Retrotransposons and the Evolution of Genome Size in Pisum


Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material and the Selection of Accessions for Analysis
2.2. Population Genetic Considerations
2.2.1. Allele Frequency Distribution
2.2.2. Age Distribution of Occupied Sites
3. Results
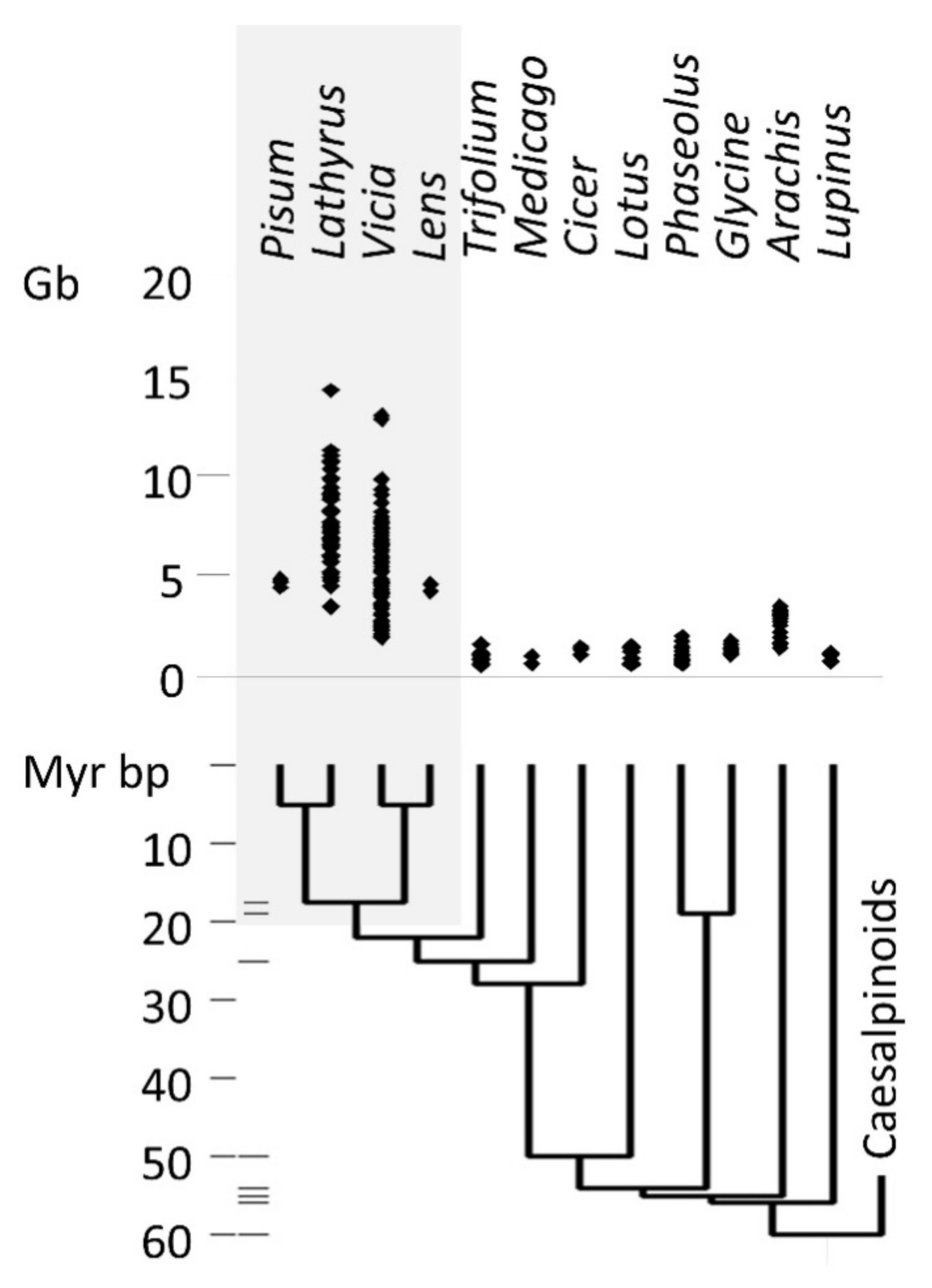
3.1. Retrotransposons in Legumes
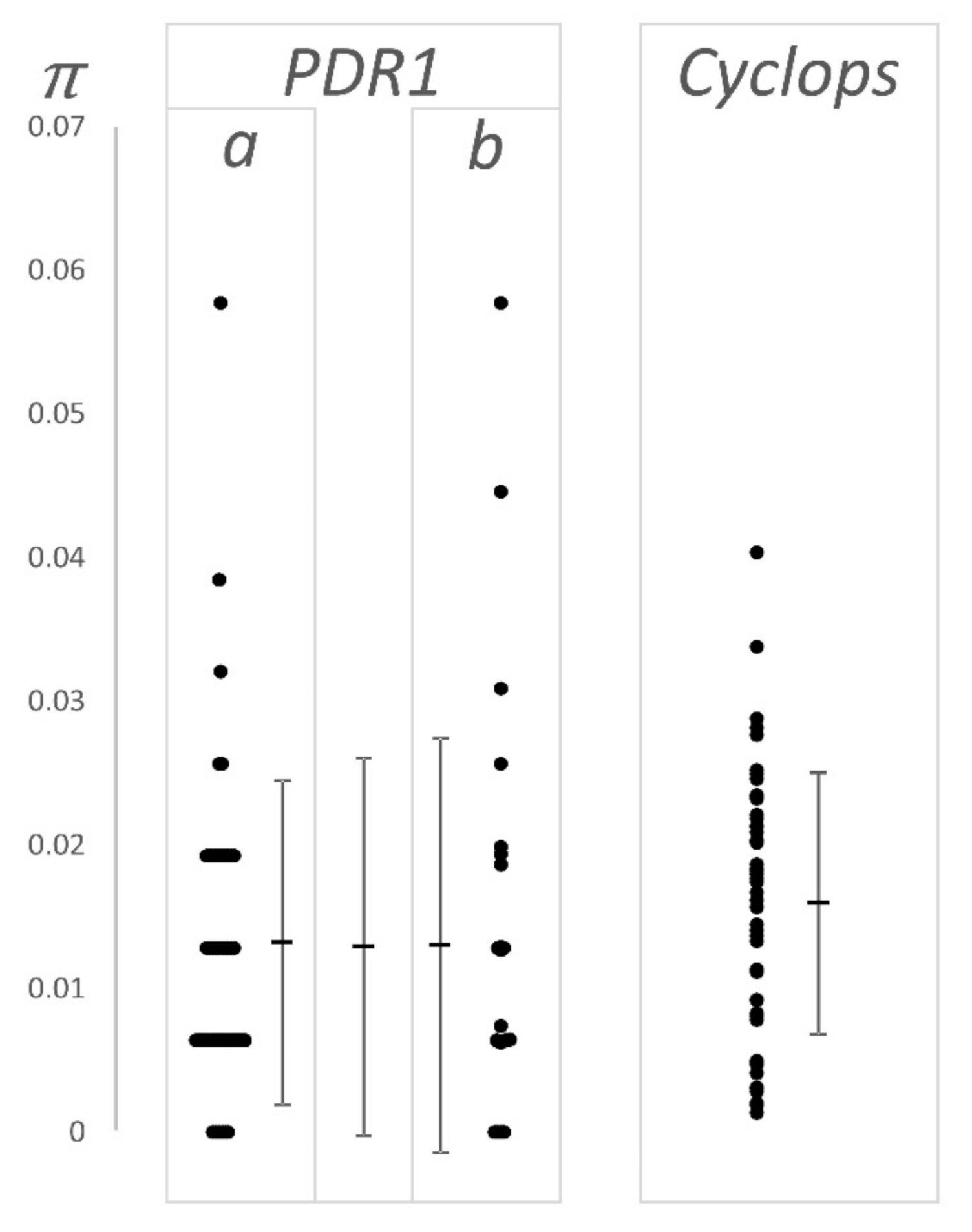
3.2. Allele Frequency Distribution
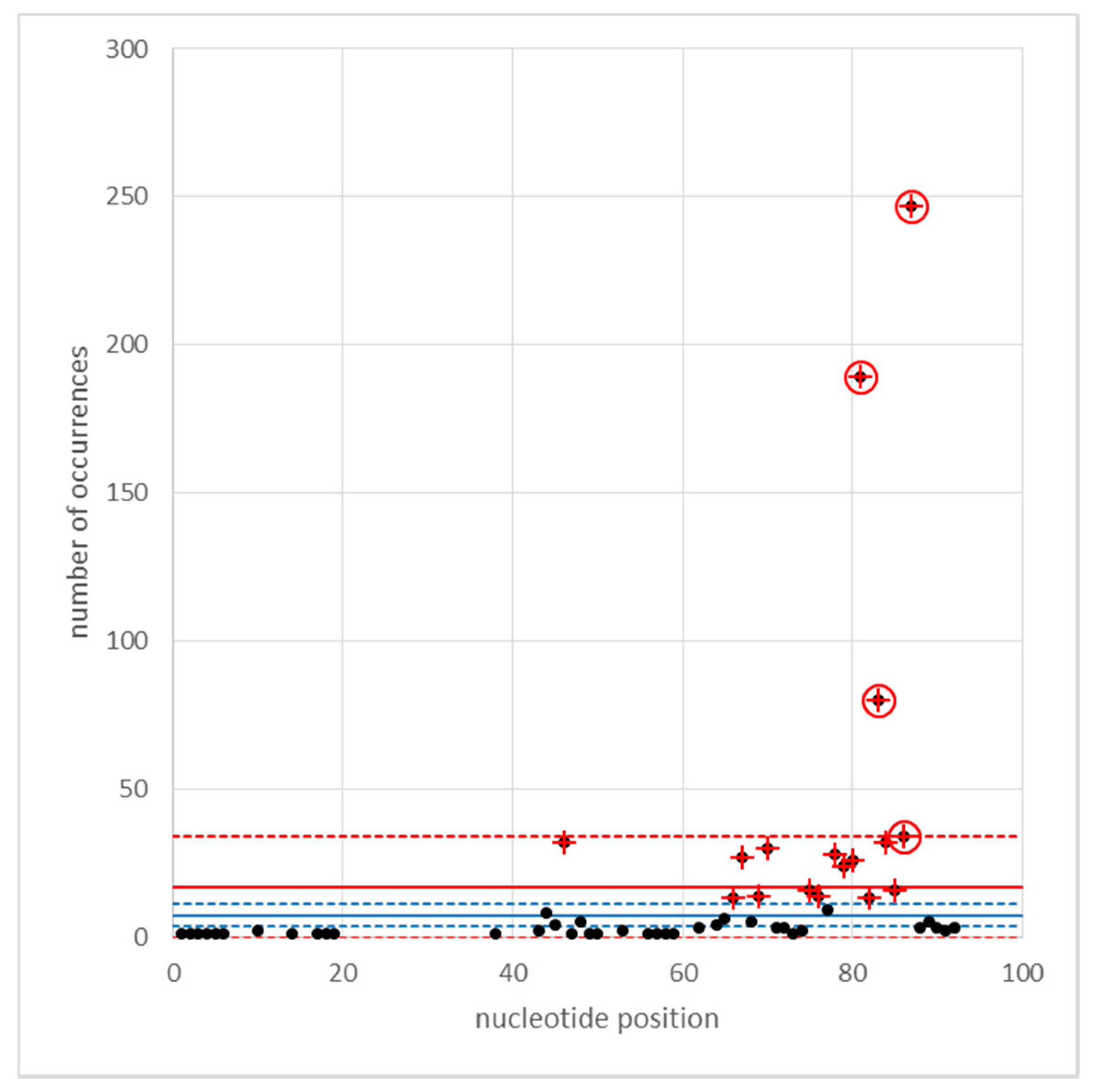
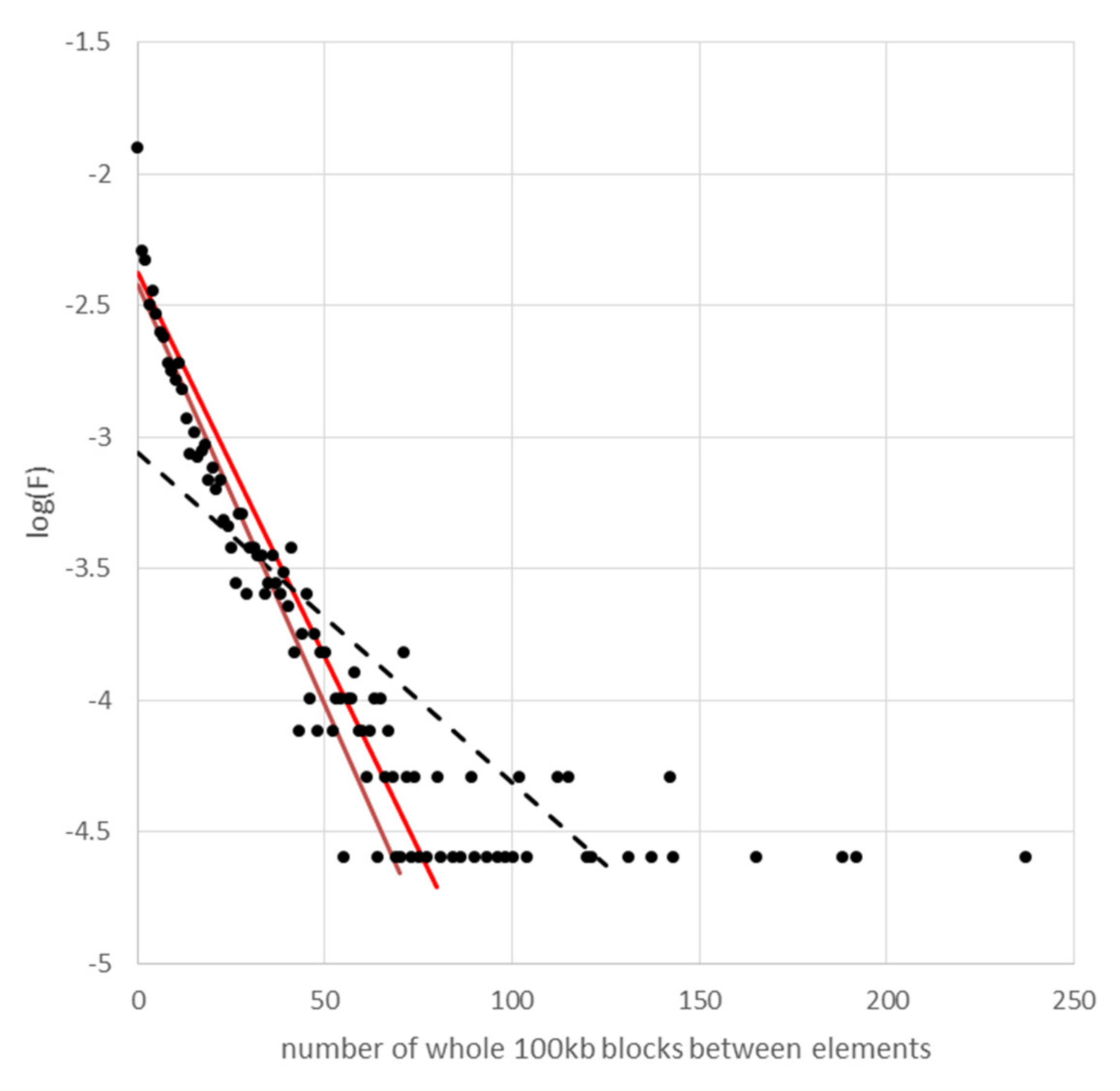
3.3. The Distribution of Cyclops Elements in the Cameor Genome Assembly
