, J.R.; Larson, D.R.; Plevak, M.F.; Melton, L.J. A Long-Term Study of Prognosis in Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2002, 346, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Mailankody, S.; Landgren, O. Monoclonal gammopathy of undetermined significance and Waldenström’s macroglobulinemia. Best Pract. Res. Clin. Haematol. 2016, 29, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Eisele, L.; Dürig, J.; Hüttmann, A.; Dührsen, U.; Assert, R.; Bokhof, B.; Erbel, R.; Mann, K.; Jöckel, K.-H.; Moebus, S. Prevalence and progression of monoclonal gammopathy of undetermined significance and light-chain MGUS in Germany. Ann. Hematol. 2012, 91, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Therneau, T.M.; Rajkumar, S.V.; Larson, D.R.; Plevak, M.F.; Offord, J.R.; Dispenzieri, A.; Katzmann, J.A.; Melton, L.J. Prevalence of Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2006, 354, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Murray, D.; Kumar, S.K.; Kyle, R.A.; Dispenzieri, A.; Dasari, S.; Larson, D.R.; Vachon, C.; Cerhan, J.R.; Rajkumar, S.V. Detection and prevalence of monoclonal gammopathy of undetermined significance: A study utilizing mass spectrometry-based monoclonal immunoglobulin rapid accurate mass measurement. Blood Cancer J. 2019, 9, 102. [Google Scholar] [CrossRef] [PubMed]
- Atkin, C.; Reddy-Kolanu, V.; Drayson, M.T.; Sapey, E.; Richter, A.G. The prevalence and significance of monoclonal gammopathy of undetermined significance in acute medical admissions. Br. J. Haematol. 2020, 189, 1127–1135. [Google Scholar] [CrossRef]
- Kristinsson, S.Y.; Rögnvaldsson, S.; Thorsteinsdottir, S.; Reed, E.R.; Oskarsson, J.T.T.; Petursdottir, I.; Sigurdardottir, G.A.; Vidarsson, B.; Onundarson, P.T.; Agnarsson, B.A.; et al. Screening for Monoclonal Gammopathy of Undetermined Significance: A Population-Based Randomized Clinical Trial. First Results from the Iceland Screens, Treats, or Prevents Multiple Myeloma (iStopMM) Study. Blood 2021, 138 (Suppl. 1), 156. [Google Scholar] [CrossRef]
- Fermand, J.P.; Bridoux, F.; Dispenzieri, A.; Jaccard, A.; Kyle, R.A.; Leung, N.; Merlini, G. Monoclonal gammopathy of clinical significance: A novel concept with therapeutic implications. Blood 2018, 132, 1478–1485. [Google Scholar] [CrossRef]
- Khwaja, J.; D’Sa, S.; Minnema, M.C.; Kersten, M.J.; Wechalekar, A.; Vos, J.M. IgM monoclonal gammopathies of clinical significance: Diagnosis and management. Haematologica 2022, 107, 2037–2050. [Google Scholar] [CrossRef]
- Castillo, J.J.; Callander, N.S.; Baljevic, M.; Sborov, D.W.; Kumar, S. The evaluation and management of monoclonal gammopathy of renal significance and monoclonal gammopathy of neurological significance. Am. J. Hematol. 2021, 96, 846–853. [Google Scholar] [CrossRef]
- Kyle, R.A.; Therneau, T.M.; Rajkumar, S.V.; Remstein, E.D.; Offord, J.R.; Larson, D.R.; Plevak, M.F.; Melton, L.J. Long-term follow-up of IgM monoclonal gammopathy of undetermined significance. Blood 2003, 102, 3759–3764. [Google Scholar] [CrossRef]
- Hanewinckel, R.; van Oijen, M.; Ikram, M.A.; van Doorn, P.A. The epidemiology and risk factors of chronic polyneuropathy. Eur. J. Epidemiol. 2016, 31, 5–20. [Google Scholar] [CrossRef]
- Baldereschi, M.; Inzitari, M.; di Carlo, A.; Farchi, G.; Scafato, E.; Inzitari, D. Epidemiology of distal symmetrical neuropathies in the Italian elderly. Neurology 2007, 68, 1460–1467. [Google Scholar] [CrossRef] [PubMed]
- Nobile-Orazio, E.; Barbieri, S.; Baldini, L.; Marmiroli, P.; Carpo, M.; Premoselli, S.; Manfredini, E.; Scarlato, G. Peripheral neuropathy in monoclonal gammopathy of undetermined significance: Prevalence and immunopathogenetic studies. Acta Neurol. Scand. 1992, 85, 383–390. [Google Scholar] [CrossRef]
- Vrethem, M.; Cruz, M.; Wen-Xin, H.; Malm, C.; Holmgren, H.; Ernerudh, J. Clinical, neurophysiological and immunological evidence of polyneuropathy in patients with monoclonal gammopathies. J. Neurol. Sci. 1993, 114, 193–199. [Google Scholar] [CrossRef]
- Steiner, N.; Schwärzler, A.; Göbel, G.; Löscher, W.; Wanschitz, J.; Gunsilius, E. Are neurological complications of monoclonal gammopathy of undetermined significance underestimated? Oncotarget 2017, 8, 5081–5091. [Google Scholar] [CrossRef]
- Rögnvaldsson, S.; Steingrímsson, V.; Turesson, I.; Björkholm, M.; Landgren, O.; Kristinsson, S.Y. Peripheral neuropathy and monoclonal gammopathy of undetermined significance: A population-based study including 15,351 cases and 58,619 matched controls. Haematologica 2020, 105, 2679–2681. [Google Scholar] [CrossRef]
- Kelly, J.J.; Kyle, R.A.; O’Brien, P.C.; Dyck, P.J. Prevalence of monoclonal protein in peripheral neuropathy. Neurology 1981, 31, 1480. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, S.; Kyle, R.A.; Dyck, P.J. Neuropathy associated with monoclonal gammopathies of undetermined significance. Ann. Neurol. 1991, 30, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Baldini, L.; Nobile-Orazio, E.; Guffanti, A.; Barbieri, S.; Carpo, M.; Cro, L.; Cesana, B.; Damilano, I.; Maiolo, A.T. Peripheral neuropathy in IgM monoclonal gammopathy and Wäldenstrom’s macroglobulinemia: A frequent complication in elderly males with low MAG-reactive serum monoclonal component. Am. J. Hematol. 1994, 45, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Latov, N.; Sherman, W.H.; Nemni, R.; Galassi, G.; Shyong, J.S.; Penn, A.S.; Chess, L.; Olarte, M.R.; Rowland, L.P.; Osserman, E.F. Plasma-cell dyscrasia and peripheral neuropathy with a monoclonal antibody to peripheral-nerve myelin. N. Engl. J. Med. 1980, 303, 618–621. [Google Scholar] [CrossRef] [PubMed]
- Lunn, M.P. Neuropathies and paraproteins. Curr. Opin. Neurol. 2019, 32, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, E.; Marks, D.; Raza, S. Diagnosis and management of neuropathies associated with plasma cell dyscrasias. Hematol. Oncol. 2018, 36, 3–14. [Google Scholar] [CrossRef]
- Vallat, J.M.; Duchesne, M.; Corcia, P.; Richard, L.; Ghorab, K.; Magy, L.; Mathis, S. The Wide Spectrum of Pathophysiologic Mechanisms of Paraproteinemic Neuropathy. Neurology 2021, 96, 214–225. [Google Scholar] [CrossRef]
- Latov, N. Antibody testing in neuropathy associated with anti-Myelin-Associated Glycoprotein antibodies: Where we are after 40 years. Curr. Opin. Neurol. 2021, 34, 625–630. [Google Scholar] [CrossRef]
- Dalakas, M.C. Advances in the diagnosis, immunopathogenesis and therapies of IgM-anti-MAG antibody-mediated neuropathies. Ther. Adv. Neurol. Disord. 2018, 11, 1756285617746640. [Google Scholar] [CrossRef]
- Steck, A.J. Anti-MAG neuropathy: From biology to clinical management. J. Neuroimmunol. 2021, 361, 577725. [Google Scholar] [CrossRef]
- Varettoni, M.; Zibellini, S.; Capello, D.; Arcaini, L.; Rossi, D.; Pascutto, C.; Rattotti, S.; Mangiacavalli, S.; Pochintesta, L.; Gotti, M.; et al. Clues to pathogenesis of Waldenström macroglobulinemia and immunoglobulin M monoclonal gammopathy of undetermined significance provided by analysis of immunoglobulin heavy chain gene rearrangement and clustering of B-cell receptors. Leuk. Lymphoma 2013, 54, 2485–2489. [Google Scholar] [CrossRef]
