
Genome-Based Medicine for Acute Myeloid Leukemia: Study and Targeting of Molecular Alterations and Use of Minimal Residual Disease as a Biomarker



Abstract
:1. Introduction
2. Molecular Abnormalities of AMLs
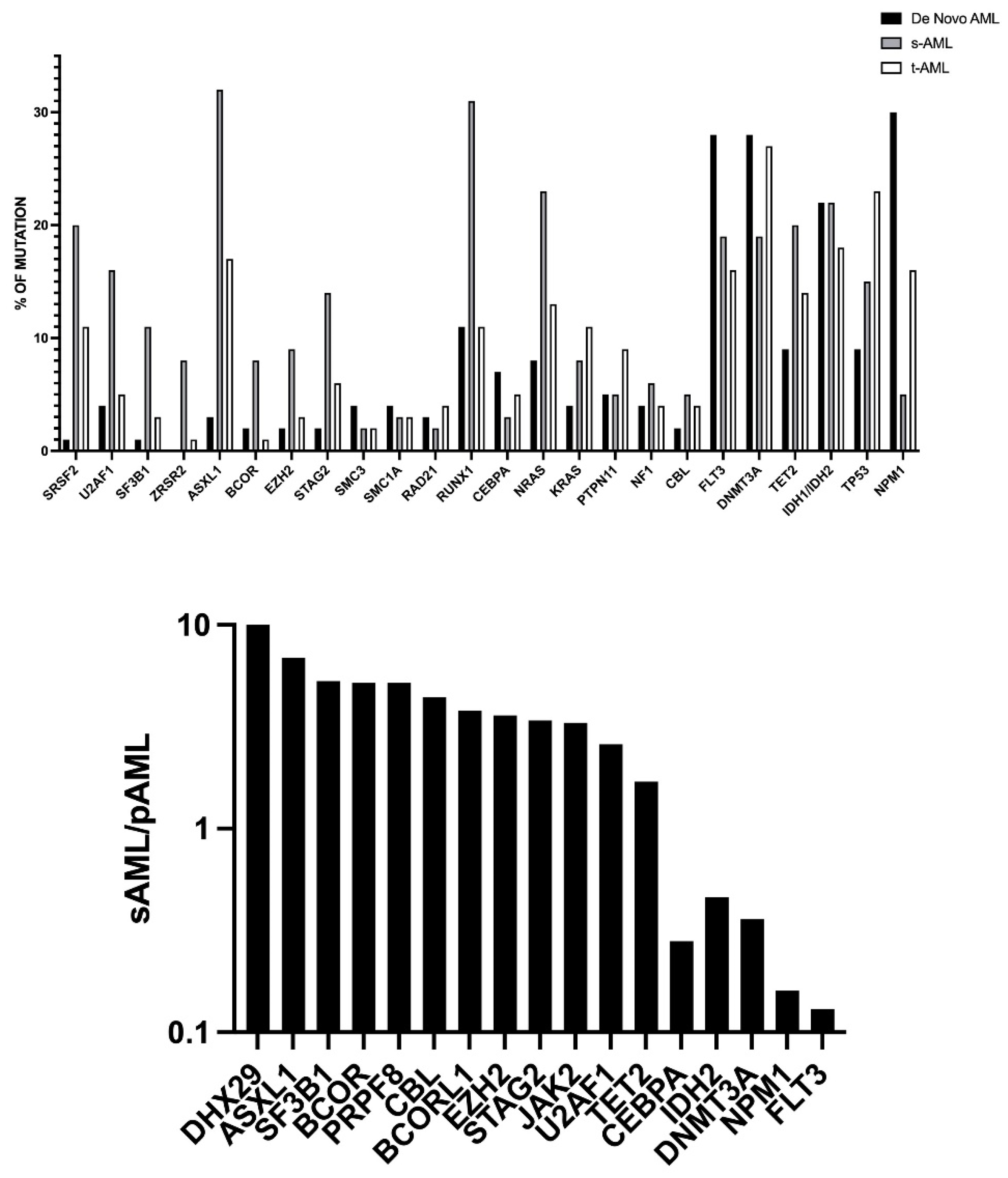
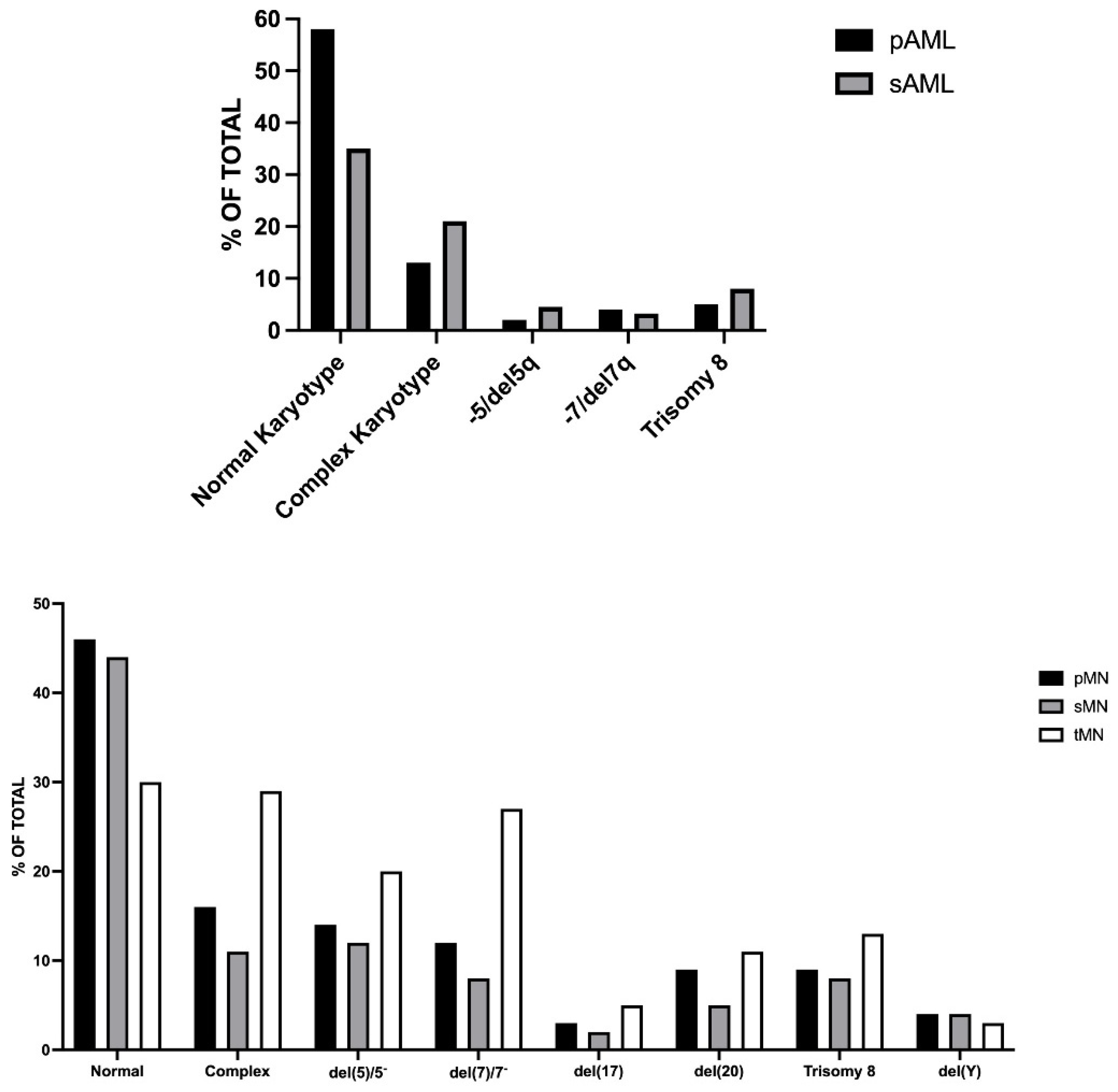
2.1. De Novo, Secondary and Therapy-Related AMLs
2.2. Molecular Classification of AML
2.3. Genetic Alterations in Relapsed AMLs
2.4. Clonal Hematopoiesis of Undetermined Potential (CHIP) and tAML Development
3. Genetic Heterogeneity and Clonal Evolution of AML
4. MRD Evaluation in AML
4.1. Methodology
4.2. MRD in AML Patients after Induction Chemotherapy and in Pre-Transplantation
Some Studies Were Based on the MRD Assessment by MFC
4.3. Clonal Hematopoiesis and MRD Evaluation Post-Chemotherapy
4.4. MRD in AML Patients in Post-Transplantation
4.5. MRD in Refractory/Relapsing AML (R/R AML)
4.6. MRD Evaluation in Elderly AML Patients Undergoing Reduced-Intensity Treatments
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X.; Zeidan, A.M. Epidemiology of acute myeloid leukemia: Recent progress and enduring challenges. Blood Rev. 2019, 36, 70–87. [Google Scholar] [CrossRef] [PubMed]
- Juliusson, G.; Abrahamsson, J.; Lazarevic, V.; Antonovic, P.; Derolf, A.; Gorelius, H.; Lehmanns, S.; Myhr-Eriksson, K.; Mollgord, L.; Uggla, B.; et al. Prevalence and characteristics of survivors from acute myeloid leukemia in Sweden. Leukemia 2017, 31, 728–731. [Google Scholar] [CrossRef] [PubMed]
- Roman, E.; Smith, A.; Appleton, S.; Crouch, S.; Kelly, R.; Kinsey, S.; Corgo, C.; Patmore, R. Myeloid malignancies in the real-world: Occurrence, progression and survival in the UK’s population-based Haematological Malignancy Research Network 20094-15. Cancer Epidemiol. 2016, 42, 186–198. [Google Scholar] [CrossRef]
- Vardiman, J.W.; Harris, N.L.; Brunning, R.D. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood 2002, 100, 2292–2302. [Google Scholar] [CrossRef] [PubMed]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitrz, M.T.; Porwit, A.; Harris, N.L.; LeBeau, M.M.; Hallstrom-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar]
- Swerdlow, S.H.; Campo, E.; Harris, N.L. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; WHO: Geneva, Switzerland, 2008. [Google Scholar]
- Ostgard, L.S.; Medeiros, B.C.; Sengelov, H.; Nargaard, M.; Andersen, M.K.; Dufva, I.K.; Friis, L.S.; Kjeldsen, E.; Werenbeg Marcher, C.; Preiss, B.; et al. Epidemiology and clinical significance of secondary and therapy-related acute myeloid leukemia: A national population-based cohort study. J. Clin. Oncol. 2015, 33, 3641–3649. [Google Scholar] [CrossRef]
- Lindsley, R.C.; Mar, B.G.; Mazzola, E.; Grauman, P.V.; Shareef, S.; Allen, S.L.; Pigneux, A.; Wetzler, M.; Stuart, R.K.; Erba, H.P.; et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015, 125, 1367–1376. [Google Scholar] [CrossRef]
- Nazha, A.S.; Zarzour, A.; Al-Issa, K.; Radivoyevitch, T.; Carraway, H.E.; Hirsch, C.M.; Przychodzen, B.; Patel, B.J.; Clemente, M.; Sanikommu, S.R.; et al. The complexity of interpreting genomic data in patients with acute myeloid leukemia. Blood Cancer J. 2016, 6, e510. [Google Scholar] [CrossRef]
- Kayser, S.; Dohner, K.; Krauter, J.; Kohne, C.H.; Horst, H.A.; Held, G.; von Lillienfeld-Tost, M.; Wilhelm, S.; Kundgen, A.; Gotze, K.; et al. The impact of therapy-related acute myeloid leukemia (AML) on outcome in 2853 adult patients with newly diagnosed AML. Blood 2011, 117, 2137–2145. [Google Scholar] [CrossRef]
- Kuzmanovic, T.; Patel, B.J.; Sankommu, S.R.; Nagata, Y.; Awada, H.; Kerr, C.M.; Przychodzen, B.P.; Iha, B.K.; Hiwasa, D.; Singhal, D.; et al. Genomics of therapy-related myeloid neoplasms. Haematologica 2020, 105, e98–e101. [Google Scholar] [CrossRef]
- Metzler, K.H.; Herold, T.; Rothenberg-Thurley, M.; Amler, S.; Sauerland, M.C.; Gorlich, D.; Schneider, S.; Kostandin, N.P.; Dufour, A.; Braundl, K.; et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood 2016, 128, 686–698. [Google Scholar]
- McNermey, M.E.; Godley, L.A.; LeBeau, M.M. Therapy-related neoplasms: When genetics and environment collide. Nat. Rev. Cancer 2017, 17, 503–527. [Google Scholar]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Blumfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Cree, I.A. The WHO classification of hematolymphoid tumors. Leukemia 2022, in press. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.; et al. The 5th edition of the world health organization classification of hematolymphoid tumors: Myeloid and histiocytic/dendritic neoplasms. Leukemia 2022, in press. [Google Scholar]