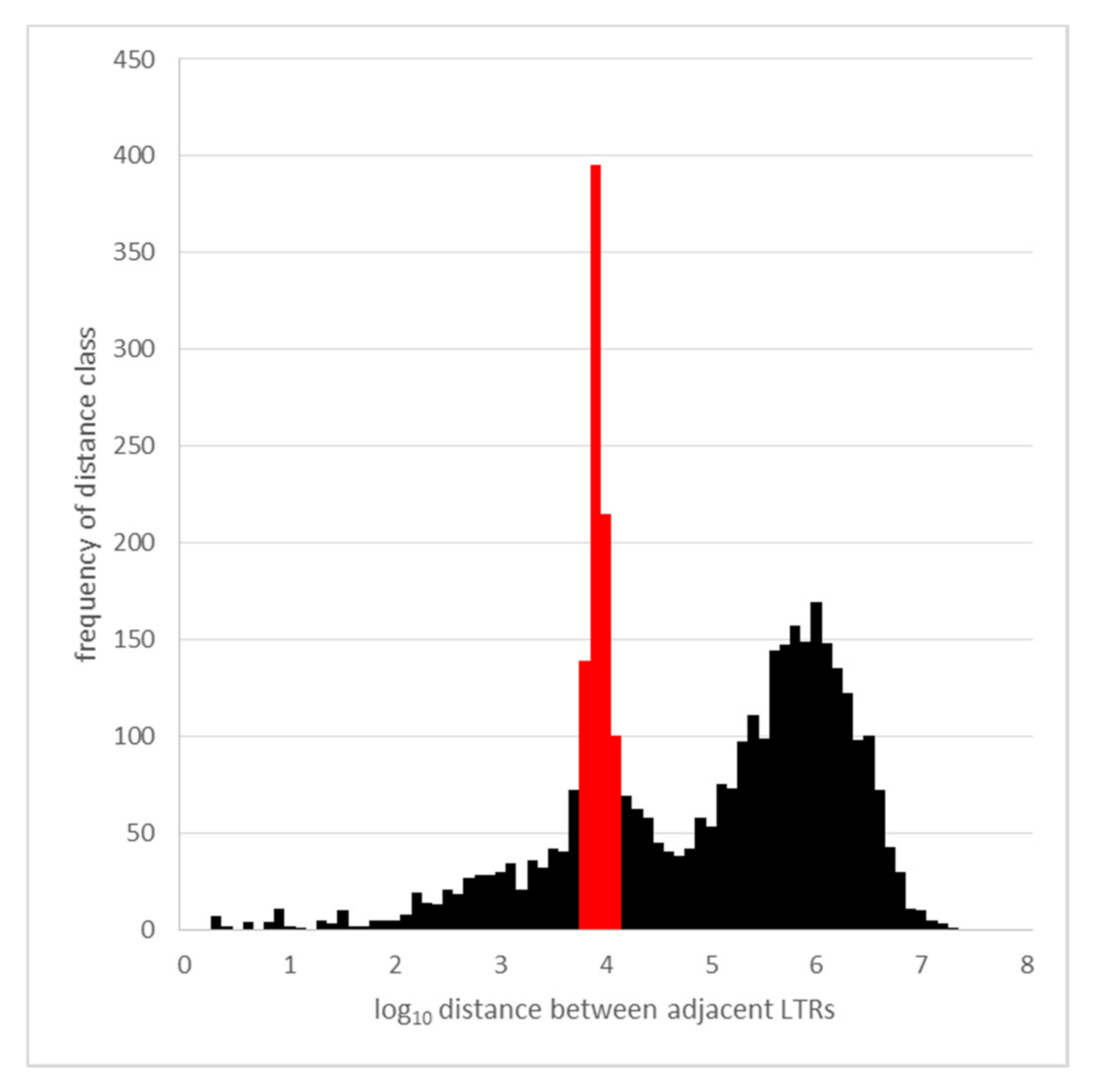
3.4. Occupied Site Allele Age Distribution
4. Discussion
4.1. Nucleotide Diversity and Effective Population Size
4.2. Age Distribution of LTR Pairs and Effective Population Size
4.3. Gain and Loss
4.4. Comparison with Other Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. The Taxonomic Distribution of Genome Sizes in the Viceae

Appendix B. Estimating 4Neμ from Φ(x)

Appendix C. Identification of Cyclops LTRs



Appendix D. Population Structure and Taxonomy of Natural and Domesticated Pisum
References
- Eddy, S.R. The C-value paradox, junk DNA and ENCODE. Curr. Biol. 2012, 22, R898-9. [Google Scholar] [CrossRef]
- Lynch, M. The Origins of Genome Architecture; Sinauer Associates, Inc.: Sunderland, MA, USA, 2007. [Google Scholar]
- LIS—Legume Information System. Available online: https://legumeinfo.org/species (accessed on 23 September 2020).