- Paiva, B.; Corchete, L.A.; Vidriales, M.B.; García-Sanz, R.; Perez, J.J.; Aires-Mejia, I.; Sanchez, M.-L.; Barcena, P.; Alignani, D.; Jimenez, C.; et al. The cellular origin and malignant transformation of Waldenström macroglobulinemia. Blood 2015, 125, 2370–2380. [Google Scholar] [CrossRef]
- García-Sanz, R.; Dogliotti, I.; Zaccaria, G.M.; Ocio, E.M.; Rubio, A.; Murillo, I.; Escalante, F.; Aguilera, C.; García-Mateo, A.; de Coca, A.G.; et al. 6q deletion in Waldenström macroglobulinaemia negatively affects time to transformation and survival. Br. J. Haematol. 2021, 192, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Treon, S.P.; Xu, L.; Yang, G.; Zhou, Y.; Liu, X.; Cao, Y.; Sheehy, P.; Manning, R.J.; Patterson, C.J.; Tripsas, C.; et al. MYD88 L265P Somatic Mutation in Waldenström’s Macroglobulinemia. N. Engl. J. Med. 2012, 367, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, W.; Deng, Q.; Li, L.; Hsi, E.D.; Young, K.H.; Zhang, M.; Li, Y. MYD88 L265P Mutation in Lymphoid Malignancies. Cancer Res. 2018, 78, 2457–2462. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Hunter, Z.R.; Yang, G.; Zhou, Y.; Cao, Y.; Liu, X.; Morra, E.; Trojani, A.; Greco, A.; Arcaini, L.; et al. MYD88 L265P in Waldenström macroglobulinemia, immunoglobulin M monoclonal gammopathy, and other B-cell lymphoproliferative disorders using conventional and quantitative allele-specific polymerase chain reaction. Blood 2013, 121, 2051–2058. [Google Scholar] [CrossRef]
- Cao, X.X.; Meng, Q.; Cai, H.; He, T.H.; Zhang, C.-L.; Su, W.; Sun, J.; Li, Y.; Xu, W.; Zhou, D.-B.; et al. Detection of MYD88 L265P and WHIM-like CXCR4 mutation in patients with IgM monoclonal gammopathy related disease. Ann. Hematol. 2017, 96, 971–976. [Google Scholar] [CrossRef]
- Ferrante, M.; Furlan, D.; Zibellini, S.; Borriero, M.; Candido, C.; Sahnane, N.; Uccella, S.; Genuardi, E.; Alessandria, B.; Bianchi, B.; et al. MYD88L265P Detection in IgM Monoclonal Gammopathies: Methodological Considerations for Routine Implementation. Diagnostics 2021, 11, 779. [Google Scholar] [CrossRef]
- Drandi, D.; Decruyenaere, P.; Ferrante, M.; Offner, F.; Vandesompele, J.; Ferrero, S. Nucleic Acid Biomarkers in Waldenström Macroglobulinemia and IgM-MGUS: Current Insights and Clinical Relevance. Diagnostics 2022, 12, 969. [Google Scholar] [CrossRef]
- Jiménez, C.; Sebastián, E.; Chillón, M.C.; Giraldo, P.; Mariano Hernández, J.; Escalante, F.; González-López, T.J.; Aguilera, C.; de Coca, A.G.; Murillo, I.; et al. MYD88 L265P is a marker highly characteristic of, but not restricted to, Waldenström’s macroglobulinemia. Leukemia 2013, 27, 1722–1728. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Kuzu, I.; Dogan, A.; Dirnhofer, S.; Chan, J.K.C.; Sander, B.; Ott, G.; Xerri, L.; Quintanilla-Martinez, L.; Campo, E. The many faces of small B cell lymphomas with plasmacytic differentiation and the contribution of MYD88 testing. Virchows Arch. 2016, 468, 259–275. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen Recognition and Innate Immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate Immune Pattern Recognition: A Cell Biological Perspective. Annu. Rev. Immunol. 2015, 33, 257–290. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.C.; Lo, Y.C.; Wu, H. Helical assembly in the MyD88–IRAK4–IRAK2 complex in TLR/IL-1R signalling. Nature 2010, 465, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Motshwene, P.G.; Moncrieffe, M.C.; Grossmann, J.G.; Kao, C.; Ayaluru, M.; Sandercock, A.M.; Sandercock, A.M.; Robinson, C.V.; Latz, E.; Gay, N.J. An Oligomeric Signaling Platform Formed by the Toll-like Receptor Signal Transducers MyD88 and IRAK-4. J. Biol. Chem. 2009, 284, 25404–25411. [Google Scholar] [CrossRef]
- Balka, K.R.; Nardo, D. Understanding early TLR signaling through the Myddosome. J. Leukoc. Biol. 2019, 105, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Visentin, A.; Pravato, S.; Castellani, F.; Campagnolo, M.; Angotzi, F.; Cavarretta, C.A.; Cellini, A.; Ruocco, V.; Salvalaggio, A.; Tedeschi, A.; et al. From Biology to Treatment of Monoclonal Gammopathies of Neurological Significance. Cancers 2022, 14, 1562. [Google Scholar] [CrossRef]
- Panwalkar, A.; Verstovsek, S.; Giles, F. Nuclear factor-KappaB modulation as a therapeutic approach in hematologic malignancies. Cancer 2004, 100, 1578–1589. [Google Scholar] [CrossRef]
- Leleu, X.; Eeckhoute, J.; Jia, X.; Roccaro, A.M.; Moreau, A.S.; Farag, M.; Sacco, A.; Ngo, H.T.; Runnels, J.; Melhem, M.R.; et al. Targeting NF-κB in Waldenstrom macroglobulinemia. Blood 2008, 111, 5068–5077. [Google Scholar] [CrossRef]
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119. [Google Scholar] [CrossRef]
- Yang, G.; Buhrlage, S.J.; Tan, L.; Liu, X.; Chen, J.; Xu, L.; Tsakmaklis, N.; Chen, J.G.; Patterson, C.J.; Brown, J.R.; et al. HCK is a survival determinant transactivated by mutated MYD88, and a direct target of ibrutinib. Blood 2016, 127, 3237–3252. [Google Scholar] [CrossRef]
- Pontoriero, M.; Fiume, G.; Vecchio, E.; de Laurentiis, A.; Albano, F.; Iaccino, E.; Mimmi, S.; Pisano, A.; Agosti, V.; Giovannone, E.; et al. Activation of NF-κB in B cell receptor signaling through Bruton’s tyrosine kinase-dependent phosphorylation of IκB-α. J. Mol. Med. 2019, 97, 675–690. [Google Scholar] [CrossRef]
- Treon, S.P.; Tripsas, C.K.; Meid, K.; Warren, D.; Varma, G.; Green, R.; Argyropoulos, K.V.; Yang, G.; Cao, Y.; Xu, L.; et al. Ibrutinib in Previously Treated Waldenström’s Macroglobulinemia. N. Engl. J. Med. 2015, 372, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Castellani, F.; Visentin, A.; Campagnolo, M.; Salvalaggio, A.; Cacciavillani, M.; Candiotto, C.; Bertorelle, R.; Trentin, L.; Briani, C. The Bruton tyrosine kinase inhibitor ibrutinib improves anti-MAG antibody polyneuropathy. Neurol.-Neuroimmunol. Neuroinflammat. 2020, 7, e720. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Chien, E.Y.T.; Mol, C.D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Bi, F.C.; et al. Structures of the CXCR4 Chemokine GPCR with Small-Molecule and Cyclic Peptide Antagonists. Science 2010, 330, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.J.; Xu, L.; Gustine, J.N.; Keezer, A.; Meid, K.; Dubeau, T.E.; Liu, X.; Demos, M.G.; Kofides, A.; Tsakmaklis, N.; et al. CXCR4 mutation subtypes impact response and survival outcomes in patients with Waldenström macroglobulinaemia treated with ibrutinib. Br. J. Haematol. 2019, 187, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Allain, J.S.; Thonier, F.; Pihan, M.; Boulland, M.L.; de Guibert, S.; Launay, V.; Doncker, A.-V.; Ganard, M.; Aliouat, A.; Pangault, C.; et al. IGHV segment utilization in immunoglobulin gene rearrangement differentiates patients with anti-myelin-associated glycoprotein neuropathy from others immunoglobulin M-gammopathies. Haematologica 2018, 103, e207–e210. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Fricker, S.P. CXCL12 (SDF-1)/CXCR4 Pathway in Cancer. Clin. Cancer Res. 2010, 16, 2927–2931. [Google Scholar] [CrossRef]