- Kuendgen, A.; Nomdedeu, M.; Tuechler, H.; Garcia-Manero, G.; Korokji, R.S.; Sekeres, M.A.; Della Porta, M.G.; Cazzola, M.; DeZern, A.E.; Roboz, G.J.; et al. Therapy-related myelodysplastic syndromes deserve specific diagnostic sub-classification and risk-stratification—An approach to classification of patients with t-MDS. Leukemia 2021, 35, 835–849. [Google Scholar]
- Eisfeld, A.K.; Kohlschidt, J.; Mims, A.; Nicolet, D.; Walker, C.J.; Blachly, J.S.; Carroll, A.J.; Papaioannou, D.; Kolitz, J.E.; Powell, B.E.; et al. Additional gene mutations may refine the 2017 European Leukemia Net classification in adult patients with de novo acute myeloid leukemia aged <60 years. Leukemia 2020, 34, 3215–3227. [Google Scholar] [CrossRef]
- Tsai, C.H.; Hou, H.A.; Tang, J.L.; Liu, C.Y.; Lin, C.C.; Chou, W.C.; Tseng, M.H.; Chiang, Y.C.; Kuo, Y.Y.; Liu, M.C.; et al. Genetic alterations and their clinical implications in older patients with acute myeloid leukemia. Leukemia 2016, 30, 1485–1492. [Google Scholar] [CrossRef]
- Silva, P.; Neumann, M.; Schroeder, M.P.; Vosberg, S.; Schlee, C.; Isaakidis, K.; Ortiz-Tranchez, J.; Fransecky, L.R.; Hartung, T.; Turmen, S.; et al. Acute myeloid leukemia in elderly is characterized by a distinct genetic and epigenetic landscape. Leukemia 2017, 31, 1640–1644. [Google Scholar] [CrossRef]
- Eisfeld, A.K.; Kohlschmidts, J.; Mrozek, K.; Blanchly, J.S.; Walker, C.J.; Nicolet, D.; Orwick, S.; Maharry, S.E.; Carroll, A.J.; Stone, R.M.; et al. Mutation patterns identify adult patients with de novo acute myeloid leukemia aged 60 years or older who respond favorably to standard chemotherapy: An analysis of Alliance studies. Leukemia 2018, 3260, 1338–1348. [Google Scholar] [CrossRef]
- Prassek, V.V.; Rothenberg-Thurley, M.; Sauerland, M.C.; Herold, T.; Janke, H.; Kaslezyh, B.; Kostandin, N.P.; Goerlich, D.; Krug, U.; Faldum, A.; et al. Genetics of acute myeloid leukemia in the elderly: Mutation spectrum and clinical impact in intensively treated patients aged 75 years or older. Haeamtologica 2018, 103, 1853–1861. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mingall, A.J.; Robertson, A.G.; Hoadley, A.S.K.; Triche, T.J.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 20959–22074. [Google Scholar]
- Hou, H.A.; Lin, C.C.; Chou, W.C.; Liu, C.Y.; Chen, C.Y.; Tang, J.L.; Lai, Y.J.; Tseng, M.H.; Huang, C.F.; Chiang, Y.C.; et al. Integration of cytogenetic and molecular alterations in risk stratification of 318 patients with de novo non-M3 acute myeloid leukemia. Leukemia 2014, 28, 50–58. [Google Scholar] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, K.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Hauser, M.; Thol, F.; Bolli, N.; et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Bullinger, L.; Dohner, K.; Dohner, H. Genomics of acute myeloid leukemia diagnosis and pathways. J. Clin. Oncol. 2017, 35, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Kishtagari, A.; Levine, R.L.; Viny, A.D. Driver mutations in acute myeloid leukemia. Curr. Opin. Hematol. 2020, 27, 49–57. [Google Scholar]
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Borthakur, G. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar]
- Duncavage, E.J.; Schroeder, M.C.; O’Loughlin, M.; Wilson, R.; MacMillan, S.; Bohannon, A.; Kruchowski, S.; Garza, J.; Du, F.; Highes, A.E.O.; et al. Genome sequencing as an alternative to cytogenetic analysis in myeloid cancers. N. Engl. J. Med. 2021, 384, 925–935. [Google Scholar] [CrossRef]
- Greif, P.A.; Hartmann, L.; Vosberg, S.; Stief, S.M.; Mattes, R.; Helmann, I.; Metzler, K.H.; Herold, T.; Bamopoulos, S.A.; Kerbs, P.; et al. Evolution of cytogenetically normal acute myeloid leukemia during therapy and relapse: An exome sequencing study of 50 patients. Clin. Cancer Res. 2018, 24, 1716–1726. [Google Scholar]
- Rapaport, F.; Neelaraju, Y.; Baslan, T.; Hassane, D.; Gruszczynska, A.; de Massy, M.R.; Farnoud, N.; Haddox, S.; Lee, T.; Medina-Martinez, J.; et al. Genomic and evolutionary portraits of disease relapse in acute myeloid leukemia. Leukemia 2021, 35, 2688–2692. [Google Scholar] [CrossRef]
