1. Introduction
Microbial lipolytic enzymes are of biotechnological interest due to their versatility in bioconversions processes such as transesterification (acidolysis, aminolysis, alcoholysis, and interesterification) and esterification. They have multiple applications in diverse industries (food, textile and leather, pharmaceuticals, cosmetics, and detergents). In addition, lipases fulfill multiple needs of the biotechnological industry such as the synthesis of flavors or biodiesel production [
1,
2].
The most relevant features of lipases are their stereo-, regio-, and chemo-specificity [
2,
3], associated with α/β hydrolase canonical folding that exposes the serine, aspartic acid, and histidine residues in a catalytic triad. This tertiary structure of lipases possesses domains (the cap or lid, the binding pocket, and the oxyanionic hole) recognized as essentials for their interaction with a substrate [
3]. Two different lipolytic enzymes have been distinguished: lipases (E.C. 3.1.1.3), which hydrolyze short-, medium-, and long-chain triacylglycerides and act at the water-oil interface, and carboxylesterases (E.C. 3.1.1.1), which hydrolyze short chain substrates and cannot act on triglycerides [
4].
Extracellular bacterial lipolytic enzymes are key for the hydrolysis of triacylglycerides and act as a pathogenic factor [
5]. Bacterial lipases are classified according to their origin and conserved motifs in their amino acid sequences. Hence, their initial classification as family I enzymes for true lipases and family II to VIII enzymes for carboxilesterases has been increased to XXXV families, with new members being added up to the present day [
4,
6]. The classification of new lipolytic enzymes and the study and description of their catalytic performance will allow us to better exploit their capabilities for innovative applications.
The
Neobacillus thermocopriae species is classified based on the characteristics of the Niacini clade [
7]. As members of the genus
Neobacillus, they are rod-shaped, Gram-positive or Gram-negatives variables, and aerobic or facultatively anaerobic; they form endospores under adverse nutrient or environmental conditions, can be motile or non-motile, and have a maximum growth temperature ranging from 50 to 55 °C, with the optimum temperature falling between 25 and 37 °C. Eleven protein sequences, specific for the clade, can be used as molecular markers.
This study aimed to clone, express, and characterize a lipolytic enzyme produced by the thermophilic microorganism
Neobacillus thermocopriae C255. This strain was isolated from an ash sample collected from the soil surface in the foothills of the Popocatépetl volcano at a site known as Paso de Cortés, coordinates 19° 04′ 49.7” N–98° 33′ 21.6” W, with an altitude of 2880 m.a.s.l. The sample site is located in an open area in an oak and coniferous forest with an average temperature of 21 °C on the day of sampling. This zone is located within the Trans Mexican Volcanic Belt, which crosses the country from the south of the Gulf of California to the Gulf of México. It was formed by of the subduction of the Rivera and Cocos Plates and is located on the southwestern margin of the North American Plate. This region includes volcanic centers, geothermal soils, and subterranean thermal water currents [
8].
2. Materials and Methods
2.1. Bacterial Strain Isolation and Screening for Lipolytic Activity
A sample of collected ash was added as an inoculum and incubated in Luria Bertani medium (10 g/L of casein tryptone, 5 g/L of NaCl, 5 g/L of yeast extract, pH 6.5) at 55 °C, 125 rpm, for 12 h. An aliquot of the grown culture was taken for subsequent reseeding in Luria Bertani plates until an axenic culture was obtained. The strains obtained were seeded on rhodamine plates (2.5% (
v/
v) high oleic safflower oil, 8 g/L of nutritive oil, 4 g/L of NaCl, 20 g/L of agar, 0.001%
w/
v rhodamine B) and incubated at 55 °C for 18 h to detect lipolytic activity [
9].