- Bianchi, M.E.; Mezzapelle, R. The Chemokine Receptor CXCR4 in Cell Proliferation and Tissue Regeneration. Front. Immunol. 2020, 11, 2109. [Google Scholar] [CrossRef]
- Kryczek, I.; Wei, S.; Keller, E.; Liu, R.; Zou, W. Stroma-derived factor (SDF-1/CXCL12) and human tumor pathogenesis. Am. J. Physiol.-Cell Physiol. 2007, 292, C987–C995. [Google Scholar] [CrossRef]
- Xu, L.; Hunter, Z.R.; Tsakmaklis, N.; Cao, Y.; Yang, G.; Chen, J.; Liu, X.; Kanan, S.; Castillo, J.J.; Tai, Y.-T.; et al. Clonal architecture of CXCR4 WHIM-like mutations in Waldenström Macroglobulinaemia. Br. J. Haematol. 2016, 172, 735–744. [Google Scholar] [CrossRef]
- Cao, Y.; Hunter, Z.R.; Liu, X.; Xu, L.; Yang, G.; Chen, J.; Patterson, C.J.; Tsakmaklis, N.; Kanan, S.; Rodig, S.; et al. The WHIM-like CXCR4S338X somatic mutation activates AKT and ERK, and promotes resistance to ibrutinib and other agents used in the treatment of Waldenstrom’s Macroglobulinemia. Leukemia 2015, 29, 169–176. [Google Scholar] [CrossRef]
- Garbay, B.; Heape, A.M.; Sargueil, F.; Cassagne, C. Myelin synthesis in the peripheral nervous system. Prog. Neurobiol. 2000, 61, 267–304. [Google Scholar] [CrossRef]
- Kidd, G.J.; Ohno, N.; Trapp, B.D. Chapter 5-Biology of Schwann cells. In Handbook of Clinical Neurology; Said, G., Krarup, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 55–79. [Google Scholar]
- Saher, G.; Simons, M. Cholesterol and Myelin Biogenesis. In Cholesterol Binding and Cholesterol Transport Proteins: Structure and Function in Health and Disease; Harris, J.R., Ed.; Springer: Dordrecht, The Netherlands, 2010; pp. 489–508. [Google Scholar]
- Patzig, J.; Jahn, O.; Tenzer, S.; Wichert, S.P.; de Monasterio-Schrader, P.; Rosfa, S.; Kuharev, J.; Yan, K.; Bormuth, I.; Bremer, J.; et al. Quantitative and Integrative Proteome Analysis of Peripheral Nerve Myelin Identifies Novel Myelin Proteins and Candidate Neuropathy Loci. J. Neurosci. 2011, 31, 16369. [Google Scholar] [CrossRef] [PubMed]
- Salzer, J.L. Polarized Domains of Myelinated Axons. Neuron 2003, 40, 297–318. [Google Scholar] [CrossRef]
- D’Sa, S.; Kersten, M.J.; Castillo, J.J.; Dimopoulos, M.; Kastritis, E.; Laane, E.; Leblond, V.; Merlini, G.; Treon, S.P.; Vos, J.M.; et al. Investigation and management of IgM and Waldenström-associated peripheral neuropathies: Recommendations from the IWWM-8 consensus panel. Br. J. Haematol. 2017, 176, 728–742. [Google Scholar] [CrossRef]
- Dellagi, K.; Dupouey, P.; Brouet, J.C.; Billecocq, A.; Gomez, D.; Clauvel, J.P.; Seligmann, M. Waldenström’s macroglobulinemia and peripheral neuropathy: A clinical and immunologic study of 25 patients. Blood 1983, 62, 280–285. [Google Scholar] [CrossRef]
- Braun, P.E.; Frail, D.E.; Latov, N. Myelin-associated glycoprotein is the antigen for a monoclonal IgM in polyneuropathy. J. Neurochem. 1982, 39, 1261–1265. [Google Scholar] [CrossRef]
- Leibowitz, S.; Gregson, N.A.; Kennedy, M.; Kahn, S.N. IgM paraproteins with immunological specificity for a Schwann cell component and peripheral nerve myelin in patients with polyneuropathy. J. Neurol. Sci. 1983, 59, 153–165. [Google Scholar] [CrossRef]
- Kahn, S.N.; Smith, I.A.; Eames, R.A.; Thomas, P.K.; Lacey, B.W. IgM paraproteinemia and autoimmune peripheral neuropathy. N. Engl. J. Med. 1981, 304, 1430–1431. [Google Scholar]
- Ilyas, A.A.; Quarles, R.H.; Dalakas, M.C.; Brady, R.O. Polyneuropathy with monoclonal gammopathy: Glycolipids are frequently antigens for IgM paraproteins. Proc. Natl. Acad. Sci. USA 1985, 82, 6697–6700. [Google Scholar] [CrossRef]
- Ilyas, A.A.; Li, S.C.; Chou, D.K.; Li, Y.T.; Jungalwala, F.B.; Dalakas, M.C.; Quarles, R.H. Gangliosides GM2, IV4GalNAcGM1b, and IV4GalNAcGC1a as antigens for monoclonal immunoglobulin M in neuropathy associated with gammopathy. J. Biol. Chem. 1988, 263, 4369–4373. [Google Scholar] [CrossRef]
- Kelly, J.J.; Adelman, L.S.; Berkman, E.; Bhan, I. Polyneuropathies associated with IgM monoclonal gammopathies. Arch. Neurol. 1988, 45, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Dispenzieri, A. POEMS syndrome: 2021 Update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2021, 96, 872–888. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.R.; Warren, J.D.; Jacobs, J.M.; Groves, M.J.; Yong, K.; Honan, W.P.; Thomas, P.K.; Reilly, A.M.M. Microvasculitic paraproteinaemic polyneuropathy and B-cell lymphoma. J. Peripher. Nerv. Syst. 2003, 8, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A. Acute hyperviscosity: Syndromes and management. Blood 2018, 132, 1379–1385. [Google Scholar] [CrossRef]
- Ilyas, A.A.; Quarles, R.H.; MacIntosh, T.D.; Dobersen, M.J.; Trapp, B.D.; Dalakas, M.C.; Brady, R.O. IgM in a human neuropathy related to paraproteinemia binds to a carbohydrate determinant in the myelin-associated glycoprotein and to a ganglioside. Proc. Natl. Acad. Sci. USA 1984, 81, 1225–1229. [Google Scholar] [CrossRef]
- Quarles, R.H.; Ilyas, A.A.; Willison, H.J. Antibodies to glycolipids in demyelinating diseases of the human peripheral nervous system. Chem. Phys. Lipids 1986, 42, 235–248. [Google Scholar] [CrossRef]
- Burger, D.; Simon, M.; Perruisseau, G.; Steck, A.J. The epitope(s) recognized by HNK-1 antibody and IgM paraprotein in neuropathy is present on several N-linked oligosaccharide structures on human P0 and myelin-associated glycoprotein. J. Neurochem. 1990, 54, 1569–1575. [Google Scholar] [CrossRef]
- Willison, H.J.; O’Leary, C.P.; Veitch, J.; Blumhardt, L.D.; Busby, M.; Donaghy, M.; Fuhr, P.; Ford, H.; Hahn, A.; Renaud, S.; et al. The clinical and laboratory features of chronic sensory ataxic neuropathy with anti-disialosyl IgM antibodies. Brain 2001, 124 Pt 10, 1968–1977. [Google Scholar] [CrossRef]
- Ilyas, A.A.; Quarles, R.H.; Dalakas, M.C.; Fishman, P.H.; Brady, R.O. Monoclonal IgM in a patient with paraproteinemic polyneuropathy binds to gangliosides containing disialosyl groups. Ann. Neurol. 1985, 18, 655–659. [Google Scholar] [CrossRef]
- Carpo, M.; Meucci, N.; Allaria, S.; Marmiroli, P.; Monaco, S.; Toscano, A.; Simonetti, S.; Scarlato, G.; Nobile-Orazio, E. Anti-sulfatide IgM antibodies in peripheral neuropathy. J. Neurol. Sci. 2000, 176, 144–150. [Google Scholar] [CrossRef]
- Pestronk, A.; Li, F.; Griffin, J.; Feldman, E.L.; Cornblath, D.; Trotter, J.; Zhu, S.; Yee, W.C.; Phillips, D.; Peeples, D.M.; et al. Polyneuropathy syndromes associated with serum antibodies to sulfatide and myelin-associated glycoprotein. Neurology 1991, 41, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Dabby, R.; Weimer, L.H.; Hays, A.P.; Olarte, M.; Latov, N. Antisulfatide antibodies in neuropathy: Clinical and electrophysiologic correlates. Neurology 2000, 54, 1448–1452. [Google Scholar] [CrossRef] [PubMed]
- Petratos, S.; Turnbull, V.J.; Papadopoulos, R.; Ayers, M.; Gonzales, M.F. High-titre IgM anti-sulfatide antibodies in individuals with IgM paraproteinaemia and associated peripheral neuropathy. Immunol. Cell Biol. 2000, 78, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Boso, F.; Ruggero, S.; Giannotta, C.; Benedetti, L.; Marfia, G.A.; Ermani, M.; Campagnolo, M.; Salvalaggio, A.; Gallia, F.; De Michelis, C.; et al. Anti-sulfatide/galactocerebroside antibodies in immunoglobulin M paraproteinemic neuropathies. Eur. J. Neurol. 2017, 24, 1334–1340. [Google Scholar] [CrossRef]