- Vosberg, S.; Greif, P.A. Clonal evolution of acute myeloid leukemia from diagnosis to relapse. Genes Chrom. Cancer 2020, 58, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J. Age-related clonal hematopoiesis associated with diverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- Abelson, S.; Collord, G.; Ng, S.; Weissbrod, O.; Cohen, M.N.; Niemer, E.; Barda, N.; Zuzarte, P.C.; Heisler, R.; Sundravadanam, Y.; et al. Prediction of acute myeloid leukemia risk in healthy individuals. Nature 2018, 559, 400–404. [Google Scholar] [PubMed]
- Desai, P.; Mencia-Trinchant, N.; Savenkov, O.; Simon, M.S.; Cheang, G.; Lee, S.; Samuel, M.; Ritchie, E.K.; Guzman, M.L.; Baliman, K.V.; et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat. Med. 2018, 24, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Young, A.L.; Tong, R.S.; Birmann, B.M.; Druley, T.E. Clonal hematopoiesis and risk of acute myeloid leukemia. Haematologica 2019, 104, 2010–2017. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.J.; Papula, A.; Poon, Y.; Wong, W.H.; Young, A.L.; Druley, T.E.; Fisher, D.S.; Blundell, J.R. The evolutionary dynamics and fitness landscape of clonal hematopoiesis. Science 2020, 367, 1449–1454. [Google Scholar] [CrossRef]
- Skead, K.; Houle, A.A.; Abelson, S.; Agbessi, M.; Bruat, V.; Lin, B.; Soave, D.; Shlush, L.; Wright, S.; Dick, J.; et al. Interacting evolutionary pressures drive mutation dynamics and health outcomes in aging blood. Nat. Commun. 2021, 2, 4921. [Google Scholar] [CrossRef]
- Gillis, N.K.; Ball, M.; Zhang, Q.; Ma, Z.; Zhao, Y.; Yoder, S.J.; Balasis, M.E.; Masa, T.E.; Sallman, D.A.; Lancet, J.E.; et al. Clonal haemopoiesis and therapy-related myeloid malignancies in elderly patients: A proof-of-concept, case-control study. Lancet Oncol. 2017, 18, 112–121. [Google Scholar] [CrossRef]
- Takahashi, K.; Wang, F.; Kantarjian, H.; Doss, D.; Khanna, K.; Thompson, E.; Zhao, L.; Patel, K.; Neelapu, S.; Gumbs, C.; et al. Pre-leukemic clonal hematopoiesis and the risk of therapy-related myeloid neoplasms: A case-control study. Lancet Oncol. 2017, 18, 100–111. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Wang, F.; Kantarjian, H.; Song, X.; Patel, K.; Neelapu, S.; Gumbs, C.; Little, L.; Tippen, S.; Thornton, R.; et al. Copy number alterations detected as clonal hematopoiesis of indeterminate potential. Blood Adv. 2017, 1, 1031–1036. [Google Scholar]
- Coombs, C.C.; Zehir, A.; Devlin, S.M.; Kishtagari, A.; Syed, A.; Jonsson, P.; Hyman, D.M.; Solit, D.B.; Robson, M.E.; Baselga, J.; et al. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem. Cell 2017, 21, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.I.; Dayaram, T.; Tovy, A.; De Braekeleer, E.; Jeong, M.; Wang, F.; Zhang, J.; Heffernan, T.; Gera, S.; Kovacs, J.J.; et al. PPM1D mutations drive clonal hematopoiesis in response to cytotoxic chemotherapy. Cell Stem. Cell 2018, 23, 700–713. [Google Scholar] [CrossRef] [PubMed]
- Bolton, K.L.; Ptaskin, R.N.; Gao, T.; Braunstein, L.; Devlin, S.M.; Kelly, D.; Patel, M.; Barthon, A.; Syed, A.; Yabe, M.; et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat. Genet. 2020, 52, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukemia revealed by whole-genome sequencing. Nature 2012, 481, 506–511. [Google Scholar] [CrossRef]
- Hirsch, P.; Zhang, Y.; Tang, R.; Joulin, V.; Boutroux, H.; Pronier, E.; Moatti, H.; Flandrin, P.; Marzac, C.; Bories, D.; et al. Genetic hierarchy and temporal variegation in the clonal history of acute myeloid leukemia. Nat. Commun. 2016, 7, 12475. [Google Scholar] [CrossRef]
- Paguirigan, A.L.; Smith, J.; Meshinchi, S.; Carroll, M.; Maley, C.; Radich, J.P. Single-cell genotyping demonstrates complex clonal diversity in acute myeloid leukemia. Sci. Transl. Med. 2015, 7, 281re2. [Google Scholar] [CrossRef] [PubMed]
- Van Galen, P.; Hovestadt, V.; Wadsworth, M.H.; Hugehs, T.K.; Griffin, G.K.; Battaglia, S.; Verga, G.A.; Stephansky, J.; Pastika, T.J.; Story, J.L.; et al. Single-cell RNA-seq reevals AML hierarchies relevant to disease progression and immunity. Cell 2019, 176, 1255–1281. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; Wang, F.; Jahn, K.; Hu, T.; Tanaka, T.; Sasaki, Y.; Kuipers, J.; Loghavi, S.; Wang, S.A.; Yan, Y.; et al. Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics. Nat. Commun. 2020, 11, 5327. [Google Scholar] [CrossRef]