2.2. Genomic DNA Extraction
The genomic DNA of the selected strain, C255, was obtained using traditional methods described elsewhere. In brief, a cell pellet retrieved from a well grown culture (5 mL) was resuspended in 1 mL of resuspension buffer (50 mM Tris-EDTA pH8), ice-incubated for 10 min, mixed with 100 μL of lysozyme solution (250 mM of Tris pH8, 10 mg/mL of lysozyme), and then iced-incubated again for 45 min. Lysis was achieved using 200 μL of lysis solution (0.5% SDS, 50 mM Tris pH 7.5, 0.4 M EDTA, 1 mg/mL Proteinase K), incubated at 50 °C for 60 min, and mixed via inversion. At this point, the sample was split into two microtubes, and 600 μL of phenol saturated with 50 mM of Tris was added to each sub-sample, iced-incubated for 10 min, and centrifugated at 10,000× g at 4 °C for 15 min. The supernatant was recovered, and 3 M sodium acetate was added at a 1/10 (v/v) proportion, mixed via inversion, ice-incubated for 10 min, centrifugated at 15,000× g at 4 °C for 15 min, and the supernatant was recovered. An equal volume of isopropanol was added at an ambient temperature, mixed via inversion, iced-incubated for 25 min, and centrifugated at 13,000× g for 15 min at 4 °C. Then, isopropanol was decanted, and DNA that adhered to the bottom of the microtube was washed with 75% (v/v) cold ethanol and centrifugated at 16,000× g at 4 °C for 10 min. The DNA recovered was evaporated using SpeedVac for 20 min and resuspended in nuclease-free water. DNA was quantified using NanoDrop and verified in electrophoresis 1% agarose gel.
2.3. Genome Sequencing, Assembly, Annotation and Lipolytic Enzyme Selection
The genome sequencing of the selected strain (C255) was carried out using Illumina-MiSeq. The genome was assembled using SPAdes [
10] and annotated using RAST [
11]. The small subunit ribosomal RNA 16S gene and Flagellar M-ring protein FliF sequence were used for taxonomic identification [
7]. Enzymes identified as lipases or carboxylesterases were manually searched for using the annotation file. The potential sequences were processed using InterPro [
12]. The nucleotide FASTA file from RAST was loaded using EGGNOG-Mapper [
13] to obtain the KO numbers of the annotated genes. The list was loaded using KEGG Mapper [
14] to identify metabolic pathways where the selected Mgl-C255 enzyme was involved.
2.4. Mgl-C255 In Silico Sequence Analysis and Model Prediction
The nucleotides sequence of the selected enzyme gene was translated into amino acids using ExPASy Translate. The primary structure was used to calculate physicochemical parameters using ExPASy ProtParam; these parameters included the amino acid composition, molecular weight, isoelectric point, and molar extinction coefficient. Also, we identify conserved domains, PFAM conserved domains classification, and the presence of signal peptide using InterPro and transmembrane regions using TMHMM [
15]. Moreover, we predicted the tertiary structure model using CoLab Fold [
16], and this model was useful for locating the catalytic triad, hydrophobic patches, and composition of secondary structures of the α/β hydrolase canonical fold.
2.5. Cloning, Expression and Purification of Mgl-C255
The Mgl-C255 gene (mgl) was amplified via PCR and cloned with the NZYEasy Cloning and Expression I kit (MB282, NZYTech, Lisbon, Portugal) using the pHTP1 plasmid, which added a His6 Tag to the recombinant protein. The vector size was 5332 bp, and the vector had a T7 promoter that controlled gene expression. The cloning region was flanked by 16 bp, which annealed extensions added to the primers. Primers were designed with extensions according to the manufacturer’s instructions, allowing us to perform a ligase-independent cloning reaction with the plasmid. The PCR gene amplification program was as follows: DNA denaturation (95 °C, 3 min), then 30 cycles (95 °C, 30 s; 55 °C, 30 s; 72 °C, 60 s), followed by elongation (72 °C, 7 min). The reaction for genetic construction (pHTP-mgl) was prepared with enzymes and buffers supplied by the manufacturer and these elements were incubated at 37 °C for 60 min. The transformation was developed with E. coli BL21 made competent through the CaCl2 chemical process. Transformed cells were incubated in 1 mL of liquid LB medium at 37 °C for 45 min and transferred to a LB plate paired with kanamycin at a 0.05 mg/mL final concentration. Cloning was confirmed via gene amplification from the plasmid (pHTP-mgl).
A 200 mL culture of recombinant Mgl-C255 at OD600 = 0.8 was added to IPTG with a final concentration of 0.8 mM and incubated at 25 °C for 12 h to induce Mgl-C255 expression. Cells were recovered via centrifugation at 8000× g at 4 °C for 10 min. The supernatant was discarded. The pellet was washed twice with binding buffer (50 mM of sodium phosphate, 0.3 M of NaCl, 5 mM of imidazole, pH 8.0) and resuspended with 2 mL of binding buffer. Resuspended cells were lysed with ultrasonic pulses (20 cycles/20 s) at a 65% amplitude in an ice bath. Cellular lysate was separated via centrifugation at 15,000× g at 4 °C for 20 min. Soluble protein was recovered from the supernatant and filtered through a 0.45 μM nitrocellulose membrane. This is how the Mgl-C255 crude extract was obtained.