- Willison, H.J. Anti-ganglioside Antibodies in Peripheral Nerve Pathology. In Gangliosides: Methods and Protocols; Sonnino, S., Prinetti, A., Eds.; Springer: New York, NY, USA, 2018; pp. 173–188. [Google Scholar]
- Briani, C.; Berger, J.S.; Latov, N. Antibodies to chondroitin sulfate C: A new detection assay and correlations with neurological diseases. J. Neuroimmunol. 1998, 84, 117–121. [Google Scholar] [CrossRef]
- Matà, S.; Torricelli, S.; Barilaro, A.; Grippo, A.; Forleo, P.; del Mastio, M.; Sorbi, S. Polyneuropathy and monoclonal gammopathy of undetermined significance (MGUS); update of a clinical experience. J. Neurol. Sci. 2021, 423, 117335. [Google Scholar] [CrossRef]
- Nobile-Orazio, E.; Manfredini, E.; Carpo, M.; Meucci, N.; Monaco, S.; Ferrari, S.; Bonetti, B.; Cavaletti, G.; Gemignani, F.; Durelli, L.; et al. Frequency and clinical correlates of anti-neural IgM antibodies in neuropathy associated with IgM monoclonal gammopathy. Ann. Neurol. 1994, 36, 416–424. [Google Scholar] [CrossRef]
- Ellie, E.; Vital, A.; Steck, A.; Boiron, J.M.; Vital, C.; Julien, J. Neuropathy associated with “benign” anti-myelin-associated glycoprotein IgM gammopathy: Clinical, immunological, neurophysiological pathological findings and response to treatment in 33 cases. J. Neurol. 1996, 243, 34–43. [Google Scholar] [CrossRef]
- Chassande, B.; Léger, J.M.; Younes-Chennoufi, A.B.; Bengoufa, D.; Maisonobe, T.; Bouche, P.; Baumann, N. Peripheral neuropathy associated with IgM monoclonal gammopathy: Correlations between M-protein antibody activity and clinical/electrophysiological features in 40 cases. Muscle Nerve 1998, 21, 55–62. [Google Scholar] [CrossRef]
- Katz, J.S.; Saperstein, D.S.; Gronseth, G.; Amato, A.A.; Barohn, R.J. Distal acquired demyelinating symmetric neuropathy. Neurology 2000, 54, 615–620. [Google Scholar] [CrossRef]
- Bardel, B.; Molinier-Frenkel, V.; le Bras, F.; Ayache, S.S.; Nordine, T.; Lefaucheur, J.P.; Planté-Bordeneuve, V. Revisiting the spectrum of IgM-related neuropathies in a large cohort of IgM monoclonal gammopathy. J. Neurol. 2022, 269, 4955–4960. [Google Scholar] [CrossRef] [PubMed]
- Nobile-Orazio, E.; Francomano, E.; Daverio, R.; Barbieri, S.; Marmiroli, P.; Manfredini, E.; Carpo, M.; Moggio, M.; Legname, G.; Baldini, L.; et al. Anti-myelin-associated glycoprotein IgM antibody titers in neuropathy associated with macroglobulinemia. Ann. Neurol. 1989, 26, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Kuijf, M.L.; Eurelings, M.; Tio-Gillen, A.P.; van Doorn, P.A.; van den Berg, L.H.; Hooijkaas, H.; Stork, J.; Notermans, N.C.; Jacobs, B.C. Detection of anti-MAG antibodies in polyneuropathy associated with IgM monoclonal gammopathy. Neurology 2009, 73, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Jaskowski, T.D.; Martins, T.B.; Litwin, C.M.; Hill, H.R. Immunoglobulin (Ig) M antibody against myelin associated glycoprotein (MAG): A comparison of methods. J. Clin. Lab. Anal. 2004, 18, 247–250. [Google Scholar] [CrossRef]
- Jaskowski, T.D.; Prince, H.E.; Greer, R.W.; Litwin, C.M.; Hill, H.R. Further comparisons of assays for detecting MAG IgM autoantibodies. J. Neuroimmunol. 2007, 187, 175–178. [Google Scholar] [CrossRef]
- Matà, S.; Ambrosini, S.; Saccomanno, D.; Biagioli, T.; Carpo, M.; Amantini, A.; Giannini, F.; Barilaro, A.; Toscani, L.; Del Mastio, M.; et al. Anti-MAG IgM: Differences in antibody tests and correlation with clinical findings. Neurol. Sci. 2020, 41, 365–372. [Google Scholar] [CrossRef]
- Liberatore, G.; Giannotta, C.; Sajeev, B.P.; Morenghi, E.; Terenghi, F.; Gallia, F.; Doneddu, P.E.; Manganelli, F.; Cocito, D.; Filosto, M.; et al. Sensitivity and specificity of a commercial ELISA test for anti-MAG antibodies in patients with neuropathy. J. Neuroimmunol. 2020, 345, 577288. [Google Scholar] [CrossRef]
- Svahn, J.; Petiot, P.; Antoine, J.C.; Vial, C.; Delmont, E.; Viala, K.; Steck, A.J.; Magot, A.; Cauquil, C.; Zarea, A.; et al. Anti-MAG antibodies in 202 patients: Clinicopathological and therapeutic features. J. Neurol. Neurosurg. Psychiatry 2018, 89, 499–505. [Google Scholar] [CrossRef]
- Magy, L.; Kaboré, R.; Mathis, S.; Lebeau, P.; Ghorab, K.; Caudie, C.; Vallat, J.-M. Heterogeneity of Polyneuropathy Associated with Anti-MAG Antibodies. J. Immunol. Res. 2015, 2015, 450391. [Google Scholar] [CrossRef]
- Hafler, D.A.; Johnson, D.; Kelly, J.J.; Panitch, H.; Kyle, R.; Weiner, H.L. Monoclonal gammopathy and neuropathy: Myelin-associated glycoprotein reactivity and clinical characteristics. Neurology 1986, 36, 75–78. [Google Scholar] [CrossRef]
- Smith, I.S. The natural history of chronic demyelinating neuropathy associated with benign IgM paraproteinaemia. A clinical and neurophysiological study. Brain 1994, 117 Pt 5, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Galassi, G.; Tondelli, M.; Ariatti, A.; Benuzzi, F.; Nichelli, P.; Valzania, F. Long-term disability and prognostic factors in polyneuropathy associated with anti-myelin-associated glycoprotein (MAG) antibodies. Int. J. Neurosci. 2017, 127, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Saifee, T.A.; Schwingenschuh, P.; Reilly, M.M.; Lunn, M.P.; Katschnig, P.; Kassavetis, P.; Pareés, I.; Manji, H.; Bhatia, K.; Rothwell, J.C.; et al. Tremor in inflammatory neuropathies. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1282–1287. [Google Scholar] [CrossRef] [PubMed]
- Ahlskog, M.C.; Kumar, N.; Mauermann, M.L.; Klein, C.J. IgM-monoclonal gammopathy neuropathy and tremor: A first epidemiologic case control study. Park. Relat. Disord. 2012, 18, 748–752. [Google Scholar] [CrossRef]
- Bain, P.G.; Britton, T.C.; Jenkins, I.H.; Thompson, P.D.; Rothwell, J.C.; Thomas, P.K.; Brooks, D.; Marsden, C.D. Tremor associated with benign IgM paraproteinaemic neuropathy. Brain 1996, 119 Pt 3, 789–799. [Google Scholar] [CrossRef]
- Nobile-Orazio, E.; Meucci, N.; Baldini, L.; di Troia, A.; Scarlato, G. Long-term prognosis of neuropathy associated with anti-MAG IgM M-proteins and its relationship to immune therapies. Brain 2000, 123 Pt 4, 710–717. [Google Scholar] [CrossRef]
- Joint Task Force of the EFNS and The PNS. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of multifocal motor neuropathy. Report of a Joint Task Force of the European Federation of Neurological Societies and the Peripheral Nerve Society-first revision. J. Peripher. Nerv. Syst. 2010, 15, 295–301. [Google Scholar] [CrossRef]
- Kelly, J.J., Jr. The electrodiagnostic findings in polyneuropathies associated with IgM monoclonal gammopathies. Muscle Nerve 1990, 13, 1113–1117. [Google Scholar] [CrossRef]
- Goedee, H.S.; Notermans, N.C.; Visser, L.H.; van Asseldonk, J.H.; Franssen, H.; Vrancken, A.; Nikolakopoulos, S.; Berg, L.H.V.D.; Pol, W.L. Neuropathy associated with immunoglobulin M monoclonal gammopathy: A combined sonographic and nerve conduction study. Muscle Nerve 2019, 60, 263–270. [Google Scholar] [CrossRef]