- Cannon, S. (Iowa State University, Ames, IA, USA). Personal communication, 2016.
- Plant DNA C-values Database. Available online: https://cvalues.science.kew.org/ (accessed on 23 September 2020).
- Baranyi, M.; Greilhuberv, J.; Święcicki, W.W. Genome size in wild Pisum species. Theor. Appl. Genet. 1996, 93, 717–721. [Google Scholar] [CrossRef]
- Vershinin, A.V.; Allnutt, T.R.; Knox, M.R.; Ambrose, M.J.; Ellis, T.H.N. Transposable elements reveal the impact of introgression, rather than transposition, in Pisum diversity, evolution and domestication. Mol. Biol. Evol. 2003, 20, 2067–2075. [Google Scholar] [CrossRef] [PubMed]
- Hill, P.; Burford, D.; Martin, D.M.; Flavell, A.J. Retrotransposon populations of Vicia species with varying genome size. Mol. Genet. Genom. 2005, 273, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Macas, J.; Novák, P.; Pellicer, J.; Čížková, J.; Koblížková, A.; Neumann, P.; Fuková, I.; Doležel, J.; Kelly, L.J.; Leitch, I.J. In Depth Characterization of repetitive DNA in 23 plant genomes reveals sources of genome size variation in the legume tribe Fabeae. PLoS ONE 2015, 10, e0143424. [Google Scholar] [CrossRef] [PubMed]
- Vondrak, T.; Robledillo, L.Á.; Novák, P.; Koblížková, A.; Neumann, P.; Macas, J. Characterization of repeat arrays in ultra-long Nanopore reads reveals frequent origin of satellite DNA from retrotransposon-derived tandem repeats. Plant J. 2020, 101, 484–500. [Google Scholar] [CrossRef] [PubMed]
- Boeke, J.D.; Chapman, K.B. Retrotransposition mechanisms. Curr. Opin. Cell Biol. 1991, 3, 502–507. [Google Scholar] [CrossRef]
- Bennetzen, J.L.; Kellogg, E.A. Do plants have a one-way ticket to genomic obesity? Plant Cell 1997, 9, 1509–1514. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Bennetzen, J.L. Rapid recent growth and divergence of rice nuclear genomes. Proc. Natl. Acad. Sci. USA 2004, 101, 12404–12410. [Google Scholar] [CrossRef] [PubMed]
- Bennetzen, J.L.; Ma, J.X.; Devos, K. Mechanisms of recent genome size variation in flowering plants. Ann. Bot. 2005, 95, 127–132. [Google Scholar] [CrossRef]
- Vitte, C.; Panaud, O. LTR retrotransposons and flowering plant genome size: Emergence of the increase/decrease model. Cytogenet. Genome Res. 2005, 110, 91–107. [Google Scholar] [CrossRef] [PubMed]
- Vitte, C.; Panaud, O.; Quesneville, H. LTR retrotransposons in rice (Oryza sativa, L.): Recent burst amplifications followed by rapid DNA loss. BMC Genom. 2007, 8, 218. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, J.S.; Proulx, S.R.; Rapp, R.A.; Wendel, J.F. Rapid DNA loss as a counterbalance to genome expansion through retrotransposon proliferation in plants. Proc. Natl. Acad. Sci. USA 2009, 106, 17811–17816. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.X.; Rizzon, C.; Du, J.C.; Zhu, L.C.; Bennetzen, J.L.; Jackson, S.A.; Gaut, B.S.; Ma, J.X. Do genetic recombination and gene density shape the pattern of DNA elimination in rice long terminal repeat retrotransposons? Genome Res. 2009, 19, 2221–2230. [Google Scholar] [CrossRef]
- Jedlicka, P.; Lexa, M.; Keinovsky, E. What can Long Terminal Repeats tell us about the age of LTR retrotransposons, gene conversion and ectopic recombination? Front. Plant Sci. 2020, 11, 644. [Google Scholar] [CrossRef]