Mgl-C255 purification was performed via affinity chromatography (Profinity™ IMAC. Cat. No. 1560123). Uncharged resin was pre-treated and charged with 0.2 M of NiSO4 at pH 7.0 according to the manufacturer’s instructions. Charged resin (1 mL) was placed in a column and equilibrated with the binding buffer. The column was loaded with 1 mL of Mgl-C255 crude extract up to 10 C.V. with binding buffer. The column was placed in cold, gentle agitation for 1 h, and then the column was washed with 15 C.V. of binding buffer. Subsequently, the Mgl-C255 extract was eluted with 0.5 mL of elution buffer (50 mM of sodium phosphate, 0.3 M of NaCl, 400 mM of imidazole, pH 8.0) until no protein was detected at A280. The recovered fractions were analyzed for Mgl-C255 presence and dialyzed against either 50 mM of sodium phosphate at pH 7.4 or 50 mM of HEPES at pH 8.0. This Mgl-C255 solution was named enzymatic extract.
2.6. Lipolytic Activity Assay and Protein Quantification
The enzymatic activity of Mgl-C255 was quantified using a modified Nawani assay [
17]. In brief, 0.4 mL of 50 mM sodium phosphate at pH 7.4 and 0.05 mL of enzymatic extract at 0.315–0.345 mg/mL were incubated at 35 °C for 5 min. Then, 0.05 mL of substrate solution (0.01 M
p-nitrophenyl laurate in ethanol) was added, and it was incubated for 30 min under the same conditions and 0.125 mL of 0.1 M Na
2CO
3 was added. The solution was centrifugated at 15,000×
g at 4 °C for 15 min. A blank without enzyme was placed and the absorbance was measured in a spectrophotometer at 405 nm.
For lipolytic activity quantification, the p-nitrophenol molar absorption coefficient 0.001159 μmol−1 cm−1 was considered. One unit of lipase activity was calculated as the μmol of p-nitrophenol released per mg of enzyme under the assay conditions.
For protein quantification, the absorption at 280 nm was measured using a NanoDrop™ 2000 spectrophotometer (Thermo Fisher Scientific, Inc., Waltham, MA, USA). (Thermofisher).
2.7. Zymogram
An in situ activity assay [
18] was performed on a 12% polyacrylamide gel under denaturing conditions. After electrophoresis, the gel was incubated for 30 min with constant agitation in a 2.5% (
v/
v) Triton X-100 solution and washed twice for 20 min with 50 mM of sodium phosphate at pH 7.0; fluorescence was detected after incubation for 3 min with 10 mL of substrate solution (50 mM of sodium phosphate at pH 7.0 with 0.2 mL of 25 mM MUF-butyrate in ethylene glycol monomethyl ether). A fluorescent band indicated lipolytic activity. After zymogram, the gel was stained with Coomasie blue.
2.8. Biochemical Characterization of Mgl-C255
2.8.1. Optimum Temperature and pH
To determine the optimum pH, the spectrophotometric assay described above was performed, with a varying buffer pH, for all cases at 50 mM: sodium phosphate (pHs 6.0 and 6.5), HEPES (pHs 7.0, 7.5 and 8.0), TRIS-HCl (pH 8.5), and CHES (pHs 9.0, 9.5, and 10.0). To determine the optimum temperature of the enzyme, the spectrophotometric assay was carried out at pH 8.0 by varying the incubation temperature from 20 to 60 °C. For determining both the optimum pH and optimum temperature, the highest activity was considered to be 100% relative activity.
2.8.2. Substrate Chain Length Preference
Substrate preference was evaluated with short- (C:2, C:4 and C:5), medium-, and long-chain (C:8, C:10, C:12, C:14 y C:16) p-nitrophenyl substrates. A lipolytic assay was performed with 50 mM of HEPES buffer at pH 8.0 at 35 °C. The release of p-nitrophenol was measured in all cases. The highest activity was considered to be 100% of relative activity.