- Franssen, H.; Notermans, N.C. Length dependence in polyneuropathy associated with IgM gammopathy. Ann. Neurol. 2006, 59, 365–371. [Google Scholar] [CrossRef]
- Kaku, D.A.; England, J.D.; Sumner, A.J. Distal accentuation of conduction slowing in polyneuropathy associated with antibodies to myelin-associated glycoprotein and sulphated glucuronyl paragloboside. Brain 1994, 117, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Lozeron, P.; Ribrag, V.; Adams, D.; Brisset, M.; Vignon, M.; Baron, M.; Malphettes, M.; Theaudin, M.; Arnulf, B.; Kubis, N. Is distal motor and/or sensory demyelination a distinctive feature of anti-MAG neuropathy? J. Neurol. 2016, 263, 1761–1770. [Google Scholar] [CrossRef] [PubMed]
- Faber, C.G.; Notermans, N.C.; Wokke, J.H.J.; Franssen, H. Entrapment in anti myelin-associated glycoprotein neuropathy. J. Neurol. 2009, 256, 620. [Google Scholar] [CrossRef] [PubMed]
- Bourque, P.R.; Masson-Roy, J.; Warman-Chardon, J.; Massie, R.; Melanson, M.; Brooks, J.; Breiner, A. Temporal evolution of nerve conduction study abnormalities in anti-myelin-associated glycoprotein neuropathy. Muscle Nerve 2021, 63, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Trapp, B.D.; Quarles, R.H. Presence of the myelin-associated glycoprotein correlates with alterations in the periodicity of peripheral myelin. J. Cell Biol. 1982, 92, 877–882. [Google Scholar] [CrossRef]
- Erb, M.; Flueck, B.; Kern, F.; Erne, B.; Steck, A.J.; Schaeren-Wiemers, N. Unraveling the differential expression of the two isoforms of myelin-associated glycoprotein in a mouse expressing GFP-tagged S-MAG specifically regulated and targeted into the different myelin compartments. Mol. Cell. Neurosci. 2006, 31, 613–627. [Google Scholar] [CrossRef]
- Lopez, P.H.H. Role of Myelin-Associated Glycoprotein (Siglec-4a) in the Nervous System. In Glycobiology of the Nervous System; Yu, R.K., Schengrund, C.L., Eds.; Springer: New York, NY, USA, 2014; pp. 245–262. [Google Scholar]
- Yin, X.; Crawford, T.O.; Griffin, J.W.; Tu, P.H.; Lee, V.M.Y.; Li, C.; Order, J.; Trapp, B.D. Myelin-Associated Glycoprotein Is a Myelin Signal that Modulates the Caliber of Myelinated Axons. J. Neurosci. 1998, 18, 1953. [Google Scholar] [CrossRef]
- Nguyen, T.; Mehta, N.R.; Conant, K.; Kim, K.J.; Jones, M.; Calabresi, P.A.; Melli, G.; Hoke, A.; Schnaar, R.; Ming, G.-L.; et al. Axonal Protective Effects of the Myelin-Associated Glycoprotein. J. Neurosci. 2009, 29, 630. [Google Scholar] [CrossRef]
- Weiss, M.D.; Luciano, C.A.; Quarles, R.H. Nerve conduction abnormalities in aging mice deficient for myelin-associated glycoprotein. Muscle Nerve 2001, 24, 1380–1387. [Google Scholar] [CrossRef]
- Quarles, R.H. Myelin-associated glycoprotein (MAG): Past, present and beyond. J. Neurochem. 2007, 100, 1431–1448. [Google Scholar] [CrossRef]
- Kelm, S.; Pelz, A.; Schauer, R.; Filbin, M.T.; Tang, S.; de Bellard, M.E.; Schnaar, R.L.; Mahoney, J.A.; Hartnell, A.; Bradfield, P.; et al. Sialoadhesin, myelin-associated glycoprotein and CD22 define a new family of sialic acid-dependent adhesion molecules of the immunoglobulin superfamily. Curr. Biol. 1994, 4, 965–972. [Google Scholar] [CrossRef]
- Crocker, P.R.; Clark, E.A.; Filbin, M.; Gordon, S.; Jones, Y.; Kehrl, J.H.; Kelm, S.; Le Douarin, N.; Powell, L.; Roder, J.; et al. Siglecs: A family of sialic-acid binding lectins. Glycobiology 1998, 8, 5–6. [Google Scholar]
- Chester, M.A. IUPAC-IUB Joint Commission on Biochemical Nomenclature (JCBN) Nomenclature of glycolipids. Eur. J. Biochem. 1998, 257, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Pronker, M.F.; Lemstra, S.; Snijder, J.; Heck, A.J.R.; Thies-Weesie, D.M.E.; Pasterkamp, R.J.; Janssen, B.J.C. Structural basis of myelin-associated glycoprotein adhesion and signalling. Nat. Commun. 2016, 7, 13584. [Google Scholar] [CrossRef]
- Takatsu, M.; Hays, A.P.; Latov, N.; Abrams, G.M.; Nemni, R.; Sherman, W.H.; Nobile-Orazio, E.; Saito, T.; Freddo, L. Immunofluorescence study of patients with neuropathy and IgM M proteins. Ann. Neurol. 1985, 18, 173–181. [Google Scholar] [CrossRef]
- Vital, A.; Vital, C.; Julien, J.; Baquey, A.; Steck, A.J. Polyneuropathy associated with IgM monoclonal gammopathy. Acta Neuropathol. 1989, 79, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, J.M.; Erne, B.; Bernasconi, L.; Tosi, C.; Probst, A.; Landmann, L.; Steck, A.J. Confocal microscopic localization of anti-myelin-associated glycoprotein autoantibodies in a patient with peripheral neuropathy initially lacking a detectable IgM gammopathy. Acta Neuropathol. 1998, 95, 540–546. [Google Scholar] [CrossRef]
- Kawagashira, Y.; Koike, H.; Tomita, M.; Morozumi, S.; Iijima, M.; Nakamura, T.; Katsuno, M.; Tanaka, F.; Sobue, G. Morphological Progression of Myelin Abnormalities in IgM-Monoclonal Gammopathy of Undetermined Significance Anti-Myelin-Associated Glycoprotein Neuropathy. J. Neuropathol. Exp. Neurol. 2010, 69, 1143–1157. [Google Scholar] [CrossRef]
- Vital, A.; Lagueny, A.; Julien, J.; Ferrer, X.; Barat, M.; Hermosilla, E.; Rouanet-Larrivière, M.; Henry, P.; Bredin, A.; Louiset, P.; et al. Chronic inflammatory demyelinating polyneuropathy associated with dysglobulinemia: A peripheral nerve biopsy study in 18 cases. Acta Neuropathol. 2000, 100, 63–68. [Google Scholar] [CrossRef]
- Monaco, S.; Bonetti, B.; Ferrari, S.; Moretto, G.; Nardelli, E.; Tedesco, F.; Mollnes, T.E.; Nobile-Orazio, E.; Manfredini, E.; Bonazzi, L.; et al. Complement-mediated demyelination in patients with IgM monoclonal gammopathy and polyneuropathy. N. Engl. J. Med. 1990, 322, 649–652. [Google Scholar] [CrossRef]
- Lunn, M.P.; Crawford, T.O.; Hughes, R.A.; Griffin, J.W.; Sheikh, K.A. Anti-myelin-associated glycoprotein antibodies alter neurofilament spacing. Brain 2002, 125 Pt 4, 904–911. [Google Scholar] [PubMed]
- Madrid, R.; Bradley, W.G. The pathology of neuropathies with focal thickening of the myelin sheath (tomaculous neuropathy): Studies on the formation of the abnormal myelin sheath. J. Neurol. Sci. 1975, 25, 415–448. [Google Scholar] [CrossRef]
- Kawagashira, Y.; Koike, H.; Takahashi, M.; Ohyama, K.; Iijima, M.; Katsuno, M.; Niwa, J.-I.; Doyu, M.; Sobue, G. Aberrant Expression of Nodal and Paranodal Molecules in Neuropathy Associated With IgM Monoclonal Gammopathy With Anti-Myelin-Associated Glycoprotein Antibodies. J. Neuropathol. Exp. Neurol. 2020, 79, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, R.; Erne, B.; Lauria, G.; Pareyson, D.; Borgna, M.; Morbin, M.; Arnold, A.; Czaplinski, A.; Fuhr, P.; Schaeren-Wiemers, N.; et al. IgM deposits on skin nerves in anti-myelin-associated glycoprotein neuropathy. Ann. Neurol. 2005, 57, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Tatum, A.H. Experimental paraprotein neuropathy, demyelination by passive transfer of human IgM anti-myelin-associated glycoprotein. Ann. Neurol. 1993, 33, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Willison, H.J.; Trapp, B.D.; Bacher, J.D.; Dalakas, M.C.; Griffin, J.W.; Quarles, R.H. Demyelination induced by intraneural injection of human antimyelin-associated glycoprotein antibodies. Muscle Nerve 1988, 11, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Ilyas, A.A.; Gu, Y.; Dalakas, M.C.; Quarles, R.H.; Bhatt, S. Induction of experimental ataxic sensory neuronopathy in cats by immunization with purified SGPG. J. Neuroimmunol. 2008, 193, 87–93. [Google Scholar] [CrossRef]