- Miles, L.A.; Bowman, R.L.; Merlinsky, T.R.; Csete, I.S.; Ooi, A.T.; Durruthy-Durruthy, R.; Bowman, M.; Famulare, C.; Patel, M.A.; Mendez, P.; et al. Single cell mutation analysis of clonal evolution in myeloid malignancies. Nature 2020, 587, 477–482. [Google Scholar] [CrossRef]
- Dix, C.; Lo, T.H.; Clark, G.; Abadir, E. Measurable residual disease in acute myeloid leukemia using flow cytometry: A review of where we are and where we are going. J. Clin. Med. 2020, 9, 1714. [Google Scholar] [CrossRef]
- Zeijlemaker, W.; Kelder, A.; Cloos, J.; Schuurhuis, G.J. Immunophenotypic detection of measurable residual (stem cell) disease using LAIP approach in acute myeloid leukemia. Curr. Prot. Cytom. 2019, 91, e66. [Google Scholar] [CrossRef] [PubMed]
- Baer, M.; Stewart, C.; Dodge, R.; Laget, G.; Sule, N.; Mrozek, K.; Schiffer, C.; Powell, B.; Kolitz, J.; Moore, J.; et al. High frequency of immunophenotype changes in acute myeloid leukemia at relapse: Implications for residual disease detection (Cancer and Leukemia Group B Study B361). Blood 2001, 97, 3574–3580. [Google Scholar] [CrossRef] [PubMed]
- Voso, M.T.; Ottone, T.; Lavorgna, S.; Venditti, A.; Maurillo, L.; Lo-Coco, F.; Buccisano, F. MRD in AML: The role of new techniques. Front. Oncol. 2019, 9, 655. [Google Scholar] [CrossRef]
- Hiatt, J.B.; Pritchard, C.D.; Salipante, S.J.; O’Roak, B.J.; Shendure, J. Single molecule molecular inversion probes for targeted, high-accuracy detection of low-frequency variation. Genome Res. 2013, 23, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Newmann, A.M.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated digital error suppression for improved decision of circulating tumor DNA. Nat. Biotechnol. 2016, 34, 547–555. [Google Scholar] [CrossRef]
- Schmitt, M.W.; Kennedy, S.R.; Salk, J.J.; Fox, E.J.; Hiatt, J.B.; Loeb, L.A. Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 14508–14513. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Harvey, L.; Martincorena, I. Somatic mutation landscapes at single-molecule resolution. Nature 2021, 593, 405–410. [Google Scholar] [CrossRef]
- Cohen, J.D.; Douville, C.; Dudley, J.C.; Mog, B.J.; Popoli, M.; Ptak, M.; Dobbyn, L.; Silliman, N.; Schaefer, J.; Tie, J.; et al. Detection of low-frequency DNA variants by targeted sequencing of the Watson and Crick strands. Nat. Biotechnol. 2021, 39, 1220–1227. [Google Scholar] [CrossRef]
- Dai, P.; Wu, L.R.; Chen, S.X.; Wang, M.X.; Cheng, L.Y.; Zhang, J.X.; Hao, P.; Yao, W.; Zarka, J.; Issa, G.C.; et al. Calibration-free NGS quantitation of mutations below 0.01% VAF. Nat. Commun. 2021, 12, 6123. [Google Scholar] [CrossRef]
- Thol, F.; Gabdoulline, R.; Liebich, A.; Klement, P.; Schiller, J.; Kandziora, C. Measurable residual disease monitoring by NGS before allogeneic hematopoietic cell transplantation in AML. Blood 2018, 132, 1703–1713. [Google Scholar] [CrossRef] [Green Version]
- Patkar, N.; Kakirde, C.; Shaikh, A.F.; Salve, R.; Bhanshe, P.; Chatterjee, G.; Rajpal, S.; Joshi, S.; Chaudhary, S.; Kodgule, R.; et al. Clinical impact of panel-based error-corrected next generation sequencing versus flow cytometry to detect measurable residual disease (MRD) in acute myeloid leukemia (AML). Leukemia 2021, 35, 1392–1404. [Google Scholar] [CrossRef] [PubMed]
- Walter, R.B.; Ofran, Y.; Wiezbowska, A.; Ravandi, F.; Hourigan, C.S.; Ngai, L.L.; Venditti, A.; Buccisano, F.; Ossenkoppele, G.J.; Roboz, G.J. Measurable residual disease as a biomarker in acute myeloid leukemia: Theoretical and practical considerations. Leukemia 2021, 35, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Aitken, M.; Ravandi, F.; Patel, K.P.; Short, N.J. Prognostic and therapeutic implications of measurable residual disease in acute myeloid leukemia. J. Hematol. Oncol. 2021, 14, 137. [Google Scholar] [CrossRef]
- Ngai, L.L.; Kelder, A.; Janssen, J.; Ossenkoppele, G.J. MRD tailored therapy in AML: What we have learned so far. Front. Oncol. 2021, 10, 603636. [Google Scholar] [CrossRef]
- Heuser, M.; Freeman, S.D.; Ossenkoppele, G.J.; Bussisano, F.; Hourigan, C.S.; Ngai, L.L.; Tettero, J.M.; Bachas, C.; Baer, C.; Béné, M.C.; et al. 2021 update on MDR in acute myeloid leukemia: A consensus document from the European LeukemiaNet MRD Working Party. Blood 2021, 138, 2753–2773. [Google Scholar] [CrossRef] [PubMed]