2.8.3. Effect of Metal ions on Enzymatic Activity
Mgl-C255 was incubated at 35 °C for 1 h in 50 mM of HEPES at pH 8.0 with the metal ion maintained at a concentration of 1 mM. After that, the activity assay described previously was performed with p-nitrophenyl laurate used as the substrate to measure residual activity. The salts tested were: KCl, BaCl2, LiCl, MnCl2, NaCl, CaCl2, SrCl2, MgCl2, CuCl2, and HgCl2. The control sample consisted of the enzyme with buffer only, which was considered to have 100% relative activity.
2.8.4. Effect of Detergents and Inhibitors on Enzymatic Activity
Mgl-C255 was incubated at 35 °C for 1 h in 50 mM of HEPES at pH 8.0 with the following compounds at varying final concentrations: PMSF (1 mM), β-mercaptoethanol (1%), Triton X-100 (1%), Tween 20 (1%), SDS (0.1%), and EDTA (1 mM). The control sample consisted of the enzyme with buffer only, which had 100% relative activity. A spectrophotometric assay was performed, as described above, with p-nitrophenyl laurate being the substrate.
2.8.5. Solvent Stability of the Enzyme
To evaluate the stability of Mgl-C255 in the presence of solvents, the purified enzyme dissolved in 50 mM of HEPES pH 8.0 was lyophilized. Then, the enzyme was incubated at 30 °C for 1 h, with the indicated solvent composed as follows: DMSO, acetonitrile, ethanol, acetone, 1-propanol, 2-propanol, tert-butanol, 1-butanol, hexane, and octanol. At the end of the incubation period, residual activity was measured in a 100 μL aliquot, according to activity assay previously described, except that p-nitrophenyl butyrate was used as the substrate. The control sample was incubated in 50 mM of HEPES at pH 8.0, which was considered to have 100% relative activity.
2.8.6. Transesterification and Aminolysis Reactions
Based on Mgl-C255 biochemical characterization results, selected reactions were performed. Vinyl propionate was used as the acyl donor, with tert-butanol, β-citronellol, D-glucose, D-xylose, or D-fructose used as the acceptors. Reactions were performed at 35 °C for 24 h at 300 rpm. In each case, another reaction without enzymes was prepared as a control.
For reactions with tert-butanol or β-citronellol, 1 mmol of each substrate, 900 μL of acetone as the reaction medium, 0.04 g molecular sieves, and 0.01 g of lyophilized Mgl-C255 were used.
For D-fructose, 1 mmol was dissolved in 1 mL of ethanol with 0.5 mL of DMSO. For D-xylose or D-fructose, 1 mmol was dissolved in 1 mL of ethanol with 0.8 mL of DMSO. Every dissolved substrate volume was divided into two vials: one for the enzyme reaction and one for the control (without Mgl-C255). No other solvent was added as a reaction medium. Thus, reactions were prepared with 0.5 mmol of monosaccharide, 2 mmol of vinyl propionate for the reaction with D-xylose, 2.5 mmol of vinyl propionate for the reactions with D-glucose or D-fructose, 0.04 g molecular sieves, and 0.01 g of lyophilized Mgl-C255.
An aminolysis reaction was assayed with benzylamine as the acyl group acceptor and methyl acetate as the donor in the following form: 1 mmol of each substrate, 900 μL of acetone as the reaction medium, 0.04 g molecular sieves, and 0.01 g of lyophilized Mgl-C255. A control was placed without an enzyme. The reaction proceeded at 35 °C for 24 h at 300 rpm.
2.8.7. TLC Analysis
The presence of the product was verified via thin layer chromatography using silica gels plates and a mixture of heptane, methyl acetate, and ethanol as the mobile phase. A proportion of 1:2:2 was used for alcohol transesterification and aminolysis, while a proportion of 1:1:3 was used for monosaccharides transesterifications. The plates were developed with an iodine chamber.