- Maeda, Y.; Bigbee, J.W.; Maeda, R.; Miyatani, N.; Kalb, R.G.; Yu, R.K. Induction of demyelination by intraneural injection of antibodies against sulfoglucuronyl paragloboside. Exp. Neurol. 1991, 113, 221–225. [Google Scholar] [CrossRef]
- Ritz, M.F.; Erne, B.; Ferracin, F.; Vital, A.; Vital, C.; Steck, A.J. Anti-MAG IgM penetration into myelinated fibers correlates with the extent of myelin widening. Muscle Nerve 1999, 22, 1030–1037. [Google Scholar] [CrossRef]
- Hays, A.P.; Lee, S.S.L.; Latov, N. Immune reactive C3d on the surface of myelin sheaths in neuropathy. J. Neuroimmunol. 1988, 18, 231–244. [Google Scholar] [CrossRef]
- Stork, A.C.; Cats, E.A.; Vlam, L.; Heezius, E.; Rooijakkers, S.; Herpers, B.; De Jong, B.A.; Rijkers, G.; Van Strijp, J.; Notermans, N.C.; et al. Classical and lectin complement pathway activity in polyneuropathy associated with IgM monoclonal gammopathy. J. Neuroimmunol. 2016, 290, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Amaador, K.; Wieske, L.; Koel-Simmelink, M.J.A.; Kamp, A.; Jongerius, I.; de Heer, K.; Teunissen, C.E.; Minnema, M.C.; Notermans, N.C.; Eftimov, F.; et al. Serum neurofilament light chain, contactin-1 and complement activation in anti-MAG IgM paraprotein-related peripheral neuropathy. J. Neurol. 2022, 269, 3700–3705. [Google Scholar] [CrossRef] [PubMed]
- Vos, J.M.; Notermans, N.C.; D’Sa, S.; Lunn, M.P.; van der Pol, W.L.; Kraan, W.; Reilly, M.M.; Chalker, J.; Gupta, R.; Kersten, M.-J.; et al. High prevalence of the MYD88 L265P mutation in IgM anti-MAG paraprotein-associated peripheral neuropathy. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1007–1009. [Google Scholar] [CrossRef]
- Hänggi, P.; Aliu, B.; Martin, K.; Herrendorff, R.; Steck, A.J. Decrease in Serum Anti-MAG Autoantibodies Is Associated With Therapy Response in Patients With Anti-MAG Neuropathy: Retrospective Study. Neurol. Neuroimmunol. Neuroinflamm. 2022, 9, e1109. [Google Scholar] [CrossRef] [PubMed]
- Niermeijer, J.M.; Fischer, K.; Eurelings, M.; Franssen, H.; Wokke, J.H.; Notermans, N.C. Prognosis of polyneuropathy due to IgM monoclonal gammopathy: A prospective cohort study. Neurology 2010, 74, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Dyck, P.J.; Low, P.A.; Windebank, A.J.; Jaradeh, S.S.; Gosselin, S.; Bourque, P.; Smith, B.E.; Kratz, K.M.; Karnes, J.L.; Evans, B.A.; et al. Plasma exchange in polyneuropathy associated with monoclonal gammopathy of undetermined significance. N. Engl. J. Med. 1991, 325, 1482–1486. [Google Scholar] [CrossRef]
- Oksenhendler, E.; Chevret, S.; Léger, J.M.; Louboutin, J.P.; Bussel, A.; Brouet, J.C. Plasma exchange and chlorambucil in polyneuropathy associated with monoclonal IgM gammopathy. IgM-associated Polyneuropathy Study Group. J. Neurol. Neurosurg. Psychiatry 1995, 59, 243–247. [Google Scholar] [CrossRef]
- Gorson, K.C.; Ropper, A.H.; Weinberg, D.H.; Weinstein, R. Treatment experience in patients with anti–myelin-associated glycoprotein neuropathy. Muscle Nerve 2001, 24, 778–786. [Google Scholar] [CrossRef]
- Baron, M.; Lozeron, P.; Harel, S.; Bengoufa, D.; Vignon, M.; Asli, B.; Malphettes, M.; Parquet, N.; Brignier, A.; Fermand, J.P.; et al. Plasma exchanges for severe acute neurological deterioration in patients with IgM anti-myelin-associated glycoprotein (anti-MAG) neuropathy. J. Neurol. 2017, 264, 1132–1135. [Google Scholar] [CrossRef]
- Siciliano, G.; Moriconi, L.; Gianni, G.; Richieri, E.; Vignocchi, M.G.; Rossi, B. Selective techniques of apheresis in polyneuropathy associated with monoclonal gammopathy of undetermined significance. Acta Neurol. Scand. 1994, 89, 117–122. [Google Scholar] [CrossRef]
- Dalakas, M.C.; Rakocevic, G.; Salajegheh, M.; Dambrosia, J.M.; Hahn, A.F.; Raju, R.; McElroy, B. Placebo-controlled trial of rituximab in IgM anti–myelin-associated glycoprotein antibody demyelinating neuropathy. Ann. Neurol. 2009, 65, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Léger, J.M.; Viala, K.; Nicolas, G.; Créange, A.; Vallat, J.M.; Pouget, J.; Clavelou, P.; Vial, C.; Steck, A.; Musset, L.; et al. Placebo-controlled trial of rituximab in IgM anti-myelin-associated glycoprotein neuropathy. Neurology 2013, 80, 2217–2225. [Google Scholar] [CrossRef] [PubMed]
- Campagnolo, M.; Zambello, R.; Nobile-Orazio, E.; Benedetti, L.; Marfia, G.A.; Riva, N.; Castellani, F.; Bianco, M.; Salvalaggio, A.; Garnero, M.; et al. IgM MGUS and Waldenstrom-associated anti-MAG neuropathies display similar response to rituximab therapy. J. Neurol. Neurosurg. Psychiatry 2017, 88, 1094–1097. [Google Scholar] [CrossRef]
- Niermeijer, J.M.; Eurelings, M.; van der Linden, M.W.; Lokhorst, H.M.; Franssen, H.; Fischer, K.; Teunissen, L.L.; Berg, L.H.V.D.; Schobben, F.; Wokke, J.H.J.; et al. Intermittent cyclophosphamide with prednisone versus placebo for polyneuropathy with IgM monoclonal gammopathy. Neurology 2007, 69, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Hamidou, M.A.; Belizna, C.; Wiertlewsky, S.; Audrain, M.; Biron, C.; Grolleau, J.Y.; Mussini, J.-M. Intravenous cyclophosphamide in refractory polyneuropathy associated with IgM monoclonal gammopathy: An uncontrolled open trial. Am. J. Med. 2005, 118, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Wilson, H.C.; Lunn, M.P.T.; Schey, S.; Hughes, R.A.C. Successful treatment of IgM paraproteinaemic neuropathy with fludarabine. J. Neurol. Neurosurg. Psychiatry 1999, 66, 575. [Google Scholar] [CrossRef]
- Niermeijer, J.M.; Eurelings, M.; Lokhorst, H.; Franssen, H.; Fijnheer, R.; Wokke, J.H.; Notermans, N.C.; Halperin, J.J.; Landis, D.M.; Kleinman, G.M. Neurologic and hematologic response to fludarabine treatment in IgM MGUS polyneuropathy. Neurology 2006, 67, 2076–2079. [Google Scholar] [CrossRef]
- Ghosh, A.; Littlewood, T.; Donaghy, M. Cladribine in the treatment of IgM paraproteinemic polyneuropathy. Neurology 2002, 59, 1290. [Google Scholar] [CrossRef]
- Ernerudh, J.H.; Vrethem, M.; Andersen, O.; Lindberg, C.; Berlin, G. Immunochemical and clinical effects of immunosuppressive treatment in monoclonal IgM neuropathy. J. Neurol. Neurosurg. Psychiatry 1992, 55, 930–934. [Google Scholar] [CrossRef]
- Dalakas, M.C.; Quarles, R.H.; Farrer, R.G.; Dambrosia, J.; Soueidan, S.; Stein, D.P.; Cupler, E.; Sekul, E.A.; Otero, C. A controlled study of intravenous immunoglobulin in demyelinating neuropathy with IgM gammopathy. Ann. Neurol. 1996, 40, 792–795. [Google Scholar] [CrossRef]
- Comi, G.; Roveri, L.; Swan, A.; Willison, H.; Bojar, M.; Illa, I.; Karageorgiou, C.; Nobile-Orazio, E.; Bergh, P.V.D.; Hughes, R.; et al. A randomised controlled trial of intravenous immunoglobulin in IgM paraprotein associated demyelinating neuropathy. J. Neurol. 2002, 249, 1370–1377. [Google Scholar] [CrossRef] [PubMed]