- Kimura, M. The Neutral Theory of Molecular Evolution; Cambridge University Press: Cambridge, UK, 1983. [Google Scholar]
- Ellis, T.H.N.; Poyser, S.J.; Knox, M.R.; Vershinin, A.V.; Ambrose, M.J. Polymorphism of insertion sites of Ty1-copia class retrotransposons and its use for linkage and diversity analysis in pea. Mol. Gen. Genet. 1998, 260, 9–19. [Google Scholar] [CrossRef]
- Kreplak, J.; Madoui, M.-A.; Cápal, P.; Novák, P.; Labadie, K.; Aubert, G.; Bayer, P.E.; Gali, K.K.; Syme, R.A.; Main, D.; et al. A reference genome for pea provides insight into legume genome evolution. Nat. Genet. 2019, 51, 1411–1422. [Google Scholar] [CrossRef]
- Lee, D.; Ellis, T.H.N.; Turner, L.; Hellens, R.P.; Cleary, W.G. A copia-like element in Pisum demonstrates the uses of dispersed repeated sequences in genetic analysis. Plant Molec. Biol. 1990, 15, 707–722. [Google Scholar] [CrossRef]
- Chavanne, F.; Zhang, D.-X.; Liaud, M.-F.; Cerff, R. Structure and evolution of Cyclops: A novel giant retrotransposon of the Ty3/Gypsy family highly amplified in pea and other legume species. Plant Mol. Biol. 1998, 37, 363–375. [Google Scholar] [CrossRef]
- Jing, R.C.; Knox, M.R.; Lee, J.M.; Vershinin, A.V.; Ambrose, M.; Ellis, T.H.N.; Flavell, A.J. Insertional polymorphism and antiquity of PDR1 retrotransposon insertions in Pisum species. Genetics 2005, 171, 741–752. [Google Scholar] [CrossRef]
- SeedStor Homepage. Available online: https://www.seedstor.ac.uk/ (accessed on 23 September 2020).
- Kimura, M.; Crow, J.F. The number of alleles that can be maintained in a finite population. Genetics 1964, 49, 725–738. [Google Scholar] [PubMed]
- Kimura, M.; Ohta, T. Theoretical Aspects of Population Genetics; Monographs in Population Biology. 4; Princeton University Press: Princeton, NJ, USA, 1971. [Google Scholar]
- Kimura, M. Rare variant alleles in the light of the neutral theory. Mol. Biol. Evol. 1983, 1, 84–93. [Google Scholar] [PubMed]
- Garfinkel, D.J.; Stefanisko, K.M.; Nyswaner, K.M.; Moore, S.P.; Oh, J.; Stephen, H.; Hughes, S.H. Retrotransposon Suicide: Formation of Ty1 Circles and Autointegration via a Central DNA Flap. J. Virol. 2006, 80, 11920–11934. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schulman, A.H. Retrotransposon replication in plants. Curr. Opin. Virol. 2013, 3, 604–614. [Google Scholar] [CrossRef]
- SanMiguel, P.; Gaut, B.S.; Tikhonov, A.; Nakijama, Y.; Bennetzen, J.L. The paleontology of intergene retrotransposons of maize. Nat. Genet. 1998, 20, 43–45. [Google Scholar] [CrossRef]
- Kimura, M.; Ohta, T. The age of a neutral mutant persisting in a finite population. Genetics 1973, 75, 199–5312. [Google Scholar]
- Lavin, M.; Herendeen, P.S.; Wojciechowski, M.F. Evolutionary rates analysis of Leguminosae implicates a rapid diversification of lineages during the Tertiary. Syst. Biol. 2005, 54, 575–594. [Google Scholar] [CrossRef]
- Bertioli, D.J.; Cannon, S.B.; Froenicke, L.; Huang, G.; Farmer, A.D.; Cannon, E.K.S.; Liu, X.; Gao, D.; Clevenger, J.; Dash, S.; et al. The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat. Genet. 2016, 48, 438–446. [Google Scholar] [CrossRef]
- Mood, A.M. The Distribution Theory of Runs. Ann. Mat. Stat. 1940, 11, 367–392. [Google Scholar] [CrossRef]