- Araki, D.; Wood, B.L.; Othus, M.; Radich, J.P.; Halpern, A.B.; Zhou, Y.; Mielcarek, M.; Estey, E.H.; Appelbaum, F.R.; Walter, R.B. Allogeneic hematopoietic cell transplantation for acute myeloid leukemia: Time to move toward a minimal residual disease-based definition of complete remission? J. Clin. Oncol. 2016, 34, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xie, H.; Wood, B.L.; Walter, R.B.; Pagel, J.M.; Becker, P.S.; Sandhu, V.K.; Abkowitz, J.L.; Appelbaum, F.R. Relation of clinical response and minimal residual disease and their prognostic impact on outcome in acute myeloid leukemia. J. Clin. Oncol. 2015, 33, 1258–1264. [Google Scholar] [CrossRef]
- Freeman, S.D.; Hills, R.K.; Virgo, P.; Khan, N.; Couzens, S.; Dillon, R.; Gilkes, A.; Gilkes, A.; Upton, L.; Nielsen, O.J.; et al. Measurable residual disease at induction redefines partial response in acute myeloid leukemia and stratifies outcomes in patients at standard risk without NPM1 mutations. J. Clin. Oncol. 2018, 38, 1486–1497. [Google Scholar] [CrossRef]
- Yu, S.; Fan, Z.; Ma, L.; Wang, Y.; Huang, F.; Zhang, Q.; Huang, J.; Wang, S.; Wang, S.; Xu, N.; et al. Association between measurable residual disease in patients with intermediate-risk acute myeloid leukemia and first remission, treatment, and outcomes. JAMA Netw. Open 2021, 4, e2115991. [Google Scholar] [CrossRef]
- Klco, J.M.; Miller, C.A.; Griffith, M.; Petti, A.; Spencer, D.H.; Ketkar-Kulkarni, S.; Wartman, L.D.; Christopher, M.; Lamprecht, T.L.; Helton, N.M.; et al. Association between mutation clearance after induction therapy and outcomes in acute myeloid leukemia. JAMA 2015, 314, 811–822. [Google Scholar] [CrossRef] [Green Version]
- Morita, K.; Kantarjian, H.M.; Wang, F.; Yan, Y.; Bueso-Ramos, C.; Sasaki, K.; Issa, G.C.; Wang, S.; Jorgensen, J.; Song, X.; et al. Clearance of somatic mutations at remission and the risk of relapse in acute myeloid leukemia. J. Clin. Oncol. 2018, 36, 1788–1797. [Google Scholar] [CrossRef] [PubMed]
- Hourigan, C.S.; Dillon, L.W.; Gui, G.; Logan, B.R.; Fei, M.; Ghannam, J.; Li, Y.; Licon, A.; Alyea, E.P.; Bashey, A.; et al. Impact of conditioning intensity of allogeneic transplantation for acute myeloid leukemia with genomic evidence of residual disease. J. Clin. Oncol. 2019, 38, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.H.; Tang, J.L.; Tien, F.M.; Kuo, T.Y.; Lin, C.C.; Tseng, M.H.; Peng, Y.L.; Hou, M.F.; Chuang, Y.K.; Liu, M.C.; et al. Clinical implications of sequential MRD monitoring by NGS at 2 time points after chemotherapy in patients with AML. Blood Adv. 2021, 5, 2456–2464. [Google Scholar] [CrossRef]
- Ivey, A.; Hills, R.K.; Simpson, M.A.; Jovanovic, J.V.; Gilkes, A.; Grech, A.; Patel, Y.; Bhudia, N.; Farah, H.; Mason, J.; et al. Assessment of minimal residual disease in standard-risk AML. N. Engl. J. Med. 2016, 374, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Bill, M.; Grimm, J.; Jentzsch, M.; Kloss, L.; Goldmann, K.; Schutz, J.; Beinicke, S.; Hantschel, J.; Cross, M.; Vucinic, V.; et al. Digital droplet PCR-based absolute quantification of pre-transplant NPM1 mutation burden predicts relapse in acute myeloid leukemia patients. Ann. Hematol. 2018, 97, 1757–1765. [Google Scholar] [CrossRef]
- Dillon, R.; Hills, R.; Freeman, S.; Potter, N.; Jovanovic, J.; Ivey, A.; Kanda, A.S.; Runglall, M.; Foot, N.; Valganon, M.; et al. Molecular MRD status and outcome after transplantation in NPM1-mutated AML. Blood 2020, 135, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Jentzsch, M.; Bischof, L.; Backaus, D.; Brauer, D.; Schilz, J.; Georg-Nikolaus, F.; Vucinic, V.; Niedeweiser, D.; Platzbecker, U.; Schwind, S. Impact of the MDR status in AML patients undergoing allogeneis stem cell transplantation in first vs. second remission. Blood Adv. 2022, in press. [Google Scholar] [CrossRef] [PubMed]
- Getta, B.M.; Devlin, S.M.; Levine, R.L.; Arcila, M.E.; Mohanty, A.S.; Zehir, A.; Tallman, M.S.; Giralt, S.A.; Roshal, M. Multicolor flow cytometry and multigene next-generation sequencing are complementary and highly predictive for relapse in acute myeloid leukemia after allogeneic transplantation. Biol. Blood Marrow Transplant. 2017, 23, 1064–1071. [Google Scholar] [CrossRef]
- Hasserjian, R.P.; Steensma, D.P.; Graubert, T.A.; Ebert, B.L. Clonal hematopoiesis and measurable residual disease assessment in AML. Blood 2020, 135, 1729–1738. [Google Scholar] [CrossRef]