- Mariette, X.; Brouet, J.C.; Chevret, S.; Léger, J.M.; Clavelou, P.; Pouget, J.; Vallat, J.; Vial, C. A randomised double blind trial versus placebo does not confirm the benefit of α-interferon in polyneuropathy associated with monoclonal IgM. J. Neurol. Neurosurg. Psychiatry 2000, 69, 279. [Google Scholar] [CrossRef] [PubMed]
- Notermans, N.C.; Vermeulen, M.; Lokhorst, H.M.; van Doorn, P.A.; van den Berg, L.H.; Teunissen, L.L. Pulsed high-dose dexamethasone treatment of polyneuropathy associated with monoclonal gammopathy. J. Neurol. 1997, 244, 462–463. [Google Scholar] [CrossRef][Green Version]
- Gorson, K.C.; Amato, A.A.; Ropper, A.H. Efficacy of mycophenolate mofetil in patients with chronic immune demyelinating polyneuropathy. Neurology 2004, 63, 715–717. [Google Scholar] [CrossRef]
- Lunn, M.P.; Nobile-Orazio, E. Immunotherapy for IgM anti-myelin-associated glycoprotein paraprotein-associated peripheral neuropathies. Cochrane Database Syst. Rev. 2016, 10, Cd002827. [Google Scholar] [CrossRef] [PubMed]
- Pruppers, M.H.J.; Merkies, I.S.J.; Notermans, N.C. Recent advances in outcome measures in IgM-anti-MAG+ neuropathies. Curr. Opin. Neurol. 2015, 28, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Pruppers, M.H.J.; Merkies, I.S.J.; Lunn, M.P.T.; Notermans, N.C.; van den Bergh, P.; Blomkwist-Markens, P. 230th ENMC International Workshop: Improving future assessment and research in IgM anti-MAG peripheral neuropathy: A consensus collaborative effort, Naarden, The Netherlands, 24–26 February 2017. Neuromuscul. Disord. 2017, 27, 1065–1072. [Google Scholar] [CrossRef]
- Smith, T.; Sherman, W.; Olarte, M.R.; Lovelace, R.E. Peripheral neuropathy associated with plasma cell dyscrasia: A clinical and electrophysiological follow-up study. Acta Neurol. Scand. 1987, 75, 244–248. [Google Scholar] [CrossRef]
- van Meerten, T.; Hagenbeek, A. CD20-Targeted Therapy: The Next Generation of Antibodies. Semin. Hematol. 2010, 47, 199–210. [Google Scholar] [CrossRef]
- Leget, G.A.; Czuczman, M.S. Use of rituximab, the new FDA-approved antibody. Curr. Opin. Oncol. 1998, 10, 548–551. [Google Scholar] [CrossRef]
- van Sorge, N.M.; van der Pol, W.L.; van de Winkel, J.G.J. FcγR polymorphisms: Implications for function, disease susceptibility and immunotherapy. Tissue Antigens 2003, 61, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Levine, T.; Pestronk, A.; Florence, J.; Al-Lozi, M.T.; Lopate, G.; Miller, T.; Ramneantu, I.; Waheed, W.; Stambuk, M.; Stone, M.J.; et al. Peripheral neuropathies in Waldenström’s macroglobulinaemia. J. Neurol. Neurosurg. Psychiatry 2006, 77, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M. Rituximab in anti-MAG neuropathy: More evidence for efficacy and more predictive factors. J. Neurol. Sci. 2017, 377, 224–226. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, L.; Briani, C.; Grandis, M.; Vigo, T.; Gobbi, M.; Ghiglione, E.; Carpo, M.; Cocito, D.; Caporale, C.M.; Sormani, M.P.; et al. Predictors of response to rituximab in patients with neuropathy and anti–myelin associated glycoprotein immunoglobulin M. J. Peripher. Nerv. Syst. 2007, 12, 102–107. [Google Scholar] [CrossRef]
- Gazzola, S.; Delmont, E.; Franques, J.; Boucraut, J.; Salort-Campana, E.; Verschueren, A.; Sagui, E.; Hubert, A.-M.; Pouget, J.; Attarian, S. Predictive factors of efficacy of rituximab in patients with anti-MAG neuropathy. J. Neurol. Sci. 2017, 377, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Kawagashira, Y.; Koike, H.; Ohyama, K.; Hashimoto, R.; Iijima, M.; Adachi, H.; Katsuno, M.; Chapman, M.; Lunn, M.; Sobue, G. Axonal loss influences the response to rituximab treatment in neuropathy associated with IgM monoclonal gammopathy with anti-myelin-associated glycoprotein antibody. J. Neurol. Sci. 2015, 348, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Stork, A.C.; Notermans, N.C.; van den Berg, L.H.; Schellevis, R.D.; Niermeijer, J.M.; Nederend, M.; Leusen, J.H.W.; van der Pol, W. Fcγ receptor IIIA genotype is associated with rituximab response in antimyelin-associated glycoprotein neuropathy. J. Neurol. Neurosurg. Psychiatry 2014, 85, 918–920. [Google Scholar] [CrossRef]
- Sala, E.; Robert-Varvat, F.; Paul, S.; Camdessanché, J.P.; Antoine, J.C. Acute neurological worsening after Rituximab treatment in patients with anti-MAG neuropathy. J. Neurol. Sci. 2014, 345, 224–227. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Zervas, C.; Zomas, A.; Kiamouris, C.; Viniou, N.A.; Grigoraki, V.; Karkantaris, C.; Mitsouli, C.; Gika, D.; Christakis, J.; et al. Treatment of Waldenström’s Macroglobulinemia With Rituximab. J. Clin. Oncol. 2002, 20, 2327–2333. [Google Scholar] [CrossRef]
- Ghobrial, I.M.; Fonseca, R.; Greipp, P.R.; Blood, E.; Rue, M.; Vesole, D.H.; Gertz, M.A. Initial immunoglobulin M ‘flare’ after rituximab therapy in patients diagnosed with Waldenstrom macroglobulinemia. Cancer 2004, 101, 2593–2598. [Google Scholar] [CrossRef]
- Vo, M.L.; Martin, P.; Latov, N. Correlation of Changes in Gait Parameters, With Phenotype, Outcome Measures, and Electrodiagnostic Abnormalities in a Patient With Anti-MAG Neuropathy After Exacerbation and Improvement. J. Clin. Neuromuscul. Dis. 2015, 17, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.J.; Advani, R.H.; Branagan, A.R.; Buske, C.; Dimopoulos, M.A.; D’Sa, S.; Kersten, M.J.; Leblond, V.; Minnema, M.C.; Owen, R.G.; et al. Consensus treatment recommendations from the tenth International Workshop for Waldenström Macroglobulinaemia. Lancet Haematol. 2020, 7, e827–e837. [Google Scholar] [CrossRef]
- Colchester, N.T.H.; Allen, D.; Katifi, H.A.; Burt, T.; Lown, R.N.; Pinto, A.A.; Duncombe, A.S. Chemoimmunotherapy with rituximab, cyclophosphamide and prednisolone in IgM paraproteinaemic neuropathy: Evidence of sustained improvement in electrophysiological, serological and functional outcomes. Haematologica 2021, 106, 302–305. [Google Scholar] [CrossRef]
- Gruson, B.; Ghomari, K.; Beaumont, M.; Garidi, R.; Just, A.; Merle, P.; Merlusca, L.; Marolleau, J.P.; Royer, B. Long-term response to rituximab and fludarabine combination in IgM anti-myelin-associated glycoprotein neuropathy. J. Peripher. Nerv. Syst. 2011, 16, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Massa, F.; Zuppa, A.; Pesce, G.; Demichelis, C.; Bergamaschi, M.; Garnero, M.; Briani, C.; Ferrari, S.; Schenone, A.; Benedetti, L. Bendamustine-rituximab (BR) combined therapy for treatment of immuno-mediated neuropathies associated with hematologic malignancy. J. Neurol. Sci. 2020, 413, 116777. [Google Scholar] [CrossRef]
- Hospital, M.A.; Viala, K.; Dragomir, S.; Levy, V.; Cohen-Aubart, F.; Neil, J.; Musset, L.; Choquet, S.; Leger, J.-M.; Leblond, V. Immunotherapy-based regimen in anti-MAG neuropathy: Results in 45 patients. Haematologica 2013, 98, e155–e157. [Google Scholar] [CrossRef]