3. Results
3.1. Strain Screening and Isolation
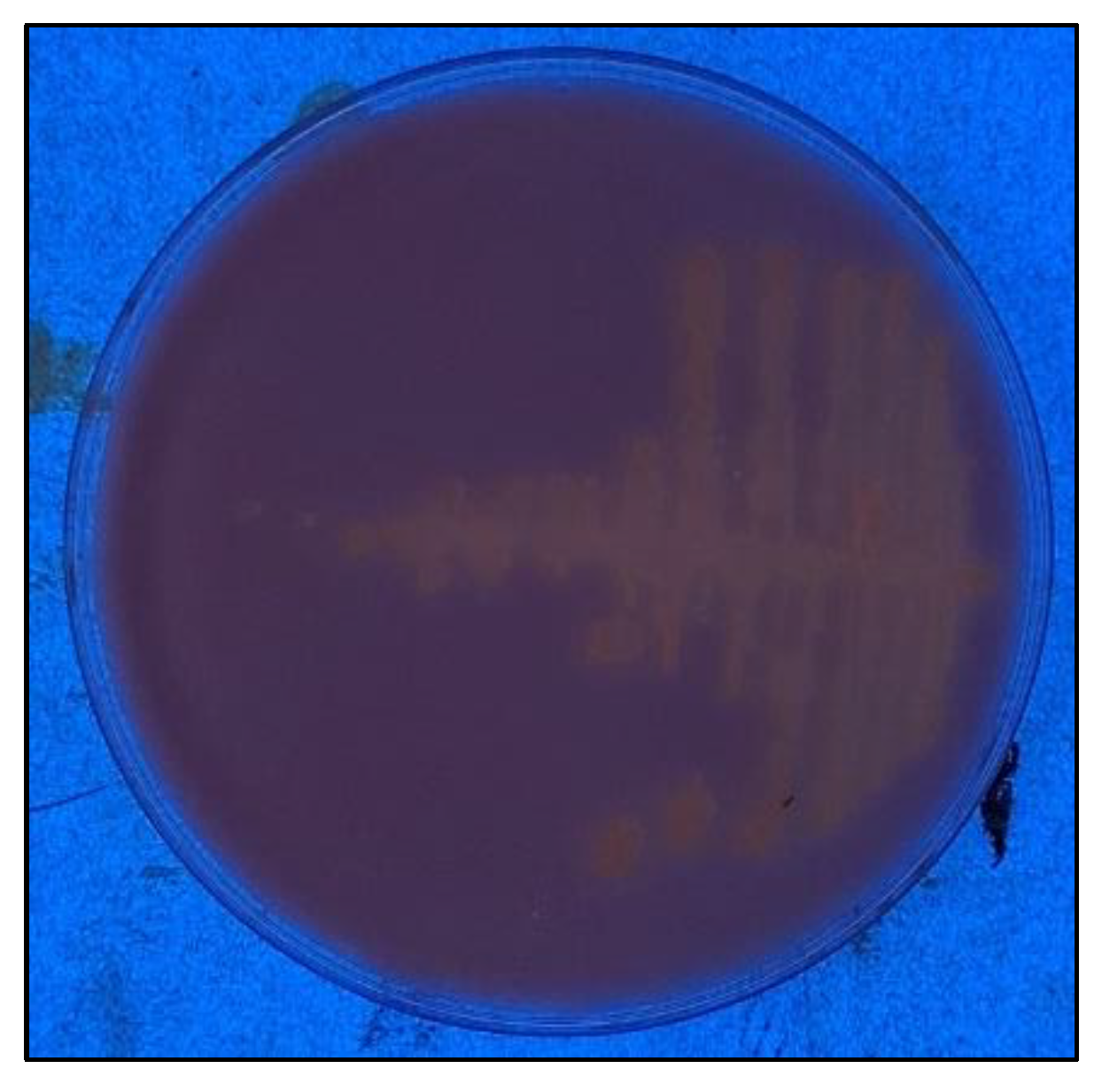
After inoculating LB medium with an ash sample, we obtained an axenic culture of a Gram-positive strain composed of the short bacillus C255, which was subsequently identified as
Neobacillus thermocopriae C255. The strain was grown at 55 °C and 125 rpm in hydrolyzed rhodamine-b (
Figure 1) on a plate with high-oleic safflower oil.
3.2. Genome Sequencing, Assembly and Annotation
Genome sequencing through Illumina-Miseq yielded 373,967 sequences with lengths of 35–151 and 37% GC. Genome assembly through SPAdes resulted in 153 contigs, 46 of which were greater than or equal to 1000 nt; the largest contig was 288,690 nt, the GC content was confirmed as being 37.40%, and the following metrics were obtained for contig length: N50 150,002, N75 78,462, L50 8, and L75 16.