- Murphy, T.; Zou, J.; Daher-Reyes, G.S.; Arruda, A.; Gupta, V.; McNamara, C.J.; Mindeu, M.D.; Schimmer, A.D.; Sibai, H.; Yee, K.W.; et al. Impact of preleukemic mutations and their persistence on hematologic recovery after induction chemotherapy for AML. Blood Adv. 2019, 3, 2307–2311. [Google Scholar] [CrossRef]
- Cappelli, L.V.; Meggendorfer, M.; Baer, C.; Nadarajah, N.; Hutter, S.; Jeromin, S.; Dicker, F.; Kern, W.; Haferlach, T.; Haferlach, C.; et al. Indeterminate and oncogenic potential: CHIP vs. CHOP mutations in AML with NPM1 alteration. Leukemia 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Morita, K.; Loghavi, S.; Wang, F.; Furudate, K.; Sasaki, Y.; Little, L.; Gumbs, C.; Matthws, J.; Daver, N.; et al. Clonal dynamics and clinical implications of postremission clonal hematopoiesis in acute myeloid leukemia. Blood 2021, 138, 1733–1739. [Google Scholar] [CrossRef] [PubMed]
- Onate, G.; Bataller, A.; Garrido, A.; Hoyos, M.; Arnan, M.; Vives, S.; Coll, R.; Tormo, M.; Sampol, M.A.; Escoda, L.; et al. Prognostic impact of DNMT3A mutation in acute myeloid leukemia with mutated NPM1. Blood Adv. 2021, in press. [Google Scholar]
- Assi, R.; Masri, N.; Dalli, I.A.; El-Cheikh, J.; Zarbachi, A. Post-transplant maintenance therapy for patients with acute myeloid leukemia: Current approaches and the need for more trials. J. Blood Med. 2021, 12, 21–32. [Google Scholar] [CrossRef]
- Schuler, E.; Boughoufala, S.; Rautenberg, C.; Nachtkamp, K.; Dienst, A.; Fenk, R.; Haas, R.; Kondakci, M.; Germing, U.; Schroeder, T.R.; et al. Relapse patterns and treatment strategies in patients receiving allogeneic hematopoietic stem cell transplantation for myeloid malignancies. Ann. Hematol. 2019, 98, 1225–1235. [Google Scholar] [CrossRef]
- Zhou, Y.; Othus, M.; Araki, D.; Wood, B.L.; Radich, J.P.; Halpern, A.B.; Mielcarek, M.; Estey, E.H.; Appelbaum, F.R.; Walter, R.B. Pre- and post-transplant quantification of measurable (“minimal”) residual disease via multiparameter flow cytometry in adult acute myeloid leukemia. Leukemia 2016, 30, 1456–1464. [Google Scholar] [CrossRef]
- Shah, M.V.; Jorgensen, J.L.; Saliba, R.M.; Wang, S.A.; Alousi, A.M.; Andersson, B.S.; Bashir, Q.; Ciurea, S.O.; Kebriaei, P.; Marin, D.; et al. Early post-transplant minimal residual disease assessment improves risk stratification in acute myeloid leukemia. Biol. Blood Marrow Transpl. 2018, 24, 1514–1520. [Google Scholar] [CrossRef]
- Heuser, M.; Heida, B.; Buttner, K.; Wienecke, C.P.; Teich, K.; Franke, C.; Brandes, M.; Klement, P.; Liebich, A.; Wichmann, M.; et al. Posttransplantation MRD monitoring in patients with AML by next-generation sequencing using DTA and non-DTA mutations. Blood Adv. 2021, 5, 2294–2304. [Google Scholar]
- Thol, F.; Heida, B.; Buettner, K.; Wienecke, C.; Teich, K.; Funke, C.; Maximilian, B.; Klement, P.; Liebich, A.; Schiller, J.; et al. Post transplantation measurable residual disease (MRD) monitoring using next-generation sequencing is highly predictive for relapse after allogeneic stem cell transplantation. Blood 2019, 134 (Suppl. S1), 184. [Google Scholar] [CrossRef]
- Martin-Rojas, R.; Oarbeascoa, G.; Bailén, R.; Gomez-Centuriòn, I.; Juarez, L.M.; Dorado, N.; Martinez-Lapierche, C.; Pérez-Corral, A.; Buno, I.; Anguita, J.; et al. Impact of minimal residual disease and chimerism monitoring at different timepoints after allogeneic stem cell transplantation for acute myeloid leukemia. Blood 2020, 136 (Suppl. S1), abst.3353. [Google Scholar] [CrossRef]
- Short, N.J.; Rafei, H.; Dever, N.; Hwang, H.; Ninh, J.; Jorgensen, J.L.; Kadia, T.M.; Di Nardo, C.D.; Wang, S.A.; Jabbour, E.; et al. Prognostic impact of complete remission with MRD negativity in patients with relapsed or refractory AML. Blood Adv. 2020, 4, 6117–6126. [Google Scholar] [CrossRef] [PubMed]
- Hao, Q.; Liu, X.; Zhang, Y.; Zhang, D.; Li, B.; Wang, J. MRD abnormal expression predict poor outcomes for refractory or relapsed acute myeloid leukemia after allogeneic hematopoietic stem cell transplantation. J. Clin. Lab. Anal. 2021, e23974, in press. [Google Scholar] [CrossRef] [PubMed]
- Klyuchnov, E.; Badbaran, A.; Massoud, R.; Fritsche-Friedland, U.; Janson, D.; Ayuk, F.; Christopeit, M.; Woslchke, C.; Backer, U.; Kroger, N. Post-transplantation multicolored flow cytometry-minimal residual disease status on day 100 predicts outcomes for patients with refractory acute myeloid leukemia. Transplant. Cell Ther. 2022, 28, 267.e1–267.e7. [Google Scholar]