- Nivet, T.; Baptiste, A.; Belin, L.; Ghillani-Dalbin, P.; Algrin, C.; Choquet, S.; Lamy, T.; Morel, V.; Musset, L.; Roos-Weil, D.; et al. Immunochemotherapy versus rituximab in anti-myelin-associated glycoprotein neuropathy: A report of 64 patients. Br. J. Haematol. 2022, 198, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Pratt, G.; El-Sharkawi, D.; Kothari, J.; D’Sa, S.; Auer, R.; McCarthy, H.; Krishna, R.; Miles, O.; Kyriakou, C.; Owen, R. Diagnosis and management of Waldenström macroglobulinaemia—A British Society for Haematology guideline. Br. J. Haematol. 2022, 197, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Kastritis, E.; Leblond, V.; Dimopoulos, M.A.; Kimby, E.; Staber, P.; Kersten, M.J.; Tedeschi, A.; Buske, C. Waldenström’s macroglobulinaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv41–iv50. [Google Scholar] [CrossRef]
- McDonald, C.; Xanthopoulos, C.; Kostareli, E. The role of Bruton’s tyrosine kinase in the immune system and disease. Immunology 2021, 164, 722–736. [Google Scholar] [CrossRef]
- Mato, A.R.; Shah, N.N.; Jurczak, W.; Cheah, C.Y.; Pagel, J.M.; Woyach, J.A.; Fakhri, B.; Eyre, T.A.; Lamanna, N.; Patel, M.R.; et al. Pirtobrutinib in relapsed or refractory B-cell malignancies (BRUIN): A phase 1/2 study. Lancet 2021, 397, 892–901. [Google Scholar] [CrossRef]
- Minnema, M.C.; Vos, J.; Eftimov, F.; Vrancken, A. P-034: MAGNAZ trial-A prospective phase II study in patients with monoclonal gammopathy of unknown significance (MGUS) and anti-Myelin Associated Glycoprotein (MAG) Neuropathy and Zanubrutinib Treatment. Clin. Lymphoma Myeloma Leuk. 2021, 21, S57. [Google Scholar] [CrossRef]
- Castillo, J.J.; Allan, J.N.; Siddiqi, T.; Advani, R.H.; Meid, K.; Leventoff, C.; White, T.P.; Flynn, C.A.; Sarosiek, S.; Branagan, A.R.; et al. Venetoclax in Previously Treated Waldenström Macroglobulinemia. J. Clin. Oncol. 2021, 40, 63–71. [Google Scholar] [CrossRef]
- Treon, S.P.; Meid, K.; Hunter, Z.R.; Flynn, C.A.; Sarosiek, S.R.; Leventoff, C.R.; White, T.P.; Cao, Y.; Roccaro, A.M.; Sacco, A.; et al. Phase 1 study of ibrutinib and the CXCR4 antagonist ulocuplumab in CXCR4-mutated Waldenström macroglobulinemia. Blood 2021, 138, 1535–1539. [Google Scholar] [CrossRef] [PubMed]
- Kastritis, E.; Dimopoulos, M.A. Proteasome Inhibitors in Waldenström Macroglobulinemia. Hematol. Oncol. Clin. North Am. 2018, 32, 829–840. [Google Scholar] [CrossRef]
- Casan, J.M.L.; Wong, J.; Northcott, M.J.; Opat, S. Anti-CD20 monoclonal antibodies: Reviewing a revolution. Hum. Vaccines Immunother. 2018, 14, 2820–2841. [Google Scholar] [CrossRef]
- Tobinai, K.; Klein, C.; Oya, N.; Fingerle-Rowson, G. A Review of Obinutuzumab (GA101), a Novel Type II Anti-CD20 Monoclonal Antibody, for the Treatment of Patients with B-Cell Malignancies. Adv. Ther. 2017, 34, 324–356. [Google Scholar] [CrossRef]
- Rakocevic, G.; Martinez-Outschoorn, U.; Dalakas, M.C. Obinutuzumab, a potent anti–B-cell agent, for rituximab-unresponsive IgM anti-MAG neuropathy. Neurol.-Neuroimmunol. Neuroinflammat. 2018, 5, e460. [Google Scholar] [CrossRef]
- Briani, C.; Visentin, A.; Salvalaggio, A.; Cacciavillani, M.; Trentin, L. Obinutuzumab, a new anti-CD20 antibody, and chlorambucil are active and effective in anti-myelin-associated glycoprotein antibody polyneuropathy. Eur. J. Neurol. 2019, 26, 371–375. [Google Scholar] [CrossRef]
- Briani, C.; Visentin, A.; Castellani, F.; Cacciavillani, M.; Trentin, L. The BCL2 Inhibitor Venetoclax Plus Rituximab Is Active in MYD88 Wild-Type Polyneuropathy With Anti-MAG Antibodies. Neurol.-Neuroimmunol. Neuroinflammation 2022, 9, e1181. [Google Scholar] [CrossRef]
- Howard, J.F., Jr. Myasthenia gravis: The role of complement at the neuromuscular junction. Ann. N. Y. Acad. Sci. 2018, 1412, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Misawa, S.; Kuwabara, S.; Sato, Y.; Yamaguchi, N.; Nagashima, K.; Katayama, K.; Sekiguchi, Y.; Iwai, Y.; Amino, H.; Suichi, T.; et al. Safety and efficacy of eculizumab in Guillain-Barré syndrome: A multicentre, double-blind, randomised phase 2 trial. Lancet Neurol 2018, 17, 519–529. [Google Scholar] [CrossRef]
- Querol, L.A.; Hartung, H.P.; Lewis, R.A.; van Doorn, P.A.; Hammond, T.R.; Atassi, N.; Alonso-Alonso, M.; Dalakas, M.C. The Role of the Complement System in Chronic Inflammatory Demyelinating Polyneuropathy: Implications for Complement-Targeted Therapies. Neurotherapeutics 2022, 19, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Vlam, L.; Cats, E.A.; Harschnitz, O.; Jansen, M.D.; Piepers, S.; Veldink, J.H.; Franssen, H.; Stork, A.C.; Heezius, E.; Rooijakkers, S.H.; et al. Complement activity is associated with disease severity in multifocal motor neuropathy. Neurol.-Neuroimmunol. Neuroinflammation 2015, 2, e119. [Google Scholar] [CrossRef] [PubMed]
- Romano, R.; Giardino, G.; Cirillo, E.; Prencipe, R.; Pignata, C. Complement system network in cell physiology and in human diseases. Int. Rev. Immunol. 2021, 40, 159–170. [Google Scholar] [CrossRef]
- Varga, L.; Farkas, H. rhC1INH: A new drug for the treatment of attacks in hereditary angioedema caused by C1-inhibitor deficiency. Expert Rev. Clin. Immunol. 2011, 7, 143–153. [Google Scholar] [CrossRef]
- van de Walle, I.; Silence, K.; Budding, K.; van de Ven, L.; Dijkxhoorn, K.; de Zeeuw, E.; Yildiz, C.; Gabriels, S.; Percier, J.-M.; Wildemann, J.; et al. ARGX-117, a therapeutic complement inhibiting antibody targeting C2. J. Allergy Clin. Immunol. 2021, 147, 1420–1429. [Google Scholar] [CrossRef]
- Mastellos, D.C.; Yancopoulou, D.; Kokkinos, P.; Huber-Lang, M.; Hajishengallis, G.; Biglarnia, A.R.; Lupu, F.; Nilsson, B.; Risitano, A.M.; Ricklin, D.; et al. Compstatin: A C3-targeted complement inhibitor reaching its prime for bedside intervention. Eur. J. Clin. Investig. 2015, 45, 423–440. [Google Scholar] [CrossRef]
- Patriquin, C.J.; Kuo, K.H.M. Eculizumab and Beyond: The Past, Present, and Future of Complement Therapeutics. Transfus. Med. Rev. 2019, 33, 256–265. [Google Scholar] [CrossRef]
- Dalakas, M.C.; Alexopoulos, H.; Spaeth, P.J. Complement in neurological disorders and emerging complement-targeted therapeutics. Nat. Rev. Neurol. 2020, 16, 601–617. [Google Scholar] [CrossRef]
- Cardozo, T.; Cardozo, L.; Boutjdir, M. Autoantibody:Autoantigen Competitor Decoys: Application to Cardiac Phenotypes. Front. Immunol. 2022, 13, 812649. [Google Scholar] [CrossRef] [PubMed]
- Herrendorff, R.; Hänggi, P.; Pfister, H.; Yang, F.; Demeestere, D.; Hunziker, F.; Frey, S.; Schaeren-Wiemers, N.; Steck, A.J.; Ernst, B. Selective in vivo removal of pathogenic anti-MAG autoantibodies, an antigen-specific treatment option for anti-MAG neuropathy. Proc. Natl. Acad. Sci. USA 2017, 114, E3689–E3698. [Google Scholar] [CrossRef] [PubMed]
- Aliu, B.; Demeestere, D.; Seydoux, E.; Boucraut, J.; Delmont, E.; Brodovitch, A.; Oberholzer, T.; Attarian, S.; Théaudin, M.; Tsouni, P.; et al. Selective inhibition of anti-MAG IgM autoantibody binding to myelin by an antigen-specific glycopolymer. J. Neurochem. 2020, 154, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J. Complement activation-related pseudoallergy: A stress reaction in blood triggered by nanomedicines and biologicals. Mol. Immunol. 2014, 61, 163–173. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).