Genome annotation was performed with RAST. Molecular identification was carried out with the nucleotide sequence of the small subunit of 16S ribosomal RNA and amino acid sequence of the flagellar M-ring protein FliF, which were compared in the NCBI database, demonstrating similarities of 100% (1301 bps) with the partial sequence of the 16S gene (Accession: PP813672.1) of Neobacillus thermocopriae strain MAILOU-3 and 100% (536 aa) with the FliF protein (Accession: WP_327989547.1) of Neobacillus thermocopriae.
The information obtained from annotation was used to manually search for sequences annotated as lipases and carboxylesterases, identifying a monoacylglycerol lipase of 267 amino acids, a carboxylesterase of 248 amino acids, and a GDSL-SGNH lipase of 274 amino acids; the monoacylglycerol lipase selected for this work was named MGL-C255. This enzyme possessed the α/β hydrolase fold, without a signal peptide or transmembrane region, and was classified as PFAM 12146.
A FASTA file of nucleotide sequences obtained with annotation was analyzed with EGGNOG-Mapper; as a result, 1944 products with KO numbers and 1030 without KO numbers were obtained. Subsequently, KEGG Mapper analysis located Mgl-C255 and 19 other enzymes in glycerophospholipid metabolism (Pathway 00564), with the function designated as E.C. 3.1.1.5 for the hydrolysis of the acyl chains of both 1- or 2-acyl-sn-glycero-3-phosphocholine and 1- or 2-acyl-sn-glycero-3-phosphoethanolamine.
3.3. In Silico Analysis of the Sequence and Model Prediction of the Selected Enzyme
The Mgl-C255 nucleotide sequence was translated into 267 amino acids, with a molecular weight of 31 kDa, isoelectric point of 6.38, and molar extinction coefficient of 58,900 M
−1 cm
−1. The sequence had no signal peptide or transmembrane regions. Tertiary structure model prediction was conducted and a catalytic triad (S90, D207 and H237) was located (
Figure 2a), as well as a cap domain. The extent of enzyme surface hydrophobicity indicated a high proportion of hydrophobic residues in the enzyme cap domain (
Figure 2b). Although this was determined via an in silico approximation, the estimation of these values was relevant as a reference, with the expected behavior of the enzyme in the purification and characterization process shown below.
The enzyme possessed a canonical hydrolase α/β fold (
Figure 3) formed by seven β-sheets (β2 to β8) and six α-helixes (αA to αF), in addition to the five structures that formed the cap (β1′, β2′, α1, α2, and α3).
The sequence alignment of Mgl-C255 was performed with other monoglyceride lipase enzymes whose structures had been experimentally resolved, revealing 90% coverage and 30% identity with a monoglyceride lipase from
Homo sapiens (PDB:3HJU) [
19], 92% coverage and 28% identity with a monoglyceride lipase from
Mycobacterium tuberculosis (PDB:7P0Y) [
20], 89% coverage and 32% identity with a monoglyceride lipase from
Paleococcus ferrophilus (PDB:6QE2) [
21], and 90% coverage and 25% identity with a monoglyceride lipase from
Saccharomyces cerevisiae (PDB:4ZWN) [
22]. With this alignment, the catalytic triad and three conserved blocks were located within the structure and sequences (
Figure 4), which allowed for the classification of Mgl-C255 as a family V carboxylesterase, with Ser in block II and Asp and His in block III [
6].
3.4. Cloning and Expression of Mgl-C255
To obtain the recombinant Mgl-C255 enzyme, the following primers were designed: FmglC255: 5′-TCAGCAAGGGCTGAGG
ATGTGGAAGTGGGAAGC and RmglC255: 5′-TCAGCGGAAGCTGAGG
TTACACAATATATCCGATTGCC. These primers included the extensions needed for the cloning kit, as well as the zones for hybridization with the enzyme gene (underlined). The PCR program described in the Materials and Methods Section was used, having a T
m of 55 °C. Once
E. coli BL21 cells were transformed, an LB agar plate was inoculated. Two colonies grew at 37 °C after 12 h of incubation. Gene insertion was verified via PCR (
Figure 5a).
Mgl-C255 gene expression was induced with 0.8 mM of IPTG, and the protein was partially purified (
Figure 5b). Purification was performed using the crude extract obtained, as described above, via affinity chromatography. The specific activity increased by 5.04 (purification fold), and the enzyme recovery rate was 88% (yield).