- Di Nardo, C.D.; Lachowiez, C.A.; Takahashi, K.; Loghavi, S.; Xiao, L.; Kadia, T.; Daver, N.; Adeoti, M.; Short, N.J.; Sasaki, K.; et al. Venetoclax combined with FLAG-IDA induction and consolidation in newly diagnosed and relapsed or refractory acute myeloid leukemia. J. Clin. Oncol. 2021, 39, 2768–2778. [Google Scholar] [CrossRef] [PubMed]
- Shahswar, R.; Beutel, G.; Gabdoulline, R.; Konecke, K.; Stadler, M.; Gohring, G.; Schlegelerger, B.; Li, Z.; Dallmann, L.K.; Wienecke, C.P.; et al. FLAVIDA chemotherapy induces MRD-negative remission in patients with relapsed/refractory acute myeloid leukemia. In Proceedings of the EHA 2021 Virtual Congress, Virtual, 9–17 June 2021. Abst. #S139. [Google Scholar]
- Boddu, P.; Jorgensen, J.; Kantarjian, H.; Borthakur, G.; Kadia, T.; Daver, N.; Alvarado, Y.; Pemmaraju, N.; Bose, P.; Naqvi, K.; et al. Achievement of a negative minimal residual disease state after hypomethylating agent therapy in older patients with AML reduces the risk of relapse. Leukemia 2018, 32, 241–244. [Google Scholar] [CrossRef]
- Simoes, C.; Paiva, B.; Martinez-Cudròn, D.; Bargna, J.M.; Vives, S.; Algarra, L.; Tormo, M.; Martinez, P.; Serrano, J.; Herrera, P.; et al. Measurable residual disease in elderly acute myeloid leukemia: Results from the PETHEMA-FLUGAZA phase 3 clinical trial. Blood Adv. 2021, 5, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Maiti, A.; DiNardo, C.D.; Wang, S.A.; Jorgensen, J.; Kadia, T.M.; Daver, N.G.; Short, N.J.; Yilmaz, M.; Pemmaraju, N.; Borthakur, G.; et al. Prognostic value of measurable residual disease after venetoclax and decitabine in acute myeloid leukemia. Blood Adv. 2021, 5, 1875–1884. [Google Scholar] [CrossRef]
- Pratz, K.; Jonas, B.A.; Pullarkat, V.; Recher, C.; Schuh, A.C.; Thirman, M.J.; Suen Garcia, J.; Dinardo, C.D.; Vorobyev, V.; Fracchiolla, N.; et al. Measurable residual disease response in acute myeloid leukemia treated with venetoclax and azacitidine. J. Clin. Oncol. 2021, 39 (Suppl. S15), 7018. [Google Scholar] [CrossRef]
- Vazquez, R.; Breal, C.; Zalmai, L.; Friedrich, C.; Almire, C.; Contejean, A.; Barreau, S.; Grignano, E.; Willems, L.; Deau-Fischer, B.; et al. Venetoclax combination therapy induces deep AML remission with eradication of leukemic stem cells and remodeling of clonal hematopoiesis. Blood Cancer J. 2021, 11, 62. [Google Scholar] [CrossRef]
- Tiong, I.S.; Dillon, R.; Ivey, A.; Teh, T.C.; Nguyen, P.; Cummings, N.; Taussig, D.C.; Latif, A.L.; Potter, N.E.; Runglall, M.; et al. Venetoclax induces rapid elimination of NPM1 mutant measurable residual disease in combination with low-intensity chemotherapy in acute myeloid leukemia. Brit. J. Haematol. 2021, 192, 1026–1030. [Google Scholar] [CrossRef]
- Ragon, B.K.; Daver, N.; Garcia-Manero, G.; Ravandi, F.; Cortes, J.; Kadia, T.; Oran, B.; Ohanian, M.; Ferrajoli, A.; Pemmaraju, N.; et al. Minimal residual disease eradication with epigenetic therapy in core binding factor acute myeloid leukemia. Am. J. Hematol. 2017, 92, 845–850. [Google Scholar] [CrossRef] [PubMed]
- Platzbecker, U.; Middeke, J.M.; Sockel, K.; Herbst, R.; Wolf, D.; Baldus, C.D.; Oelschlagel, U.; Mutherig, A.; Fransecky, L.; Noppeney, R.; et al. Measurable residual disease-guided treatment with azacitidine to prevent haematological relapse in patients with myelodysplastic syndrome and acute myeloid leukemia (RELAZA2): An open-label, multicentre, phase 2 trial. Lancet Oncol. 2018, 19, 1668–1679. [Google Scholar] [CrossRef]
- Wei, A.H.; Dohner, H.; Pocock, C.; Montesinos, P.; Afananyev, B.; Dombret, H.; Ravandi, F.; Sayar, H.M.; Jang, H.; Porka, K.; et al. Oral azacitidine maintenance therapy for acute myeloid leukemia in first remission. N. Engl. J. Med. 2020, 383, 2526–2537. [Google Scholar] [CrossRef] [PubMed]
- Roboz, G.J.; Ravandi, F.; Wei, A.H.; Dombret, H.; Dohner, H.; Thol, F.; Voso, M.T.; Schuh, A.C.; Porkka, K.; La Torre, I.; et al. Oral azacitidine prolongs survival of patients with AML in remission independently of measurable residual disease status. Blood 2022, 139, 2145–2154. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Name | MFC-LAIP | MFC-DfN | RT-qPCR | ddPCR | NGS |
|---|---|---|---|---|---|