- Jing, R.; Johnson, R.; Seres, A.; Kiss, G.; Ambrose, M.J.; Knox, M.R.; Ellis, T.H.N.; Flavell, A.J. Gene-based sequence diversity analysis of field pea (Pisum). Genetics 2007, 177, 2263–2275. [Google Scholar] [CrossRef]
- Sulima, A.S.; Zhukov, V.A.; Afonin, A.A.; Zhernakov, A.I.; Tikhonovich, I.A.; Lutova, L.A. Selection signatures in the first exon of paralogous receptor kinase genes from the Sym2 region of the Pisumsativum, L. genome. Front. Plant Sci. 2017, 8, 1957. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, M.A.; Shaw, M.; Cooper, R.D.; Frew, T.J.; Butler, R.C.; Murray, S.R.; Moya, L.; Coyne, C.J.; Timmerman-Vaughan, G.M. Association mapping of starch chain length distribution and amylose content in pea (Pisum sativum L.) using carbohydrate metabolism candidate genes. BMC Plant Biol. 2017, 17, 132. [Google Scholar] [CrossRef] [PubMed]
- Casacuberta, J.M.; Vernhettes, S.; Audeon, C.; Grandbastien, M.-A. Quasispecies in retrotransposons: A role for sequence variability in Tnt1 evolution. Genetica 1997, 100, 109–117. [Google Scholar] [CrossRef]
- Hahn, M.W. Toward a selection theory of molecular evolution. Evolution 2008, 62, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, H.; Hechenleitner, P.; Santos-Guerra, A.; Menezes de Sequeira, M.; Pennington, R.T.; Kenicer, G.; Carine, M.A. Systematics, biogeography, and character evolution of the legume tribe Fabeae with special focus on the middle-Atlantic island lineages. BMC Evol. Biol. 2012, 12, 250. [Google Scholar] [CrossRef]
- Pea Genome Project. Available online: https://urgi.versailles.inra.fr/Species/Pisum/Pea-Genome-project (accessed on 25 September 2020).
- FAOSTAT. Available online: http://www.fao.org/faostat/en/#data/QC (accessed on 25 September 2020).
- Smýkal, P.; Hradilová, I.; Trněný, O.; Brus, J.; Rathore, A.; Bariotakis, M.; Das, R.R.; Bhattacharyya, D.; Richards, C.; Coyne, C.J.; et al. Genomic diversity and macroecology of the crop wild relatives of domesticated pea. Sci. Rep. 2017, 7, 17384. [Google Scholar] [CrossRef]
- Mumtaz, A.S.; Shehadeh, A.; Ellis, T.H.N.; Ambrose, M.J.; Maxted, N. The collection and ecogeography of non-cultivated peas (Pisum, L.) from Syria. PGR Newslet. 2006, 146, 3–8. [Google Scholar]
- Blixt, S. Mutation genetics in Pisum. Agri. Hort. Genet. 1972, 30, 1–293. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | μ ± SD, n 1 | Estimated Age 2 | Source |
|---|---|---|---|
| PDR1 | 0.013 ± 0.011, 49 | 1.89 ± 0.33 | [25] |
| PDR1 | 0.013 ± 0.014, 25 | 1.86 ± 0.55 | This work, [22] |
| PDR1 | 0.013 ± 0.013, 74 | 1.88 ± 0.30 | combined |
| Cyclops | 0.016 ± 0.009, 49 | 2.20 ± 0.30 | This work, [22] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ellis, T.H.N.; Vershinin, A.V. Retrotransposons and the Evolution of Genome Size in Pisum. BioTech 2020, 9, 24. https://doi.org/10.3390/biotech9040024
Ellis THN, Vershinin AV. Retrotransposons and the Evolution of Genome Size in Pisum. BioTech. 2020; 9(4):24. https://doi.org/10.3390/biotech9040024
Chicago/Turabian StyleEllis, T. H. Noel, and Alexander V. Vershinin. 2020. "Retrotransposons and the Evolution of Genome Size in Pisum" BioTech 9, no. 4: 24. https://doi.org/10.3390/biotech9040024
APA StyleEllis, T. H. N., & Vershinin, A. V. (2020). Retrotransposons and the Evolution of Genome Size in Pisum. BioTech, 9(4), 24. https://doi.org/10.3390/biotech9040024