The activity assay performed in situ indicated that substrate hydrolysis (MUF-butyrate) occurred at a 65 kDa band, suggesting that a false dimer [
21] or artifact of the enzyme (31.1 kDa) was generated by interactions between the hydrophobic residues in the cap.
3.5. Biochemical Characterization
A spectrophotometric assay was employed to determine Mgl-C255 lipolytic activity at different pHs and temperatures, as well as in the presence of metal ions, inhibitors, and solvents.
Our results indicate that Mgl-C255 had the highest activity at a slightly alkaline pH (
Figure 6a) between 7.5 and 8.5, while at more acidic (6.0) or alkaline (9.5 and 10) extremes, its activity declined. pH 8 was used to evaluate the effect of temperature on the enzyme (
Figure 6b). The activity assay was performed using
p-nitrophenyl laurate as the substrate, and the amount of enzyme was standardized to 0.017 mg for each assay. The enzyme retained its activity between 30 and 45 °C, while at 60 °C, it retained half of its highest activity (40 °C).
In terms of substrate preference, the enzyme showed a higher affinity for short-chain substrates (
Figure 7a), as well as for the monoacylglycerol lipase from
Paleococcus ferrophilus [
21]. We observed that the addition of sodium carbonate at the end of the assay caused immediate short-chain substrate hydrolysis, so this step was replaced by the addition of 0.125 mL of a 50 mM HEPES buffer at pH 8.0. The amount of enzyme and the reaction time for the short-chain substrates assays were also halved. The units of activity obtained with the medium- and long-chain substrates were only 3 and 1% (
Figure 7b) with respect to
p-nitrophenyl acetate (C-2). These results show that Mgl-C255 behaved similarly to carboxylesterases.
Regarding the catalytic performance of the enzyme in the presence of metal ions, inhibitors, and solvents (
Table 1), we found that at the concentration evaluated, lithium, sodium, potassium, calcium, and barium ions had no effect on Mgl-C255 activity, while strontium, magnesium, manganese, copper, and mercury ions reduced this activity. Some metal ions can be considered as cofactors that enhance catalytic activity. This occurs by stabilization of the enzyme conformation and its active site. Calcium ions increased the activity of some lipases. On the other hand, the mercury ions bound with the thiol group of cysteine close to the catalytic triad, preventing the substrate from accessing the active site.
The enzyme reaction mixture was incubated with every metal ion, detergent, inhibitor, or organic solvent at 35 °C for 1 h. The enzyme was lyophilized for organic solvent testing. In each case, a control was placed with buffer only, representing 100% activity.
With respect to the effect of inhibitors (
Table 1), we observed that β-mercaptoethanol, Triton X-100, Tween 20, and EDTA had no significant effects on Mgl-C255 activity, while SDS and PMSF reduced its activity the former through a denaturation effect and the loss of its tertiary structure and the latter by inhibiting the action of enzymes that have serine at their active site, in this case Ser 90. On the other hand, the absence of disulfide bonds, through to the in-silico analysis of the sequence, was experimentally confirmed by the unchanged activity of β-mercaptoethanol, which is known for breaking disulfide bonds.
Concerning the activity in the presence of solvents (
Table 1), in all cases, there was a decrease in activity with respect to the control; however, we observed that acetone, DMSO, and acetonitrile, but not hexane, could be used as reaction media for Mgl-C255.
3.6. Reactions
Alcohol transesterification reactions, such as the reaction of vinyl propionate with tert-butanol or β-citronellol, were qualitatively verified through thin layer chromatography (TLC). Silica gel plates were used as the stationary phase and heptane/methyl acetate/ethanol (1:2:2) was used as the mobile phase. In both cases (tert-butanol or β-citronellol), the product was more clearly observed (
Figure 8, lanes 1 and 3) compared to the reaction without the enzyme (lanes 2 and 4); however, since β-citronellol was used as a racemic mixture, further tests should be conducted to verify the enantioselectivity of the enzyme.
Monosaccharide transesterification reactions (vinyl propionate with D-glucose, D-fructose, or D-xylose) were evaluated (
Figure 8) via thin layer chromatography (mobile phase heptane/methyl acetate/ethanol in 1:1:3 ratio), and the product is clearly seen (lanes 5, 7, and 9), in contrast to the reaction without the enzyme (lanes 6, 8, and 10).