| Sensitivity | 10−3–10−5 | 10−3–10−5 | 10−4–10−6 | 10−4–10−6 | 10−4–10−7 |
| Applicability | >90% | >90% | 50–60% | 50–60% | 80–90% |
| Principle | Flow cytometry evaluation of membrane immuno-phenotype Leukemia Associated Immunophenotype (LAIP). The technique defines individual-specific surface markers at diagnosis and evaluates these markers at various times during and after the end of treatment. | Flow cytometry evaluation of membrane immuno-phenotype Different from Normal (DfN). The technique is based on the detection of aberrant surface marker expression at follow-up. | Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) measures the amount of a specific mRNA. | Digital droplet polymerase chain reaction (RT-qPCR) measures the amount of a specific mRNA. | Next-generation sequencing (NGS), a DNA sequencing technology that rapidly sequences the whole genome. Error-corrected NGS involves the physical incorporation of random oligonucleotides or unique molecular identifiers (UMI) at the library preparation prior to amplification of DNA, reduces the errors of standard NGS, and thus increases the capacity to detect gene mutation at low–very low VAF. |
| Main Characteristics | Major advantages: It is widely available given the diffusion of flow cytometry; It is widely applicable to >90% of AMLs; Its evaluation is relatively fast. Main limitations: It requires standardization and harmonization between laboratories; It requires a relatively high number of cells; It requires technical expertise for the analysis and interpretation of the results. | Major advantages: It is widely available given the diffusion of flow cytometry; It is widely applicable to >90% of AMLs; Its evaluation is relatively fast. Main limitations: It requires standardization and harmonization between laboratories; It requires a relatively high number of cells; It requires technical expertise for the analysis and interpretation of the results. | It is used for the detection of the following gene alterations: NPM1 mutations; PML-RARA fusion; RUNX1-RUNXT1 fusion; CBFB-MYH11 fusion. Major advantages: It is a sensitive technique; Well-standardized; It is a semi-quantitative technique; It allows an easy interpretation of the results. Major limitations: It requires a standard curve; Single gene assessed per assay; The capacity to detect a gene alteration is limited to the primer-spanning regions. | It is used for the detection of the following gene alterations: NPM1 mutations; PML-RARA fusion; RUNX1-RUNXT1 fusion; CBFB-MYH11 fusion. Major advantages: It is a very sensitive technique; Higher sensitivity than RT-qPCR; No requirement for a standard curve; It provides an absolute quantitation. Major limitations: The capacity to detect a gene alteration is limited to the primer-spanning regions; It requires technical experience. | Major advantages: It is widely applicable to about 80–90% of AMLs; It can simultaneously examine multiple genes; Sensitivity very high with error-corrected NGS. Main limitations: Availability only in state-of-the-art and well-funded centers; Bioinformatics required for the interpretation of the results; Relatively expensive; It requires considerable technical expertise in the analysis and interpretation of data. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Testa, U.; Castelli, G.; Pelosi, E. Genome-Based Medicine for Acute Myeloid Leukemia: Study and Targeting of Molecular Alterations and Use of Minimal Residual Disease as a Biomarker. Hemato 2022, 3, 543-568. https://doi.org/10.3390/hemato3030038
Testa U, Castelli G, Pelosi E. Genome-Based Medicine for Acute Myeloid Leukemia: Study and Targeting of Molecular Alterations and Use of Minimal Residual Disease as a Biomarker. Hemato. 2022; 3(3):543-568. https://doi.org/10.3390/hemato3030038
Chicago/Turabian StyleTesta, Ugo, Germana Castelli, and Elvira Pelosi. 2022. "Genome-Based Medicine for Acute Myeloid Leukemia: Study and Targeting of Molecular Alterations and Use of Minimal Residual Disease as a Biomarker" Hemato 3, no. 3: 543-568. https://doi.org/10.3390/hemato3030038
APA StyleTesta, U., Castelli, G., & Pelosi, E. (2022). Genome-Based Medicine for Acute Myeloid Leukemia: Study and Targeting of Molecular Alterations and Use of Minimal Residual Disease as a Biomarker. Hemato, 3(3), 543-568. https://doi.org/10.3390/hemato3030038