Similarly, for the aminolysis reaction (
Figure 8, lanes 11 and 12), using benzylamine as an acyl group acceptor and methyl acetate as a donor, TLC was performed on the same mobile phase as the alcohol and vinyl propionate reactions. At the bottom of the plate (lane 11), we can see a product of the reaction, which is not present in lane 12.
4. Discussion
In this study, the strain Neobacillus thermocopriae C255 was isolated from volcanic ash deposited in an open space, surrounded by a coniferous forest, on the foothills of Popocatépetl, which is considered an active volcano. In the laboratory, the strain grew in conditions of 55 °C, pH 6.5, 0.5% NaCl, and orbital agitation at 125 rpm.
This species was first reported through the isolation of the strain SgZ-7 from compost [
23]. Named
Bacillus thermocopriae, it was described as being an anaerobic facultative, rod-shaped, and Gram-positive, with motility, endospore formation, growth between 40 and 60 °C, a pH between 6 and 10, and 0 to 3% NaCl, with the optimum values being 50 °C, a pH of 7.2, and 1% NaCl. After that,
Bacillus thermocopriae was located in the Niacini clade [
7] and named the
Neobacillus genus.
The isolation of thermophilic microorganisms as a source of enzymes of biotechnological interest has benefited from genomic sequencing. Thus, with the annotation of the genome of Neobacillus thermocopriae C255, it was possible to obtain a lipolyitic enzyme, specifically a monoacyl glycerol lipase (267 amino acids, 31.1 kDa), with a canonical α/β hydrolase fold named Mgl-C255.
Monoacyl glycerol lipases have been studied due to their role in the degradation of endocannabinoids, specifically substrate interaction and the inhibition of human MGL [
19,
24,
25]. Structural analysis, such as the cap plasticity, changing from an open to a closed conformation, and the hydrophobicity of the substrate binding pocket, have also been used to study matter [
20,
21]. In this study, a new monoacylglycerol lipase of bacterial origin is presented for further biotechnological application. To achieve this, Mgl-C255 was cloned, expressed, biochemically characterized, and tested via preliminary transesterification reactions.
The biochemical characterization of Mgl-C255 indicated that its highest activity occurred at 40 °C. It was slightly alkalophilic (pH 7.5 to 8.5) and had a marked preference for short-chain substrates, obtaining its highest activity with p-nitrophenyl acetate.
Metal ions that function as cofactors that increase the activity of lipolytic enzymes have no significant effect on Mgl-C255 or the chelating EDTA; even the presence of β-mercaptoethanol had no effect, indicating that it is an enzyme that does not form disulfide bridges. On the other hand, Cu
+2 and Hg
+2 ions (which bind the thiol group of cysteines close to the active site [
26]), PMSF (which is a typical inhibitor of serine-catalytic enzymes), and SDS (which denatures proteins) did diminish its activity.
Stability assays in organic solvents demonstrated that Mgl-C255 retained at least 50% of its activity in the presence of DMSO, ethanol, acetone, and 2-propanol; hence, these solvents were candidates to be the reaction medium in synthesis reactions but not in more non-polar solvents such as hexane and 1-octanol, where the enzyme lost its activity. In the presence of acetonitrile, 1-propanol, 2-propanol, and tert-butanol, the enzyme retained at least 40% of its activity.
Transesterification reactions performed with tert-butanol or β-citronellol formed tert-butyl propionate and β-citronellyl propionate, which are more non-polar than reactant alcohols [
27]. Tert-butanol is a tertiary alcohol, and β-citronellol was added as a racemic mixture R-S-β-citronellol. Reactants’ access to the active site is conditioned by the open or closed conformation of the cap and the binding pocket [
21], as shown in
Figure 2b. Gas chromatography can identify the products by using a chiral column to separate the racemic mixture from β-citronellol and its products. This could be beneficial for determining if Mgl-C255 is enantioselective. Further analysis is needed to identify and quantify the reaction products.
On the other hand, transesterification reactions with monosaccharides lead to the formation of glucopyranosyl-, fructofuranosyl-, and xylopyranosyl-propionate. D-glucose has one secondary hydroxil group, while D-fructose has two and D-xylose has none. Thus, if Mgl-C255 is regioselective, fructose can produce mono- or di- esters, while glucose and xylose only monoesters [
28,
29]. To corroborate the products shown in
Figure 8 (lanes 5, 7, and 9), HPLC will be needed [
30].


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}